


Abstract
Estrogen receptors (ERs), which mediate the proliferative action of estrogens in breast cancer cells, are ligand-dependent transcription factors that regulate expression of their primary target genes through several mechanisms. In addition to direct binding to cognate DNA sequences, ERs can be recruited to DNA through other transcription factors (tethering), or affect gene transcription through modulation of signaling cascades by non-genomic mechanisms of action. To better characterize the mechanisms of gene regulation by estrogens, we have identified more than 700 putative primary and about 1300 putative secondary target genes of estradiol in MCF-7 cells through microarray analysis performed in the presence or absence of the translation inhibitor cycloheximide. Although siRNA-mediated inhibition of ERα expression antagonized the effects of estradiol on up- and down-regulated primary target genes, estrogen response elements (EREs) were enriched only in the vicinity of up-regulated genes. Binding sites for several other transcription factors, including proteins known to tether ERα, were enriched in up- and/or down-regulated primary targets. Secondary estrogen targets were particularly enriched in sites for E2F family members, several of which were transcriptionally regulated by estradiol, consistent with a major role of these factors in mediating the effects of estrogens on gene expression and cellular growth.
INTRODUCTION
The pleiotropic effects of estrogens in its numerous target tissues, including the reproductive, skeletal, cardiovascular and central nervous systems (1–5) are mediated in large part via ERs (6), which are members of the superfamily of nuclear receptors and function as hormone-dependent transcription factors (7–9). ERs bind DNA directly through their central, conserved DNA-binding domains composed of two zinc fingers of the C4 type (10,11). Cognate DNA-binding motifs, also called estrogen response elements (EREs), have been characterized in estrogen-responsive promoters (12–15). Consensus EREs derived by compiling natural response elements are 15 bp palindromes of PuGGTCA motifs with a 3 bp spacer and correspond to the highest affinity binding sites for ERs in vitro (16,17). However, natural response elements often deviate from the consensus at one or several positions (14,15). Both estrogen receptors share similar in vitro DNA-binding patterns (18), but their transcriptional activation properties differ (6,19,20), possibly due to differential recruitment of transcriptional coactivator complexes responsible for histone acetylation, chromatin remodeling and enhanced recruitment of the basal transcription machinery (21–27). ERα is thought to mediate the proliferative effects of estrogens in breast cancer cells. Indeed, its expression is preserved or increased in two-thirds of breast tumors, correlating with sensitivity of tumors to antiestrogenic treatment (3–5). On the other hand, the expression of ERβ, which was reported to play a role in terminal differentiation of breast epithelial cells (28), appears to be reduced during tumorigenesis (29,30).
In addition to mediating gene regulation through direct binding to DNA, ERs can regulate gene expression through protein–protein interaction with other transcription factors (tethering). Several transcription factors were shown to mediate positive or negative transcriptional regulation by ERs in the absence of EREs, including AP1, Sp1, NF-κB (15,31–33). In addition, interference between estrogen signaling and other intracellular signaling pathways including the MAPK and PI3K pathways have been widely reported and may result from interactions between ERs and components of these signaling cascades (15,34–38). Finally, it has recently been suggested that estrogens may act also through a membrane receptor member of the GPCR family, GPR30, although the importance of these receptors in breast tumorigenesis remains to be established (39–43). These so-called non-genomic mechanisms of action can lead to rapid kinase-mediated activation of transcription factors and thus modulate gene expression in response to estrogens. Primary gene regulation by estrogen (i.e. genes regulated in the absence of de novo protein synthesis) can therefore result from at least three different mechanisms, including tethering and non-genomic action in addition to classical, ERE-mediated transcriptional regulation.
Better understanding of the mechanisms of action of estrogens in breast tumorigenesis necessitates large-scale identification of estrogen target genes and comprehensive analysis of the mechanisms of target gene regulation to assess the contribution of different regulatory mechanisms and specific targets to the proliferative effects of estrogens. Genome-wide microarray analysis of estradiol (E2) target genes has been performed in ERα-positive breast cancer cell lines such as MCF-7, T47D and ZR75 cells, leading to the identification of a large number of target genes (44–50). It is however not always clear to which degree target identification is affected by cell culture conditions, choice of microarray platform and statistical analysis tools. In addition, few studies have used conditions that distinguish between primary and secondary target genes. This may explain why enrichment in EREs in estrogen target genes identified through microarray analysis was not reported in most studies. On the other hand, the promoter regions of 89 E2 target genes regulated in the presence of CHX in T47D cells were found enriched in EREs (46). However, the number of primary E2 target genes identified in that study remains low and the question of how far from the transcriptional start sites of primary target genes enrichment in EREs can be observed remains open given the relatively narrow window used. High-affinity EREs located distally from the start sites of estrogen target genes are functional ER-binding sites in chromatin immunoprecipitation (ChIP) experiments (50,51). Although the role of ERs bound to distal sites in transcriptional regulation remains to be systematically analyzed, chromatin conformation capture assays revealed that ERα-bound chromatin regions can act at large distances from regulated genes (48,52). In addition to EREs, binding sites for several other transcription factors were enriched in ERα-bound ChIP fragments, suggesting roles for these factors in ERα tethering (48).
In order to better study the mechanisms of target gene regulation by estrogens, we aimed to discriminate between primary and secondary E2 target genes in MCF-7 cells through microarray analysis of gene expression patterns in response to E2 either in the presence or the absence of the protein synthesis inhibitor cycloheximide. We verified the validity of this approach by demonstrating enrichment in EREs in primary, but not secondary up-regulated genes, and identified other transcription factors involved in the regulation of primary and/or secondary estrogen targets. Our results are discussed in the context of previous studies based on large-scale microarray analysis or genome-wide ChIP-on-chip mapping of ERα-associated chromatin regions.
MATERIALS AND METHODS
Cell culture and treatments
MCF-7 breast carcinoma cells were maintained in α-minimal Eagle's medium (α-MEM) (Wisent, St-Bruno, QC, Canada) supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich, Oakville, ON, Canada). Three days before experiments, cells were switched to phenol red-free Dulbecco's modified Eagle's medium (DMEM) (Wisent) containing 10% charcoal-treated FBS, 1% sodium pyruvate (Wisent), 1% penicillin/streptomycin (Wisent) and 1% l-glutamine (Wisent). Cells were seeded in 10 cm plates at a density such that near-confluency is obtained at the end of the treatment. The day before hormonal stimulation, the medium was changed to phenol red-free DMEM supplemented with 0.5% charcoal-treated FBS. Pre-treatments with cycloheximide (10 μg/ml, Sigma) were initiated 1 h before hormonal treatment with 17β-estradiol (E2, 25 nM, Sigma), ICI 182,780 (ICI, 100 nM, Sigma) or vehicle (0.1% ethanol) for variable periods of time as indicated in figure legends. Selected siGenome siRNAs (Dharmacon, Chicago, IL, USA) were transfected using siLentFect lipid reagent according to the instruction of the manufacturer (Bio-Rad Laboratories, Hercules, CA, USA). Medium was changed 24 h after transfection with fresh phenol red-free DMEM supplemented with 0.5% charcoal-treated FBS and hormonal treatments were initiated 48 h after transfection. Cells were harvested 24 h after (i.e. 72 h after siRNA transfection).
Western analysis
Whole cell extracts were prepared as described previously (53) using antibodies directed against ERα (mouse monoclonal B10 antibody, a kind gift from Prof. P. Chambon) or against β-actin mouse monoclonal antibody (AC-15, Sigma Diagnostics).
Growth assays
MCF-7 breast cancer cells were seeded to low density and treated every 2–3 days with vehicle (0, 0.1% ethanol), 17β-estradiol (E2, 25 nM) or ICI 182-720 (ICI, 100 nM) in 5% charcoal-stripped FBS. Protein concentrations were measured after 9 days as previously described (54). When siRNA were used, cells were re-plated 24 h after transfection at low density and treated twice over a 5-day period with vehicle (0, 0.1% ethanol), 17β-estradiol (E2, 25 nM) or ICI 182-720 (ICI, 100 nM) prior to quantification of total protein concentrations. Experiments shown are representative of two to three independent experiments performed with duplicate samples.
Cell cycle analysis
MCF-7 cells were transfected as previously described. After 24 h, medium was changed for phenol red-free DMEM supplemented with 0.5% charcoal-treated FBS and 24 h later, stimulated with either vehicle (0, 0.1% ethanol) or estradiol (E2, 25 nM) for a last 24 h. Cell cycle values were obtained after ethanol fixation and propidium iodide staining followed by data acquisition on a FACS ‘BD LSR II’ and analysis using the ModFit LT cell cycle software. Experiments were performed at least twice with duplicate samples.
RNA purification and DNA microarrays
After treatment of MCF-7 cells with estradiol or vehicle in the presence or absence of cycloheximide, medium was completely removed and cells were collected in 1 ml of TRI-Reagent (Sigma). Total RNA was extracted as recommended by the manufacturer and further purified with RNeasy MinElute Cleanup Kit (QIAgen, Mississauga, ON, Canada). cRNA synthesis from total RNA, labeling and hybridization to Affymetrix HG-U133 2.0 Plus gene chips were performed at the Genome Quebec and McGill University Innovation Center using standard protocols (http://www.genomequebec.mcgill.ca/). A total of 16 chips were hybridized, analyzing quadruplicate samples for each of the four treatment conditions. The microarray analysis was performed with the affylmGUI Bioconductor package (55) after normalization of probe-level data with RMA (56). Genes deemed significantly regulated were those with ≥1.4-fold change between vehicle and E2 treatments, average log2-expression levels greater than 5 across all samples (A-value) and a P-value for moderated t-statistics (57) smaller than 0.01. Regulated genes were ranked according to |fold|*A*(1 – P-value). The 2144 regulated genes were classified into different subsets as follows. UP-regulated: 1214, DOWN-regulated: 955, UP −CHX: 858, UP only −CHX: 670, UP + and −CHX: 188, UP only +CHX: 356, UP +CHX: 544, DOWN −CHX: 838, DOWN only −CHX: 719, DOWN + and −CHX: 119, DOWN only +CHX: 117, DOWN +CHX: 236. The microarray data is accessible through the GEO accession number GSE8597.
Screening for transcription factor binding sites
The reference human genome sequences (hg17, May 2004) were downloaded from the UCSC Genome Browser database (58). Genomic sequences in windows centered around the transcription start sites (TSS) of all annotated gene in the RefSeq Genes track (59) were extracted from the genome. These sequences were screened with 299 matrices for transcription factor binding sites using a base score cutoff of 65% and 5% increments. The program used for screening implements the formulas described by Wasserman and Sandelin (60); other programs developed for genome-wide screen of nuclear receptor response elements (51,61,62) were used to calculate the distance from the TSS and identify binding sites within transposable elements or regions bound in ChIP-on-chip experiments. The majority of matrices used was from the public version of TRANSFAC (63) or represented variation of TRANSFAC matrices (i.e. E2F matrix M00516 where only the portion matching TTTSGCGC was used). Selected matrices were compiled from the literature including matrices for estrogen response elements (51,64,65) and RXR/RAR (66). Searches were also performed with the same sequences from which transposable elements were removed, or with only sequences present in conserved regions, as described in the ‘PhastCons Conserved Elements’ track from the UCSC (67), within the same windows. For each transcription factor, four cutoffs were chosen with frequencies in all gene promoters closest to those of EREs (70%: 0.32 ERE/gene, 75%: 0.16 ERE/gene, 80%: 0.06 ERE/gene, 85%: 0.02 ERE/gene). Cutoffs were required to be between 2 sites/gene and 0.01 site/gene to avoid overly abundant sites or rare sites. Z-scores and P-values were determined for each of the selected cutoffs to assess the statistical significance of the observed enrichment in promoters of different sets of regulated genes versus those of all annotated genes. The Z-scores and P-values were calculated with programs adapted from oPOSSUM perl application programming interface (API) using the cutoff recommended by the authors (68). The P-value indicates if the proportion of genes is greater than would be expected by chance in a Fisher exact test. The Z-score evaluates the significance of the rate of occurrence of sites in the test set of regulated genes versus the expected rate estimated from the background set composed of all genes in the genome using a binomial distribution model. Stars indicate the significance of both parameters (Z-scores >10, P-value <0.01). The ERα ChIP-on-chip dataset (50) was downloaded from the Brown Lab website (http://research.dfci.harvard.edu/brownlab/datasets/index.php) and the E2F1 ChIP-on-chip dataset (69) was downloaded from the NCBI (GSE5175).
Gene expression quantification
Total RNAs were extracted as described earlier and aliquots of 2 μg were reverse transcribed using the RevertAid H first minus strand cDNA synthesis kit (MBI Fermentas, Burlington, ON, Canada) as recommended by the manufacturer. Reverse transcription products were diluted 10 times in pure water prior to real-time quantitative PCR. Gene expression levels were determined using primer and probe sets from the Universal Probe Library (https://www.roche-applied-science.com). PCR reactions were performed in 384-well plates using 2 μl of cDNA sample, 5 μl of TaqMan PCR Master Mix (Applied Biosystems, CA, USA), 2 μM of each primer (sequences available upon request) and 1 μM of the Universal TaqMan probe in a total volume of 10 μl.
The ABI PRISM® 7900HT Sequence Detection System (Applied Biosystems) was used to detect amplification levels and was programmed for an initial step at 95°C for 10 min, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. All reactions were run in duplicates or triplicates and average values were calculated. Quantification was performed with at least two independent experiments. The housekeeping RPLP0 (ribosomal protein, large, P0) gene was used as endogenous control. Relative expression levels of target genes and SD values were determined using the ΔΔCT method.
Chromatin immunoprecipitation
For chromatin immunoprecipitation (ChIP) assays, chromatin was cross-linked by treating cells with 1.5% formaldehyde for 10 min at room temperature and fragmented by sonication as previously reported (51,52), yielding fragments of average size ∼400 bp. Immunoprecipitation were conducted as previously described (51,52). Antibodies against a C-terminal epitope of hERα (SC-543) and against β-actin (SC-8432) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Detailed conditions for immunoprecipitation, PCR amplification and primer sequences (synthesized by Sigma Genosis) used in ChIP assays are available upon request. Chromatin immunoprecipitation experiments were performed twice with similar results. A representative set of results is shown.
RESULTS
Identification of primary target genes of estrogen receptor alpha in MCF-7 cells
MCF-7 cells are one of the most widely used cell models to study estrogen signaling as they are strongly growth-stimulated by 17β-estradiol (E2) in vitro (Figure 1A) and in xenograft experiments (70), and express high levels of estrogen receptor alpha (ERα). Transfection of siRNAs directed against ERα, but not GAPDH or Luciferase (LUC), strongly suppressed MCF-7 cell growth (Figure 1B), indicating that ERa expression levels are rate limiting for growth of these cells. In the absence of E2, MCF-7 cells cultured in 0.5% charcoal-treated serum were mostly arrested in the G0–G1 phase (>80%), while addition of E2 for 24 h augmented significantly the proportion of cells in S phase (Figure 1C). On the other hand, transfection of siRNAs against ERα reduced this proportion both under basal and induced conditions. Thus, it is likely that genes regulated by estrogens under these conditions include a large proportion of cell cycle-regulated genes, some of which may not be primarily regulated by estrogens. Indeed, E2 treatment leads to regulation of transcription factors, cofactors and signaling proteins, which amplifies the genomic effects of estrogens but complicates the analysis of the mechanisms of target gene regulation.
Figure 1.

Role of ERα in mediating the effect of estradiol in MCF-7 breast cancer cells. (A) MCF-7 breast cancer cells were treated every 2 days with vehicle (0, 0.1% ethanol), 17β-estradiol (E2, 25 nM) or ICI 182 720 (ICI, 100 nM) and protein concentrations were measured after 9 days. (B) Proliferation assays of MCF-7 transfected without (0) or with siRNA against ERα, GAPDH or Luciferase (LUC). Treatments were initiated 24 h after transfection and protein concentrations were measured 6 days post-transfection. Inset: western blot analysis of ERα and β-actin expression levels in cells transfected with siRNAs directed against ERα or in control cells (T: transfection control). (C) Cell cycle analysis performed with cells transfected or not with siRNAs for 72 h and stimulated with vehicle or hormone during the last 24 h.
To identify primary estrogen target genes in MCF-7 cells, we performed treatments with E2 for 24 h with or without pre-treatment of cells with the protein synthesis inhibitor cycloheximide (CHX). Gene expression patterns were analyzed using Affymetrix GeneChips HG-U133 2.0 Plus chips representing over 47 000 human transcripts. Differential expression was assessed as the ratio between the average of the four replicates in the presence of estrogen versus its absence, either in the presence of CHX or in its absence. A cutoff of 1.4-fold change in expression was applied, based on the observed regulation of known ER targets such as TFF1 under these conditions. A total of 2144 significantly regulated genes were identified and ranked according to their combined P-value, fold-regulation and intensity of expression (see the lists of the top 100 genes regulated in the absence or presence of CHX in Table 1). There was a significant overlap in regulated genes with other microarray studies, which was optimal when using similar filtering parameters for the analysis of all the datasets [922/2144 with Carroll et al. (50) and 357/2144 with Rae et al. (71); see Figure 2A]. Statistical analysis by a Fisher exact test indicates that all overlaps between two datasets are significant (P-value lower than 2.2E−16 for each pair of microarrays); in addition, much smaller overlaps are found in random simulations performed 1 million times (percentages or number of genes identified by a star in Figure 2A). The large majority (>90%) of genes in the overlapping sets were regulated similarly in pairwise comparisons (see legend of Figure 2A). A total of 203 target genes were identified as estrogen target genes in all three studies. This partial overlap may reflect differences in experimental conditions (e.g. time of treatment, culture medium) or in the type of microarrays (Affymetrix U133A versus U133 Plus 2.0).
Table 1.
Top hundred genes regulated by E2 in the presence or absence of cycloheximide
| Without CHX | With CHX | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| RANK | Symbol | Fold | A | P-value | Validated | RANK | Symbol | Fold | A | P-value | Validated |
| 1 | MYBL1 | 13.85 | 7.23 | 4.07E−10 | yes | 1 | KRT13 | 15.90 | 6.81 | 3.46E−13 | – |
| 2 | FLJ30058 | 14.85 | 5.66 | 5.16E−12 | – | 2 | LOC283551 | 15.04 | 5.81 | 3.26E−13 | – |
| 3 | PKIB | 7.09 | 10.22 | 5.01E−13 | yes | 3 | FLJ10847 | 11.11 | 5.12 | 1.46E−16 | – |
| 4 | SYTL4 | 5.79 | 7.86 | 4.39E−10 | – | 4 | CYP26B1 | 7.46 | 7.05 | 5.91E−12 | yes (DNS) |
| 5 | SGK3 | 5.99 | 6.93 | 2.40E−08 | – | 5 | SGK3 | 6.89 | 6.93 | 1.86E−08 | yes (DNS) |
| 6 | UGT2B15 | 6.19 | 6.45 | 3.14E−09 | – | 6 | FLJ30058 | 7.60 | 5.66 | 4.93E−11 | – |
| 7 | PCP4 | 5.03 | 7.28 | 1.14E−11 | – | 7 | SLC26A2 | 5.40 | 7.85 | 1.22E−11 | yes |
| 8 | TMPRSS3 | 5.42 | 6.72 | 9.36E−12 | – | 8 | MYBL1 | 5.27 | 7.23 | 3.45E−07 | yes |
| 9 | MCM10 | 4.96 | 6.97 | 8.87E−11 | yes | 9 | RET | 4.68 | 8.12 | 5.30E−12 | yes |
| 10 | RRM2 | 3.44 | 10.03 | 9.07E−12 | – | 10 | C8orf46 | 6.10 | 5.85 | 5.78E−11 | – |
| 11 | MLF1IP | 3.89 | 8.76 | 1.56E−11 | – | 11 | RBP7 | 4.53 | 7.75 | 5.91E−12 | yes |
| 12 | ATAD2 | 3.47 | 9.45 | 6.83E−10 | – | 12 | RBM24 | 4.95 | 7.02 | 7.32E−12 | – |
| 13 | DCC1 | 4.78 | 6.64 | 1.34E−10 | – | 13 | TMEM64 | 3.26 | 10.13 | 3.50E−10 | – |
| 14 | UHRF1 | 3.49 | 9.04 | 2.63E−11 | – | 14 | CA12 | 3.43 | 9.42 | 5.66E−13 | yes |
| 15 | TK1 | 3.58 | 8.80 | 4.97E−11 | – | 15 | XBP1 | 3.13 | 10.27 | 4.50E−11 | yes |
| 16 | IL17RB | 4.24 | 7.38 | 1.87E−11 | – | 16 | KCNK6 | 4.33 | 7.32 | 5.30E−12 | – |
| 17 | GREB1 | 3.14 | 9.85 | 5.09E−11 | yes | 17 | DEPDC6 | 3.75 | 8.35 | 8.37E−12 | – |
| 18 | PBK | 3.93 | 7.85 | 8.91E−10 | – | 18 | IL17RB | 4.08 | 7.33 | 2.12E−11 | – |
| 19 | BRIP1 | 3.08 | 9.84 | 2.39E−07 | – | 19 | OACT1 | 4.68 | 6.32 | 8.37E−12 | – |
| 20 | TYMS | 3.28 | 9.18 | 5.91E−11 | – | 20 | CDH26 | 4.96 | 5.82 | 1.26E−07 | yes |
| 21 | DTL | 4.49 | 6.66 | 4.39E−10 | – | 21 | CALCR | 2.82 | 9.97 | 3.18E−11 | yes |
| 22 | EXO1 | 4.54 | 6.58 | 9.07E−12 | – | 22 | CELSR2 | 3.22 | 8.66 | 1.41E−12 | yes (DNS) |
| 23 | LOC221981 | −2.89 | 10.32 | 3.88E−10 | – | 23 | KRT4 | −3.91 | 7.01 | 1.53E−08 | yes |
| 24 | ANLN | 3.34 | 8.90 | 8.05E−09 | – | 24 | PAPSS2 | 2.60 | 10.44 | 1.03E−10 | – |
| 25 | CCNE2 | 3.69 | 7.89 | 1.95E−09 | – | 25 | DSCAM | 4.12 | 6.57 | 1.05E−10 | yes |
| 26 | CDC2 | 3.10 | 9.34 | 1.98E−10 | – | 26 | CARD10 | 4.04 | 6.59 | 1.59E−11 | yes (DNS) |
| 27 | AREG | 3.32 | 8.69 | 2.35E−11 | yes | 27 | TPD52L1 | 2.55 | 10.14 | 5.69E−11 | – |
| 28 | RLN2 | 3.38 | 8.36 | 1.11E−09 | – | 28 | CXCL12 | 3.62 | 6.99 | 8.64E−10 | – |
| 29 | MGP | 3.79 | 7.37 | 4.63E−11 | – | 29 | LRIG1 | 3.82 | 6.54 | 5.91E−12 | yes |
| 30 | CXCL12 | 3.97 | 6.99 | 2.28E−10 | – | 30 | RLN2 | 2.97 | 8.36 | 9.91E−09 | – |
| 31 | CHEK1 | 4.18 | 6.61 | 7.62E−09 | Yes (DNS) | 31 | ANKH | 3.77 | 6.58 | 3.18E−11 | – |
| 32 | TOP2A | 2.97 | 9.24 | 1.60E−09 | – | 32 | C14orf139 | 4.71 | 5.08 | 5.30E−12 | – |
| 33 | KIAA0101 | 2.63 | 10.41 | 1.38E−10 | – | 33 | C22orf19 | 2.90 | 8.23 | 2.37E−11 | – |
| 34 | RERG | 3.33 | 8.15 | 9.07E−12 | – | 34 | RP13-360B22.2 | 2.88 | 8.21 | 4.39E−11 | – |
| 35 | CYP26B1 | 3.85 | 7.05 | 6.70E−10 | yes | 35 | PXK | 3.59 | 6.53 | 4.33E−09 | – |
| 36 | FSHPRH1 | 4.41 | 6.15 | 1.30E−10 | – | 36 | TUBA3 | 2.18 | 10.62 | 3.25E−09 | – |
| 37 | Pfs2 | 3.31 | 8.19 | 1.24E−10 | – | 37 | MYB | 2.62 | 8.66 | 5.91E−12 | yes |
| 38 | BRCA1 | 4.15 | 6.49 | 2.17E−10 | yes | 38 | C2orf23 | 3.04 | 7.43 | 5.91E−12 | – |
| 39 | CDKN3 | 2.90 | 9.28 | 1.33E−09 | – | 39 | UGT2B15 | 3.47 | 6.45 | 1.20E−06 | – |
| 40 | SFXN2 | 3.15 | 8.51 | 1.87E−11 | – | 40 | STC2 | 2.38 | 9.31 | 2.50E−11 | – |
| 41 | FLJ10719 | 3.69 | 7.25 | 1.60E−09 | – | 41 | P4HA2 | 2.62 | 8.29 | 5.59E−11 | – |
| 42 | CDC45L | 3.82 | 6.86 | 1.14E−11 | yes | 42 | FLJ33718 | 2.73 | 7.92 | 1.03E−10 | – |
| 43 | PCNA | 2.61 | 10.01 | 2.54E−10 | – | 43 | SMOX | 3.20 | 6.72 | 2.81E−11 | – |
| 44 | CDH2 | −2.68 | 9.71 | 2.48E−11 | – | 44 | GPSM1 | 2.62 | 8.13 | 8.27E−11 | – |
| 45 | HEY2 | 3.46 | 7.51 | 2.48E−11 | – | 45 | GREB1 | 2.15 | 9.85 | 1.62E−08 | yes |
| 46 | C10orf3 | 3.47 | 7.47 | 4.06E−08 | – | 46 | ITGA2 | 3.52 | 5.99 | 9.34E−10 | – |
| 47 | G30 | −2.74 | 9.40 | 1.21E−11 | – | 47 | FKBP4 | 1.90 | 10.90 | 2.01E−09 | – |
| 48 | GMNN | 2.99 | 8.60 | 4.97E−11 | – | 48 | PKIB | 2.00 | 10.22 | 1.22E−07 | yes |
| 49 | DEPDC6 | 3.05 | 8.35 | 6.81E−11 | – | 49 | RAB27B | −2.33 | 8.70 | 1.78E−08 | yes |
| 50 | CDC6 | 4.21 | 6.05 | 3.51E−09 | – | 50 | KIAA1324 | 2.10 | 9.64 | 1.79E−10 | – |
| 51 | KLK11 | 2.74 | 9.28 | 2.48E−11 | – | 51 | PCP4 | 2.78 | 7.28 | 4.14E−09 | – |
| 52 | FKSG14 | 4.34 | 5.82 | 1.10E−08 | – | 52 | UGCG | 2.64 | 7.56 | 2.44E−08 | – |
| 53 | FHL1 | 2.93 | 8.63 | 4.39E−10 | – | 53 | RHOD | 2.03 | 9.83 | 1.11E−09 | – |
| 54 | MAD2L1 | 3.14 | 8.04 | 6.55E−09 | – | 54 | ANXA9 | 2.00 | 9.92 | 2.47E−09 | – |
| 55 | PRIM1 | 3.22 | 7.84 | 4.63E−11 | – | 55 | WFS1 | 2.43 | 8.16 | 7.02E−10 | – |
| 56 | GLA | 2.78 | 9.06 | 1.14E−11 | – | 56 | SLC29A1 | 3.22 | 6.14 | 2.05E−10 | – |
| 57 | CDT1 | 2.98 | 8.42 | 1.49E−11 | – | 57 | SOX9 | 2.47 | 8.02 | 1.70E−09 | – |
| 58 | MYBL2 | 3.15 | 7.89 | 9.57E−11 | yes | 58 | PLEKHH1 | 2.88 | 6.87 | 8.27E−11 | – |
| 59 | BCMP11 | 3.27 | 7.57 | 2.37E−09 | – | 59 | SH2BP1 | 2.33 | 8.45 | 9.91E−09 | yes |
| 60 | HCAP-G | 3.54 | 6.99 | 8.72E−10 | – | 60 | WISP2 | 2.04 | 9.65 | 8.39E−10 | yes |
| 61 | RAD51AP1 | 3.97 | 6.20 | 3.32E−08 | – | 61 | SLC9A3R1 | 1.76 | 11.14 | 7.70E−09 | – |
| 62 | CDH26 | 4.20 | 5.82 | 2.32E−07 | yes | 62 | RERG | 2.41 | 8.15 | 2.90E−10 | Yes (DNS) |
| 63 | ZNF367 | 3.30 | 7.33 | 1.38E−10 | – | 63 | IGFBP4 | 1.97 | 9.88 | 7.97E−09 | yes |
| 64 | CDCA5 | 3.06 | 7.91 | 4.85E−11 | – | 64 | CD33L3 | 2.82 | 6.89 | 4.17E−10 | – |
| 65 | ASF1B | 2.97 | 8.10 | 1.38E−11 | – | 65 | RNF150 | 3.57 | 5.41 | 3.97E−10 | – |
| 66 | UBE2C | 2.38 | 10.08 | 4.02E−10 | – | 66 | TGM2 | 2.97 | 6.48 | 1.85E−08 | – |
| 67 | UBE2T | 2.63 | 9.11 | 2.48E−11 | – | 67 | SOX2 | −2.21 | 8.69 | 1.83E−08 | – |
| 68 | KNTC2 | 3.32 | 7.16 | 2.71E−07 | – | 68 | SCARB1 | 2.27 | 8.44 | 1.86E−10 | – |
| 69 | MELK | 3.07 | 7.70 | 1.19E−10 | – | 69 | ABHD2 | 2.44 | 7.85 | 1.10E−09 | – |
| 70 | SPBC25 | 4.36 | 5.42 | 3.51E−09 | – | 70 | BLVRB | 1.97 | 9.71 | 8.39E−10 | – |
| 71 | LOC440687 | 2.90 | 8.12 | 3.59E−09 | – | 71 | IDH1 | −2.00 | 9.54 | 2.09E−10 | yes (DNS) |
| 72 | E2F8 | 3.33 | 7.09 | 9.46E−10 | yes | 72 | SYNE2 | 2.31 | 8.25 | 9.16E−09 | – |
| 73 | KIAA1212 | −2.34 | 10.05 | 3.07E−11 | – | 73 | SYTL5 | 1.98 | 9.55 | 3.26E−09 | yes |
| 74 | RAB31 | 2.77 | 8.48 | 3.01E−11 | – | 74 | COL18A1 | 2.34 | 8.02 | 2.34E−10 | – |
| 75 | FANCD2 | 3.41 | 6.87 | 1.02E−09 | – | 75 | ISG20 | 2.15 | 8.65 | 2.53E−08 | – |
| 76 | KIF11 | 3.32 | 7.04 | 1.33E−07 | – | 76 | SYTL4 | 2.36 | 7.86 | 1.06E−05 | – |
| 77 | BRCA2 | 3.90 | 5.99 | 1.48E−09 | yes | 77 | SEPT9 | 1.72 | 10.71 | 1.81E−08 | – |
| 78 | MCM2 | 2.64 | 8.84 | 7.18E−10 | yes | 78 | CCND1 | 1.83 | 9.95 | 2.55E−07 | yes |
| 79 | HELLS | 3.49 | 6.67 | 7.95E−11 | – | 79 | SLC35E2 | 1.87 | 9.75 | 5.74E−09 | – |
| 80 | MCM6 | 2.37 | 9.79 | 1.10E−09 | yes | 80 | SLC25A24 | 1.72 | 10.58 | 2.16E−07 | – |
| 81 | TTK | 3.19 | 7.26 | 8.41E−09 | – | 81 | MYO1B | −1.69 | 10.73 | 6.67E−08 | yes (DNS) |
| 82 | E2F7 | 3.94 | 5.88 | 6.39E−09 | yes | 82 | FER1L3 | 1.63 | 11.04 | 9.86E−08 | – |
| 83 | PRC1 | 2.42 | 9.55 | 4.49E−10 | – | 83 | ADAMTS19 | −2.08 | 8.64 | 2.51E−10 | – |
| 84 | RET | 2.84 | 8.12 | 4.49E−10 | yes | 84 | ARHGAP26 | 2.84 | 6.29 | 8.64E−10 | – |
| 85 | PSF1 | 2.77 | 8.31 | 2.48E−11 | – | 85 | KLF4 | 1.61 | 10.94 | 1.69E−07 | – |
| 86 | XBP1 | 2.24 | 10.27 | 2.59E−09 | yes | 86 | TPBG | 1.55 | 11.39 | 4.87E−07 | – |
| 87 | PLAC1 | 4.21 | 5.42 | 2.06E−11 | – | 87 | HIPK2 | 2.13 | 8.25 | 2.16E−09 | – |
| 88 | LOC441168 | −2.11 | 10.79 | 2.54E−10 | – | 88 | NPY1R | 2.01 | 8.71 | 2.56E−08 | – |
| 89 | CDCA1 | 3.51 | 6.46 | 1.78E−07 | – | 89 | EFEMP1 | −1.92 | 9.09 | 1.68E−08 | – |
| 90 | HIST1H4C | 2.29 | 9.88 | 6.50E−09 | – | 90 | C6orf141 | 1.81 | 9.67 | 1.85E−08 | – |
| 91 | RAD51 | 3.78 | 6.00 | 2.06E−11 | – | 91 | TST | 1.80 | 9.72 | 1.25E−08 | – |
| 92 | KIF23 | 2.89 | 7.84 | 2.78E−07 | – | 92 | MCM6 | 1.77 | 9.79 | 6.11E−07 | yes |
| 93 | PLK4 | 3.32 | 6.79 | 9.88E−09 | yes (DNS) | 93 | GLA | 1.90 | 9.06 | 4.33E−09 | – |
| 94 | FEN1 | 2.63 | 8.56 | 1.04E−10 | yes | 94 | THRAP4 | 2.51 | 6.84 | 6.29E−09 | – |
| 95 | EGR3 | 3.95 | 5.70 | 1.92E−09 | yes (DNS) | 95 | RBBP8 | 2.06 | 8.34 | 4.62E−07 | yes (DNS) |
| 96 | DUT | 2.36 | 9.52 | 4.49E−10 | – | 96 | FLJ20366 | 2.61 | 6.57 | 6.62E−09 | – |
| 97 | OFCC1 | −2.02 | 11.10 | 5.46E−10 | – | 97 | LAPTM4A | 1.47 | 11.64 | 7.83E−07 | – |
| 98 | POLE2 | 3.32 | 6.74 | 3.00E−10 | yes | 98 | SFXN2 | 2.01 | 8.51 | 1.39E−08 | – |
| 99 | RNASEH2A | 2.60 | 8.61 | 4.82E−10 | – | 99 | PGR | 3.21 | 5.34 | 1.48E−07 | yes |
| 100 | DHFR | 2.86 | 7.78 | 3.23E−09 | – | 100 | BRP44L | 2.10 | 8.14 | 5.41E−09 | – |
Genes were ranked based on a combination of three parameters, fold-change between E2 and vehicle treatments, amplitude of array signals across all conditions and P-value based on moderated t-statistics. Genes in bold are those found significantly regulated both in the absence and presence of cycloheximide (CHX). Validated genes are those for which regulation was observed in Q-PCR with at least two independent experiments (see also Figures 3–5 and Supplementary Figure 2; DNS, data not shown; –, not tested).
Figure 2.

Cycloheximide-sensitive and resistant E2 target genes in MCF-7 breast cancer cells. (A) Comparative analysis of regulated genes in different large-scale microarray studies. The number of genes in common between this study and those described in Carroll et al. (50) and Rae et al. (71) or unique to each study is indicated. Regulation of genes common to two different studies is convergent (up- or down-regulation) in 90% of the genes for the overlap between the datasets in Refs (50) and (71), 96% for the overlap between this study and Ref. (71), and 92% for the overlap between this study and Ref. (50). Average overlaps obtained in random simulations performed 1 million times are identified by a star and are expressed as percentages of total genes for pairwise comparisons or as the average number of genes common to all studies. (B) Identification of primary and secondary estrogen target genes in MCF-7 cells. RNA samples were collected 24 h after treatment of MCF-7 cells with vehicle or 17β-estradiol (25 nM). Cells were pre-treated 1 h before E2 stimulation with cycloheximide (CHX, 10 μg/ml). Microarray analysis was performed with four biological replicates for each condition (see Material and Methods section). Numbers of genes significantly regulated (≥1.4-fold change between vehicle and E2 treatments, ≥5 amplitude of array signals across all conditions and ≤0.01 P-value based on moderated t-statistics) are indicated for each category.
Regulated genes included nearly equivalent numbers of up- and down-regulated genes in the absence of CHX (858 up-regulated and 838 down-regulated genes, Figure 2B), but more than twice as many up- versus down-regulated genes in the presence of CHX (544 up-regulated and 236 down-regulated genes, Figure 2B). Only 25 genes had opposite regulation in the absence or presence of CHX. The proportion of genes regulated in the absence (78%) versus the presence (35%) of CHX in this study was similar when considering only genes that were not identified as E2 targets in the two previous large-scale studies in MCF-7 cells (50,71). This indicates that treatment with the protein synthesis inhibitor is unlikely to account for the partial overlap with these studies. Moreover, although it is possible that CHX treatment may mask regulation of some target genes, CHX treatment did not have a pronounced global effect on expression of the group of genes induced only in the absence of CHX (Supplementary Figure 1). Regulation of target gene expression by E2 in the absence or presence of CHX was confirmed using Q-PCR for 41 putative primary and 22 putative secondary target genes (Supplementary Figure 2).
Surprisingly, the overlap between genes regulated in the presence and absence of CHX was only partial (35% for up-, 50% for down-regulated genes). Regulation of gene expression only in the presence of CHX may be due to several factors, including regulation below the cutoff and/or lack of detection of transcripts in the −CHX category, or suppression of negative feedback mechanisms on ER-mediated gene expression by CHX treatment. Several well-known estrogen target genes were represented in the set regulated only in the presence of CHX, including CCND1, IGFBP4, C3 and MYC. Regulation by E2 of several genes in this category could be detected at earlier times (peak around 8 h after E2 stimulation, Figure 3B), and was also observed in the presence of CHX at 8 h and at earlier time points (data not shown), confirming their status as primary target genes. On the other hand, CHX treatment had an attenuating effect on regulation of other genes such as the immediate early FOS gene (Figure 4A), although regulation of the FOS gene was still clearly observed in the presence of CHX treatment at 1–2 h (data not shown), as previously reported (72).
Figure 3.

Expression of several genes induced only in the presence of CHX at 24 h is regulated in its absence at earlier times. Q-PCR analysis of the effects of E2 (25 nM) with or without CHX (10 μg/ml) as a function of time on the expression levels of selected genes identified through microarray analysis. Data is representative of at least two independent experiments and is presented as the ratio between +E2 and −E2 at all time points. (A) Pattern of expression of a gene stably induced by estradiol. (B) Patterns of expression of several genes induced only in the presence of cycloheximide at 24 h in the microarray analysis.
Figure 4.
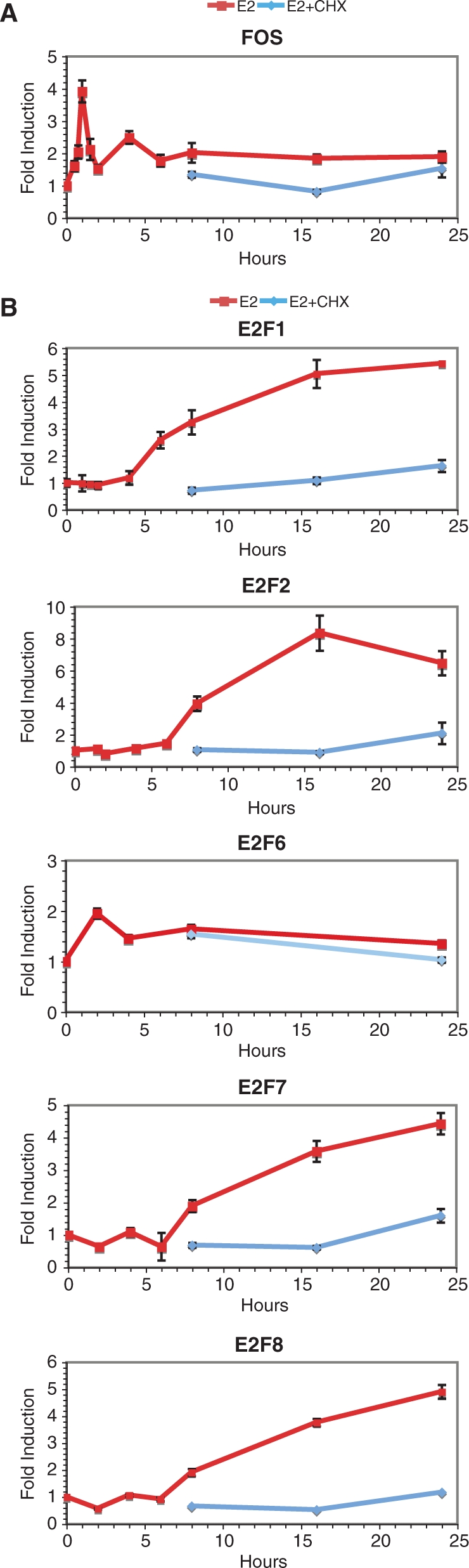
Several transcription factors in the E2F family are target genes of estradiol in MCF-7 breast cancer cells. Q-PCR analysis of the effects of E2 (25 nM) with or without CHX (10 μg/ml) as a function of time on FOS (A) and on E2F transcription factors whose expression was found to be regulated by E2 in the microarray analysis (B). This data is representative of at least two independent experiments and is presented as the ratio between +E2 and −E2 at all time points.
Representation of genes involved in cell cycle control, including cyclins and other cell cycle regulators, is strikingly different in genes regulated in the absence or presence of CHX (data not shown). This correlates with the fact that CHX treatment led to loss of progression into S phase (Supplementary Figure 3). The group of genes whose induction by E2 was repressed by CHX includes genes associated with the proliferation signature of breast cancer cells, such as members of the E2F family of transcription factors (Figure 4B), of the cell division cycle (CDC) and minichromosome maintenance (MCM) gene families (Supplementary Figure 2). Their regulation reflects the proliferative effects of estrogens and the need for synthesis of cell cycle regulatory proteins that are controlled between cycles by degradation (73).
ERα participates in both up- and down-regulation of gene expression, but EREs are enriched only in the flanking sequences of up-regulated primary target genes
To investigate whether ERα plays a similar role in the up- and down-regulation of primary target genes, we selected a sample of five primary up- and five down-regulated target genes and assessed the effect of siRNA-mediated ERα depletion (Figure 5). Two different siRNAs directed against ERα both markedly reduced levels of expression of the receptor and levels of cyclin D1 (CCND1) mRNAs (Figure 5A and B), consistent with their anti-proliferative effects (Figure 1B and C). Modulation of both up- (Figure 5C) or down-regulated (Figure 5D) primary estrogen target genes was markedly attenuated by depletion of ERα. Transfection of siRNAs directed against GAPDH or LUC on the other hand had generally little impact on expression of ER target genes in the absence or presence of estradiol, although we observed a reproducible effect of the GAPDH siRNA on lowering TFF1 expression and of the LUC siRNA on increasing expression of ERBB2, possibly due to effects of these siRNAs on unknown factors regulating expression of these genes. Together these results indicate that expression levels of the receptor are a limiting factor for overall target gene regulation in MCF-7 cells. These results also suggest that overexpression of ERα during tumorigenesis likely plays an important role in potentiating estrogen signaling.
Figure 5.

Regulation by estradiol of both up- and down-regulated primary target genes is dependent on ERα. Two siRNAs against ERα and control siRNAs against GAPDH or Luciferase (LUC) were transiently transfected in MCF-7 cells 48 h before stimulation with E2 (25 nM) or vehicle (0.1% ethanol) for 24 h, and levels of expression of ERα (A), CCND1 (B), up-regulated (C) or down-regulated (D) primary target genes were evaluated by using Q-PCR of reverse-transcribed mRNAs.
The classical mechanism of action of ERα signaling involves direct binding to cognate DNA sequences, the EREs, whose consensus sequence is a palindrome of PuGGTCA motifs (13,16,17). Surprisingly, there was no difference in the percentage of EREs identified between groups of up-, down- or non-regulated genes in ZR75 cells (47). On the other hand, enrichment in EREs was reported in the promoter sequences of target genes identified in the presence of cycloheximide in T47D cells (46); however, the number of primary target genes identified was small (89 genes) and enrichment was assessed only in a 3.5 kb window of regulatory sequences. Here, we compared ERE frequencies in genes regulated by estradiol in the presence and/or absence of the protein synthesis inhibitor cycloheximide (CHX) in MCF-7 cells. EREs were identified at variable distances from the transcriptional start sites of genes using a customized position weight matrix, derived by compiling binding motifs from the literature and by symmetrical complementation reflecting the palindromic nature of this site (Figure 6A; note that this matrix yielded higher enrichment in EREs than the TRANSFAC matrix in control datasets corresponding to lists of genes with known EREs or ERα-bound ChIP fragments, data not shown). Windows of 20 kb centered on the transcriptional start sites (TSS) of target genes were used with different cutoffs corresponding to frequencies of sites in the genome comprised between 0.1 and 2 sites per gene. This approach detected a significant enrichment in EREs in a test list of genes found to contain EREs in a genome-wide search for high-affinity EREs (51) compared to the whole genome (data not shown). The ratio between the numbers of sites detected in regulated genes versus all genes in the genome remained close to one at all cutoffs when both up- and down-regulated genes were considered together, but increased with higher cutoffs in up-regulated genes while it decreased in down-regulated genes (Figure 6B). However statistical significance (P-value <0.01 and Z-score >10) was reached only at one point in total up-regulated genes in this window. On the other hand, ERE enrichments in E2 target genes up-regulated in the presence of CHX (+CHX group), but not in its absence (−CHX group), were statistically significant. Further subdivision of the +CHX group into only+CHX and −and+CHX indicated statistically significant enrichment in both the only+CHX group and the −and+CHX groups (Figure 6B). No enrichment was seen in the only–CHX group, consistent with the interpretation that most of the genes in this group are secondary target genes. Similarly, while we did not find cutoff-dependent enrichment in EREs in the datasets of up-regulated genes regulated in Carroll et al. (50) or in Rae et al. (71), enrichment in EREs can be observed in any intersection between our set of primary up-regulated genes and these other sets of regulated genes (Supplementary Figure 4). In addition, our analysis of another published dataset of genes regulated in MCF-7 cells by E2 and CHX at 48 h (74) also indicates enrichment in EREs in up-regulated, but not down-regulated primary targets (Supplementary Figure 5). Thus, the observation that EREs are enriched in primary up-regulated target genes is not dataset dependent. Of note, the observation that enrichment was higher with higher cutoffs indicates that sequences closer to the consensus are more likely to mediate transcriptional regulation, indicating a statistical relationship between in vitro affinity and in vivo binding to EREs.
Figure 6.

Estrogen response elements are enriched in the vicinity of primary up-regulated genes. (A) Position weight matrix used for identification of putative EREs in the vicinity of target genes. The frequency of nucleotides at each position of the ERE in the matrix is shown. (B) The ratios of the number of EREs per gene in 20 kb windows (±10 kb) around the transcriptional start sites of regulated genes to the number of EREs per gene in the same windows of all annotated genes in the human genome are shown for various groups of estrogen target genes. (C) The ratios of EREs found in ERα-bound chromatin regions (Ref. 50) located within a 20 kb window of the regulated genes identified in our study over EREs found in ERα-bound chromatin regions at ±10 kb of all genes in the genome are shown for various groups of estrogen target genes. ChIP regions were standardized to 1 kb on either side of the center of the region for this analysis. The numbers of genes in each category analyzed in B and C are indicated in Materials and Methods section.
Distribution of EREs in sliding 2.5 kb windows with 500 bp increments between −25 and +25 kb of the TSS indicates that enrichment of EREs (identified at a 75% cutoff) in genes up-regulated in the presence of CHX, but not in those up-regulated in its absence, can be observed within −20 to +20 kb of the TSS (Figure 7A). Finally, enrichment in EREs was higher when considering only ERα-associated chromatin regions (50) found within a 20 kb window around the TSS of different groups of genes, reflecting the capacity of ChIP to accurately pinpoint the regulatory regions in target gene flanking sequences (Figure 6C). However, no enrichment was observed in the only–CHX group of up-regulated genes even when considering only ERα-bound fragments (Figure 6C). Together, these results validate the use of CHX to identify primary E2 target genes.
Figure 7.

Distribution of EREs in a 50 kb window around the TSS of estrogen target genes. (A) Number of EREs per gene identified at a 75% cutoff rate using the matrix described in Figure 6A in a 2.5 kb sliding window within 50 kb of genomic regions centered around the transcriptional start sites (TSS) of primary (+CHX) or total (−CHX) up-regulated genes. (B) Number of EREs per gene identified at a 75% cutoff rate in a 2.5 kb sliding window within 50 kb genomic regions centered around the transcriptional start sites (TSS) of primary (+CHX) or total (−CHX) down-regulated genes. Note that all described alternative TSS were considered in this analysis.
Surprisingly, no enrichment but rather a statistically significant depletion in EREs was observed in a 20 kb region around the TSS of genes repressed by estrogens (Figures 6B and 7B). We examined whether enrichment in EREs was observed in ERα-associated chromatin regions (50) found near genes down-regulated in the absence or presence of CHX (Figure 6C). No enrichment in ERE sequences was observed in the genes down-regulated either in the presence or the absence of CHX. This suggests that binding sites for other transcription factors mediate the down-regulation of genes by E2 in the presence of CHX (see also the next section).
To test whether predicted EREs can bind ERα in vivo, selected regions containing EREs identified using a 65% cutoff and present at ±25 kb of the TSS of target genes up-regulated in the presence of CHX were amplified by PCR after ChIP. Binding of ERα was observed on all predicted elements with the exception of the ERE with two mismatches present at +2.3 kb of the EGR3 TSS (Figure 8). Only two of the nine validated elements (the WISP2 −452 bp ERE and CA12 −6 kb ERE) were detected previously in genome-scale ChIP experiments (50). The intensity of binding to individual elements did not necessarily reflect their degree of similarity to the consensus sequence, as one of the strongest sites bound [at −452 bp of the WISP2 TSS, also observed in Ref. (50)], diverges from the consensus ERE by two mismatches. This may be due to the sequence context of the EREs, which is likely to affect in vivo affinity through cooperativity with other transcription factors bound close to the EREs. In addition, the occurrence of several EREs in the flanking regions of primary target genes suggests potential cooperativity between EREs for DNA binding and transcriptional activation through formation of chromatin loops as described for the GREB1 gene (52).
Figure 8.

Identification of functional EREs in the flanking regions of primary target genes. Chromatin immunoprecipitation was performed using primer pairs specific for selected EREs identified through bioinformatic screens of the flanking regions of primary E2 target genes. The ChIP assay was performed after 45 min stimulation by vehicle (0, 0.1% ethanol), 17β-estradiol (E2, 25 nM) or ICI 182 720 (ICI, 100 nM) using a polyclonal antibody against ERα or a negative control antibody against β-actin. NTC: non-template control for PCR amplification. The number of mismatches (m) with the consensus ERE is indicated for each element along with distances (bp) to the most upstream TSS. These results are representative of two independent experiments.
Binding sites for other transcription factors are enriched in up- and down-regulated primary estrogen target genes
The lack of enrichment of EREs in down-regulated target genes suggests transcriptional regulation via other mechanisms than ERa binding to EREs. Several transcription factors have been reported to mediate indirect recruitment of ERα to DNA via protein–protein interactions [see refs (15,31–33) for reviews]. In addition, other transcription factors may also mediate estrogenic regulation via non-genomic mechanisms (15,34–38). Binding sites for transcription factors participating in either mechanism may be enriched in the flanking sequences of primary regulated genes, while only enrichment in tethering transcription factors is expected in ERα-associated regions identified through ChIP.
To identify transcription factor binding sites enriched in either up- or down-regulated primary targets, we screened TRANSFAC matrices for >1.5-fold enrichment in frequencies within 5 or 10 kb windows around the TSS of different classes of regulated genes versus all annotated genes. Due to the variable representation of different transcription factor binding sites in low-complexity regions, this analysis was performed considering either whole genomic sequences, sequences without repeated elements or sequences conserved through evolution. Enrichments with Z-scores higher than 10 and P-values <0.01 for at least two different cut-offs of the same matrix were retained as significant. This analysis was performed using either all significantly regulated genes (Supplementary Table 1) or the top 100 regulated genes (Supplementary Table 2) in each category.
Binding sites enriched in primary up-regulated target genes (regulated in the presence of CHX) included, in addition to EREs, sites for AHR/ARNT, AP1, CCAAT box factors, C/EBP homologous protein (CHOP), CHX10 homeobox factor, EGR factors, Maf, GCbox factors and ERE-like sequences with different arrangements of the motifs (Supplementary Tables 1 and 2). On the other hand, binding sites enriched in primary down-regulated genes include sites for the forkhead transcription factor FREAC3 (FOXC1), CHX10 and PAX homeobox factors, EVI1, Ikaros and SRY transcription factors (Supplementary Tables 1 and 2).
E2F transcription factors are a major relay for transcriptional regulation by estrogens of secondary target genes in MCF-7 cells
No enrichment of EREs was observed in genes regulated only in the absence of CHX, consistent with their regulation by other transcription factors. Screening of TRANSFAC matrices indicated enrichment of E2F-binding sites in up-regulated secondary targets, as well as of sites for CCAAT box factors, CREB and Forkhead (HNF-3B) transcription factors (Supplementary Tables 1 and 2). In addition, sites for AP1, CREB, C/EBP, CCAAT-box factors, forkhead factors (FOXA, FOXD3, FOXO4, HFH8), E2F, GATA factors, GC-box factors, MAZR and PAX factors were enriched in down-regulated secondary target genes (Supplementary Tables 1 and 2).
Because secondary target genes are enriched in cell cycle-associated proteins and E2F factors play well-characterized roles in the regulation of cell proliferation, we examined more closely the possible contribution of these factors in the networks of gene regulation by estrogens. Enrichment in E2F sites within a 10 kb window around transcriptional start sites (±5 kb) was strong in genes up-regulated in the absence of CHX (Figure 9A). It was also observed in genes up-regulated in the presence of CHX, but not in the only+CHX group, suggesting that the −and+CHX group of regulated genes was responsible for this over-representation. A similar enrichment in secondary, but not primary target genes was observed when restricting the analysis for each group to genes found to contain E2F-binding sites in their promoter in ChIP experiments (69), although enrichment in the −and+CHX group was not observed. Enrichment in down-regulated secondary genes was not observed when considering all regulated genes (Figure 9, Supplementary Table 1), but was detected in the top 100 regulated genes (Supplementary Table 2). Several E2F family members in addition to E2F1 were regulated by E2, including E2F2, 6, 7 and 8 (Figure 4B), which may explain the prevalence of the E2F signature in regulated secondary genes.
Figure 9.

E2F response elements are enriched in secondary up-regulated genes. (A) Relative frequencies of E2F-binding sites within conserved regions in a 10 kb window (±5 kb) around the transcriptional start sites of regulated genes as a function of the cutoff rates used for matrix-based identification of sites (see Materials and Methods section). Enrichment is observed in up-regulated secondary target genes (not in the only+CHX category). Enrichment in the +CHX group likely reflects overlap with the −CHX group. (B) Enrichment of E2F sites in a 10 kb window around the transcriptional start sites of genes regulated by E2 in the absence and/or presence of CHX in the current study and reported to contain an E2F1-bound chromatin region in their promoters in Ref. (69). The numbers of genes in each category analyzed in A and B are indicated in Materials and Methods section.
Genes down-regulated in the absence of CHX were enriched in binding sites for NF-κB (Supplementary Table 1), consistent with the repressive role of estrogens on NF-κB signaling (15,33) although enrichment of these sites in the absence rather than the presence of CHX was unexpected. Similarly, binding sites for GATA factors were enriched in down-regulated secondary target genes (Supplementary Table 2). Repression of GATA1 transcriptional activity by estrogens has been shown previously (75,76). More surprisingly in view of the reported stimulatory effects of estrogens on Sp1- and AP1-mediated transcription, sites for these transcription factors were also found enriched in down-regulated secondary target genes (Supplementary Tables 1 and 2). These results suggest that AP1 sites may be involved in mediating gene repression as well as activation (see above for enrichment in up-regulated primary target genes).
Unexpectedly, c-Myc-binding sites were not found enriched in any of the target gene categories, whether considering all sequences, low-complexity sequences or conserved sequences. This is puzzling in view of the observation that induction or repression of MYC expression recapitulates the effects of estrogen and antiestrogens, respectively, on the cell cycle of ERα-positive breast cancer cells (77,78). However, comparison of E2 target genes with those of c-Myc target genes (79) and with genes regulated by siRNA-mediated suppression of MYC expression (80) indicates a statistically significant enrichment of c-Myc target genes in genes regulated by E2 (data not shown). C-Myc target genes were distributed in different groups of regulation (up versus down, primary versus secondary), which may explain the lack of significant enrichment in a specific group. Poor performance of the TRANSFAC matrices in identifying c-Myc-binding sites could also play a role in the absence of detected over-representation of these sites in estrogen target genes. Finally, we also monitored whether c-Myc-binding sites may be enriched in primary target genes, consistent with a mechanism of action through non-genomic activation by estrogens. Indeed, estradiol has been shown to stabilize the c-Myc protein for at least 12 h through modulation of its phosphorylation status (81). However, we did not observe significant enrichment in this category of regulated genes either. It remains possible however that identification of E2 target genes at earlier time points may reveal enrichment in c-Myc-binding sites, since MYC is one of the earliest known estrogen target genes (82). Of interest, TIAM1, a repressor of c-Myc that prevents its apoptotic effects (83), is also a primary estrogen target gene and may limit the duration of the induction of some c-Myc target genes.
Together, these data indicate that E2F is a major mediator of propagation of estrogen signaling in MCF-7 breast cancer cells for both up- and down-regulated secondary target genes, and suggest that AP1 may play a role in the down-regulation of secondary target genes.
DISCUSSION
Microarray analyses of estrogen target genes have been performed previously in various ERα-positive cell lines. However, only a limited number of genes have been found to be commonly regulated so far (46,71). Although this may be due to intrinsic differences between responses in these cells (71), variability between studies may also take place due to differences in cell culture conditions, microarray platform and selection criteria for significantly regulated genes. In this study, we chose to perform microarray analysis in 0.5% charcoal-treated serum, to minimize unstimulated levels of cellular growth; other studies were performed in 5% (45,50) or 10% serum (70,71). Our control was provided by cells treated with vehicle for the same time as with E2, whereas other studies have compared induction by E2 at different times with the absence of treatment at time 0 (50,70,71). We used the HG-U133 2.0 Plus GeneChips, while most other studies used anterior versions of Affymetrix GeneChips (45,70,71). Finally, we used a combination of three parameters, i.e. fold-change, average expression levels and P-value, to rank regulated genes, with set minima for each parameter (see Materials and Methods section). We identified 2144 significantly regulated genes, of which more than 50% overlapped with at least one of the two previous large-scale microarray studies also performed in MCF-7 cells (50,71). As we did not observe a higher overlap between sets of regulated genes described in these two other studies, we speculate that this partial overlap results from a combination of differences in experimental design and/or microarray platforms, rather than from the use of cycloheximide specific to our study.
While enrichment in different transcription factors has been reported in E2 targets in different studies (47,70), only one study based on microarray analysis exclusively has reported enrichment in EREs in the promoter sequences (−3 to +0.5 kb) of 89 genes identified as primary targets based on lack of effect of cycloheximide on regulation (46). Here, we identified 780 genes significantly regulated by E2 in the presence of CHX (544 up-regulated and 236 down-regulated genes). Overlap with genes regulated in the absence of CHX was partial. This can be explained in part by the fact that regulation of several genes found significantly modulated in the presence of CHX was also observed in its absence at earlier times, but became non-significant at 24 h. Genes in this category may include G1 phase-specific genes, as cycloheximide inhibited the increase in S phase induced by E2. In addition, possible inhibition by cycloheximide of negative feedback regulatory loops may also result in increased gene regulation at 24 h. Because CHX has major effects on gene expression (more than 4000 genes were either up- or down-regulated in our study, data not shown), it remains possible that CHX treatment may mask regulation of some primary target genes. However, we did not find that CHX treatment had a significant global effect on expression of the group of genes induced only in the absence of CHX (Supplementary Figure 1).
Enrichment in EREs was observed only in genes up-regulated in the presence of CHX, including both genes regulated in the absence and presence of CHX and those regulated in the presence of CHX only. These observations validate the characterization of these genes as primary E2 targets, and explain the lack of reported enrichment of EREs in previous microarray studies that did not discriminate between primary and secondary target genes. Enrichment in EREs in genes up-regulated in the presence of CHX was detectable in a +/−20 kb window centered on the TSS of regulated genes, consistent with a role of distal EREs in transcriptional regulation. This enrichment is not data-dependent, as we observed similar results using the subsets of our primary target genes that are in common with other microarray analyses, or using a dataset of 130 primary estrogen target genes regulated at 48 h in MCF-7 cells (74) (Supplementary Figures 4 and 5).
Regulatory sequences of up-regulated primary target genes often contained several EREs in a 20 kb window around the TSS. For instance, two functional EREs were validated in ChIP experiments in several selected primary target genes (CDH26, WISP2, CA12). Of interest, only two of the EREs validated in ChIP assays were previously reported in genome-wide ChIP-on-chip studies (50), likely due to the stringency used in this study to minimize false positive rates. While no correlation between identity with the consensus sequence and apparent strength of binding was observed for individual binding sites, general enrichment in EREs were more pronounced for sequences more similar to the consensus sequence, indicating that sequences that are high-affinity binding sites in vitro have a higher statistical chance of mediating E2 regulation in vivo, although the genomic context of the response element influences affinity at the individual level. The presence of multiple EREs in the vicinity of regulated genes may result in the formation of multiple chromatin loops associating these EREs and the TSS of regulated primary target genes as previously described (52).
Intriguingly, genes down-regulated in the presence of CHX were not enriched in EREs. Rather, a statistically significant depletion in EREs was observed. We also observed a lack of enrichment in EREs in ERα-bound chromatin regions located in the vicinity of genes repressed by E2 in Ref. (50). This may indicate that the presence of EREs in the flanking regions of genes confers a likelihood of positive rather than negative regulation. However, down-regulation of gene expression via binding of ERα to strategically positioned EREs remains possible, as EREs are still found, although less represented, in the vicinity of repressed primary E2 target genes. Similarly to what was observed in our dataset, we find that EREs were not enriched in down-regulated primary genes identified after 48 h stimulation with E2 (74). However, it will be of interest in the future to assess the frequency of EREs in the flanking regions of sets of primary genes down-regulated at other, including earlier, time points.
Binding sites for other transcription factors were enriched in primary up- and down-regulated genes. Binding sites enriched in down-regulated primary target genes have not before been associated with E2 action, but include two zinc finger transcription factors participating in corepressor complexes, EVI1 (84) and Ikaros (IKZF1) (85). On the other hand, binding sites enriched in up-regulated target genes include AP1, which have been previously demonstrated to mediate tethering of ERs. In addition, the ARNT gene encodes a protein that forms a complex with the ligand-bound Ah receptor (AhR). Activated AhR/Arnt was shown to act as a coactivator of estrogen receptor-mediated signaling, while ERα-AhR/Arnt protein–protein interactions mediate estradiol-dependent transrepression of dioxin-inducible gene transcription (86,87). Enrichment of AhR/Arnt sites in primary up-regulated genes suggests that AhR/Arnt-ERα complexes also mediate estradiol-dependent up-regulation of some AhR/Arnt target genes. Finally, EGR genes are, like AP1 components JUN and FOS, immediate early genes activated by a variety of signaling molecules (88,89). It is possible that these factors may contribute to gene regulation via non-genomic activation by estrogen, as suggested before (90).
Sites enriched in primary target genes may mediate regulation either through tethering of ERα or through non-genomic effects. Comparison with sites enriched in ERα-bound chromatin regions may help to discriminate between the two mechanisms, since only transcription factors mediating ERα recruitment via tethering should have sites within ChIP fragments. Enrichment of AP1, Maf, C/EBP, GATA and SOX-binding sites in ERα-bound chromatin regions may thus indicate recruitment of ERα to these sites. However, choice of background datasets is more complicated in the case of ChIP-on-chip data, and may affect site enrichment. For instance, enrichment in Egr or Sp1 factors in ERα-associated ChIP fragments was dependent on the choice of the reference background (promoter sequences versus flanking sequences of ChIP regions, data not shown). It should also be noted that, since cooperativity between ERα–ERE complexes and transcription factors bound to the same promoter may result in indirect association of ERα with these transcription factors through chromatin looping, low levels of enrichment of sites for a variety of transcription factors may be expected in ERα-associated regions.
Several other transcription factors were found enriched in secondary target genes. Surprisingly, NF-κB, AP1, GATA and Sp1 sites were all enriched in secondary down-regulated genes. This observation is consistent with the previous report that AP1 sites were present in ER-associated chromatin regions found close to genes repressed by E2 at late time points, this regulation being dependent on the induction of a co-repressor component such as the NRIP1 ERα co-repressor (50). It will be of interest to determine whether NRIP1 induction is also required for regulation of genes repressed by E2 and containing binding sites for NF-κB, GATA or Sp1 sites.
One of the most enriched binding sites in up-regulated secondary target genes is that for E2F transcription factors. Enrichment in E2F-binding sites is consistent with the well-known role of this family of transcription factors in the control of cell cycle, with known targets including several minichromosome maintenance (MCM) genes, which are essential for cell cycling due to the function of their products as replication-licensing factors (91). Other common E2F and ER targets involved in cell cycle regulation include the CDC6 and CDC25A cell division cycle genes, the PCNA and POLA2 genes, the replication factor C subunit RFC4, the structural maintenance of chromosome 2 (SMC2) and the protein regulator of cytokinesis one (PRC1) genes. Notably, we observed that not only E2F1, but also E2F2, E2F7 and E2F8 transcription factors were up-regulated by E2. E2F7 and E2F8 can repress E2F-activated transcription (92,93). E2F6 is also an inhibitory E2F family member (94,95) that can repress c-Myc target genes (96), but its induction profile was different from that of other E2F factors. The regulation of this gene may be explained by the fact that it is transcribed in the opposite direction from GREB1, a primary E2 target gene containing three consensus EREs in its flanking sequences (51, 52). Additional studies will be necessary to investigate the mechanisms of the transcriptional regulation of E2F family members by E2, and their exact individual roles in gene expression and cell cycle regulation by E2.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We are thankful to Pierre Chagnon and Raphaëlle Lambert of the IRIC Genomics platform for help with the TaqMan Q-PCR analysis of estrogen target gene expression. We also thank Patrick Gendron at the IRIC Bioinformatics platform for help with the IRIC computer cluster. This work was supported by grant MOP13147 from the Canadian Institutes for Health Research and by grant 194583-01 from the Natural Sciences and Engineering Council of Canada to S.M. V.B. has been supported by a CIHR post-doctoral fellowship and JD by an FRSQ studentship. S.M. and J.H.W. are holders of FRSQ Chercheur-Boursier National awards, and S.M. holds the CIBC Breast Cancer Research Chair at Université de Montréal. Funding to pay the Open Access publication charges for this article was provided by grant MOP13147 from the Canadian Institutes for Health Research.
Conflict of interest statement. None declared.
REFERENCES
- 1.Couse JF, Korach KS. Estrogen receptor null mice: what have we learned and where will they lead us? Endocrine Rev. 1999;20:358–417. doi: 10.1210/edrv.20.3.0370. [DOI] [PubMed] [Google Scholar]
- 2.McEwen BS, Alves SE. Estrogen actions in the central nervous system. Endocrine Rev. 1999;20:279–307. doi: 10.1210/edrv.20.3.0365. [DOI] [PubMed] [Google Scholar]
- 3.Jordan VC. Estrogen, selective estrogen receptor modulation, and coronary heart disease: something or nothing. J. Natl Canc. Inst. 2001;93:2–4. doi: 10.1093/jnci/93.1.2. [DOI] [PubMed] [Google Scholar]
- 4.Ali S, Coombes RC. Endocrine-responsive breast cancer and strategies for combating resistance. Nat. Rev. Cancer. 2002;2:101–112. doi: 10.1038/nrc721. [DOI] [PubMed] [Google Scholar]
- 5.Simpson ER. Sources of estrogen and their importance. J. Steroid Biochem. Mol. Biol. 2003;86:225–230. doi: 10.1016/s0960-0760(03)00360-1. [DOI] [PubMed] [Google Scholar]
- 6.Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, et al. Mechanisms of estrogen action. Physiol. Rev. 2001;81:1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 7.Robinson-Rechavi M, Escriva Garcia H, Laudet V. The nuclear receptor superfamily. J. Cell Sci. 2003;116:585–586. doi: 10.1242/jcs.00247. [DOI] [PubMed] [Google Scholar]
- 8.Kishimoto M, Fujiki R, Takezawa S, Sasaki Y, Nakamura T, Yamaoka K, Kitagawa H, Kato S. Nuclear receptor mediated gene regulation through chromatin remodeling and histone modifications. Endocr. J. 2006;53:157–172. doi: 10.1507/endocrj.53.157. [DOI] [PubMed] [Google Scholar]
- 9.Metivier R, Reid G, Gannon F. Transcription in four dimensions: nuclear receptor-directed initiation of gene expression. EMBO Rep. 2006;7:161–167. doi: 10.1038/sj.embor.7400626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mader S, Chambon P, White JH. Defining a minimal estrogen receptor DNA binding domain. Nucleic Acids Res. 1993;21:1125–1132. doi: 10.1093/nar/21.5.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwabe JW, Chapman L, Finch JT, Rhodes D. The crystal structure of the estrogen receptor DNA-binding domain bound to DNA: how receptors discriminate between their response elements. Cell. 1993;75:567–578. doi: 10.1016/0092-8674(93)90390-c. [DOI] [PubMed] [Google Scholar]
- 12.Walker P, Germond J-E, Brown-Luedi M, Givel F, Wahli W. Sequence homologies in the region preceding the transcription initiation site of the liver estrogen-responsive vitellogenin and apo-VLDLII gene. Nucleic Acids Res. 1984;12:8611–8626. doi: 10.1093/nar/12.22.8611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klein-Hitpass L, Ryffel GU, Heitlinger E, Cato AC. A 13 bp palindrome is a functional estrogen responsive element and interacts specifically with estrogen receptor. Nucleic Acids Res. 1988;16:647–663. doi: 10.1093/nar/16.2.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klinge CM. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001;29:2905–2919. doi: 10.1093/nar/29.14.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanchez R, Nguyen D, Rocha W, White JH, Mader S. Diversity in the mechanisms of gene regulation by estrogen receptors. Bioessays. 2002;24:244–254. doi: 10.1002/bies.10066. [DOI] [PubMed] [Google Scholar]
- 16.Driscoll MD, Sathya G, Muyan M, Klinge CM, Hilf R, Bambara RA. Sequence requirements for estrogen receptor binding to estrogen response elements. J. Biol. Chem. 1998;273:29321–29330. doi: 10.1074/jbc.273.45.29321. [DOI] [PubMed] [Google Scholar]
- 17.Kulakosky PC, McCarty MA, Jernigan SC, Risinger KE, Klinge CM. Response element sequence modulates estrogen receptor alpha and beta affinity and activity. J. Mol. Endocrinol. 2002;29:137–152. doi: 10.1677/jme.0.0290137. [DOI] [PubMed] [Google Scholar]
- 18.Nguyen D, Bail M, Pesant G, Dupont VN, Rouault E, Deschenes J, Rocha W, Melancon G, Steinberg SV, et al. Rational design of an estrogen receptor mutant with altered DNA-binding specificity. Nucleic Acids Res. 2007;35:3465–3477. doi: 10.1093/nar/gkm241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bai Y, Giguere V. Isoform-selective interactions between estrogen receptors and steroid receptor coactivators promoted by estradiol and ErbB-2 signaling in living cells. Mol. Endocrinol. 2003;17:589–599. doi: 10.1210/me.2002-0351. [DOI] [PubMed] [Google Scholar]
- 20.Gustafsson JA. ERbeta scientific visions translate to clinical uses. Climacteric. 2006;9:156–160. doi: 10.1080/14689360600734328. [DOI] [PubMed] [Google Scholar]
- 21.Leo C, Chen JD. The SRC family of nuclear receptor coactivators. Gene. 2000;245:1–11. doi: 10.1016/s0378-1119(00)00024-x. [DOI] [PubMed] [Google Scholar]
- 22.Rosenfeld MG, Glass CK. Coregulator codes of transcriptional regulation by nuclear receptors. J. Biol. Chem. 2001;276:36865–36868. doi: 10.1074/jbc.R100041200. [DOI] [PubMed] [Google Scholar]
- 23.Rachez C, Freedman LP. Mediator complexes and transcription. Curr. Opin. Cell Biol. 2001;13:274–280. doi: 10.1016/s0955-0674(00)00209-x. [DOI] [PubMed] [Google Scholar]
- 24.Dilworth FJ, Chambon P. Nuclear receptors coordinate the activities of chromatin remodeling complexes and coactivators to facilitate initiation of transcription. Oncogene. 2001;20:3047–3054. doi: 10.1038/sj.onc.1204329. [DOI] [PubMed] [Google Scholar]
- 25.McDonnell DP, Norris JD. Connections and regulation of the human estrogen receptor. Science. 2002;296:1642–1644. doi: 10.1126/science.1071884. [DOI] [PubMed] [Google Scholar]
- 26.McKenna NJ, O'Malley BW. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell. 2002;108:465–474. doi: 10.1016/s0092-8674(02)00641-4. [DOI] [PubMed] [Google Scholar]
- 27.Belandia B, Parker MG. Nuclear receptors: a rendezvous for chromatin remodeling factors. Cell. 2003;114:277–280. doi: 10.1016/s0092-8674(03)00599-3. [DOI] [PubMed] [Google Scholar]
- 28.Forster C, Makela S, Warri A, Kietz S, Becker D, Hultenby K, Warner M, Gustafsson JA. Involvement of estrogen receptor beta in terminal differentiation of mammary gland epithelium. Proc. Natl Acad. Sci. USA. 2002;99:15578–15583. doi: 10.1073/pnas.192561299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leygue E, Dotzlaw H, Watson PH, Murphy LC. Altered estrogen receptor alpha and beta messenger RNA expression during human breast tumorigenesis. Cancer Res. 1998;58:3197–3201. [PubMed] [Google Scholar]
- 30.Roger P, Sahla ME, Makela S, Gustafsson JA, Baldet P, Rochefort H. Decreased expression of estrogen receptor beta protein in proliferative preinvasive mammary tumors. Cancer Res. 2001;61:2537–2541. [PubMed] [Google Scholar]
- 31.Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, Webb P. Estrogen receptor pathways to AP-1. J. Steroid Biochem. Mol. Biol. 2000;74:311–317. doi: 10.1016/s0960-0760(00)00108-4. [DOI] [PubMed] [Google Scholar]
- 32.Safe S. Transcriptional activation of genes by 17 beta-estradiol through estrogen receptor-Sp1 interactions. Vitam. Horm. 2001;62:231–252. doi: 10.1016/s0083-6729(01)62006-5. [DOI] [PubMed] [Google Scholar]
- 33.Kalaitzidis D, Gilmore TD. Transcription factor cross-talk: the estrogen receptor and NF-kappaB. Trends Endocrinol. Metab. 2005;16:46–52. doi: 10.1016/j.tem.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 34.Migliaccio A, Di Domenico M, Castoria G, de Falco A, Bontempo P, Nola E, Auricchio F. Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cells. EMBO J. 1996;15:1292–1300. [PMC free article] [PubMed] [Google Scholar]
- 35.Sun M, Paciga JE, Feldman RI, Yuan Z, Coppola D, Lu YY, Shelley SA, Nicosia SV, Cheng JQ. Phosphatidylinositol-3-OH Kinase (PI3K)/AKT2, activated in breast cancer, regulates and is induced by estrogen receptor alpha (ERalpha) via interaction between ERalpha and PI3K. Cancer Res. 2001;61:5985–5991. [PubMed] [Google Scholar]
- 36.Pedram A, Razandi M, Aitkenhead M, Hughes CC, Levin ER. Integration of the non-genomic and genomic actions of estrogen. Membrane-initiated signaling by steroid to transcription and cell biology. J. Biol. Chem. 2002;277:50768–50775. doi: 10.1074/jbc.M210106200. [DOI] [PubMed] [Google Scholar]
- 37.Simoncini T, Fornari L, Mannella P, Varone G, Caruso A, Liao JK, Genazzani AR. Novel non-transcriptional mechanisms for estrogen receptor signaling in the cardiovascular system. Interaction of estrogen receptor alpha with phosphatidylinositol 3-OH kinase. Steroids. 2002;67:935–939. doi: 10.1016/s0039-128x(02)00040-5. [DOI] [PubMed] [Google Scholar]
- 38.Pedram A, Razandi M, Levin ER. Nature of functional estrogen receptors at the plasma membrane. Mol. Endocrinol. 2006;20:1996–2009. doi: 10.1210/me.2005-0525. [DOI] [PubMed] [Google Scholar]
- 39.Filardo EJ, Quinn JA, Bland KI, Frackelton AR., Jr Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol. Endocrinol. 2000;14:1649–1660. doi: 10.1210/mend.14.10.0532. [DOI] [PubMed] [Google Scholar]
- 40.Maggiolini M, Vivacqua A, Fasanella G, Recchia AG, Sisci D, Pezzi V, Montanaro D, Musti AM, Picard D, et al. The G protein-coupled receptor GPR30 mediates c-fos up-regulation by 17beta-estradiol and phytoestrogens in breast cancer cells. J. Biol. Chem. 2004;279:27008–27016. doi: 10.1074/jbc.M403588200. [DOI] [PubMed] [Google Scholar]
- 41.Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- 42.Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. 2005;146:624–632. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- 43.Funakoshi T, Yanai A, Shinoda K, Kawano MM, Mizukami Y. G protein-coupled receptor 30 is an estrogen receptor in the plasma membrane. Biochem. Biophys. Res. Commun. 2006;346:904–910. doi: 10.1016/j.bbrc.2006.05.191. [DOI] [PubMed] [Google Scholar]
- 44.Soulez M, Parker MG. Identification of novel oestrogen receptor target genes in human ZR75-1 breast cancer cells by expression profiling. J. Mol. Endocrinol. 2001;27:259–274. doi: 10.1677/jme.0.0270259. [DOI] [PubMed] [Google Scholar]
- 45.Frasor J, Danes JM, Komm B, Chang KC, Lyttle CR, Katzenellenbogen BS. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology. 2003;144:4562–4574. doi: 10.1210/en.2003-0567. [DOI] [PubMed] [Google Scholar]
- 46.Lin CY, Strom A, Vega VB, Kong SL, Yeo AL, Thomsen JS, Chan WC, Doray B, Bangarusamy DK, et al. Discovery of estrogen receptor alpha target genes and response elements in breast tumor cells. Genome Biol. 2004;5:R66. doi: 10.1186/gb-2004-5-9-r66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cicatiello L, Scafoglio C, Altucci L, Cancemi M, Natoli G, Facchiano A, Iazzetti G, Calogero R, Biglia N, et al. A genomic view of estrogen actions in human breast cancer cells by expression profiling of the hormone-responsive transcriptome. J. Mol. Endocrinol. 2004;32:719–775. doi: 10.1677/jme.0.0320719. [DOI] [PubMed] [Google Scholar]
- 48.Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43. doi: 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 49.Vega VB, Lin CY, Lai KS, Kong SL, Xie M, Su X, Teh HF, Thomsen JS, Yeo AL, et al. Multiplatform genome-wide identification and modeling of functional human estrogen receptor binding sites. Genome Biol. 2006;7:R82. doi: 10.1186/gb-2006-7-9-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, et al. Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- 51.Bourdeau V, Deschenes J, Metivier R, Nagai Y, Nguyen D, Bretschneider N, Gannon F, White JH, Mader S. Genome-wide identification of high-affinity estrogen response elements in human and mouse. Mol. Endocrinol. 2004;18:1411–1427. doi: 10.1210/me.2003-0441. [DOI] [PubMed] [Google Scholar]
- 52.Deschenes J, Bourdeau V, White JH, Mader S. Regulation of GREB1 transcription by estrogen receptor alpha through a multipartite enhancer spread over 20 KB of upstream flanking sequences. J. Biol. Chem. 2007;282:17335–17339. doi: 10.1074/jbc.C700030200. [DOI] [PubMed] [Google Scholar]
- 53.Rocha W, Sanchez R, Deschenes J, Auger A, Hebert E, White JH, Mader S. Opposite effects of histone deacetylase inhibitors on glucocorticoid and estrogen signaling in human endometrial Ishikawa cells. Mol. Pharmacol. 2005;68:1852–1862. doi: 10.1124/mol.105.014514. [DOI] [PubMed] [Google Scholar]
- 54.Dayan G, Lupien M, Auger A, Anghel SI, Rocha W, Croisetiere S, Katzenellenbogen JA, Mader S. Tamoxifen and raloxifene differ in their functional interactions with aspartate 351 of estrogen receptor alpha. Mol. Pharmacol. 2006;70:579–588. doi: 10.1124/mol.105.021931. [DOI] [PubMed] [Google Scholar]
- 55.Wettenhall JM, Simpson KM, Satterley K, Smyth GK. affylmGUI: a graphical user interface for linear modeling of single channel microarray data. Bioinformatics. 2006;22:897–899. doi: 10.1093/bioinformatics/btl025. [DOI] [PubMed] [Google Scholar]
- 56.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
- 58.Karolchik D, Baertsch R, Diekhans M, Furey TS, Hinrichs A, Lu YT, Roskin KM, Schwartz M, Sugnet CW, et al. The UCSC Genome Browser Database. Nucleic Acids Res. 2003;31:51–54. doi: 10.1093/nar/gkg129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pruitt KD, Tatusova T, Maglott DR. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005;33:D501–D504. doi: 10.1093/nar/gki025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wasserman WW, Sandelin A. Applied bioinformatics for the identification of regulatory elements. Nat. Rev. Genet. 2004;5:276–287. doi: 10.1038/nrg1315. [DOI] [PubMed] [Google Scholar]
- 61.Wang TT, Tavera-Mendoza LE, Laperriere D, Libby E, MacLeod NB, Nagai Y, Bourdeau V, Konstorum A, Lallemant B, et al. Large-scale in silico and microarray-based identification of direct 1,25-dihydroxyvitamin D3 target genes. Mol. Endocrinol. 2005;19:2685–2695. doi: 10.1210/me.2005-0106. [DOI] [PubMed] [Google Scholar]
- 62.Laperriere D, Wang TT, White JH, Mader S. Widespread Alu repeat-driven expansion of consensus DR2 retinoic acid response elements during primate evolution. BMC Genomics. 2007;8:23. doi: 10.1186/1471-2164-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:D108–D110. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jin VX, Sun H, Pohar TT, Liyanarachchi S, Palaniswamy SK, Huang TH, Davuluri RV. ERTargetDB: an integral information resource of transcription regulation of estrogen receptor target genes. J. Mol. Endocrinol. 2005;35:225–230. doi: 10.1677/jme.1.01839. [DOI] [PubMed] [Google Scholar]
- 65.O’Lone R, Frith MC, Karlsson EK, Hansen U. Genomic targets of nuclear estrogen receptors. Mol. Endocrinol. 2004;18:1859–1875. doi: 10.1210/me.2003-0044. [DOI] [PubMed] [Google Scholar]
- 66.Balmer JE, Blomhoff R. A robust characterization of retinoic acid response elements based on a comparison of sites in three species. J. Steroid Biochem. Mol. Biol. 2005;96:347–354. doi: 10.1016/j.jsbmb.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 67.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ho Sui SJ, Mortimer JR, Arenillas DJ, Brumm J, Walsh CJ, Kennedy BP, Wasserman WW. oPOSSUM: identification of over-represented transcription factor binding sites in co-expressed genes. Nucleic Acids Res. 2005;33:3154–3164. doi: 10.1093/nar/gki624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jin VX, Rabinovich A, Squazzo SL, Green R, Farnham PJ. A computational genomics approach to identify cis-regulatory modules from chromatin immunoprecipitation microarray data – a case study using E2F1. Genome Res. 2006;16:1585–1595. doi: 10.1101/gr.5520206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Creighton CJ, Cordero KE, Larios JM, Miller RS, Johnson MD, Chinnaiyan AM, Lippman ME, Rae JM. Genes regulated by estrogen in breast tumor cells in vitro are similarly regulated in vivo in tumor xenografts and human breast tumors. Genome Biol. 2006;7:R28. doi: 10.1186/gb-2006-7-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rae JM, Johnson MD, Scheys JO, Cordero KE, Larios JM, Lippman ME. GREB 1 is a critical regulator of hormone dependent breast cancer growth. Breast Cancer Res. Treat. 2005;92:141–149. doi: 10.1007/s10549-005-1483-4. [DOI] [PubMed] [Google Scholar]
- 72.van der Burg B, van Selm-Miltenburg AJ, de Laat SW, van Zoelen EJ. Direct effects of estrogen on c-fos and c-myc protooncogene expression and cellular proliferation in human breast cancer cells. Mol. Cell. Endocrinol. 1989;64:223–228. doi: 10.1016/0303-7207(89)90149-4. [DOI] [PubMed] [Google Scholar]
- 73.Gutierrez GJ, Ronai Z. Ubiquitin and SUMO systems in the regulation of mitotic checkpoints. Trends Biochem. Sci. 2006;31:324–332. doi: 10.1016/j.tibs.2006.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Scholtens D, Miron A, Merchant FM, Miller A, Miron PL, Iglehart JD, Gentleman R. Analyzing factorial designed microarray experiments. J. Multivariate Anal. 2004;90:19–43. [Google Scholar]
- 75.Blobel GA, Sieff CA, Orkin SH. Ligand-dependent repression of the erythroid transcription factor GATA-1 by the estrogen receptor. Mol. Cell. Biol. 1995;15:3147–3153. doi: 10.1128/mcb.15.6.3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Holth LT, Sun JM, Coutts AS, Murphy LC, Davie JR. Estrogen receptor diminishes DNA-binding activities of chicken GATA-1 and CACCC-binding proteins. DNA Cell Biol. 1997;16:1477–1482. doi: 10.1089/dna.1997.16.1477. [DOI] [PubMed] [Google Scholar]
- 77.Prall OW, Rogan EM, Musgrove EA, Watts CK, Sutherland RL. c-Myc or cyclin D1 mimics estrogen effects on cyclin E-Cdk2 activation and cell cycle reentry. Mol. Cell. Biol. 1998;18:4499–4508. doi: 10.1128/mcb.18.8.4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Doisneau-Sixou SF, Sergio CM, Carroll JS, Hui R, Musgrove EA, Sutherland RL. Estrogen and antiestrogen regulation of cell cycle progression in breast cancer cells. Endocr. Relat. Cancer. 2003;10:179–186. doi: 10.1677/erc.0.0100179. [DOI] [PubMed] [Google Scholar]
- 79.Zhao F, Xuan Z, Liu L, Zhang MQ. TRED: a Transcriptional Regulatory Element Database and a platform for in silico gene regulation studies. Nucleic Acids Res. 2005;33:D103–D107. doi: 10.1093/nar/gki004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cappellen D, Schlange T, Bauer M, Maurer F, Hynes NE. Novel c-MYC target genes mediate differential effects on cell proliferation and migration. EMBO Rep. 2007;8:70–76. doi: 10.1038/sj.embor.7400849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rodrik V, Gomes E, Hui L, Rockwell P, Foster DA. Myc stabilization in response to estrogen and phospholipase D in MCF-7 breast cancer cells. FEBS Lett. 2006;580:5647–5652. doi: 10.1016/j.febslet.2006.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Foster JS, Henley DC, Ahamed S, Wimalasena J. Estrogens and cell-cycle regulation in breast cancer. Trends Endocrinol. Metab. 2001;12:320–327. doi: 10.1016/s1043-2760(01)00436-2. [DOI] [PubMed] [Google Scholar]
- 83.Otsuki Y, Tanaka M, Kamo T, Kitanaka C, Kuchino Y, Sugimura H. Guanine nucleotide exchange factor, Tiam1, directly binds to c-Myc and interferes with c-Myc-mediated apoptosis in rat-1 fibroblasts. J. Biol. Chem. 2003;278:5132–5140. doi: 10.1074/jbc.M206733200. [DOI] [PubMed] [Google Scholar]
- 84.Hirai H, Izutsu K, Kurokawa M, Mitani K. Oncogenic mechanisms of Evi-1 protein. Cancer Chemother. Pharmacol. 2001;48(Suppl. 1):S35–S40. doi: 10.1007/s002800100303. [DOI] [PubMed] [Google Scholar]
- 85.Koipally J, Renold A, Kim J, Georgopoulos K. Repression by Ikaros and Aiolos is mediated through histone deacetylase complexes. EMBO J. 1999;18:3090–3100. doi: 10.1093/emboj/18.11.3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brunnberg S, Pettersson K, Rydin E, Matthews J, Hanberg A, Pongratz I. The basic helix-loop-helix-PAS protein ARNT functions as a potent coactivator of estrogen receptor-dependent transcription. Proc. Natl Acad. Sci. USA. 2003;100:6517–6522. doi: 10.1073/pnas.1136688100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ohtake F, Takeyama K, Matsumoto T, Kitagawa H, Yamamoto Y, Nohara K, Tohyama C, Krust A, Mimura J, et al. Modulation of oestrogen receptor signalling by association with the activated dioxin receptor. Nature. 2003;423:545–550. doi: 10.1038/nature01606. [DOI] [PubMed] [Google Scholar]
- 88.Sukhatme VP. Early transcriptional events in cell growth: the Egr family. J. Am. Soc. Nephrol. 1990;1:859–866. doi: 10.1681/ASN.V16859. [DOI] [PubMed] [Google Scholar]
- 89.Khachigian LM, Collins T. Early growth response factor 1: a pleiotropic mediator of inducible gene expression. J. Mol. Med. 1998;76:613–616. doi: 10.1007/s001090050258. [DOI] [PubMed] [Google Scholar]
- 90.Chen CC, Lee WR, Safe S. Egr-1 is activated by 17beta-estradiol in MCF-7 cells by mitogen-activated protein kinase-dependent phosphorylation of ELK-1. J. Cell. Biochem. 2004;93:1063–1074. doi: 10.1002/jcb.20257. [DOI] [PubMed] [Google Scholar]
- 91.Kato K, Toki T, Shimizu M, Shiozawa T, Fujii S, Nikaido T, Konishi I. Expression of replication-licensing factors MCM2 and MCM3 in normal, hyperplastic, and carcinomatous endometrium: correlation with expression of Ki-67 and estrogen and progesterone receptors. Int. J. Gynecol. Pathol. 2003;22:334–340. doi: 10.1097/01.pgp.0000092129.10100.5e. [DOI] [PubMed] [Google Scholar]
- 92.Di Stefano L, Jensen MR, Helin K. E2F7, a novel E2F featuring DP-independent repression of a subset of E2F-regulated genes. EMBO J. 2003;22:6289–6298. doi: 10.1093/emboj/cdg613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Christensen J, Cloos P, Toftegaard U, Klinkenberg D, Bracken AP, Trinh E, Heeran M, Di Stefano L, Helin K. Characterization of E2F8, a novel E2F-like cell-cycle regulated repressor of E2F-activated transcription. Nucleic Acids Res. 2005;33:5458–5470. doi: 10.1093/nar/gki855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Trimarchi JM, Fairchild B, Verona R, Moberg K, Andon N, Lees JA. E2F-6, a member of the E2F family that can behave as a transcriptional repressor. Proc. Natl Acad. Sci. USA. 1998;95:2850–2855. doi: 10.1073/pnas.95.6.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cartwright P, Muller H, Wagener C, Holm K, Helin K. E2F-6: a novel member of the E2F family is an inhibitor of E2F-dependent transcription. Oncogene. 1998;17:611–623. doi: 10.1038/sj.onc.1201975. [DOI] [PubMed] [Google Scholar]
- 96.Ogawa H, Ishiguro K, Gaubatz S, Livingston DM, Nakatani Y. A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G0 cells. Science. 2002;296:1132–1136. doi: 10.1126/science.1069861. [DOI] [PubMed] [Google Scholar]