

Abstract
Protein folding, natural conformational changes, or interaction between partners involved in recognition phenomena brings about differences in the solvent-accessible surface area (SASA) of the polypeptide chain. This primary event can be monitored by the differential chemical reactivity of functional groups along the protein sequence. Diazirine (DZN), a photoreactive gas similar in size to water, generates methylene carbene (:CH2). The extreme chemical reactivity of this species allows the almost instantaneous and indiscriminate modification of its immediate molecular cage. 3H-DZN was successfully used in our laboratory for studying protein structure and folding. Here we address for the first time the usefulness of this probe to examine the area of interaction in protein–protein complexes. For this purpose we chose the complex formed between hen egg white lysozyme (HEWL) and the monoclonal antibody IgG1 D1.3. :CH2 labeling of free HEWL or complexed with IgG1 D1.3 yields 2.76 and 2.32 mmol CH2 per mole protein at 1 mM DZN concentration, respectively. This reduction (15%) becomes consistent with the expected decrement in the SASA of HEWL occurring upon complexation derived from crystallographic data (11%), in agreement with the known unspecific surface labeling reaction of :CH2. Further comparative analysis at the level of tryptic peptides led to the identification of the sites involved in the interaction. Remarkably, those peptides implicated in the contact area show the highest differential labeling: H15GLDNYR21, G117TDVQAWIR125, andG22YSLGNWVCAAK33. Thus, protein footprinting with DZN emerges as a feasible methodology useful for mapping contact regions of protein domains involved in macromolecular assemblies.
Keywords: diazirine, methylene carbene, protein interfaces, protein complexes, solvent-accessible surface area, protein footprinting, epitope mapping, hen egg white lysozyme
Understanding protein–protein interactions remains an unsolved problem underlying the fabric of life (Kennedy and Norman 2005). The ability of proteins to form specific, stable complexes with other proteins is crucial to all biological functions, as it becomes increasingly evident in the light of results from proteomic studies that are revealing key molecular interactions (Pandey and Mann 2000; Vidal 2005). In this realm, to fully grasp the molecular basis of protein–protein recognition, one needs to characterize in detail the contact surfaces. To this end, both experimental and theoretical efforts are currently underway (Ofran and Rost 2003; Janin 2005; Mendez et al. 2005).
In all instances, the binding event elicits a reduction in the solvent-accessible surface area (SASA) associated to each component occurring upon complex formation. The area of the interface (B), defined as the sum of the SASA of the two free partners of a complex less that of the complex, is a convenient measure of the extent of the interaction. A main group of complexes exhibits a standard size interface (on average 1600 ± 400 Å2) that suffices to provide the interaction with specificity and stability. In general, the building of these complexes brings about only minor conformational changes, such as displacements of surface loops or short stretches of the polypeptide chain and rotation of side chains of surface amino acid residues. Many antigen–antibody interactions fit into this category (Lo Conte et al. 1999).
Several biophysical and biochemical techniques converge to shed light on the study of protein interfaces. Typically, X-ray crystallography and NMR are the source of rich structural information. In addition, the use of cross- linking reagents, and in particular photoaffinity labeling methods, has been useful for elucidating chemical features and geometrical constraints of the interacting surfaces. Here, the choice of reagent requires previous knowledge on the kind of amino acid residues putatively involved in the interaction (Bayley 1983; Lundblad 2005). On the other hand, results are most often limited to defined areas around the location of the modified side chains. By contrast, a separate group of chemical techniques points to the use of reagents with the ability to modify a protein surface regardless of its chemical nature. Footprinting reagents belong to this class, providing information on topological features of the system. In this regard, apart from its wide applicability to protein folding, proton-deuteron (H/D) exchange has also been used with profit to address the identification and characterization of interfaces (Williams et al. 1996, 1997; Mandell et al. 1998). In spite of its large dynamic range, H/D exchange data are intrinsically limited to amide protons belonging to the backbone chain, therefore becoming strongly dependent on secondary structure integrity. Besides, one main drawback arises from the labile nature of the label, a factor that greatly restricts further analytical processing of the sample. Other footprinting techniques include the use of hydroxy radical reactions (1) to identify protein binding sites in nucleic acids (Tullius et al. 1987), (2) to assess conformational aspects of the native state and folding intermediates (Ermacora et al. 1992, 1994), (3) to label peptides and amino acids (Goshe and Anderson 1999; Goshe et al. 2000), and (4) to investigate a peptide binding site (Wong et al. 2005). Here, although products are generally stable, most often radical triggered reactions give rise to ill-characterized products that complicate the ensuing analytical procedures.
In this work we introduce the photoreagent diazirine (DZN) in a novel application as a footprinting reagent useful for investigating protein interfaces. DZN is a three-member ring heterocycle (CH2N2), a gas at room temperature and standard pressure, that serves as the source of methylene carbene (:CH2). The extreme chemical reactivity of this species allows the almost instantaneous and indiscriminate modification of its immediate molecular cage, inserting into any X–H bond (Turro et al. 1987). Unlike the radical reactions described above, here no fragmentation of the polypeptide chain is expected. Due to its small size, DZN might exert solvent mimicry, therefore accessing to a surface similar to that probed by water. Both the virtual absence of chemical specificity of the reactive species and the minimal size of the precursor molecule would allow this technique to provide an experimental approach to the surface theoretically defined as SASA. Unlike H/D exchange, labeling with DZN would target side chains as well as backbone exposure. Significantly, the modification with methylene carbene involves the formation of strong covalent bonds, giving rise to stable products that broaden the range of subsequent analytical procedures (Richards et al. 2000; Craig et al. 2002).
In previous works of our laboratory the tritiated form of DZN (3H-DZN) allowed us to accurately measure the global extent of modification of the polypeptide chain as well as to identify the locale of labeling. 3H-DZN has been successfully employed to study structure and folding of two model proteins: bovine α-lactalbumin (Craig et al. 2002) and Bacillus licheniformis β-lactamase (Ureta et al. 2001). Therein, we demonstrated that the expected increase in SASA occurring upon protein denaturation correlates well with the extent of methylene carbene modification. Other researchers took advantage of DZN labeling to identify an alternative conformation of replication protein A (Nuss and Alter 2004).
As our model system we chose the complex formed between hen egg white lysozyme (HEWL) and MAb IgG1 D1.3. HEWL is a 14.3-kDa monomeric protein with a sturdy conformation that has been defined in great detail. Most importantly, high-resolution structures complexes of this protein with Fab D1.3 and Fv D1.3 fragments are also available (Amit et al. 1986; Fischmann et al. 1991; Bhat et al. 1994). Apart from their intrinsic interest, antigen–antibody complexes represent a valuable model for the study of protein interactions (MacCallum et al. 1996) because (1) the size of the interface typically falls in the standard category (B ≈ 1290 Å2, measured in the HEWL–Fab D1.3 complex) (Fischmann et al. 1991), and (2) negligible conformational changes are expected upon complex formation (the measured RMSD between free and complexed HEWL is 0.64 Å) (Amit et al. 1986; for review, see Braden and Poljak 1995). The procedure herein described aims at establishing a novel methodology useful for the identification of contact surfaces between proteins, based on a general photochemical modification of the polypeptide chain with methylene carbene.
Results and Discussion
Rationale of the photochemical approach
Key to this approach is the argument that if surface regions could be labeled and the products analyzed at the level of small peptides or amino acid residues, then the differential pattern of label incorporation (i.e., by reacting the partners in their free vs. complexed forms) would permit the identification of the interaction site. Defined conformational changes occurring upon complex formation and/or the involvement of particularly flexible regions in the interaction surface could bring additional complexity to this picture. In each case, one should be forewarned of these points to be able to unambiguously interpret results. In this regard, initially we focused our efforts on a noncovalent complex comprising partners bearing a separate entity, i.e., constituents that are stable and fold independently. In this scenario, this is the first instance when a reagent with these features is used with the purpose of exploring interacting surfaces between proteins.
Methylene carbene labeling reveals the occlusion of area in a protein–protein complex
We chose the complex formed between HEWL and the MAb D1.3 as the working model (see Fig. 5a). All experiments were carried out in parallel with samples corresponding to the free and the complexed form of HEWL. The latter was prepared by mixing IgG1 D1.3, purified from mice ascites, and HEWL in a 2:1 molar ratio. Under our experimental conditions, we ensured that all HEWL became bound to the MAb and that 3H-DZN concentration was matched between samples. After the photolysis and cleanup procedures, the complex was dissociated and its components were separated by size-exclusion chromatography. For the sake of comparison, the free HEWL sample was treated in the same fashion (see Materials and Methods).
Figure 5.

(a) Crystallographic structure (PDB code 1VFB) of the complex formed between HEWL (white) and FvD1.3 (cyan). The amino acid residues of HEWL that are in direct contact with the MAb are colored in red. (b) Structure of free HEWL (PDB code 5LYM). Peptides A–D (see Table 1) are colored in blue, green, red, and yellow, respectively; the remainder of the protein is shown in white. (c) Structure of free HEWL (PDB code 5LYM). Amino acid residues in contact with the antibody are colored in red (epitope), peptide A is colored in blue, and cavities are illustrated as blobs colored in blue (cavity 1, 23 Å3) or green (cavity 2, 64 Å3). For the sake of comparison, a DZN molecule is shown as a CPK model (43 Å3).
In this work we focused our attention on epitope mapping, i.e., analyzing the regions in the HEWL partner in the complex, because of its more ready feasibility from an experimental standpoint due to its smaller size. Thus, HEWL isolated from each labeled sample was further processed.
The known stability of the reaction products of methylene carbene with the polypeptide chain (see introduction) opens the possibility of employing various analytical separation procedures without risk of losing the radioactive label. To ensure the thorough removal of radiolabeled by-products eventually remaining in the samples, an RP-HPLC separation step on a C4 column was routinely employed as the final stage of the clean-up procedure. To evaluate the specific radioactivity associated to each protein sample, both the absorbance at 280 nm and the 3H radioactivity on eluted fractions were monitored along the run (Fig. 1). Indeed, radioactivity appears exclusively associated to the protein peak and incorporation of the label occurs in a strictly light-dependent fashion (nonirradiated samples incorporate an insignificant label) (data not shown). Analysis of peak shape reveals a slight broadening and an increase in the retention time of the radioactive peak with respect to the mass peak, with these effects arising as the consequence of the rather more hydrophobic nature of the product and the intrinsic chemical heterogeneity caused by the methylation reaction. On the other hand, no evidence for protein fragmentation exists, since no radioactivity elutes anywhere else along the run.
Figure 1.
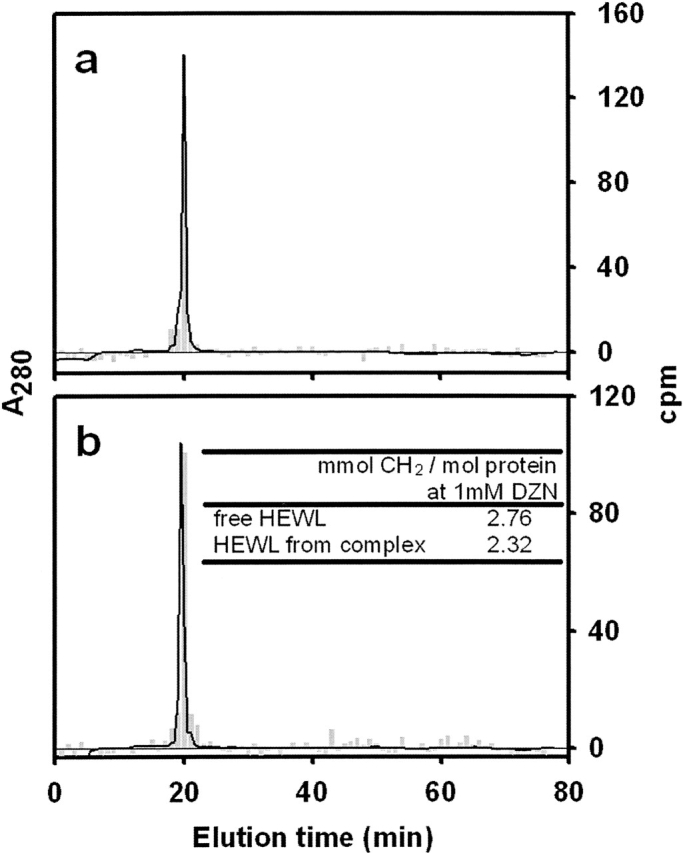
Separation of HEWL labeled with methylene carbene by RP-HPLC. Photolabeled HEWL eluted after size-exclusion chromatography (see Materials and Methods, “Photolabeling of free HEWL and HEWL complexed With IgG1 D1.3”) was sampled onto an analytical C4 column. Panels a and b correspond to HEWL labeled in its free or complexed form, respectively. Elution was monitored by UV absorption at 280 nm (solid line) and by measurement of the radioactivity associated to each collected fraction (gray bars). The inset table (b) shows the radioactivity incorporated to each HEWL sample expressed as mmol CH2/mol protein at 1 mM DZN.
Finally, the extent of modification, calculated from the specific radioactivity on pooled fractions of the protein peak, was expressed as mmoles of methylene per mole of protein at a given DZN concentration (1 mM). This last normalization allows one to make accurate comparisons among samples arising from different experiments. This is possible due to the demonstrated linear dependence of the extent of modification with DZN concentration (see Fig. 5 in Craig et al. 2002). The table inserted into Figure 1 shows the values for the extent of modification corresponding to each sample. Complexed HEWL incorporates 15% less label than the free form. This experimental value correlates well with the occluded area in the complex estimated from crystal- lographic data: The SASA of HEWL (6529 Å2, calculated with a probe radius of 1.4 Å for the water molecule) is reduced by 749 Å2 upon complex formation, yielding a decrease of ∼11%. Furthermore, the agreement between these results is consistent with the nonspecific reaction of methylene carbene with the polypeptide chain.
Methylene carbene labeling allows the identification of the sites of interaction
A further effort of our work focused on the identification of the area of interaction between HEWL and the MAb, the criterion being to seek where differences in carbene incorporation occur along the sequence. For this purpose, we pursued a comparative analysis on samples of HEWL labeled in its free or complexed form by fragmentation of the protein with trypsin. Group separation of the resulting peptides was achieved by size-exclusion chromatography on a Superdex Peptide HR 10/30. The radioactivity associated to each fraction was accurately quantitated along the run (Fig. 2a,b). All radioactivity was found to be associated to any given peptide fraction, an observation consistent with a general photolabeling reaction with carbene, which minimally increases the molecular weight of the product. In addition, a general trend exists to have more labels incorporated as the molecular mass of the peptide increases, a result in agreement with the known lack of specificity of the photolabeling reaction. Nevertheless, although qualitatively similar, the labeling patterns of each sample showed small but clear quantitative differences. To express this differential labeling event in a straightforward fashion we chose to plot the relative differential labeling for each peptide peak: an experimental parameter that represents the numerical difference between the label incorporation for free and complexed HEWL (Table 1), expressed relative to the value for the complexed form (Fig. 2c; for details, see also Materials and Methods). By comparison of this parameter along the run, peak C emerges as that showing the largest value. In addition, the peptide material eluting in each peak was subsequently analyzed by RP-HPLC (C18) (Fig. 3). All main peaks in each chromatogram were unambiguously identified by amino acid analysis and the data tabulated (Table 1). Remarkably, those peptides comprising amino acid residues involved in the area of interaction with the MAb happened to elute in peak C. The locations of these peptides in the known three-dimensional structure of native HEWL are highlighted in Figure 5b. On the other hand, clearly those regions of HEWL compromised in the epitope (mapped by peptides C) show reduced local mobility upon complex formation, as revealed by the comparison of normalized B factors derived from the known structures of complexed and free HEWL (data not shown). Indeed, those amino acid residues change from a surface location to a compact and hydrophobic milieu embedded at the interface. Taken together, results from this analysis and from our labeling data would support a transition to a rigid, “core-like” nature upon complexation. In fact, from independent studies on conformational equilibria of other globular proteins, we know that close packing of the core is the main impediment preventing methylene carbene labeling (Craig et al. 2002; Ureta 2003; Craig 2004). As revealed by the crystallographic structure of the complex, the epitope is discontinuous and centered around residues 20 and 120 (Fischmann et al. 1991). More precisely, the amino acid residues constituting the contact region are the following: D18, N19, G22, S24, N27, G117, T118, D119, V120, Q121, I124, and R125. Residue Q121 is essential for binding because it inserts into a hydrophobic pocket comprised by Y32 and W92 in VL, and Y101 in VH. In addition, the side chain of Q121 is hydrogen-bonded to F91 in VL, and it interacts via van der Waals contact with several residues lining the walls of this pocket. Overall, the combination site is relatively flat and decorated with low bulges and shallow wells surrounded by aromatic side chains. Although all CDRs become involved in this interaction, CDR 3 in VH makes the most extensive contact with the antigen. These structural features converge to determine a relatively high binding affinity (2.7 × 108 M–1) (Bhat et al. 1994). Our expectation is that methylene carbene labeling will be applicable to complexes spanning a wide range of affinities (presumably, dissociation constants as low as micromolar or even less), taking into account that the high protein concentration herein used will ensure negligible amounts of free forms in the equilibrium mixture.
Figure 2.
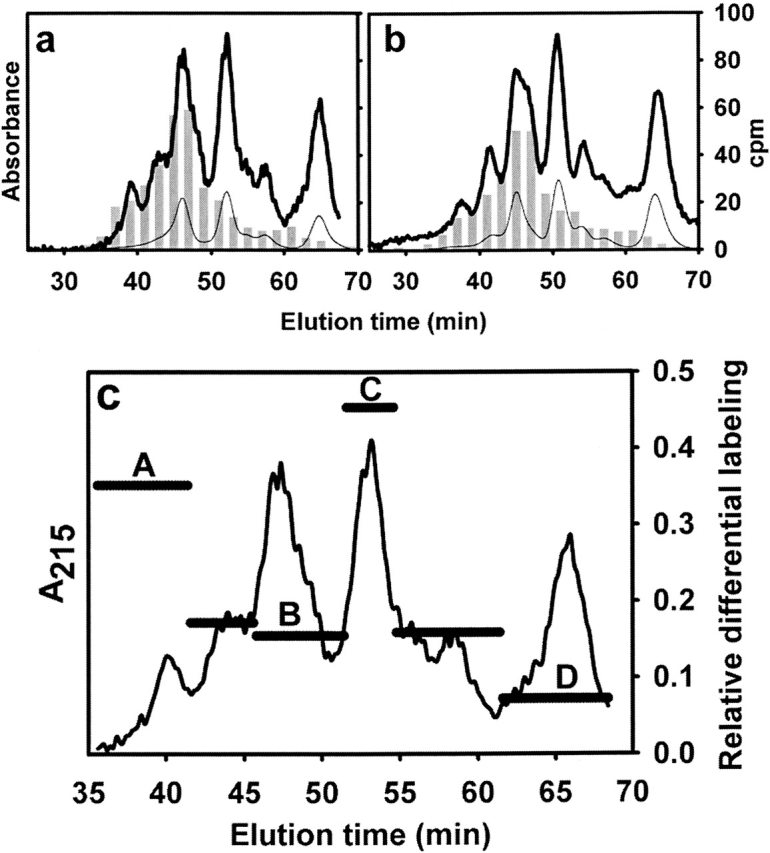
Separation by size-exclusion chromatography of tryptic peptides derived from HEWL labeled with methylene carbene. After the clean-up step described above (see Fig. 1), HEWL samples were reduced, carbamidomethylated, and digested with TPCK-trypsin (see Materials and Methods). The resulting soluble peptide mixtures corresponding to HEWL labeled in its free (a) or complexed form (b) were subjected to size-exclusion chromatography on a Superdex Peptide HR 10/30 column. Elution was monitored by the UV absorption at 280 (thin line) and 215 nm (thick line), and by measurement of the radioactivity (gray bars). In c, the horizontal bars represent the relative differential labeling of each peak (named A–D). This parameter is defined as the numerical difference between the label incorporation for free and complexed HEWL expressed relative to the value for the complexed form. The elution profile (absorbance at 215 nm) of peptides is plotted alongside.
Table 1.
Identification of tryptic peptides derived from HEWL labeled with methylene carbene

After isolation by size-exclusion chromatography followed by RP-HPLC (see Figs. 2, 3), peptides were unambiguously identified by their amino acid composition. Methylene incorporation for each peak was estimated as described in Materials and Methods. Amino acid residues that are in direct contact with the MAb D1.3 (see Figs. 4, 5) are indicated in bold.
Figure 3.

RP-HPLC analysis of tryptic peptides present in each fraction separated after size-exclusion chromatography. Pooled fractions corresponding to each peak separated after chromatography on a Superdex Peptide HR 10/30 column (A, B, C, or D; see Fig. 2c) were sampled onto a C18 column (a–d, respectively). Elution was monitored by UV absorption at 215 nm (solid line). Major peaks belonging to each pooled fraction were numbered sequentially. The aqueous acetonitrile gradient is shown as a dashed line (see Materials and Methods).
The overlap between the epitope region and the differentially labeled peptides is shown along the sequence of HEWL in lines “contacts” and “3H-DZN” (Fig. 4). In the latter, the position of peptide A is also highlighted. This site shows the second largest difference in methylene carbene labeling (see Fig. 2c). A possible explanation for this result required from us to perform a detailed analysis of cavities present in HEWL. Several high-resolution structures of HEWL in its free form (Protein Data Bank [PDB] codes 1HEW, 1HEL, 1H6M, 1LZT, 2LZT, 4LYM, 5LYM, and 1LCM) were processed with the MSP software (Connolly 1993). Particular attention was paid to ascertain the entity of the cavities found to validate that these indeed exist regardless of crystal form. The two cavities conspicuously found in all structures analyzed are shown in Figure 5c, and those amino acid residues lining their walls are shown in Figure 4 (line cavities). A parallel calculation was performed on the complexed form of lysozyme (PDB code 1VFB), where the same cavities were found. Here we chose the FvD1.3–HEWL complex, because it is available at high resolution (1.8 Å) (Bhat et al. 1994), but identical results were obtained with the Fab D1.3–HEWL complex (Fischmann et al. 1991). Indeed, although the backbone atoms of amino acids belonging to peptide A are the main contributors to the environment around the largest cavity (dubbed cavity 2), no significant difference was found between the volumes calculated for this cavity in free or complexed HEWL (64 and 60 Å3, respectively). But what is the connection between cavities and sites for methylene carbene labeling? In our laboratory it has been demonstrated that methylene carbene also labels inner surfaces (those constituting the boundaries of cavities), where DZN would dwell for a longer residence time and a different cage of reaction would be expected (Craig et al. 2002; Ureta 2003; Craig 2004). Independent evidence from others using NMR spectroscopy (Otting et al. 1997) or X-ray diffraction (Prange et al. 1998; Quillin et al. 2000) supports the location of gases with similar features to DZN (xenon, argon, methane, etc.), i.e., size and moderate hydrophobic character, within cavities and crevices present in globular proteins. In particular, Otting et al. (1997) described the binding to HEWL of cyclopropane, a molecule isosteric to DZN. In this context, one should bear in mind that—apart from providing a unique measurement of the external surface accessible to the aqueous solvent—to some extent methylene carbene labeling will also point to sites topologically involved in defining those cavities. In addition, other researchers have shown that inert gases can be lodged inside cavities even in instances where their predicted volume is smaller that the size of the gas (Quillin et al. 2000). This fitting requires subtle rearrangements allowed by the intrinsic flexibility of the polypeptide chain. In order to explain the finding regarding peptide A, our argument holds that due to rigidification brought about by the binding of the antibody to HEWL, differential methylene carbene labeling favoring the more flexible free form would be expected. This dynamic does not necessarily become evident from the picture emerging from the crystallographic data. Flexibility appears to be well correlated with enhanced methylene carbene labeling as it has been demonstrated in extremis by the heavy labeling observed for the molten globule states of both a-lactalbumin and b-lactamase (Ureta 2003; Craig 2004). On the other hand, Williams et al. (1996) measured the H/D exchange rate of the amidic protons in HEWL free and complexed with this same antibody (line H/D exchange in Fig. 4). This analysis provides interesting insights on the structural dynamics of the protein. Overall, complex formation causes a decrease in the exchange rate of several protons distributed in many sites along the sequence, suggesting that, as a consequence of the binding event, propagation to distant locations (long distance effects) can also be expected. In other words, protection from exchange occurs not only at the epitope region but also at sites far removed from that area. To explain this effect, the investigators invoked a decrease in local conformational flexibility as a consequence of complex formation. Interestingly, an evident correlation between protection factor and methylene carbene labeling occurs at the epitope region (the highest values of the protection factor map to peptides C). On the other hand, hydrogen exchange might be less sensitive to probe inner surface regions for which intrinsically low values of exchange will be expected. In this regard, by its very nature, differential methylene carbene labeling of DZN-filled cavities would lead to an “amplification” effect that might reveal slight changes in mobility in a hydrophobic milieu. It is noteworthy that peptide A maps a fracture zone in HEWL located in between the two subdomains, one predominantly a and the other b, making up the opposite walls of the active site. This location might result particularly sensitive to receive a propagation of a subtle movement occurring at a distant spot (see Fig. 5c).
Figure 4.

Linear diagram of HEWL. Numbers on the ruler represent positions along the amino acid sequence of HEWL. Horizontal bars indicate known secondary structure elements based on crystallographic data. Black boxes along the line contacts point to amino acid residues that are in direct contact with the MAb D1.3, as revealed by the structure of the complex (Fischmann et al. 1991). The line 3H-DZN shows peptides C1, C2, and C3 (black boxes), for which the largest relative differential labeling was measured, and peptide A (gray box), exhibiting the second largest value of this parameter (see Fig. 2c). The line cavities displays the positions of amino acid residues that constitute the boundaries of cavities 1 (white boxes) and 2 (gray boxes). These entities were conspicuously found in all high-resolution structures analyzed. The line H/D exchange represents graphically the experimentally measured protection factors (defined as τ complexed HEWL/τ free HEWL for each amino acid residue, τ being the lifetime of exchange of the amide proton) (see Williams et al. 1996). Boxes in shades of gray illustrate the values of this parameter according to its magnitude: black (>3), dark gray (between 2 and 3) and light gray (between 1 and 2).
Conclusions
In this work we address the applicability of methylene carbene labeling to examine the area of interaction in protein–protein complexes. For this purpose HEWL was photolabeled with DZN, either in its free form or complexed with a monoclonal antibody. The recognition phenomenon brings about a decrease in the SASA of the polypeptide chain, which reveals itself as a reduction in the extent of modification. This behavior is consistent with an unspecific surface labeling reaction. Most significantly, a comparative analysis of labeled HEWL samples led to the identification of the sites along the polypeptide sequence involved in the interaction (epitope regions). In addition, the technique proved to be sensitive enough to expose remote sites where variations in local flexibility might occur as a consequence of protein complex formation.
Overall, this work underscores the main usefulness of methylene carbene labeling to discern changes in the extent of exposure of the polypeptide chain. In a more general context, methylene carbene labeling arises as a worthy counterpart to hydrogen-exchange experiments. Unlike the latter, which depends primarily on backbone hydrogen-bonding, methylene carbene labeling would represent a valuable tool to map mainly side-chain solvent exposure, providing insights on the nature of the surfaces implicated.
Current efforts underway in our laboratory include the detection and the quantification of methylated products by ES and MALDI mass spectrometry, thus allowing us to avoid the use of a radiolabeled reagent and to reduce the amount of required protein sample (Gómez and Delfino 2005).
Materials and Methods
3H-formaldehyde (5 mCi, 10 Ci/mmol) was purchased from American Radiolabeled Chemicals, Inc.; formaldehyde (37% [w/v]) was from E. Merck; formamide, urea, and HEWL were from Sigma Chemical Co. HEWL was used without further purification, and urea was recrystallized from ethanol before use. TPCK trypsin was from Worthington. Acetonitrile from E. Merck and trifluoroacetic acid (TFA) from Riedel de Haën were of HPLC grade. All other reagents and chemicals used were of analytical grade.
HEWL concentration was determined by its UV absorption at 280 nm on a Jasco 7850 spectrophotometer using an extinction coefficient of E1% = 26 (Fasman 1989). Quantification of MAb D1.3 was also achieved by measuring its UV absorption at 280 nm, with an extinction coefficient of E1% = 15. HEWL and its tryptic peptides were separated on ÄKTA purifier (Amersham Pharmacia Biotech) and Rainin Dynamax FPLC/HPLC systems.
Obtention and purification of monoclonal antibody IgG1 D1.3
The MAb D1.3 was produced from ascites in Balb/c mice as previously described (Harper et al. 1987). Briefly, hybridoma cells (2 × 106) producing IgG1 D1.3 (gift from Dr. Roberto Poljak, Center for Advanced Research in Biotechnology, Maryland) were expanded in vivo by injecting intraperitoneally into mice (Balb/c) that had been primed ∼14 d earlier with 0.5 mL pristane (2-, 6-, 10-, 14-tetramethyl pentadecane; Sigma). The ascitic fluid was harvested 10–15 d later, and aliquots were frozen at –20°C in the presence of NaN3 (0.01% [w/v]) and stored until needed. Ascitic fluid was precipitated by adding (NH4)2SO4 (50% [w/v]) in an ice bath. After the precipitate was formed, the preparation was centrifuged during 30 min at 14,000 rpm. Then, the precipitate was dissolved in one-fifth of the initial volume and dialyzed overnight against phosphates buffer saline (PBS). IgG1 D1.3 was purified through a Sepharose 4B affinity column (Amersham Pharmacia Biotech) derivatized with HEWL. The eluate was further purified by ion-exchange through a Mono Q column (Amersham Pharmacia Biotech). Pure IgG1 D1.3 was concentrated to 10 mg/mL by using a Centricon Y-M30 centrifugation/filtration device (Millipore).
Synthesis of 3H-DZN
3H-DZN was synthesized in our laboratory by following the method described by Craig et al. (2002). 3H-DZN concentration in aqueous solution was estimated both by measuring (1) the absorbance of the dissolved gas at 320 nm using an extinction coefficient of ɛ320 = 180 M−1 cm–1 (Craig et al. 2002), and (2) the concentration of radioactivity of the solution after complete photolysis of 3H-DZN to 3H-methanol had taken place.
Photolabeling of free HEWL and HEWL complexed with IgG1 D1.3
The free HEWL sample (0.1 mM) consists of commercially available HEWL dissolved at pH 7.4 in PBS. The complexed form of HEWL was prepared by mixing HEWL and IgG1 D1.3 in PBS at a 2:1 molar ratio at least 30 min before reaction with 3H-DZN. In practice, a slight excess of MAb (∼10%) was used to ensure the complete binding of the antigen. The buffer used was degassed in advance and kept under an inert atmosphere of nitrogen.
3H-DZN (1–3 mM, 1 mCi/mmol) was dissolved by bubbling the gas into each sample placed in quartz cuvettes (4 mL volume) of 1-cm path length capped with Teflon stoppers. The concentration of the gas was equated by using a double-tipped needle to interconnect the gas phase of each vessel. Photolysis was carried out by using a UV light source (Philips HPA 1000 Halogen/Hg lamp) placed at 12 cm from the samples. Light was filtered below 300 nm with a long pass filter (Oriel 59,044) to prevent photolytic damage to protein chromophores. The cuvettes were immersed in a water bath thermostatted at 20°C, so that even at the high luminic flux used, no heating occurs. Typically, UV irradiation was extended for 45–60 min, a time that corresponds to approximately four to six half-lives of the reagent in our photolysis setup (t1/2 =10.3 min).
After photolysis, solid urea was added to both samples (8 M final concentration), and the solutions were then dialyzed against Na2CO3 (0.07 mM at pH 7.5–8.0) and freeze-dried. After these cleanup procedures, labeled protein samples were separated by size-exclusion chromatography (FPLC) on a Superdex-75 column (Amersham Pharmacia Biotech). We ensured the dissociation of the complex by dissolving this sample in diethylamine buffer (50 mM at pH 12), added with NaCl (1 M) and running the column at a 0.50 mL/min flow rate with the same buffer. For the sake of comparison, the free HEWL sample was treated in the same fashion. Elution of proteins was monitored by UV absorption at 280 nm. HEWL was collected between 28 and 35 min after sampling and immediately neutralized by mixing with Tris-HCl buffer (1 M at pH 7.0). Samples were once again dialyzed against Na2CO3 (0.07 mM at pH 7.5–8.0) and freeze-dried.
Finally, the isolated HEWL samples labeled in each condition were dissolved in aqueous GdnHCl (6 M) and separated from any remaining radioactive impurity by reversed phase HPLC on a C4 column (Vydac 214TP510, 10 mm × 250 mm), developed with three successive linear gradients of acetonitrile: water (0%–35% in 8 min, 35%–45% in 30 min, and 45%–100% in 11 min) in TFA (0.05% [v/v]), at a 3 mL/min flow rate. The elution was monitored by UV absorption at 280 nm. For analytical purposes (see Fig. 1), samples were run on a smaller C4 column (Vydac 214TP54, 4.6 mm×250 mm) with an identical gradient at a 1 mL/min flow rate. Protein samples cleaned in this fashion were either used for measuring the incorporated radioactivity or further processed into peptides as detailed in “Reduction, carbamidomethylation, and tryptic digestion of DZN-labeled HEWL” (below).
After lyophilization to remove HPLC solvents, and redissolution in sodium phosphates buffer (20 mM at pH 7.4), the radioactivity present in the samples was measured on a Pharmacia 1214 Rackbeta liquid scintillation counter. The extent of 3H-methylene carbene incorporation into HEWL was expressed as the average number of mmoles of CH2 per mole of protein, as estimated by the radioactivity measured on samples of known protein concentration by taking into account the specific radioactivity of the reagent (inset to Fig. 1).
Reduction, carbamidomethylation, and tryptic digestion of DZN-labeled HEWL
After the cleanup procedure described earlier, samples of HEWL (5 mg) labeled with 3H-methylene, on its free or complexed form, were reduced with DTT and blocked with iodoacetamide according to Waxdal et al. (1968) with the modifications described by Craig et al. (2002). Complete enzymatic digestion of these carbamidomethylated samples with TPCK trypsin was achieved in NH4CO3 H (0.1 M at pH 8.0), after 12–24 h at 37°C using a 2% (w/w) enzyme:substrate ratio.
Peptide mapping
Separation of mixtures of tryptic peptides was carried out by size-exclusion chromatography on a Superdex Peptide HR 10/30 column (Amersham Pharmacia Biotech) developed with Tris-HCl buffer (50 mM at pH 7.4) added with urea (4 M) at a flow rate of 0.25 mL/min. Elution was monitored by UV absorption at 215 nm and 280 nm and by measurement of the radioactivity. We proceeded as follows to accurately measure differences in the extent of labeling between samples of HEWL reacted in its free or complexed form. In the first place, the radioactivity collected in each peak was normalized by the amount of peptide estimated by the area under the elution profile (monitored at 215 nm). Next, these values were in turn normalized to a constant DZN concentration (arbitrarily chosen as 1 mM) and dubbed methylene incorporation (shown in Table 1). Finally, the relative differential labeling represents the numerical difference between the label incorporation for free and complexed HEWL expressed relative to the value for the complexed form (shown in Fig. 2c).
Peaks from the size-exclusion chromatography were collected and further separated by RP-HPLC on a C18 column (Vydac 218TP54, 4.6 mm × 250 mm) eluted at a 1 mL/min flow rate with aqueous TFA (0.05% [v/v]) for 10 min, followed by a linear gradient of acetonitrile: water (0%–60% in 60 min) in TFA (0.05% [v/v]). Elution was monitored by UV absorption at 215 nm. Peptide identification was achieved by amino acid analysis on an Applied Biosystems 420 A amino acid analyzer.
Molecular modeling
Calculations of SASA and analysis of cavities of free HEWL based on high-resolution crystallographic structures available for this protein (PDB codes 1HEW, 1HEL, 1H6M, 1LZT, 2LZT, 4LYM. 5LYM, and 1LCM) or the complex HEWL-Fv D1.3 (PDB code 1VFB) were carried out with MSP (Connolly 1993), Surface Racer (using F.M. Richards’ van der Waals radii set) (Tsodikov et al. 2002), and MacroModel (Mohamadi et al. 1990), with a choice of 1.4 Å probe radius for the water molecule. Figure 5 was rendered with the program Grasp (Nicholls et al. 1991). MacroModel, MSP and Grasp were run on SGI workstations (Indigo R4000 XS24Z and O2 R10000) and Surface Racer on a Pentium-based PC.
Acknowledgments
For their encouragement, helpful comments along the various stages of this work, and their critical reading of the manuscript, we thank Ms. Lucrecia M. Curto and Mr. Leonardo M. Cortez. G.E.G. is a recipient of a graduate student fellowship from the University of Buenos Aires (UBA). This research has been supported by grants to J.M.D. from UBA, the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), and the Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT).
Footnotes
Reprint requests to: José María Delfino, Departamento de Química Biológica–IQUIFIB (UBA-CONICET), Facultad de Farmacia y Bioquímica, Universidad de Buenos Aires, Junín 956, C1113AAD Buenos Aires, Argentina; e-mail: delfino@qb.ffyb.uba.ar; fax: +54-11-4962-5457.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051960406.
Abbreviations: DZN, diazirine; 3H-DZN, tritiated derivative of diazirine; CH2, methylene carbene; SASA, solvent-accessible surface area; HEWL, hen egg white lysozyme; MAb, monoclonal antibody; VL, variable domain of the heavy chain; VH, variable domain of the light chain; CDR, complementarity determining regions; RP-HPLC, reversed-phase high-performance liquid chromatography.
References
- Amit A.G., Mariuzza R.A., Phillips S.E., Poljak R.J. 1986. Three-dimensional structure of an antigen–antibody complex at 2.8 Å resolution Science 233: 747–753. [DOI] [PubMed] [Google Scholar]
- Bayley H. In Photogenerated reagents in biochemistry and molecular biology. . 1983. Elsevier, Amsterdam.
- Bhat T.N., Bentley G.A., Boulot G., Greene M.I., Tello D., Dall'Acqua W., Souchon H., Schwarz F.P., Mariuzza R.A., Poljak R.J. 1994. Bound water molecules and conformational stabilization help mediate an antigen–antibody association Proc. Natl. Acad. Sci. 91: 1089–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braden B.C. and Poljak R.J. 1995. Structural features of the reactions between antibodies and protein antigens FASEB J. 9: 9–16. [DOI] [PubMed] [Google Scholar]
- Connolly M.L. 1993. The molecular surface package J. Mol. Graph. 11: 139–141. [DOI] [PubMed] [Google Scholar]
- Craig P.O. “Diazirine: A photoreactive probe for the conformational study of proteins.“ Ph.D. dissertation 2004. University of Buenos Aires, Buenos Aires.
- Craig P.O., Ureta D.B., Delfino J.M. 2002. Probing protein conformation with a minimal photochemical reagent Protein Sci. 11: 1353–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermacora M.R., Delfino J.M., Cuenoud B., Schepartz A., Fox R.O. 1992. Conformation-dependent cleavage of staphylococcal nuclease with a disulfide-linked iron chelate Proc. Natl. Acad. Sci. 89: 6383–6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermacora M.R., Ledman D.W., Hellinga H.W., Hsu G.W., Fox R.O. 1994. Mapping staphylococcal nuclease conformation using an EDTA-Fe derivative attached to genetically engineered cysteine residues Biochemistry 33: 13625–13641. [DOI] [PubMed] [Google Scholar]
- Fasman G.D. In Practical handbook of biochemistry and molecular biology. . 1989. CRC Press, Boca Raton, FL.
- Fischmann T.O., Bentley G.A., Bhat T.N., Boulot G., Mariuzza R.A., Phillips S.E., Tello D., Poljak R.J. 1991. Crystallographic refinement of the three-dimensional structure of the FabD1.3-lysozyme complex at 2.5-Å resolution J. Biol. Chem. 266: 12915–12920. [PubMed] [Google Scholar]
- Gómez G.E. and Delfino J.M. 2005. Probing protein conformation with a general probe coupled to detection by mass spectrometry Protein Sci. 14:Suppl. 1, 62–76. [Google Scholar]
- Goshe M.B. and Anderson V.E. 1999. Hydroxyl radical–induced hydrogen/deuterium exchange in amino acid carbon-hydrogen bonds Radiat. Res. 151: 50–58. [PubMed] [Google Scholar]
- Goshe M.B., Chen Y.H., Anderson V.E. 2000. Identification of the sites of hydroxyl radical reaction with peptides by hydrogen/deuterium exchange: Prevalence of reactions with the side chains Biochemistry 39: 1761–1770. [DOI] [PubMed] [Google Scholar]
- Harper M., Lema F., Boulot G., Poljak R.J. 1987. Antigen specificity and cross-reactivity of monoclonal anti-lysozyme antibodies Mol. Immunol. 24: 97–108. [DOI] [PubMed] [Google Scholar]
- Janin J. 2005. The targets of CAPRI rounds 3–5 Proteins 60: 170–175. [DOI] [PubMed] [Google Scholar]
- Kennedy D. and Norman C. 2005. What don't we know? Science 309: 75–102. [DOI] [PubMed] [Google Scholar]
- Lo Conte L., Chothia C., Janin J. 1999. The atomic structure of protein–protein recognition sites J. Mol. Biol. 285: 2177–2198. [DOI] [PubMed] [Google Scholar]
- Lundblad R.L. In Chemical reagents for protein modification . 2005. 3d ed CRC Press, Boca Raton, FL.
- MacCallum R.M., Martin A.C., Thornton J.M. 1996. Antibody–antigen interactions: Contact analysis and binding site topography J. Mol. Biol. 262: 732–745. [DOI] [PubMed] [Google Scholar]
- Mandell J.G., Falick A.M., Komives E.A. 1998. Identification of protein–protein interfaces by decreased amide proton solvent accessibility Proc. Natl. Acad. Sci. 95: 14705–14710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez R., Leplae R., Lensink M.F., Wodak S.J. 2005. Assessment of CAPRI predictions in rounds 3–5 shows progress in docking procedures Proteins 60: 150–169. [DOI] [PubMed] [Google Scholar]
- Mohamadi F., Richards N.G.J., Guida W.C., Liskamp R., Lipton M., Caufield C., Chang G., Hendrickson T., Still W.C. 1990. Macro-model: An integrated software system for modeling organic and bioorganic molecules using molecular mechanics J. Comput. Chem. 11: 440–467. [Google Scholar]
- Nicholls A., Sharp K.A., Honig B. 1991. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons Proteins 11: 281–296. [DOI] [PubMed] [Google Scholar]
- Nuss J.E. and Alter G.M. 2004. Denaturation of replication protein A reveals an alternative conformation with intact domain structure and oligonucleotide binding activity Protein Sci. 13: 1365–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofran Y. and Rost B. 2003. Analysing six types of protein–protein interfaces J. Mol. Biol. 325: 377–387. [DOI] [PubMed] [Google Scholar]
- Otting G., Liepinsh E., Halle B., Frey U. 1997. NMR identification of hydrophobic cavities with low water occupancies in protein structures using small gas molecules Nat. Struct. Biol. 4: 396–404. [DOI] [PubMed] [Google Scholar]
- Pandey A. and Mann M. 2000. Proteomics to study genes and genomes Nature 405: 837–846. [DOI] [PubMed] [Google Scholar]
- Prange T., Schiltz M., Pernot L., Colloc'h N., Longhi S., Bourguet W., Fourme R. 1998. Exploring hydrophobic sites in proteins with xenon or krypton Proteins 30: 61–73. [PubMed] [Google Scholar]
- Quillin M.L., Breyer W.A., Griswold I.J., Matthews B.W. 2000. Size versus polarizability in protein–ligand interactions: Binding of noble gases within engineered cavities in phage T4 lysozyme J. Mol. Biol. 302: 955–977. [DOI] [PubMed] [Google Scholar]
- Richards F.M., Lamed R., Wynn R., Patel D., Olack G. 2000. Methylene as a possible universal footprinting reagent that will include hydrophobic surface areas: Overview and feasibility: Properties of diazirine as a precursor Protein Sci. 9: 2506–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsodikov O.V., Record Jr. M.T., Sergeev Y.V. 2002. Novel computer program for fast exact calculation of accessible and molecular surface areas and average surface curvature J. Comput. Chem. 23: 600–609. [DOI] [PubMed] [Google Scholar]
- Tullius T.D., Dombroski B.A., Churchill M.E., Kam L. 1987. Hydroxyl radical footprinting: A high-resolution method for mapping protein–DNA contacts Methods Enzymol. 155: 537–558. [DOI] [PubMed] [Google Scholar]
- Turro N.J., Cha Y., Gould I.R. 1987. Reactivity and intersystem crossing of singlet methylene in solution J. Am. Chem. Soc. 109: 2101–2107. [Google Scholar]
- Ureta D.B. 2003. Conformational study of β-lactamase by photolabeling with diazirine Ph.D. dissertation University of Buenos Aires, Buenos Aires.
- Ureta D.B., Craig P.O., Delfino J.M. 2001. Photochemical detection of conformational transitions in proteins Protein Sci. 10:Suppl. 1, 131. [Google Scholar]
- Vidal M. 2005. Interactome modeling FEBS Lett. 579: 1834–1838. [DOI] [PubMed] [Google Scholar]
- Waxdal M.J., Konigsberg W.H., Henley W.L., Edelman G.M. 1968. The covalent structure of a human γG-immunoglobulin, II: Isolation and characterization of the cyanogen bromide fragments Biochemistry 7: 1959–1966. [DOI] [PubMed] [Google Scholar]
- Williams Jr. D.C., Benjamin D.C., Poljak R.J., Rule G.S. 1996. Global changes in amide hydrogen exchange rates for a protein antigen in complex with three different antibodies J. Mol. Biol. 257: 866–876. [DOI] [PubMed] [Google Scholar]
- Williams Jr. D.C., Rule G.S., Poljak R.J., Benjamin D.C. 1997. Reduction in the amide hydrogen exchange rates of an anti-lysozyme Fv fragment due to formation of the Fv-lysozyme complex J. Mol. Biol. 270: 751–762. [DOI] [PubMed] [Google Scholar]
- Wong J.W., Maleknia S.D., Downard K.M. 2005. Hydroxyl radical probe of the calmodulin-melittin complex interface by electrospray ionization mass spectrometry J. Am. Soc. Mass Spectrom. 16: 225–233. [DOI] [PubMed] [Google Scholar]