

Abstract
p53 modulates a large number of cellular response pathways and is critical for the prevention of cancer. Wild-type p53, as well as tumorigenic mutants, exhibits the singular property of spontaneously losing DNA binding activity at 37°C. To understand the molecular basis for this effect, we examine the folding mechanism of the p53 DNA binding domain (DBD) at elevated temperatures. Folding kinetics do not change appreciably from 5°C to 35°C. DBD therefore folds by the same two-channel mechanism at physiological temperature as it does at 10°C. Unfolding rates, however, accelerate by 10,000-fold. Elevated temperatures thus dramatically increase the frequency of cycling between folded and unfolded states. The results suggest that function is lost because a fraction of molecules become trapped in misfolded conformations with each folding-unfolding cycle. In addition, at 37°C, the equilibrium stabilities of the off-pathway species are predicted to rival that of the native state, particularly in the case of destabilized mutants. We propose that it is the presence of these misfolded species, which can aggregate in vitro and may be degraded in the cell, that leads to p53 inactivation.
Keywords: cancer, mutation, aggregation, folding kinetics
The tumor suppressor p53 is a transcription factor that lies at the crux of the cellular damage response pathway. It coordinates a multifaceted response to DNA damage that results in cell cycle arrest, senescence, and/or apoptosis (Harris and Levine 2005). Its importance is illustrated by the finding that almost half of all human carcinomas harbor a point mutation in the p53 gene, making p53 the most frequently mutated protein associated with cancer (Olivier et al. 2002). Consequently, a major goal of cancer research is to develop therapies aimed at rescuing mutant p53. To this end, it is essential to understand how mutations affect p53’s properties at the molecular level. A clue is provided by the finding that of the ∼20,000 p53 point mutations identified to date, 97% are found in the central DNA binding domain (DBD) (Olivier et al. 2002). Efforts have therefore been directed toward determining the effects of mutation on the structure and function of the isolated DBD fragment.
The majority of previous folding studies of DBD have been carried out at 10°C. The reason is that aggregation is almost always observed upon heating to 37°C for extended periods of time. The lack of detailed information on DBD structure, function, and folding at 37°C represents a significant gap in knowledge. This point is emphasized by the fact that DBD cycles between numerous non-native forms including zinc-free, unfolded, and partially folded states (Butler and Loh 2003, 2005). None of these forms are functional, and some are suspected to be branch points for aggregation (Butler and Loh 2005). The relative populations of each state as well as their rates of interconversion potentially play a role in p53 inactivation, and both are expected to depend on temperature.
Nevertheless, studies at 10°C have established a framework for interpreting effects of mutation on DBD. Two general classes of tumorigenic mutations have emerged (Bullock et al. 1997, 2000). The first group diminishes DNA binding affinity by altering key contact residues; mutations at positions R248 and R273 fall into this category. The second class (e.g., positions R175, G245, R249, R282) impairs the stability/structure of DBD. ΔGfolding for wild-type DBD is −10.2 kcal/mol at 10°C (Bullock et al. 1997; Butler and Loh 2003) and is predicted to be −3.0 kcal/mol at 37°C, based on extrapolation of values obtained below 25°C (Bullock et al. 2000). The R175H, G245S, R249Q, and R282Q mutations reduce stability by 1.0–4.5 kcal/mol at 10°C. If ΔΔGfolding values remain constant with temperature, these data suggest that a significant fraction of mutant DBD is unfolded at physiological temperature.
The thermodynamic destabilization hypothesis, however, fails to account for the paradoxical observation that purified wild-type DBD spontaneously loses DNA binding activity at temperatures where it is folded and stable, in the absence of other proteins or regulatory factors. For example, wild-type DBD inactivates with a halftime of ∼10 min at 32°C (Foster et al. 1999), where the extrapolated value of ΔGfolding is −5.2 kcal/mol (Bullock et al. 2000). The inactivation rate is accelerated by temperature and by several tumorigenic mutations examined (Foster et al. 1999) and is decreased in a thermostable DBD variant (Matsumura and Ellington 1999). This effect may be even more pronounced in full-length p53 tetramer, as it has been reported to lose half of its DNA binding activity after heating for 5 min at 25°C (Bell et al. 2002). The extent to which this phenomenon occurs in the cell is not well established. A recent study found that the E285K p53 variant retained the active conformation for ∼2 h in cells growing at 32°C, and that the Hsp90 chaperone is required to restore function upon shift to lower temperatures (Müller et al. 2005).
We previously introduced a folding mechanism that can resolve this inconsistency (Butler and Loh 2005). The mechanism specifies two distinct and parallel folding pathways, referred to as channels, shown schematically in the right panel of ▶. The forward rate constants associated with the upper channel are greater than those of the lower channel; thus, they are designated fast and slow, respectively. The fraction of molecules that enter fast versus slow channels is determined by the k12:k13 ratio (2.5:1 at 10°C). Accordingly, ∼50% and 25% of refolding molecules partition to the fast and slow channels, respectively. The remaining 25% folds by a slower, uncharacterized route not shown in ▶.
Scheme 1.

DBD folding mechanism (left) and schematic representation of fast and slow-folding channels (right). The fast and slow channels are the upper and lower pathways, respectively, on the left, and are colored black and gray on the right. Folding tracks are depicted in the right panel as follows: track a, solid black line; track b, solid black and dashed black lines; track c, gray line. Track d is not shown. Microscopic rate constants that quantitatively reproduce kobs, a, kobs, b, and kobs, c and their associated amplitudes at 10°C are as follows (in units of sec−1): kU1 = 200, k1U = 30, k12 = 1, k21 = 0.001, k13 = 0.4, k31 = 0.01, k24 = 0.04, k42 = 0.02, k35 = 0.1, k53 = 0.02, k2N = 0.08, kN2 = 12×10−5, k36 = 0.001, k63 = 82×10−7, k6N = 0.3, kN6 = 0.005.
An important property of each channel is that they contain an off-pathway intermediate (I4 and I5). These species introduce a branch point that potentially establishes two folding “tracks” within each channel: a slower track in which molecules accumulate in I4 or I5 (and must re-enter the productive folding stream with rate constants k42 and k53, respectively), and a faster track whereby molecules bypass I4 and I5. Four folding tracks (designated a–d from fastest to slowest) are indeed observed at 10°C. Within the fast channel, track a proceeds according to the sequence U-I1-I2-N and thus represents the fastest route to the native state. Track b is identical to track a, except molecules pass through I4. Partitioning between tracks a and b is established by the k24:k2N ratio (∼0.5). Of the molecules that enter the fast channel, approximately equal amounts fold by tracks a and b. In contrast, molecules that enter the slow channel fold almost exclusively by a single track (track c). Since k35 is 10-fold greater than k36, most slow channel molecules proceed according to the sequence U-I1-(I3/I5)-I6-N. I6 is a native-like intermediate that is not populated in folding experiments. It is placed in the mechanism to account for the biphasic unfolding kinetics observed at 10°C (Butler and Loh 2005). Track d represents the uncharacterized folding phase mentioned above. It requires ∼10 h to complete and therefore appears as a “missing” phase in refolding experiments.
According to ▶, activity loss in vitro is caused by partitioning into the slower tracks (tracks b, c, and d) coupled with aggregation of I4 and I5. Inactivation would likely be more pronounced in vivo, where intracellular protein levels are high and misfolded species such as I4 and I5 can be ubiquitinated and cleared by cellular enzymes. An important conclusion is that the inactivation process is limited by structural unfolding. Several hot-spot tumorigenic mutations examined do not change folding rates or amplitudes; rather, they accelerate unfolding rates (Butler and Loh 2005). We therefore proposed that some mutations exert their effects by causing DBD to cycle unusually rapidly between unfolded and folded forms. This escalation can cause misfolded species to accumulate even under conditions where the native state is thermodynamically stable. Here, we examine the DBD folding mechanism at elevated temperatures in order to test whether the kinetic misfolding model can account for functional loss of wild-type and mutant DBD in physiological conditions.
Results
Thermal denaturation studies
There have been conflicting reports on the melting temperature (Tm) of DBD, with some studies finding that wild-type DBD is stable at 37°C (Bullock et al. 1997; Bell et al. 2002) and others concluding that it is unstable (Friedler et al. 2003). We first needed to clarify this fundamental property of DBD. A complicating factor is that the extended heating times required by these experiments typically cause aggregation and render the denaturation process irreversible. Apparent Tm values are consequently dependent on heating rate and protein concentration and can only be compared under identical conditions. Thermal denaturation experiments were performed by heating fresh, individually prepared samples (0.2 μM) at various temperatures for 10 min and monitoring increase in Trp fluorescence between 350–360 nm (Bullock et al. 1997; Friedler et al. 2003; Butler and Loh 2005). In the presence of 0.15 M KCl, wild-type DBD exhibits a cooperative transition with Tm ∼ 45°C (▶), in agreement with two previous studies (Bullock et al. 1997; Bell et al. 2002). Stability is salt dependent: In the absence of KCl, the transition is broader with Tm ∼ 32°C. This value is similar to that reported by the third study, which did not employ salt in the buffer (Friedler et al. 2003). We thus conclude that salt concentration reconciles the disparate Tm values, and that wild-type DBD does not unfold upon heating at 37°C for 10 min at physiological ionic strength. As discussed later, comparison of folding and unfolding rates verify that the native state of wild-type DBD is thermodynamically stable at 37°C. Mutants G245S, R249S, and R282Q destabilize DBD by 1.0, 2.0, and 2.1 kcal/mol, respectively, at 10°C (Butler and Loh 2003). This trend is reflected by decreased Tm values compared with wild type, although in all cases Tm ≥ 37°C.
Figure 1.

Thermal denaturation of DBD monitored by Trp fluorescence. DBD (0.2 μM) was heated at the indicated temperatures for 10 min in 25 mM HEPES (pH 7.0), 0.15 M KCl, and 1 mM TCEP unless otherwise indicated. Variants are denoted by the following symbols: closed circles, wild type; open circles, wild type with no KCl; inverted triangles, G245S; x-symbols, R249S; diamonds, R282Q. Lines are meant to guide the eye only.
Folding and unfolding experiments at 25°C
To extend our previous folding analysis at 10°C to physiological conditions, we first characterized the folding reaction at 25°C, which was chosen because it is the highest temperature at which wild-type DBD exhibits two-state, reversible behavior in equilibrium urea denaturation experiments (Bullock et al. 2000). This allows results of kinetic and equilibrium experiments to be compared directly. The refolding reaction was analyzed by direct folding (DF) and interrupted folding (IF) experiments. Both experiments employ a double-jump sequence to minimize proline isomerization in the unfolded state. In the first jump, DBD is unfolded by rapid dilution into 7 M urea. The duration of the unfolding pulse is adjusted so that refolding initiates from a state that is fully unfolded but contains native Pro isomers (all trans; see Materials and Methods). Refolding is then started by dilution into buffer. DF is monitored by Trp fluorescence at 350 nm. In IF experiments, the solution is transferred back into 3.5 M urea after various times of refolding. The unfolding reaction is then recorded by monitoring Trp fluorescence. Native molecules are identified by their characteristic unfolding rate. Intermediates are less stable and typically unfold within the mixing dead time. The fraction of native molecules formed at the time of the unfolding pulse is thus proportional to the amplitude of the observed unfolding reaction.
In strongly native conditions (0.2 M urea), DF kinetics at 25°C are similar to those at 10°C. The initial event is formation of a burst-phase intermediate of increased fluorescence (▶). The remaining fluorescence decay is fit minimally by a three-exponential equation which describes tracks a, b, and c. Track d, too slow to be measured accurately in DF experiments, appears as a missing phase. Fitted rates and amplitudes are summarized in ▶. The folding rates of tracks a, b, and c (0.13 sec−1, 0.017 sec−1, and 0.0035 sec−1, respectively) are nearly identical to the values obtained at 10°C. The fluorescence amplitudes associated with each phase are 33% (Aa), 31% (Ab), and 36% (Ac) (reported as fraction of total observed fluorescence, excluding track d). We previously established that DF amplitudes are approximately proportional to the populations that fold through each track (Butler and Loh 2005). Thus, while the overall folding mechanism is preserved at 25°C, slightly more molecules fold via track c at 25°C compared with 10°C (36% vs. 26%, respectively). The increase in flux comes mainly at the expense of track a (33% vs. 44%); track b remains unchanged.
Figure 2.
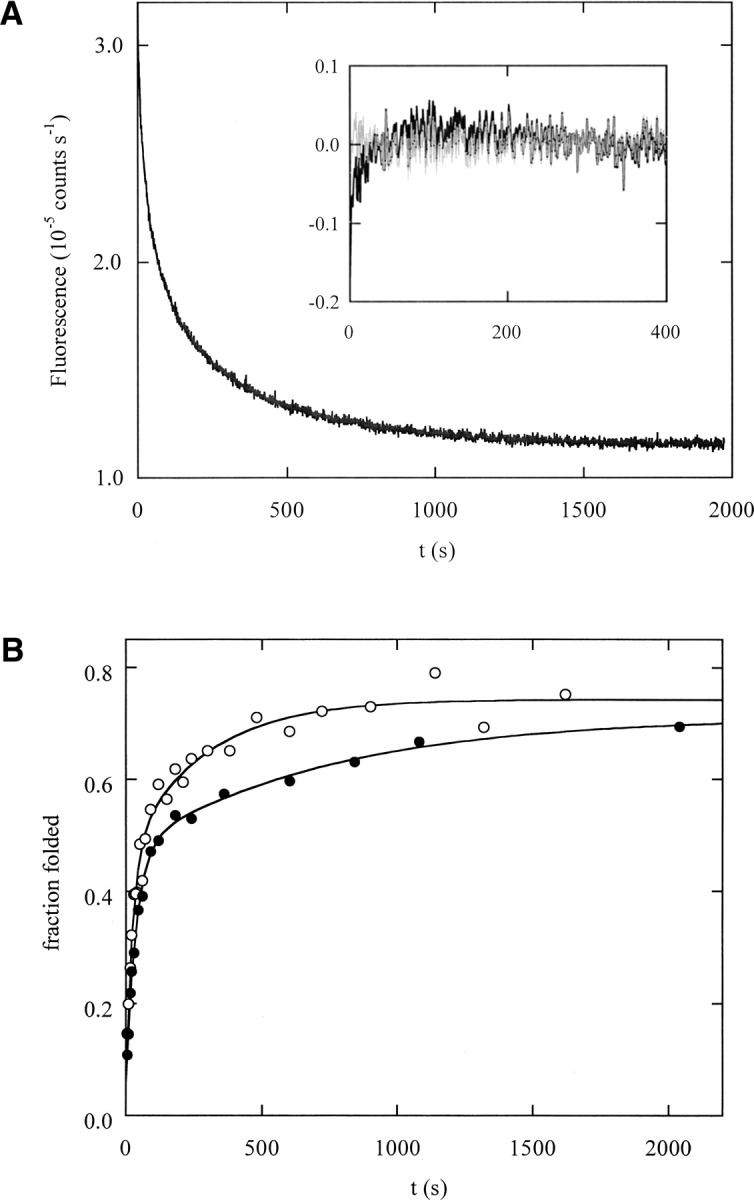
Direct folding (A) and interrupted folding (B) of wild-type DBD, monitored by Trp fluorescence. Data in panel A were obtained at 25°C (0.2 M urea). The inset shows residuals from two-exponential (black) and three-exponential (gray) curve fits, respectively. The axes of the inset have the same units as the data plot. Open and closed symbols in B represent folding at 10°C and 25°C, respectively (0.35 M urea). Lines are best fits of the data to a three-exponential function, with kobs,a fixed as described in the text.
Table 1.
Folding rates and amplitudes for wild-type DBD at 10°C and 25°C (0.35 M urea)

We performed IF experiments in order to specifically monitor formation of native molecules. IF data are adequately fit by a burst phase followed by a two-exponential decay (▶). In this case, however, the burst phase likely represents formation of native molecules via track a. The reason that track a is resolved in DF experiments but not in IF is that the mixing time and data sampling interval are considerably longer in the latter. To better estimate IF parameters, we therefore constrained kobs,a to the value obtained from DF experiments performed at the same temperature, and fit the data to a three-exponential function. IF and DF results are in good agreement given these considerations (▶). IF has the additional advantage of resolving the amplitude of track d, which is missing in DF experiments. Roughly one-fourth of molecules fold by this slowest track at 10°C and 25°C. Taken together, the data indicate that DBD folds via the same four tracks at both temperatures, but flux shifts from the fast channel to the slow channel upon raising temperature.
Unlike the folding results, unfolding kinetics change markedly with temperature. Wild-type DBD unfolding at 10°C is biphasic over all urea concentrations tested (Butler and Loh 2005). At 25°C and 3.14 M urea, however, the unfolding reaction is well fit by a single exponential (▶). It is also faster. The fitted rate constant of 0.0054 sec−1 is threefold and 30-fold higher than the fast and slow unfolding rates, respectively, observed at 10°C and the same urea concentration. Extrapolation of the native fluorescence baseline indicates that the entire unfolding amplitude is present, eliminating the possibility that an additional reaction occurs within the dead time (data not shown).
Figure 3.

Unfolding of wild-type DBD (3.14 M urea) at 10°C. The inset shows residuals from fitting the data to a one-exponential function. The axes of the inset have the same units as the data plot.
Chevron analysis at 25°C
To more fully understand how temperature affects folding and unfolding mechanisms, we recorded DF kinetics as a function of urea concentration at 25°C. The resulting chevron plots of rates (▶) and amplitudes (▶) reveal how the stabilities of intermediates and their rates of interconversion dictate the flux of molecules to the native state. Gray and black data points represent data collected at 10°C and 25°C, respectively.
Figure 4.
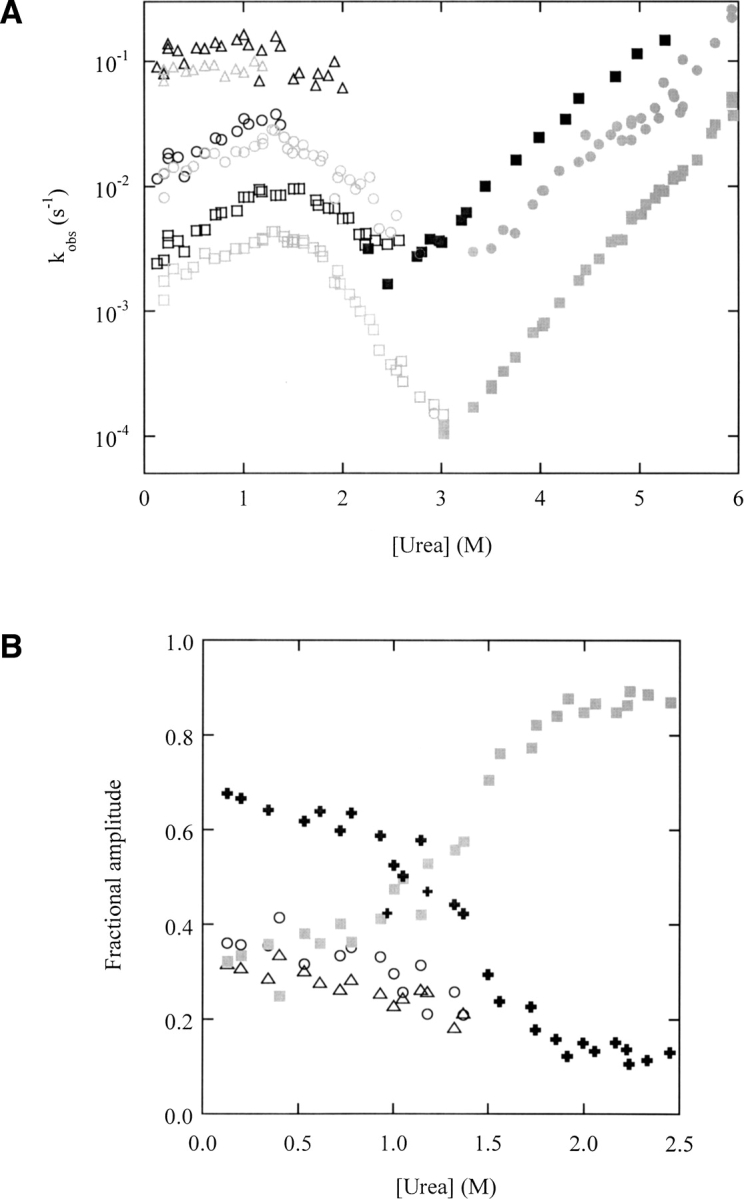
(A) Urea dependence of observed folding (open symbols) and unfolding (closed symbols) rates of wild-type DBD, obtained by fitting DF fluorescence data recorded at 10°C (gray) and 25°C (black); 10°C data are taken from (Butler and Loh 2005). Symbols are as follows: open triangles, kobs,a; open circles, kobs,b; open squares, kobs,c; closed circles, fast unfolding rate; closed squares, slow unfolding rate (only one unfolding phase is observed at ≥25°C; see text). (B) Urea dependence of direct folding fluorescence amplitudes of wild-type DBD at 25°C. Symbols are as follows: open triangles, Aa; open circles, Ab; filled squares, Ac; filled plus signs, sum of Aa and Ab. The amplitudes of the fast and slow channels are indicated by black and gray filled symbols, respectively.
The salient features of the folding reaction are conserved at both temperatures. The same folding tracks persist at 25°C, as evidenced by the three observed folding rates (open symbols). kobs,a, kobs,b, and kobs,c are slightly faster at 25°C than at 10°C, at all urea concentrations tested. The inverse rollover observed for kobs,b and kobs,c between 0 and 1.4 M urea (in which folding rates increase with urea concentration) is slightly more pronounced at 25°C. This behavior suggests that the folding rates of tracks b and c are partially limited by an unfolding event, and is the main evidence that these tracks populate off-pathway species (Butler and Loh 2005).
Fluorescence amplitudes (▶) show that approximately equal fractions of molecules fold through tracks a, b, and c in the absence of denaturant at 25°C. As urea concentration is raised from 0 to 1.0 M, Aa and Ab decrease slightly, while Ac increases. Above 1.0 M urea, kobs,a and kobs,b approach one another (▶). This similarity prevents tracks a and b from being resolved. It is therefore more instructive to combine the two tracks and treat them collectively as the fast channel. When plotted in this way, the amplitudes reveal a gradual shift from the fast channel to the slow channel between 0 and 1.0 M urea. The change becomes cooperative above 1.0 M urea so that the populations folding through the fast and slow tracks invert sharply around 1.4 M. The same shift is observed at 10°C, although it occurs with an apparent midpoint of 1.7 M urea (Butler and Loh 2005), reflecting the increased stability of DBD at the lower temperature.
In contrast to biphasic unfolding kinetics observed at 10°C, unfolding at 25°C is monophasic at all urea concentrations tested (▶). ▶ dictates that biphasic unfolding occurs because N partitions between I2 and I6 (kN2 ∼ kN6). In cases where a single exponential is observed, it is possible that kN6 may become significantly faster than kN2 so that the majority of molecules enter the slow unfolding channel. This scenario appears to be favored by conditions that destabilize DBD, particularly in the zinc binding region. Removal of zinc as well as the G245S and R249S mutations (which lie in the zinc binding loop) result in monoexponential unfolding at 10°C (Butler and Loh 2005). Elevated temperature may affect this portion of DBD in a similar fashion.
Temperature dependence of folding and unfolding kinetics
Chevron analysis at 10°C and 25°C suggest that increasing temperature does not change folding kinetics substantially, but instead accelerates the unfolding reaction. To further test this trend and to connect it to physiological conditions, we determined the dependence of folding and unfolding kinetics on temperature from 5°C to 35°C, in near-zero denaturant conditions. Folding rates were measured in 0.23 M urea (these values closely approximate those in zero denaturant) (▶). Unfolding rates were obtained by linear extrapolation of data recorded at higher urea concentrations to zero denaturant.
Kinetic partitioning between the three observed folding tracks persists over the temperature range examined. Folding rates exhibit a weak and non-Arrhenius temperature dependence (▶). kobs,a, kobs,b, and kobs,c each vary by less than fivefold over the range 5°C–35°C and exhibit broad maxima near 20°C–25°C. In contrast, the unfolding rate changes by 104-fold and displays linear Arrhenius behavior. Both of these trends have been observed for other proteins (Chen et al. 1989; Alexander et al. 1992; Schindler and Schmid 1996; Tan et al. 1996; Scalley and Baker 1997) and indicate that the activation heat capacity change is larger for unfolding than it is for refolding. An important consequence of this temperature dependence is that the wild-type DBD unfolding rate approaches the track c folding rate at 37°C. Comparison of DF amplitudes reveals a moderate change in flux from the fast to the slow channel upon raising temperature from 5°C to 25°C (▶), in agreement with ▶. Above 25°C, however, this trend reverses so that partitioning at 35°C is nearly identical to that at 10°C
Figure 5.
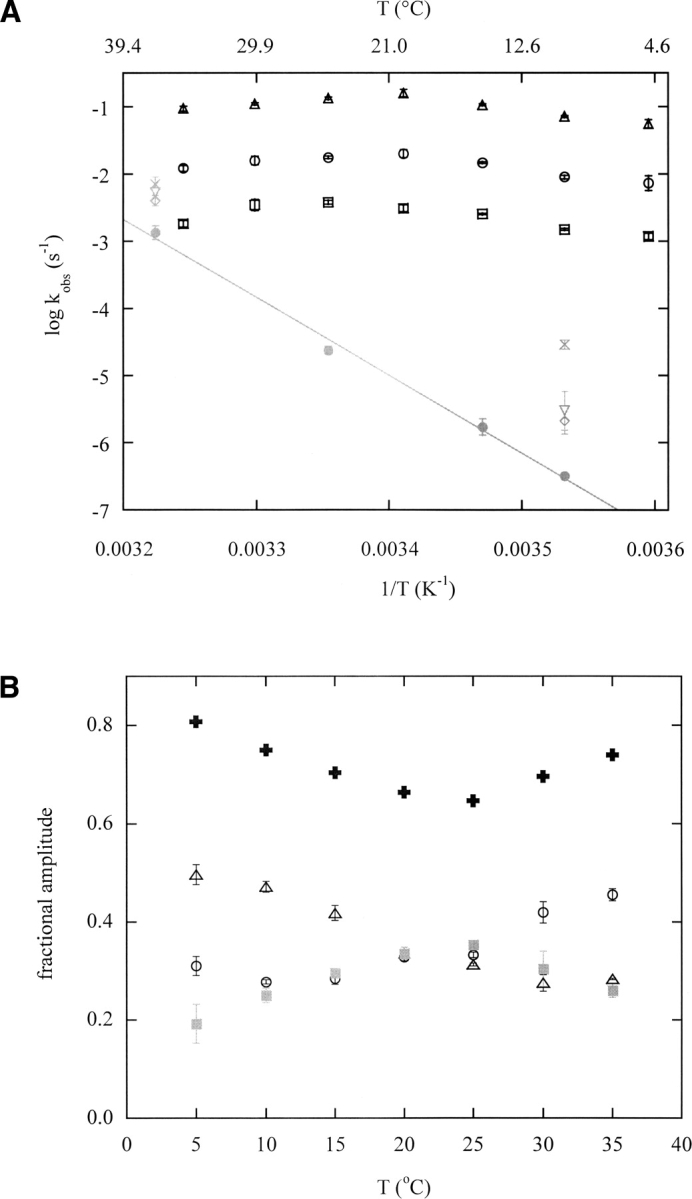
(A) Temperature dependence of observed folding rates (black symbols, 0.23 M urea) and unfolding rates (gray symbols, extrapolated to zero denaturant) for wild-type and mutant DBD, obtained by fitting DF fluorescence data. Rates of the slow unfolding phase are plotted. kobs,a, kobs,b, and kobs,c are represented by triangles, circles, and squares, respectively, as in ▶. Unfolding rates for wild type, G245S, R249S, and R282Q are filled circles, inverted triangles, crosses, and diamonds, respectively, as in ▶. The line is the linear fit of the wild-type DBD unfolding rates. (B) Temperature dependence of DF fluorescence amplitudes for wild-type DBD. Amplitudes of tracks a, b, and c are indicated by the same symbols as in A; filled plus signs represent the sum of Aa and Ab. Black and gray symbols denote fast and slow channel amplitudes, respectively. Error bars in A and B show standard errors of the mean (n = 5).
Discussion
The overall folding mechanism is conserved at elevated temperatures. Refolding rates are similar from 5°C–35°C, as is partitioning between folding tracks. In contrast, the unfolding reaction is accelerated dramatically at 37°C. How do these trends contribute to functional loss at physiological temperature? We first consider the mechanism at 10°C. According to ▶, tracks b and c populate off-pathway species (I4 and I5, respectively) whereas track a represents a direct route to the native state. Previous DBD concentration-dependent folding experiments suggested that I4 and I5 can form oligomeric dead-end species (Butler and Loh 2005). These findings led to the hypothesis that the native form of DBD becomes depleted by two related mechanisms. The first is by DBD adopting conformations I4 and/or I5 at equilibrium, caused by (for example) a mutation that selectively destabilizes native but not intermediate states. The second is irreversible aggregation of I4 and/or I5. In the latter case, I4 and I5 can be less stable than N, i.e., populated only transiently during refolding, provided that they aggregate faster than they can unfold to rejoin the productive folding stream. At 10°C, I4 and I5 are not observed in equilibrium folding experiments (Bullock et al. 1997; Butler and Loh 2005), indicating that N is more stable than either species. This scenario is illustrated by the free energy diagram of ▶ (left panel). Moreover, the slow unfolding rate exhibited by wild-type DBD at 10°C (▶) dictates that molecules cycle through I4 and I5 only infrequently. Aggregation is thus restricted to occur extremely slowly, if at all. The G245S, R249S, and R282Q mutations increase unfolding rates by fivefold to 50-fold at 10°C in kinetic experiments (Butler and Loh 2005) and destabilize N by 1.0–2.1 kcal/mol by equilibrium measurements (Butler and Loh 2003). If free energies of I4 and I5 change relatively little compared to that of U, as suggested by the result that mutations do not alter refolding kinetics, then the effect of mutation is to bring the free energy of N closer to that of I4 and I5 (▶). These changes are relatively small at 10°C, however, and are apparently not sufficient to cause inactivation by either mechanism.
Figure 6.

Schematic free energy diagrams for DBD folding at 10°C (left) and 37°C (right). On-pathway intermediates lie in the vertical line between unfolded and native states. Misfolded species I4 and I5 are placed to the left and right of that line. Bars with symbols indicate approximate free energies of mutant native states. Inverted triangles, G245S; x-symbols, R249S; diamonds, R282Q.
Raising temperature to 37°C has several consequences. First, unfolding rates increase by 104-fold (▶). In this regard, the effect of elevated temperature is similar to, but much more pronounced than, the effect of mutations at 10°C. Thus, DBD cycles through I4 and I5 rapidly at 37°C. Second, the native state is selectively destabilized compared with I4 and I5. A key observation is that the unfolding rate of wild-type DBD at 37°C approaches or exceeds the folding rate for track c but remains slower than that of track a (▶). Wild-type DBD molecules unfold nearly as fast as they fold via track c. The temperature dependencies of folding rates were not determined for G245S, R249S, and R282Q. If they exhibit the same nearly flat profile as wild type, however, then unfolding rates of the mutants are expected to be greater than kobs,c and approximately equal to kobs,b (▶). The corresponding free energy profile is depicted in the right panel of ▶. The main difference compared with the 10°C result is that, for mutant DBD, I4 and/or I5 are of comparable stability to N. The model consequently predicts that a substantial fraction of DBD—perhaps a majority in the case of some mutants—exists in states I4 and I5 at 37°C. In other words, I4 and I5 are both well-populated and kinetically accessible, making inactivation by both mechanisms possible.
It should be noted that track a folding remains faster than unfolding at physiological temperature, suggesting that the native states of wild-type and mutant DBD are thermodynamically stable relative to the unfolded state. Were I4 and I5 unable to form, DBD would be expected to remain functional at 37°C indefinitely. The model asserts that it is the propensity of DBD to misfold to off-pathway intermediates that is responsible for activity loss. ▶ does not account for the full extent of this loss, as a significant percentage of DBD is still expected to exist in the native conformation at 37°C. We suggest that irreversible processes emanating from I4 and I5 account for the difference. Available evidence indicates that this process is most likely aggregation in vitro (Butler and Loh 2005). Ubiquitination, proteolytic degradation, and association with chaperones or other proteins are additional routes that may contribute to functional loss in the cell.
The misfolding model is attractive because it resolves the primary inconsistency associated with the thermodynamic destabilization hypothesis; namely, how wild-type DBD can spontaneously inactivate at temperatures well below Tm. Even at low temperatures, where N is the most stable species present, I4 and I5 are populated fleetingly during refolding. The mechanism postulates that aggregates accumulate when the protein cycles repeatedly between native and unfolded states. Consistent with this view, tumorigenic mutations (Butler and Loh 2005), loss of the bound zinc ion (data not shown), and elevated temperature (▶) all increase cycling frequency by accelerating the unfolding rate. At physiological temperature, the data suggest that I4 and I5 are approximately as stable as N. Aggregation is anticipated to be much more pronounced because the off-pathway species are well-populated at equilibrium.
The model also provides a basis for interpreting another puzzling observation that was reported previously. The small organic compound CP-31398 has been shown to inhibit DBD activity loss in vitro and in vivo (Foster et al. 1999; Luu et al. 2002; Takimoto et al. 2002; Herbert et al. 2003; Wang et al. 2003a,b; Wischhusen et al. 2003; Demma et al. 2004; Stanhope-Bake et al. 2004), but it does not appear to bind the native state (Rippin et al. 2002). One explanation is that it might instead bind folding intermediates and block formation of I4/I5, or bind to I4/I5 and prevent their subsequent aggregation. Since most binding experiments were carried out at 20°C, where I4 and I5 are only meta-stable, such a binding event would not likely be detected by equilibrium methods. The compounds may thus act as chemical chaperones. In fact, the model offers a structural explanation for p53-chaperone interactions. p53 binds to Hsp70 and Hsp90 via DBD (Fourie et al. 1997; Rudiger et al. 2002; Müller et al. 2004). In vivo and in vitro experiments indicate that Hsp90 only binds to wild-type DBD when the latter is thermally unfolded or misfolded; no interaction was observed at temperatures <34°C (Rudiger et al. 2002). Off-path intermediates I4 and I5 are likely candidates for this interaction, as they only become significantly populated near 37°C.
What new insights into p53 inactivation and potential therapeutic strategies does the misfolding model provide? It has long been suggested that DBD adopts an alternate (“mutant”) conformation in the presence of turmorigenic mutations (Milner and Medcalf 1990, 1991; Bartek et al. 1991; Picksley et al. 1992; Stephen and Lane 1992). Treating DBD folding as largely a two-state process, Fersht and colleagues have instead focused attention on global unfolding as a principle inactivation mechanism for non-DNA contact mutations (Bullock et al. 1997, 2000). Our results suggest that both views are compatible. It is clear that strongly destabilized mutants (such as R175H) are almost completely unfolded at physiological temperature (Butler and Loh 2003). At the opposite extreme, wild-type DBD inactivates rapidly even when it is predominantly folded, presumably due to repeated folding-unfolding cycles and the corresponding accumulation of I4 and I5. Moderately destabilizing mutations likely act by a combination of the two factors. Misfolded species I4 and I5 can thus be regarded as “mutant” conformations, although they appear to be a general feature of the folding landscape of wild-type as well as mutant DBD.
Previous efforts to treat p53-related cancers have been directed toward developing small molecules that bind to and stabilize the native conformation of DBD. This endeavor remains a priority. Our results, however, suggest an alternate strategy: to identify small molecules that bind to productive folding intermediates and block formation of off-pathway states, or bind to off-pathway species and prevent their aggregation. The p53-rescue molecule CP-31398 was discovered by screening a >100,000 member library of synthetic compounds. The fact that CP-31398 does not bind native DBD suggests that preventing misfolding by binding intermediates, rather than stabilizing p53 by binding the native state, may be a more achievable goal for rescuing p53 function.
Materials and methods
Thermal denaturation experiments
DBD was expressed and purified as described (Butler and Loh 2005). Solution conditions were 25 mM HEPES (pH 7.0), 0.15 M KCl, and 1 mM TCEP. Ice-cold DBD (0.2 μM) was incubated at the indicated temperature in a cuvette placed in the thermally jacketed fluorescence sample chamber (with stirring). Temperature measurements using an external probe determined that the samples came to thermal equilibrium within 3 min. After 10 min of heating, the extent of unfolding was assayed by increase in Trp fluorescence from 300 nm to 450 nm with excitation at 280 nm (Bullock et al. 1997; Butler and Loh 2005). Fraction folded DBD was estimated by integrating the emission signal between 350 and 360 nm, subtracting the value for the native state (obtained from a linear fit of the native baseline) and adjusting the maximum observed fluorescence change to a value of unity. The fluorescence of unfolded DBD decreased sharply with increasing temperature, due to a combination of photobleaching and thermally enhanced quenching. For this reason, the unfolded baseline was not used to calculate fraction folded. All fluorescence data were collected on a Jobin-Yvon/SPEX fluoromax-3 fluorimeter.
Kinetic folding and unfolding experiments
Folding and unfolding were monitored by Trp fluorescence at 350 nm (Butler and Loh 2005). Solution conditions for all kinetic experiments were 10 mM HEPES (pH 7.0), 0.1 M NaCl, and 1 mM TCEP. DF experiments employed the double-jump method to minimize proline isomerization in the unfolded state. Native DBD was unfolded in 7 M urea for the following times (temperatures indicated in parentheses): 60 sec (5°C), 30 sec (10°C), 25 sec (15°C), 20 sec (20°C), 15 sec (25°C), 10 sec (30°C), and 5 sec (35°C). These times were determined by separate experiments to be sufficient for complete unfolding, but not long enough for proline isomerization to occur. Folding was then initiated by dilution with buffer to the final urea concentration described. Data were fit to one-, two-, or three-exponential equations of the general form y = A0 + A1exp(−k1t) + A2exp(−k2t) + A3exp(−k3t) using the Kaleidagraph software package (Synergy Software). Final urea concentrations were determined by measuring refractive indices of each sample at the end of the experiment (Pace and Scholtz 1997). Unfolding rates at zero denaturant were determined by linear extrapolation of the logarithms of unfolding rates obtained at various urea concentrations. For variable temperature unfolding experiments, wild-type DBD samples were incubated at each temperature for 10 min prior to unfolding. To avoid aggregation at higher temperatures, mutant DBD samples were unfolded by mixing a small aliquot of DBD (20°C) with preheated urea solutions. The final temperature of the solutions did not change significantly. Unfolding thus began predominantly from the native state, rather than from an equilibrium mixture of N, I4, and I5 that is expected to be present for mutants at elevated temperatures.
IF experiments were performed by first refolding DBD using the double-jump sequence described above. After variable refolding times, the protein was unfolded by diluting the solution back into 3.5 M urea. Unfolding was then monitored by the decrease in Trp fluorescence at 350 nm. The data are well-fit by a single exponential function at all urea concentrations examined (data not shown). The fraction of native molecules formed during the aging time was calculated by dividing the unfolding fluorescence amplitude by that of a control sample of native protein which had not been unfolded.
Footnotes
Reprint requests to: Stewart N. Loh, Department of Biochemistry and Molecular Biology, SUNY Upstate Medical University, 750 East Adams Street, Syracuse, NY 13210; e-mail: lohs@upstate.edu; fax: (315) 464-8750.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062324206.
References
- Alexander, P., Orban, J., and Bryan, P. 1992. Kinetic analysis of folding and unfolding the 56 amino acid IgG-binding domain of streptococcal protein G. Biochemistry 31 7243–7248. [DOI] [PubMed] [Google Scholar]
- Bartek, J., Bartkova, J., Vojtesek, B., Staskova, Z., Lukas, J., Rejthar, A., Kovarik, J., Midgley, C.A., Gannon, J.V., and Lane, D.P. 1991. Aberrant expression of the p53 oncoprotein is a common feature of a wide spectrum of human malignancies. Oncogene 6 1699–1703. [PubMed] [Google Scholar]
- Bell, S., Klein, C., Muller, L., Hansen, S., and Buchner, J. 2002. p53 contains large unstructured regions in its native state. J. Mol. Biol. 322 917–927. [DOI] [PubMed] [Google Scholar]
- Bullock, A.N., Henckel, J., DeDecker, B.S., Johnson, C.M., Nikolova, P.V., Proctor, M.R., Lane, D.P., and Fersht, A.R. 1997. Thermodynamic stability of wild-type and mutant p53 core domain. Proc. Natl. Acad. Sci. 94 14338–14342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock, A.N., Henckel, J., and Fersht, A.R. 2000. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: Definition of mutant states for rescue in cancer therapy. Oncogene 19 1245–1256. [DOI] [PubMed] [Google Scholar]
- Butler, J.S. and Loh, S.N. 2003. Structure, function, and aggregation of the zinc-free form of the p53 DNA binding domain. Biochemistry 42 2396–2403. [DOI] [PubMed] [Google Scholar]
- Butler, J.S. and Loh, S.N. 2005. Kinetic partitioning during folding of the p53 DNA binding domain. J. Mol. Biol. 350 906–918. [DOI] [PubMed] [Google Scholar]
- Chen, B., Baase, W.A., and Schellman, J.A. 1989. Low-temperature unfolding of a mutant of phage T4 lysozyme. 2. Kinetic investigations. Biochemistry 28 691–699. [DOI] [PubMed] [Google Scholar]
- Demma, M.J., Wong, S., Maxwell, E., and Dasmahapatra, B. 2004. CP-31398 restores DNA-DNA binding activity to mutant p53 in vitro but does not affect p53 homologs p63 and p73. J. Biol. Chem. 279 45887–45896. [DOI] [PubMed] [Google Scholar]
- Foster, B.A., Coffey, H.A., Morin, M.J., and Rastinejad, F. 1999. Pharmacological rescue of mutant p53 conformation and function. Science 286 2507–2510. [DOI] [PubMed] [Google Scholar]
- Fourie, A.M., Hupp, T.R., Lane, D.P., Sang, B.C., Barbosa, M.S., Sambrook, J.F., and Gething, M.J. 1997. HSP70 binding sites in the tumor suppressor protein p53. J. Biol. Chem. 272 19471–19479. [DOI] [PubMed] [Google Scholar]
- Friedler, A., Veprintsev, D.B., Hansson, L.O., and Fersht, A.R. 2003. Kinetic instability of p53 core domain mutants. J. Biol. Chem. 278 24108–24112. [DOI] [PubMed] [Google Scholar]
- Harris, S.L. and Levine, A.J. 2005. The p53 pathway: Positive and negative feedback loops. Oncogene 24 2899–2908. [DOI] [PubMed] [Google Scholar]
- Herbert, B.S., Pearce, V.P., Hynan, L.S., LaRue, D.M., Wright, W.E., Kopelovich, L., and Shay, J.W. 2003. A peroxisome proliferator-activated receptor-γ agonist and the p53 rescue drug CP-31398 inhibit the spontaneous immortalization of breast epithelial cells. Cancer Res. 63 1914–1919. [PubMed] [Google Scholar]
- Luu, Y., Bush, J., Cheung, K.J., and Li, G. 2002. The p53 stabilizing compound CP-31398 induces apoptosis by activating the intrinsic Bax/mitochondrial/caspase-9 pathway. Exp. Cell Res. 276 214–222. [DOI] [PubMed] [Google Scholar]
- Matsumura, I. and Ellington, A.D. 1999. In vitro evolution of thermostable p53 variants. Protein Sci. 8 731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner, J. and Medcalf, E.A. 1990. Temperature-dependent switching between wild-type and mutant forms of p53val135. J. Mol. Biol. 216 481–484. [DOI] [PubMed] [Google Scholar]
- Milner, J. and Medcalf, E.A. 1991. Cotranslation of activated mutant p53 with wild type drives the wild-type p53 protein into the mutant conformation. Cell 65 765–774. [DOI] [PubMed] [Google Scholar]
- Müller, L., Schaupp, A., Walerych, D., Wegele, H., and Buchner, J. 2004. Hsp90 regulates the activity of wild type p53 under physiological and elevated temperatures. J. Biol. Chem. 279 48846–48854. [DOI] [PubMed] [Google Scholar]
- Müller, P., Ceskova, P., and Vojtesek, B. 2005. Hsp90 is essential for restoring cellular functions of temperature-sensitive p53 mutant protein but not for stabilization and activation of wild-type p53. J. Biol. Chem. 280 6682–6691. [DOI] [PubMed] [Google Scholar]
- Olivier, M., Eeles, R., Hollstein, M., Khan, M.A., Harris, C.C., and Hainaut, P. 2002. The IARC TP53 database: New online mutation analysis and recommendations to users. Hum. Mutat. 19 607–614. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. and Scholtz, J.M. 1997. Protein structure: A practical approach. Oxford University Press, New York.
- Picksley, S.M., Meek, D.W., and Lane, D.P. 1992. The conformational change of a murine temperature-sensitive p53 protein is independent of a change in phosphorylation status. Oncogene 7 1649–1651. [PubMed] [Google Scholar]
- Rippin, T.M., Bykov, V.J., Freund, S.M., Selivanova, G., Wiman, K.G., and Fersht, A.R. 2002. Characterization of the p53-rescue drug CP-31398 in vitro and in living cells. Oncogene 21 2119–2129. [DOI] [PubMed] [Google Scholar]
- Rudiger, S., Freund, S.M., Veprintsev, D.B., and Fersht, A.R. 2002. CRINEPT-TROSY NMR reveals p53 core domain bound in an unfolded form to the chaperone Hsp90. Proc. Natl. Acad. Sci. 99 11085–11090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scalley, M.L. and Baker, D. 1997. Protein folding kinetics exhibit an Arrhenius temperature dependence when corrected for the temperature dependence of protein stability. Proc. Natl. Acad. Sci. 94 10636–10640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler, T. and Schmid, F.X. 1996. Thermodynamic properties of an extremely rapid protein folding reaction. Biochemistry 35 16833–16842. [DOI] [PubMed] [Google Scholar]
- Stanhope-Baker, P., Kessler, P.M., Li, W., Agarwal, M.L., and Williams, B.R. 2004. The Wilms tumor suppressor-1 target gene podocalyxin is transcriptionally repressed by p53. J. Biol. Chem. 279 33575–33585. [DOI] [PubMed] [Google Scholar]
- Stephen, C.W. and Lane, D.P. 1992. Mutant conformation of p53: Precise epitope mapping using a filamenous phage epitope library. J. Mol. Biol. 225 577–583. [DOI] [PubMed] [Google Scholar]
- Takimoto, R., Wang, W., Dicker, D.T., Rastinejad, F., Lyssikatos, J., and El-Deiry, W.S. 2002. The mutant p53-conformation modifying drug, CP-31398, can induce apoptosis of human cancer cells and can stabilize wild-type p53 protein. Cancer Biol. Ther. 1 47–55. [DOI] [PubMed] [Google Scholar]
- Tan, Y.J., Oliveberg, M., and Fersht, A.R. 1996. Titration properties and thermodynamics of the transition state for folding: Comparison of two-state and multi-state folding pathways. J. Mol. Biol. 264 377–389. [DOI] [PubMed] [Google Scholar]
- Wang, W., Rastinejad, F., and El-Deiry, W.S. 2003a. Restoring p53-dependent tumor suppression. Cancer Biol. Ther. 2 S55–S63. [PubMed] [Google Scholar]
- Wang, W., Takimoto, R., Rastinejad, F., and El-Deiry, W.S. 2003b. Stabilization of p53 by CP-31398 inhibits ubiquitination without altering phosphorylation at serine 15 or 20 or MDM2 binding. Mol. Cell. Biol. 23 2171–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wischhusen, J., Naumann, U., Ohgaki, H., Rastinejad, F., and Weller, M. 2003. CP-31398, a novel p53-stabilizing agent, induces p53-dependent and p53-independent glioma cell death. Oncogene 22 8233–8245. [DOI] [PubMed] [Google Scholar]