

Abstract
Many computational intensive bioinformatics software, such as multiple sequence alignment, population structure analysis, etc., written in C/C++ are not multicore-aware. A multicore processor is an emerging CPU technology that combines two or more independent processors into a single package. The Single Instruction Multiple Data-stream (SIMD) paradigm is heavily utilized in this class of processors. Nevertheless, most popular compilers including Microsoft Visual C/C++ 6.0, x86 gnu C-compiler gcc do not automatically create SIMD code which can fully utilize the advancement of these processors. To harness the power of the new multicore architecture certain compiler techniques must be considered. This paper presents a generic compiling strategy to assist the compiler in improving the performance of bioinformatics applications written in C/C++. The proposed framework contains 2 main steps: multithreading and vectorizing strategies. After following the strategies, the application can achieve higher speedup by taking the advantage of multicore architecture technology. Due to the extremely fast interconnection networking among multiple cores, it is suggested that the proposed optimization could be more appropriate than making use of parallelization on a small cluster computer which has larger network latency and lower bandwidth.
Keywords: multicore processor, vectorization, optimization, speedup
Background
A multicore processor is an emerging processor technology which combines two or more independent processors into a single package. For example, a dual-core processor contains only two independent processors in the same CPU package whereas a dual processor refers to a computer which two single-core CPUs. In general, multicore processors allow a computing device to perform execution at thread-level parallelism without utilizing multiple processors in separate physical packages. Most recent processors come with extensions to their instruction sets to effectively utilize the parallelism from multiple cores. Such an instruction set is commonly referred to as the Single Instruction Multiple Data-stream (SIMD) instruction set.
For example an instruction ADD, operates on a number of data items in parallel. Clearly this dramatically improves execution speed which is why these instruction set extensions were created [1]. Most compilers including Microsoft Visual C/C++ 6.0, x86 Gnu C-compiler (gcc) do not automatically generate SIMD code. Many bioinformatics applications, however, are written in C/C++ language and by default were compiled by one of these compilers. Even though the old codes can be compiled and run successfully on multicore computers, these compiled codes will not be able to fully utilize the inherent parallelism. Speedup of these codes will not noticeably increase. A considerable number of these applications is computationally intensive and, therefore, many have previously turned to parallelization solutions on cluster computing where more computers and a fast networking switch need to be invested in. Any further improvement over cluster computing depends heavily on expensive fast interconnection networking with extremely low latency. By contrast, since there are multiple cores in the same CPU package, fast interconnection is already inherently implemented and built into multicore CPUs by their makers.
As the multicore system is recently introduced to the market, utilizing multicore systems requires new tools, new algorithms, and a new way of looking at programming [2]. Amarasinghe [3] previously identified significant problems with multicore compilers and the behavior of commercial applications running on performance asymmetric systems [4]. Improving bioinformatics applications using multicore compiler will not happen overnight. In this paper, we propose a compiler optimization protocol to improve a class of bioinformatics programs written in C/C++ by utilizing multicore SIMD instructions. This idea has been successfully tested on several bioinformatics applications such as ClustalW [5] and population structure software, STRUCTURE [6].
Methodology
We introduce a generic compiler optimization framework that improves the execution time of a bioinformatics application written in C/C++ as our case study program, MT-ClustalW [7]. Figure 1 shows the multithreading strategy which contains 3 steps. First software profiling must be performed to analyze the target bioinformatics tool and identify portions which this tool can or cannot be improved. These “can not be improved” parts are the bottleneck (i.e., must be sequentially executed) of all the written codes. Then, at the non-sequential parts, the C/C++ source code should be modified by utilizing thread library and/or loop optimization technique to instruct the code portions to be run in parallel on multiple cores. Finally the optimized code must be verified to make sure that it is fully functional.
Figure 1.
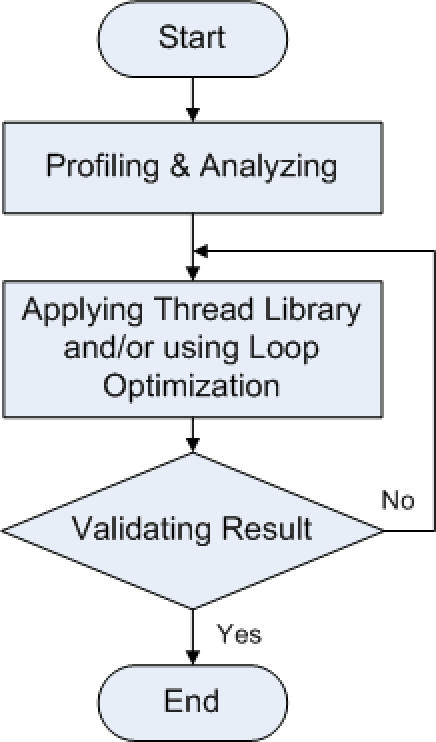
Applying multithreading and vectorizing technique flowchart.
Multithreading strategy
Profiling software for parallelization
Profile the target bioinformatics tools using Intel thread profiler [8] in order to find “bottlenecks” in the software which we can target for speedup of the program.
Applying thread library
Modify the sequential steps of the program using pthread library with MUTEX object as a synchronization object.
Validating software result
After completing the program modification, we have to verify correctness of the result and check how much speedup that the modified program can achieve. We use Beyond Compare [9] to verify the results of the modified code.
Vectorizing strategy
Analyzing the software for vectorizing strategies
We use the Intel C++ Compiler for Windows [10] to optimize the target program by enabling the /QxP option which optimizes code for Intel Core Duo processors and Intel Core Solo. processors, Intel Pentium 4 processors with Streaming SIMD Extensions 3, and compatible Intel processors with Streaming SIMD Extensions 3 (SSE3). With the /QxP option, the compiler automatically profiles the code as well as optimizes the loops. However, only the simple loops can be optimized by this approach. As another contribution of this paper, we present techniques which can assist the compiler to better optimize the loops. We used the Intel VTune Performance Analyzer to profile ClustalW in debug mode and searched for the hotspots within the functions.
Applying loop optimizing methodologies
These top-usage functions will be optimized by: loop reversal, loop fission, type casting, and procedure call reduction.
Validating resulting software
Beyond Compare™ tells us that the optimized program is able to produce correct results.
Conclusion
We presented the multithreading and vectorizing methodologies for improving bioinformatic software performance. The proposed technique was designed to fully utilize the emerging multicore processor technology. Even though, existing compilers can automatically optimize program source code, it is not able to intelligently produce good SIMD instructions. By suggesting the use of a compiler with multithreading and vectorizing instructions, the running time of computationally intensive bioinformatic applications written in C/C++ can be improved. Clearly such a collection of these techniques can be applied to speed up other bioinformatic tools written in C/C++ by promoting the inherent parallelism in multicore processors.
References
- 1. http://www.hayestechnologies.com/en/ techsimd.htm
- 2.Gorder PF. Computing in Science and Engineering. 2007;9:3. doi: 10.1109/MCSE.2010.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amarasinghe S. In CGO; 2005. p. 137. [Google Scholar]
- 4.Balakrishnan S, et al. In CGO; 2005. p. 506. [Google Scholar]
- 5.Thompson J, et al. Nucleic Acids Research. 1994;22:4673. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. http://pritch.bsd.uchicago.edu/ structure.html
- 7.Chaichoompu K, et al. HiCOMB IPDPS; Greece. 2006. [Google Scholar]
- 8. http://www.intel.com
- 9. http://www.scootersoftware.com
- 10. http://www.intel.com