

The crystal structure of the synthetase domain of human CTPS has been determined. The structure is the first of an eukaryotic CTPS and provides a starting point for drug design towards CTPS.
Keywords: cytidine triphosphate synthetase, anticancer therapy
Abstract
Cytidine triphosphate synthetase (CTPS) is a key enzyme in nucleic acid and phospholipid biosynthesis and its activity is increased in certain human cancers, making it a promising drug target. The crystal structure of the synthetase domain of human CTPS, which represents the first structure of a CTPS from an eukaryote, has been determined. The structure is homotetrameric and each active site is formed by three different subunits. Sulfate ions bound to the active sites indicate the positions of phosphate-binding sites for the substrates ATP and UTP and the feedback inhibitor CTP. Together with earlier structures of bacterial CTPS, the human CTPS structure provides an extended understanding of the structure–function relationship of CTPS-family members. The structure also serves as a basis for structure-based design of anti-proliferative inhibitors.
1. Introduction
Cytidine triphosphate synthetase (CTPS; EC 6.3.4.2) catalyses the rate-limiting step in the synthesis of CTP, a key precursor of DNA, RNA and phospholipids. In humans, CTPS exists as two isoforms having 74% identity at the amino-acid level (van Kuilenburg et al., 2000 ▶). CTPS isoform 1 is a 591-amino-acid protein with a molecular weight of 67 kDa. The enzyme consists of an N-terminal synthetase domain and a C-terminal glutaminase domain. The glutaminase domain cleaves ammonium from glutamine, generating glutamate. Ammonium transfer from the glutaminase to the synthetase domain occurs via a tunnel (Endrizzi et al., 2004 ▶; Lunn & Bearne, 2004 ▶); it can also be derived from external ammonium (Willemoes, 2004 ▶). The ammonium ion is further used by the synthetase domain to generate CTP from ATP-phosphorylated UTP. GTP is an allosteric activator of the holoenzyme (Willemoes & Sigurskjold, 2002 ▶) and CTP acts as a feedback inhibitor by binding to the synthetase domain (Long & Pardee, 1967 ▶; Endrizzi et al., 2005 ▶). The overall reaction catalysed by CTPS (Fig. 1 ▶) controls the intracellular CTP pool, which in turn affects the synthesis of nucleic acids. CTPS is regulated by phosphorylation by protein kinase A on several serine residues (Han et al., 2005 ▶). The biochemically and structurally best characterized CTPS is that from Escherichia coli (Endrizzi et al., 2004 ▶).
Figure 1.

The overall reaction catalysed by CTPS. Phosphate is transferred from ATP to the O4 of UTP and the ammonium ion obtained from glutamine is then used to replace the phosphate to generate CTP. R = ribose-5′-triphosphate.
CTPS is an actively studied target for antiviral (Dereuddre-Bosquet et al., 2004 ▶), antineoplastic (Verschuur, Van Gennip, Leen, Meinsma et al., 2000 ▶) and antiparasitic (Hofer et al., 2001 ▶) drug development. CTPS activity is also increased in a variety of human cancers (Williams et al., 1978 ▶; Verschuur, Van Gennip, Leen, Meinsma et al., 2000 ▶). Antiproliferative drugs targeted specifically towards human CTPS have been developed, with the aim of depleting cancer cells of CTP and thus slowing down tumour growth (Politi et al., 1995 ▶). Cyclopentenyl cytosine (CPEC), acivicin and 3-deazauridine are CTPS-directed drugs that slow down or arrest the proliferation of tumour cells (Hindenburg et al., 1985 ▶; Kang et al., 1989 ▶; Zhang et al., 1993 ▶; Verschuur, Van Gennip, Leen, Muller et al., 2000 ▶). Resistance to some of these drugs is caused by mutations in CTPS (Whelan et al., 1993 ▶).
The crystal structures of CTPS are known from Thermus thermophilus (Goto et al., 2004 ▶) and E. coli (Endrizzi et al., 2004 ▶), and based on these structures, together with other studies, the active site and the substrate-binding modes have been identified. Active CTPS is a homotetramer and the oligomeric state is regulated by nucleotides (Pappas et al., 1998 ▶). However, experimental three-dimensional structures have not been available of CTPS from other organisms, including mammals.
We have determined the crystal structure of the synthetase domain from human CTPS. The structure is homotetrameric and the active site has two bound sulfate ions which indicate the positions of the phosphate moieties of the substrates ATP and UTP. The structure should also provide a structural basis for drug development with the aim of effectively inhibiting CTPS activity in cancer cells.
2. Materials and methods
2.1. Cloning and expression
The cDNA encoding the synthetase domain of human CTPS (isoform 1) was subcloned into the pNIC-Bsa4 vector; the resulting construct codes for a protein with an N-terminal hexahistidine tag with an integrated TEV protease-cleavage site. The sequence of the N-terminal tag was MHHHHHHSSGVDLGTENLYFQS. The plasmid was transformed into BL21(DE3)-Gold cells containing the chaperone plasmid pG-Tf2 (Takara).
Colonies were grown in 20 ml Terrific Broth supplemented with 8 g l−1 glycerol, 50 µg ml−1 kanamycin and 34 µg ml−1 chloramphenicol at 303 K overnight. The following morning, the overnight cultures were diluted into 1500 ml Terrific Broth containing 8 g l−1 glycerol and 50 µg ml−1 kanamycin in TunAir flasks. Cells were grown at 310 K until an OD600 of 1.4 was reached. 20 min later, induction of chaperone expression was performed with tetracycline (2 ng ml−1) and cultures were cooled to 291 K. Target-protein expression was then induced overnight with 0.5 mM IPTG.
Cells were harvested by centrifugation and pellets were suspended in 50 mM sodium phosphate pH 7.5, 500 mM NaCl and 10% glycerol supplemented with one tablet of Complete EDTA-free protease-inhibitor tablet (Roche) per cell pellet and frozen at 193 K. The cells were briefly thawed in warm water and 1000 U benzonase was added. Cells were disrupted by high-pressure homogenization at 69 MPa and the samples were centrifuged for 20 min at 40 000g. The soluble fraction was filtered prior to further purification.
2.2. Purification and crystallization
Purification was conducted on an ÄKTA Xpress system. HisTrap HP and Superdex 75 columns were equilibrated with IMAC buffer 1 [50 mM sodium phosphate pH 7.5, 10 mM imidazole, 500 mM NaCl, 10% glycerol, 0.5 mM Tris(2-carboxyethyl)phosphine (TCEP)] and gel-filtration buffer (20 mM HEPES pH 7.5, 300 mM NaCl, 10% glycerol, 0.5 mM TCEP), respectively. The protein sample was loaded onto the HisTrap HP column, which was washed with IMAC buffer 1 followed by IMAC buffer 2 (50 mM sodium phosphate pH 7.5, 50 mM imidazole, 500 mM NaCl, 10% glycerol, 0.5 mM TCEP). Bound protein was eluted from the HisTrap columns with 7.5 ml IMAC elution buffer (50 mM sodium phosphate pH 7.5, 400 mM imidazole, 500 mM NaCl, 10% glycerol, 0.5 mM TCEP) and loaded onto the gel-filtration column; elution was performed using the gel-filtration buffer. The chromatogram from gel filtration showed one major protein peak, corresponding to a CTPS monomer, that consisted of highly pure human CTPS as shown by SDS–PAGE (not shown). TCEP was added to the pooled protein peak to a final concentration of 2 mM. The protein was concentrated to 5 mg ml−1 and stored at 193 K. The protein was crystallized by the hanging-drop method, equilibrating 1 µl protein solution and 1 µl of a solution containing 0.1 M Tris pH 8.8, 1.2 M (NH4)2SO4 and 50 mM malonic acid against 0.5 ml of a well solution containing 0.1 M Tris pH 8.8 and 1.2 M (NH4)2SO4. Crystals with a maximum dimension of 30 µm formed in 3 d. Prior to data collection, a crystal was briefly soaked in a cryoprotectant solution [0.1 M Tris pH 8.8, 1.4 M (NH4)2SO4, 50 mM malonic acid, 25% glycerol, 0.2 M NaCl, 2 mM TCEP].
2.3. Data collection and structure solution
Data from cryocooled (100 K) crystals were collected at ESRF beamline ID29 using an ADSC Quantum 210 detector and 1° oscillations per image. The data were processed with MOSFLM (Leslie, 1992 ▶) and SCALA (Evans, 2006 ▶). The data-processing statistics are presented in Table 1 ▶. The high R sym values can be explained by the high redundancy of the data and the weakness of the diffraction in general. The crystal used for structure solution turned out to be perfectly twinned (twinning fraction 50%) with operator k, h, −l; the apparent space group was P41212 instead of the true space group P41.
Table 1. Data-processing and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Wavelength (Å) | 0.91969 |
| Unit-cell parameters (Å) | a = 98.4, b = 98.4, c = 120.6 |
| Space group | P41 |
| Resolution range (Å) | 20–2.8 (2.95–2.8) |
| No. of observations/unique reflections | 215443/28309 |
| Rsym† (%) | 21.1 (77.2) |
| Rmeas‡ (%) | 22.7 (82.8) |
| 〈I/σ(I)〉 | 11.1 (2.8) |
| Completeness (%) | 99.7 (100) |
| Redundancy | 7.6 (7.7) |
| Rcryst§ (%) | 20.9 |
| Rfree¶ (%) | 25.7 |
| R.m.s.d. bond length†† (Å) | 0.005 |
| R.m.s.d. bond angle†† (°) | 1.2 |
| Protein atoms | 7419 |
| Water atoms | 94 |
| Sulfate ions | 5 |
| B factors (Å2) | |
| Overall | 35 |
| Protein‡‡ | 26, 41, 34, 41 |
| Main chain‡‡ | 25, 40, 34, 41 |
| Side chain‡‡ | 26, 41, 35, 42 |
| Water | 17 |
| Sulfate | 55 |
| Ramachandran plot (%) | |
| Most favoured | 79.8 |
| Additionally allowed | 18.1 |
| Generously allowed | 2.0 |
| Disallowed | 0.1 |
R
sym = 
 .
.
R
meas = 
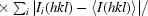 (Diederichs & Karplus, 1997 ▶).
(Diederichs & Karplus, 1997 ▶).
R
cryst = 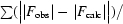
 .
.
R free was calculated like R cryst, using only a test set of 5% of the data that were not used in refinement (Brünger, 1992 ▶).
Deviations from ideal values (Engh & Huber, 1991 ▶)
Values are for chains A, B, C and D, respectively.
The structure was solved by molecular replacement using one monomer from the structure of E. coli CTPS (PDB code 1s1m; Endrizzi et al., 2004 ▶) as a model. Firstly, the data were processed in the apparent space group P41212. Molecular replacement with MOLREP (Vagin & Teplyakov, 1997 ▶) was then carried out to a high-resolution limit of 3 Å. Two monomers were found with an R factor of 51.3% and a correlation coefficient of 40.4%. The R factor fell to 46% during rigid-body refinement in REFMAC (Murshudov et al., 1997 ▶); after restrained refinement in REFMAC, the R and R free values stopped at 29.8 and 39.0%, respectively. At this point, the twinning was identified and the data were rescaled in space group P41. Molecular replacement in MOLREP with the dimeric model refined in space group P41212 was carried out, resulting in a tetramer with an R factor of 46.0% and a correlation coefficient of 56.7%. From then on, the structure was refined using CNS (Brünger et al., 1998 ▶) using the scripts for twinned data. Individual B factors for all atoms were refined and water molecules were added using the default parameters in CNS. Seven sulfate ions per asymmetric unit were clearly identified by their shape in the electron density after refinement and were included when their electron density was unambiguous.
Model building was carried out in COOT (Emsley & Cowtan, 2004 ▶). The refinement statistics are given in Table 1 ▶. Solvent-accessible areas were calculated using MSMS (Sanner et al., 1996 ▶). Superpositions were performed using SSM (Krissinel & Henrick, 2004 ▶). Figures were produced using DINO (Philippsen, 2003 ▶), POVSCRIPT+ (Fenn et al., 2003 ▶), ESPript (Gouet et al., 1999 ▶) and UCSF Chimera (Pettersen et al., 2004 ▶).
3. Results and discussion
3.1. Overall structure
The structure of the synthetase domain from human CTPS was determined to a resolution of 2.8 Å (Fig. 2 ▶ a). The synthetase domains are arranged within the asymmetric unit in a tetramer with 222 symmetry (Fig. 2 ▶ b), as previously observed for the bacterial full-length enzymes (Endrizzi et al., 2004 ▶; Goto et al., 2004 ▶), indicating that the active oligomeric state of the enzyme synthetase domain was crystallized. The structure is a dimer of dimers and three monomers contribute to ligand binding at each active site (Fig. 2 ▶ b). This explains the inactivity of the monomeric and dimeric forms, as well as the regulation of oligomerization by nucleotides (Thomas et al., 1988 ▶; Pappas et al., 1998 ▶). The tight dimers are formed by subunit pairs AB and CD in the asymmetric unit. As expected from sequence homology (Fig. 2 ▶ c), the human CTPS structure is similar to that of bacterial CTPS synthetase domains. A superposition gives r.m.s. deviations for Cα positions of 1.0 Å for the E. coli enzyme (228 aligned residues, 51% identity) and 1.1 Å for the T. thermophilus CTPS (228 aligned residues, 47% identity). Superposing the different monomers of the tetramer also gives r.m.s. deviations for Cα positions of 1.0–1.1 Å.
Figure 2.

The structure of human CTPS synthetase domain. (a) Structure of the monomer. The seven-stranded β-sheet in the middle is covered by helices on both sides. (b) Assembly into a tetramer. Surfaces of the tetramer are shown with the bound sulfates at the active site; four such active sites are present in the tetramer. Monomers are coloured as follows: A, white; B, blue; C, pink; D, orange. The view has been rotated by 90° on the right. The synthetase domains form a tetramer with 222 symmetry; the glutaminase domains would be expected to point out from the tetramer core like knobs. (c) Sequence alignment of human and E. coli CTPS synthetase domains. The secondary structures are from human CTPS and the underlined segments are disordered in human CTPS.
The total change in accessible surface area when going from four monomers to the tetramer can be calculated to be of the order of 7600 Å2 (1900 Å2 per monomer). Of this, approximately 900 Å2 per dimer is buried upon dimer (AB, CD) formation and the remaining 5800 Å2 is buried by the dimerization of the two dimers.
Regions with missing residues that could not be modelled are located on the same face of CTPS, next to the N- and C-termini. These areas (approximately residues 50–80 and 130–140, depending slightly on the subunit) are likely to form flexible loops and it is possible that they affect ligand binding. The region consisting of residues 131–138 is a loop not present in E. coli CTPS (Fig. 2 ▶ c). In the E. coli CTPS structure, the area between residues 50 and 80 of the synthetase domain forms the interface for interacting with the glutaminase domain (Endrizzi et al., 2004 ▶). The N-terminal tag of the recombinant CTPS was also disordered and could not be modelled.
A disulfide bond is present between Cys218 and Cys243 in the structure; this bond is only present in two of the four molecules, monomers A and C, in the asymmetric unit. In monomers B and D the S atoms of these residues are at a distance of 3.9 and 3.5 Å from each other, respectively. These cysteines are not conserved between species and it is uncertain if they play a role in the in vivo function of CTPS.
3.2. The active site
The active site of the CTPS synthetase domain is formed by the interaction of three subunits of the tetramer. A cavity with a diameter of 7 Å and a length of 20 Å extends from the active site towards the centre of the tetramer (Fig. 2 ▶ b) and via this channel all four active sites could be in contact with each other. In the human CTPS structure, two sulfate ions are bound within the active site and they superimpose on the predicted locations of the triphosphate groups of ATP and UTP (Fig. 3 ▶). One of the sulfates (hereafter referred to as SUL-1) is coordinated by the P-loop (residues 12–18) such that it interacts with the backbone N atoms of Gly13, Gly15, Lys16 and Gly17. In this position, binding is also supported by the helix dipole of helix α1 following the P-loop (residues 15–29). In addition, SUL-1 forms a hydrogen bond to the side chain of Lys17. The second sulfate (SUL-2) is found nearby at the interface between two subunits, where it is coordinated by Ser12 from the A subunit and Lys195 and Lys229 from the D monomer. The positions of the sulfates correspond to sites previously shown to bind sulfate in the E. coli and T. thermophilus structures (Endrizzi et al., 2004 ▶; Goto et al., 2004 ▶), further highlighting their functional relevance as markers for the substrate phosphate groups.
Figure 3.

The active site. (a) Detailed interactions of the sulfates with CTPS. Hydrogen bonds are shown by green lines. (b) Superposition of the sulfates with ADP and UTP from E. coli CTPS. Human CTPS is indicated in light grey (A subunit) and orange (D subunit) and E. coli CTPS in dark grey. ADP is in green and CTP in cyan. Note the superposition of the observed sulfates in the human CTPS structure with one phosphate of each nucleotide ligand.
CTP product feedback inhibition is a well characterized mechanism of CTPS regulation (Long & Pardee, 1967 ▶). Resistance to CTP product inhibition is conferred by mutations in the CTPS coding region in Chinese hamster ovary cells (Whelan et al., 1993 ▶). In the yeast enzyme, Glu161 is involved in this inhibitory mechanism and the E161K mutation increases resistance towards the cancer therapeutic drug CPEC (Ostrander et al., 1998 ▶). In the structure of human CTPS, Glu161 is located at the tetramer interface, making a salt bridge to Arg164. Both of these residues reside in helix α4, the N-terminus of which is involved in CTP binding (Endrizzi et al., 2005 ▶).
Interestingly, a structure of E. coli CTPS complexed with CTP and ADP has recently been solved (Endrizzi et al., 2005 ▶). A superposition of the active sites clearly shows that SUL-1 in human CTPS overlaps with the β-phosphate of ADP, while SUL-2 corresponds to the α-phosphate of CTP (Fig. 3 ▶ b). SUL-2 also marks the binding site for the phosphates in the substrate UTP, whose binding site overlaps with that of CTP. This supports a similar mode of interaction of nucleotides in the human enzyme as in the bacterial protein. However, a detailed description of nucleotide binding in the human enzymes must await the production of crystals without sulfate bound in the active site.
4. Concluding remarks
We have determined the first crystal structure of human CTPS, a key enzyme in nucleotide biosynthesis and an important drug target. This is the first structure of a CTPS from eukaryotes. In combination with previous structural studies of bacterial enzymes, our data provide a much clearer view of the structure and properties of human CTPS. This information will be of use in functional studies as well as in inhibitor design.
Supplementary Material
PDB reference: synthetase domain of human CTPS, 2c5m, r2c5msf
Acknowledgments
The Structural Genomics Consortium is a registered charity (No. 1097737) funded by the Wellcome Trust, GlaxoSmithKline, Genome Canada, the Canadian Institutes of Health Research, the Ontario Innovation Trust, the Ontario Research and Development Challenge Fund, the Canadian Foundation for Innovation, VINNOVA, The Knut and Alice Wallenberg Foundation, The Swedish Foundation for Strategic Research and Karolinska Institutet. The work was also supported by the Swedish Cancer Society and the Swedish Research Council (PN). PK is an Academy Research Fellow (Academy of Finland). We thank the ESRF for providing beamtime and Karin Wallden for data collection.
References
- Brünger, A. T. (1992). Nature (London), 355, 472–474. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed] [Google Scholar]
- Dereuddre-Bosquet, N., Roy, B., Routledge, K., Clayette, P., Foucault, G. & Lepoivre, M. (2004). Antiviral Res.61, 67–70. [DOI] [PubMed] [Google Scholar]
- Diederichs, K. & Karplus, P. A. (1997). Nature Struct. Biol.4, 269–275. [DOI] [PubMed] [Google Scholar]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Endrizzi, J. A., Kim, H., Anderson, P. M. & Baldwin, E. P. (2004). Biochemistry, 43, 6447–6463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endrizzi, J. A., Kim, H., Anderson, P. M. & Baldwin, E. P. (2005). Biochemistry, 44, 13491–13499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engh, R. A. & Huber, R. (1991). Acta Cryst. A47, 392–400. [Google Scholar]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed] [Google Scholar]
- Fenn, T. D., Ringe, D. & Petsko, G. A. (2003). J. Appl. Cryst.36, 944–947. [Google Scholar]
- Goto, M., Omi, R., Nakagawa, N., Miyahara, I. & Hirotsu, K. (2004). Structure, 12, 1413–1423. [DOI] [PubMed] [Google Scholar]
- Gouet, P., Courcelle, E., Stuart, D. I. & Metoz, F. (1999). Bioinformatics, 15, 305–308. [DOI] [PubMed] [Google Scholar]
- Han, G. S., Sreenivas, A., Choi, M. G., Chang, Y. F., Martin, S. S., Baldwin, E. P. & Carman, G. M. (2005). J. Biol. Chem.280, 38328–38336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindenburg, A. A., Taub, R. N., Grant, S., Chang, G. & Baker, M. A. (1985). Cancer Res.45, 3048–3052. [PubMed] [Google Scholar]
- Hofer, A., Steverding, D., Chabes, A., Brun, R. & Thelander, L. (2001). Proc. Natl Acad. Sci. USA, 98, 6412–6416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, G. J., Cooney, D. A., Moyer, J. D., Kelley, J. A., Kim, H. Y., Marquez, V. E. & Johns, D. G. (1989). J. Biol. Chem.264, 713–718. [PubMed] [Google Scholar]
- Krissinel, E. & Henrick, K. (2004). Acta Cryst. D60, 2256–2268. [DOI] [PubMed] [Google Scholar]
- Kuilenburg, A. B. van, Meinsma, R., Vreken, P., Waterham, H. R. & van Gennip, A. H. (2000). Biochim. Biophys. Acta, 1492, 548–552. [DOI] [PubMed] [Google Scholar]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Long, C. W. & Pardee, A. B. (1967). J. Biol. Chem.242, 4715–4721. [PubMed] [Google Scholar]
- Lunn, F. A. & Bearne, S. L. (2004). Eur. J. Biochem.271, 4204–4212. [DOI] [PubMed] [Google Scholar]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed] [Google Scholar]
- Ostrander, D. B., O’Brien, D. J., Gorman, J. A. & Carman, G. M. (1998). J. Biol. Chem.273, 18992–19001. [DOI] [PubMed] [Google Scholar]
- Pappas, A., Yang, W. L., Park, T. S. & Carman, G. M. (1998). J. Biol. Chem.273, 15954–15960. [DOI] [PubMed] [Google Scholar]
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C. & Ferrin, T. E. (2004). J. Comput. Chem.25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Philippsen, A. (2003). DINO: Visualizing Structural Biology. http://www.dino3d.org.
- Politi, P. M., Xie, F., Dahut, W., Ford, H. J., Kelley, J. A., Bastian, A., Setser, A., Allegra, C. J., Chen, A. P., Hamilton, J. M., Arbuck, S. F., Linz, P., Brammer, H. & Grem, J. L. (1995). Cancer Chemother. Pharmacol.36, 513–523. [DOI] [PubMed] [Google Scholar]
- Sanner, M. F., Olson, A. J. & Spehner, J. C. (1996). Biopolymers, 38, 305–320. [DOI] [PubMed] [Google Scholar]
- Thomas, P. E., Lamb, B. J. & Chu, E. H. (1988). Biochim. Biophys. Acta, 953, 334–344. [DOI] [PubMed] [Google Scholar]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025. [Google Scholar]
- Verschuur, A. C., Van Gennip, A. H., Leen, R., Meinsma, R., Voute, P. A. & van Kuilenburg, A. B. (2000a). Br. J. Haematol.110, 161–169. [DOI] [PubMed] [Google Scholar]
- Verschuur, A. C., Van Gennip, A. H., Leen, R., Muller, E. J., Elzinga, L., Voute, P. A. & Van Kuilenburg, A. B. (2000b). Eur. J. Cancer, 36, 627–635. [DOI] [PubMed] [Google Scholar]
- Whelan, J., Phear, G., Yamauchi, M. & Meuth, M. (1993). Nature Genet.3, 317–322. [DOI] [PubMed] [Google Scholar]
- Willemoes, M. (2004). Arch. Biochem. Biophys.424, 105–111. [DOI] [PubMed] [Google Scholar]
- Willemoes, M. & Sigurskjold, B. W. (2002). Eur. J. Biochem.269, 4772–4779. [DOI] [PubMed] [Google Scholar]
- Williams, J. C., Kizaki, H., Weber, G. & Morris, H. P. (1978). Nature (London), 271, 71–73. [DOI] [PubMed] [Google Scholar]
- Zhang, H., Cooney, D. A., Zhang, M. H., Ahluwalia, G., Ford, H. J. & Johns, D. G. (1993). Cancer Res.53, 5714–5720. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: synthetase domain of human CTPS, 2c5m, r2c5msf