

Abstract
We have analyzed structure-sequence relationships in 32 families of flavin adenine dinucleotide (FAD)-binding proteins, to prepare for genomic-scale analyses of this family. Four different FAD-family folds were identified, each containing at least two or more protein families. Three of these families, exemplified by glutathione reductase (GR), ferredoxin reductase (FR), and p-cresol methylhydroxylase (PCMH) were previously defined, and a family represented by pyruvate oxidase (PO) is newly defined. For each of the families, several conserved sequence motifs have been characterized. Several newly recognized sequence motifs are reported here for the PO, GR, and PCMH families. Each FAD fold can be uniquely identified by the presence of distinctive conserved sequence motifs. We also analyzed cofactor properties, some of which are conserved within a family fold while others display variability. Among the conserved properties is cofactor directionality: in some FAD-structural families, the adenine ring of the FAD points toward the FAD-binding domain, whereas in others the isoalloxazine ring points toward this domain. In contrast, the FAD conformation and orientation are conserved in some families while in others it displays some variability. Nevertheless, there are clear correlations among the FAD-family fold, the shape of the pocket, and the FAD conformation. Our general findings are as follows: (a) no single protein `pharmacophore' exists for binding FAD; (b) in every FAD-binding family, the pyrophosphate moiety binds to the most strongly conserved sequence motif, suggesting that pyrophosphate binding is a significant component of molecular recognition; and (c) sequence motifs can identify proteins that bind phosphate-containing ligands.
It is generally accepted that the three-dimensional structure of a polypeptide chain is determined by its amino acid sequence. Nevertheless, similar folds can have very different sequences. One of the ultimate goals in sequence analysis is to predict the structure and function of a protein based solely on its sequence. In cases where the protein of interest shares at least 30% amino acid identity with another protein, the two proteins generally exhibit similar three-dimensional structure (Doolittle 1986). Alternatively, when proteins are known to have similar structure but divergent sequences, consensus sequence motifs can be used to assess the function of unassigned sequences. These consensus motifs usually correspond to residues interacting with cofactors, substrate, or other proteins.
The increasing number of three-dimensional structures of proteins in the Protein Data Bank, complexed with appropriate ligands, provides an important tool for understanding the mechanisms of molecular recognition. In this study, we focussed on flavin adenine dinucleotide (FAD) because it and its related cofactors, nicotinamide adenine dinucleotide (NADH) and adenosine triphosphate (ATP), appear in many biological processes.
Previous comparative structural studies of mononucleotide- and dinucleotide-binding proteins reveal that some exhibit a similar three-dimensional structure with conserved sequence motifs at positions crucial for binding, although the remaining sequence can vary greatly. One of the first folds was identified by Rossmann (Rossmann et al. 1974) who discovered the correlation between the fold of dehydrogenases that bind the cofactor NADH and conserved sequence motifs. The basic structure consists of six parallel β-strands interspersed by α-helices that appear on both sides of the six-stranded β sheet. This symmetrical α/β structure is built from two halves, β1α1β2α2β3 and β4α4β5α5β6, with a crossover α-helix (α3) connecting β3 and β4 (Fig. 1 ▶). Each of these folds is known as the classical mononucleotide-binding fold or the Rossmann fold.
Fig. 1.

Topology diagram of dehydrogenases that bind NADH. This symmetrical α/β structure is composed of two halves, β1α1β2α2β3 and β4α4β5α5β6, with a crossover α-helix (α3) connecting strands β3 and β4. Each of these motifs is known as the classical mononucleotide-binding motif or the Rossmann fold. Cylinders represent α-helices and arrows denote β-strands. Dashed lines indicate elements of secondary structure below the plane of the fold. The overall pseudo two-fold rotation symmetry between the two βαβαβ motifs is shown by an arrow .
A variation of the fold observed for dehydrogenases is found in FAD-containing proteins. This fold consists of one β1α1β2α2β3 Rossmann fold, and a variation of both the second Rossmann fold and the crossover α-helix. This variation includes a three-stranded antiparallel β-sheet connecting β3 and β4, instead of the crossover α-helix observed in dehydrogenases. Moreover the sixth strand, from the second Rossmann fold, is missing whereas the fifth strand is retained but is close to the end of the sequence. Different variations in which structural elements are added or deleted were found in proteins containing other mono- and/or dinucleotides such as flavin mononucleotide (FMN) (Rao and Rossmann 1973), and nicotinamide adenine dinucleotide phosphate (NADPH) (Schulz and Schirmer 1974). Most of these proteins show a series of conserved amino acid residues at positions interacting with the mono/dinucleotide molecule.
We selected a nonredundant set of 32 protein-FAD complex structures from the Protein Data Base (PDB) for structure-sequence analysis with the goal of deepened understanding of principles of molecular recognition. On the basis of sequence-structure comparison and the interaction of cofactor atoms with the different protein residues, we identified several conserved motifs for each structural family. Some of these were previously characterized by others (Schulz and Schirmer 1974; Schulz et al. 1982; Wierenga et al. 1983; Schulz 1992; Correll et al. 1993; Lu et al. 1994; Ingelman et al. 1997; Fraaije et al. 1998) and some are newly derived. These conserved sequence motifs are called "most conserved" when they are present in all family members, and "partially conserved" when present in some but not all family members. In addition to the sequence-structure analysis, we investigated a more complete set of variables, including cofactor conformation, characteristics of the protein pocket wherein the cofactor is bound, cofactor directionality, and correlation of cofactor moieties (adenine, pyrophosphate isoalloxazine, etc.) interacting with conserved sequence motifs in the different family folds. Such fundamental discriminators may improve our understanding of protein evolution, in particular for FAD-binding proteins where many tertiary structures, often distantly related, are known. Furthermore, the presence of a variety of conserved sequence motifs in FAD families, specifically those that are pyrophosphate-binding, can be used as a tool for molecular recognition of phosphate analog-binding proteins.
Results
FAD cofactor
The dinucleotide FAD consists of adenosine monophosphate (AMP) linked to flavin mononucleotide (FMN) by a pyrophosphate bond (Fig. 2A ▶). The AMP moiety is composed of the adenine ring bonded to a ribose that is linked to a phosphate group. The FMN moiety, also known as the riboflavin moiety, is composed of the isoalloxazine-flavin, linked to a sugar, ribitol, which is coupled to a phosphate group. The catalytic function of the FAD is concentrated in the isoalloxazine ring, whereas the ribityl phosphate and the AMP moiety mainly stabilize cofactor binding to protein residues. The flavin functions mainly in a redox capacity, being able to take up two electrons from one substrate and release them two at a time to a substrate or coenzyme, or one at a time to an electron acceptor. The surrounding protein controls many of the catalytic properties of the flavin ring, such as the rate of accepting electrons, the pathway of electron flow within the flavin ring, and the flavin's oxidation-reduction potential (Mathews 1991). This control is asserted in part by the cofactor conformation, which adapts to the protein as either an elongated or bent butterfly conformation (Fig. 2A and B ▶, respectively). In the bent conformation, the AMP portion is folded back, placing the adenine and isoalloxazine rings in close proximity, whereas in the elongated conformation the adenine ring is distal from the isoalloxazine ring.
Fig. 2.

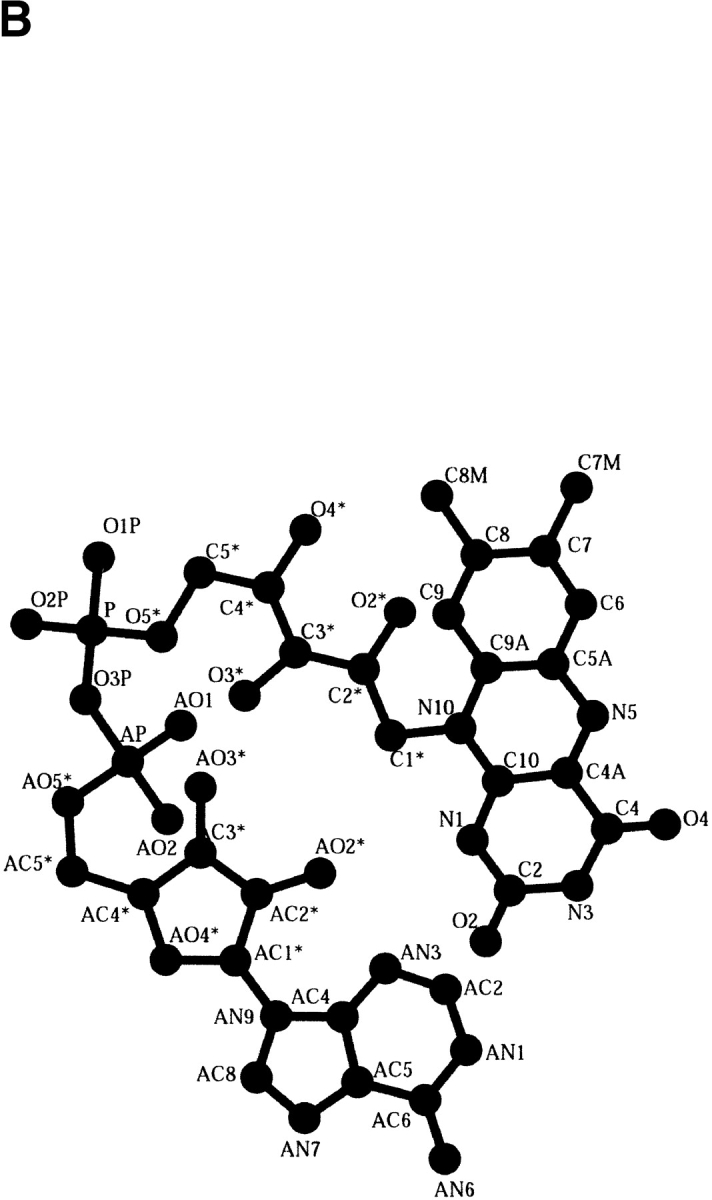
The FAD cofactor conformations. (A) An elongated conformation where the adenine ring is distal from the isoalloxazine ring. The division between the component parts FMN and AMP composing the FAD is shown. The AMP is composed of an adenine ring connected to a ribose that is connected to a phosphate group. The FMN moiety is composed of the isoalloxazine-flavin ring linked to a ribitol, which is connected to a phosphate group. (B) Bent conformation where the AMP portion is folded back, placing the adenine and isoalloxazine rings in close proximity. Variation in the proximity of the two rings determines the degree of cofactor flexibility. All atoms composing the FAD cofactor are labeled.
The glutathione reductase (GR) structural family
Structural conservation
One of the most thoroughly studied FAD-containing families is represented by the enzyme glutathione reductase (GR). The protein members of this populous family catalyze diverse reactions (Karplus and Schulz 1987; Chen et al. 1994; Mande et al. 1996; Mizutani et al. 1996; Yeh et al. 1996; Enroth et al. 1998; Eppink et al. 1998; Binda et al. 1999; Bond et al. 1999; Lennon et al. 1999; Leys et al. 1999; Trickey et al. 1999; Wohlfahrt et al. 1999; Yue et al. 1999; Ziegler et al. 1999) (Table 1). All family members adopt the Rossmann fold (β1α1β2α2β3 in Fig. 3A ▶). The topology of the GR family consists of a central five-stranded parallel β-sheet (β1, β2, β3, β7, and β8) surrounded by α-helices (α1 and α2) and an additional crossover connection composed of a three-stranded antiparallel β-sheet (β4–6) (Fig. 3A ▶).
Table 1.
Cofactors and conserved sequence motifs in the nonredundant FAD-containing protein structures retrieved from the Protein Data Bank and used in our sequence-structure analysis
| PDB Code | Family | Protein (Reference) | Cofactors | C, NCa | B, Eb | Sequence motifs |
| 1joa | GR1 | Glutathione reductase (Karplus and Schulz 1987) | FAD, NADH | NC | E | GxGxxG(x)17E, hhhxGxGxxAxE, T(x)5TxxGD, D(x)6GxxP |
| 1fcd | Sulfide dehydrogenase (Chen et al. 1994) | FAD | C | E | GxGxxG(x)19E, hhhxPxPxxPxE, S(x)6HxxGD, D(x)6PxxA | |
| 1ebd | Dihydrolipoamide dehydrogenase (Mande et al. 1996) | FAD, NADH | NC | E | GxGxxG(x)17E, hhhxGxGxxGxE, T(x)5FxxGD,D(x)6GxxP | |
| 1bzl | Trypanothione reductase (Bond et al. 1999) | FAD, NADPH | NC | E | GxGxxG(x)18D, hhhxGvxxxSxE, T(x)5YxxGD,D(x)6GxxP | |
| 3grs | NADH peroxidase (Yue et al. 1999) | FAD, NADPH | NC | E | GxGxxG(x)19E, hhhxGxGxxGxE, T(x)5FxxGD, D(x)6GxxP | |
| 1cjc | Adrenodoxin reductase (Ziegler et al. 1999) | FAD, NADPH | NC | E | GxGxxG(x)19E, hhhxGxGxxAxD, V(x)6YxxGW, GxGxxG(x)21, FxxGD, | |
| 1c10 | Thioredoxin reductase (Lennon et al. 1999) | FAD, NADPH | NC | E | D(x)8GxxP | |
| 1foh | GR2 | Phenol hydroxylase (Enroth et al. 1998) | FAD, NADPH | NC | E | GxGxxG(x)16D, S(x)5FxxGD |
| 1bf3 | p-Hydroxybenzoate hydroxylase (Eppink et al. 1998) | FAD, NADPH | NC | E | GxGxxG(x)17E, FxxGD | |
| 1cf3 | Glucose oxidase (Wohlfahrt et al. 1999) | FAD | NC | E | GxGxxG(x)18E | |
| 1b37 | Polyamine oxidase (Binda et al. 1999) | FAD, | NC | E | GxGxxG(x)18E, T(x)5FxxGD | |
| 1d4c | Fumarate reductase (Leys et al. 1999) | FAD | C | E | GxGxxG(x)17E | |
| 1b4v | Cholesterol oxidase (Yue et al. 1999) | FAD | NC | E | GxAxxG | |
| 1aa8 | D-Amino acid oxidase (Mizutani et al. 1996) | FAD | NC | E | GxGxxG(x)19D | |
| 1b3m | Sarcosine oxidase (Trickey et al. 1999) | FAD | C | E | GxGxxG(x)17D | |
| 1fdr | FR | Flavodoxin reductase (Ingelman et al. 1997) | FAD, NADPH | NC | B | RxYS, GxxSxxL(x)5G(x)8AxG, MxxxGTAIxP |
| 1a8p | Ferredoxin reductase (Sridhar Prasad et al. 1998) | FAD, NADPH | NC | B | RxYS, GxxTxxL(x)5G(x)8PxG, MxxxGTGIxP | |
| 1ndh | NADH-Cytochrome b5 reductase (Nishida et al. 1995) | FAD, NADH | NC | E | RxYT, GxxSxxL(x)5G(x)7PxG, MxxxGTGIxP | |
| 2cnd | Nitrate reductase (Lu et al. 1994) | FAD, NADH | NC | E | RxYT, GxxTxxL(x)5G(x)7PxG, MxxxGSGIxP | |
| 1amo | NADPH-P450 reductase (Wang et al. 1997) | FAD, NADPH | NC | E | RxYS, GxxTxxL(x)6G, MxxxGTGIxP | |
| 1cqx | Flavohemoglobin (Ermler et al. 1995) | FAD, NADH | NC | E | RxYS, GxxSxxL(x)6G(x)7PxG | |
| 1qlt | PCMH | p-Cresol methylhydroxylase (Cunane et al. 2000) | FAD | C | E | P(x)6GxN, G(x)7GY, K(x)6E(x)2YxxVxxG(x)8Y, R, GxxL |
| 1dii | Vanillyl-alcohol oxidase (Fraaije et al. 2000) | FAD | C | E | P(x)6GxN, G(x)7GY, R(x)6E(x)2YxxVxxG(x)8Y, R, GxxL | |
| 2mbr | Uridine diphospho-N-acetylenol- pyruvylglucosamine reductase (Benson et al. 1997) | FAD, NADPH | NC | E | P(x)6GxN, G(x)8AY, K(x)6E(x)4YxxVxxG(x)8Y, R, GxxL | |
| 1qj2 | Carbon monoxide dehydrogenase (Dobbek et al. 1999) | FAD | NC | E | P(x)8GxN, G(x)3AY, R(x)5D(x)5YxxAxxxG(x), R | |
| 1f0x | D-Lactate dehydrogenase (Dym et al. 2000) | FAD | NC | E | A(x)7AxN, G(x)7GS, R | |
| 1pow | PO | Pyruvate Oxidase (Muller et al. 1994) | FAD | NC | E | R(x)5GxG, VGxN, K(x)7IxxDP(x)9D(x)4ADxxK, KxLxxLxxxL(x)6T(x)4GxV |
| 1efv | Electron transfer (Roberts et al. 1996) | FAD | NC | E | K(x)5GxG, K(x)7VGxS, IxxDP(x)8D(x)4ADxxK, KxLxxLxxxL(x)6S(x)6GxV | |
| 1ivh | SMc | Acyl-CoA dehydrogenase (Tiffany et al. 1997) | FAD | NC | E | |
| 1dnp | DNA Photolyase (Park et al. 1995) | FAD | NC | B | ||
| 1b5t | Methylenetetrahydrofolate reductase (Guenther et al. 1999) | FAD, NADPH | NC | E | ||
| 1qr2 | Quinone oxidoreductase (Foster et al. 1999) | FAD, NADPH | NC | E |
a FAD bond type: C, covalent; NC, noncovalent.
b Conformation: B, bent; E, elongated.
c SM signifies the four single-membered families.
Fig. 3.

Topological diagram of FAD-binding domain of the four FAD-family folds. (A) Rossmann fold (β1α1β2α2β3) adopted by the glutathione reductase (GR) family members. For a full description of this fold, it must be noted that there are two subfamilies, GR1 and GR2 (see text), and that there are exceptions to the generalizations described here. For example, D-amino acid oxidase of the GR2 subfamily is an exception to the rule that the FAD-binding fold in the GR family contains a 3-strand β-meander connecting β3 and β4; instead, it has a crossover α-helix. (B) Ferredoxin reductase (FR) family fold adopting a cylindrical β-domain organized into two orthogonal sheets, β1β2β5 and β3β4β6. (C) The p-cresol methylhydroxylase (PCMH) family fold consists of two α + β subdomains; one is composed of three parallel β-strands (β1–3) and the second contains five antiparallel β-strands (β4–8) surrounded by α-helices. (D) The pyruvate oxidase (PO) family fold consists of five parallel β-strands (β1–5) interspersed by α-helices similar to the double Rossmann fold found in dehydrogenases. Cylinders represent α-helices and arrows denote β-strands. The location, indicated in dashed lines , of the conserved sequence motifs in each of the FAD-family folds is listed in Table 1.
Of the 15 GR family members listed in Table 1, two separate subfamilies were identified using the CE program for comparing a polypeptide chain to each chain in the PDB (Shindyalov and Bourne 1998). One subfamily, GR1, contains the first seven proteins listed in Table 1; the last eight proteins belong to the second subfamily, GR2. Each of the two subfamilies is obtained when any of the proteins belonging to it is used as a query in the sequence-structure search. Whereas proteins from the GR1 subfamily align well through the entire FAD-binding domain, those belonging to the GR2 subfamily align well only in their N-terminal (∼30 residues). Nevertheless, all GR family members share a similar overall three-dimensional structure in their FAD-binding domain as well as at least one conserved sequence motif (Table 1). Terminal additions as well as various insertions within the fold are present in the members of GR2, however. Specifically, insertions of several secondary structures are observed in the connections between β2 to α2 and α2 to β3 of the Rossmann fold (Fig. 3A ▶). This is in agreement with the observation that these proteins align well only in their N-terminal residues comprising β1, α1, and β2. Notably, proteins from GR1, with the exception of sulfide dehydrogenase (Van Driessche et al. 1996), comprise NAD(P)H-binding domains (between β7 and β8 in Fig. 3A ▶), adopting the Rossmann fold (Rossmann et al. 1974), in addition to the FAD-binding domain. Phenol hydroxylase (Enroth et al. 1998) and p-hydroxybenzoate hydroxylase (Eppink et al. 1998) are the only proteins from the second subfamily known to bind NADPH. However, both lack the Rossmann-type NADPH-binding fold.
FAD binding and conformation
The FAD cofactor in the GR family members adopt elongated conformations with the adenine and isoalloxazine moieties distal from each other (Table 1 and Fig. 4A ▶). The degree of extension of the cofactor can vary considerably. In addition, the cofactor may be either covalently or noncovalently bound to the protein, and there is no apparent correlation of cofactor conformation with any of the conserved sequence motifs (Table 1). In contrast, the direction of the FAD cofactor is conserved among all GR family members: the adenine ring of the cofactor points toward the FAD-binding domain, while the isoalloxazine ring points away from it (Fig. 4A ▶).
Fig. 4.

Ribbon representation of the FAD-binding domain of the four FAD-family folds complexed with FAD. (A) Rossmann fold adopted by the glutathione reductase (GR) family members. The blue shading indicates the crossover connection composed of a three-stranded antiparallel β-sheet; the red shading indicates the central five-stranded parallel β-sheet surrounded by two α-helices. The FAD cofactor adopts an elongated conformation with the adenosine moiety (gray circles) pointing toward the FAD-binding domain. (B) Ferredoxin reductase (FR) family. The two antiparallel three-stranded β-sheets are shown in red and blue. The FAD in its bent conformation is shown with the isoalloxazine ring (black circles) pointing toward the FAD-binding domain. (C) The α + β fold adopted by the p-cresol methylhydroxylase (PCMH) family members. In red are three parallel β-strands surrounded by α-helices, and in blue are five antiparallel β-strands surrounded by α-helices. The FAD molecule adopts an elongated conformation and is located in between the two subdomains with the adenine ring pointing toward them. (D) Rossmann fold adopted by pyruvate oxidase (PO) family members, shown in red. The cofactor adopts an elongated conformation and lies perpendicular to the β-strands. The stick drawing of the cofactor is depicted with gray circles for atoms in the adenine and sugar rings, with red circles for phosphate and oxygen atoms, and black circles for atoms for the isoalloxazine ring. Figure created by MOLSCRIPT (Kraulis 1991) and RASTER3D (Bacon and Anderson 1988; Merritt and Murphy 1994).
Conserved sequence motifs
The GR family members show several conserved amino acid residues at a few crucial positions, and the rest of the sequence varies. We conducted a sequence-structure alignment of the two GR subfamily members using the program CE. Conserved sequence motifs found in each of the GR family members are listed in Table 1. Specifically, four conserved motifs for FAD binding are found in GR1 members (depicted in colored boxes in Fig. 5 ▶), two of which are present in the GR2 subfamily as well (Table 1).
Fig. 5.

Structure-based amino acid alignment for proteins belonging to the GR1 family of proteins as obtained with the program CE (Shindyalov and Bourne 1998) (first seven proteins listed in Table 1). The first column gives the PDB accession code, and the second column gives two numbers that each specify the sequence position that follows. The left-hand number corresponds to the sequence and the right-hand to the PDB. The conserved sequence motifs are depicted in boxes: GxGxxGxxxhxxh(x)8hxhE(D) in blue, VVVV(I)GxGxxGxE in red, T(S)xxxxxF(Y)hhGD(E) in pink, and D(x)6GxxP in green. In yellow are all conserved residues not directly involved in binding FAD. Proteins: 1JOA, NADH peroxidase from Enterococcus faecalis; 1FCD, flavocytochrome C sulfide dehydrogenase from Chromatium vinosum; 1EBD, dihydrolipoamide dehydrogenase from Bacillus stearothermophilus; 1BZL, trypanothione reductase from Trypanosoma cruzi; 3GRS, glutathione reductase from Homo sapiens; 1CJC, adrenodoxin reductase from Bos taurus; 1CL0, thioredoxin reductase from Escherichia coli.
The most conserved and well-studied sequence motif is part of the Rossmann fold, xhxhGxGxxGxxxhxxh(x)8hxhE(D) (Fig. 5 ▶, blue box), where x is any residue and h is a hydrophobic residue (Schulz and Schirmer 1974; Schulz et al. 1982; Wierenga et al. 1983; Schulz 1992). This motif is found at the N-terminal part of the sequence, and in fact it is the only conserved sequence motif present in all GR family members (Table 1). This is in agreement with the observation that while members of GR1 align well throughout the entire FAD-binding domain, GR2 members align well mainly at the N-terminus. This consensus is known as the dinucleotide-binding motif (DBM) (Wierenga et al. 1983) or the phosphate-binding sequence signature (Moller and Amons 1985) and is a common motif among FAD- and NAD(P)H-dependent oxidoreductases. The central part of this consensus motif, GxGxxG, is part of the loop connecting the first β-strand and α-helix in the βαβαβ Rossmann fold with the N-terminal end of helix α1 pointing toward the pyrophosphate moiety for charge compensation (Fig. 3A ▶). The importance of the Gly residues in the conserved central GxGxxG is well understood (Wierenga et al. 1986): the first strictly conserved Gly allows for a tight turn of the main chain, which is important for positioning the second Gly. The second Gly, because of its missing side chain, permits close contact of the main chain to the pyrophosphate of FAD, specifically oxygen atoms OP1 or OP2. The third Gly allows close packing of the helix with the β-sheet. The hydrophobic residues provide hydrophobic interactions of the α-helix with the β-sheet. The conserved negatively charged terminal residue, Glu or Asp, hydrogen bonds to the ribose 2′ hydroxyl of the adenosine moiety. A variation of this glycine-rich sequence motif is hhhxGxGxxGxE (Fig. 5 ▶, red box), in which the central GxGxxG is common (Wierenga et al. 1986). It is part of the βαβ Rossmann NAD(P)H-binding motif and is present only in GR1 subfamily members containing the NAD(P)H-binding domain in addition to the FAD-binding domain (first seven proteins in Table 1). Sulfide dehydrogenase belongs to the GR1 subfamily; however, it lacks the NAD(P)H-binding domain, consistent with the observation that glycines are replaced by prolines in this conserved motif (Fig. 5 ▶, red box). Two enzymes of GR2, p-hydroxybenzoate (Eppink et al. 1998) and phenol hydroxylase (Enroth et al. 1998), are exceptional: they bind NADPH (Table 1) but lack a Rossmann-type NADPH-binding domain and therefore lack the conserved hhhxGxGxxGxE sequence motif typical of the NAD(P)H-binding domain (Wierenga et al. 1983).
Another highly conserved FAD-binding sequence motif was first identified from studies of rubredoxin reductase (Eggink et al. 1990), and is an 11 amino acid segment, T(S)xxxxxF(Y)hhGD(E) (Fig. 5 ▶, pink box). This motif is present in all members of GR1 and in some of GR2 (Table 1). The hydrophobic residues belong to the seventh β-stand of the FAD-binding domain, found near the C-terminus of the protein (Fig. 3A ▶). The initial Thr residue participates in the formation of the `greek key' motif found just before the seventh strand, and the invariant Gly and the terminal Asp residues are part of the loop connecting the β-strand to the α-helix, and hydrogen bonded to the OP1 or OP2 atoms of the pyrophosphate group.
There are some partially conserved sequence motifs in the GR family. Vallon (2000) has reported short GG and ATG motifs, present in only a few of the protein members. We found another partially conserved sequence motif, D(x)6GxxP (Fig. 5 ▶, green box). This motif is situated at the interface between the NAD(P)H- and FAD-binding domains and one of the x residues, located between the Gly and Pro, often an Arg residue, making a polar contact with the isoalloxazine ring. This newly derived sequence motif is absent in GR family members lacking NAD(P)H-binding domain (GR2 proteins in Table 1).
The ferredoxin reductase (FR) structural family
Structural conservation
Another well-studied FAD-family fold, represented by the enzyme ferredoxin reductase (FR) (Karplus et al. 1991), consists of six proteins listed in Table 1 (Lu et al. 1994; Ermler et al. 1995; Nishida et al. 1995; Ingelman et al. 1997; Wang et al. 1997; Sridhar Prasad et al. 1998). All family members were identified using the program CE (Shindyalov and Bourne 1998). In addition to the FAD-binding domain, all of the FR family members have the NAD(P)H-binding domain (Table 1), adopting the Rossmann fold found in the dehydrogenases (Fig. 1 ▶; Rossmann et al. 1974). However, the number of strands in the NAD(P)H-binding domain differs among the various family members. The FAD-binding fold characteristic of the FR family is a cylindrical β-domain with a flattened six-stranded antiparallel βbarrel organized into two orthogonal sheets (β1β2β5 and β4β3β6) separated by one α-helix, and is distinguished from the βαβαβ Rossmann fold (Fig. 3B ▶). The cylinder is open between strands β4 and β5. This opening of the cylinder makes space for the isoalloxazine and ribityl moieties of the FAD, to which hydrogen bonds are formed from the open edges of the strands (Fig. 4B ▶). One end of the cylinder is covered by the only helix of the domain, which is essential for the binding of the pyrophosphate groups of the FAD. The structural differences in the FAD-binding domain are manifested mainly as loops of different length and extra extending structural elements, which may be important for interactions with their redox partners. The structural core of all FR family members is highly conserved.
FAD binding and conformation
The binding pocket for the FAD cofactor is located between the FAD- and NAD(P)H-binding domains, whose relative orientations vary among the different family members. As a result, the FAD cofactor can adopt either an elongated or bent conformation (Table 1). In contrast to this variability of the general cofactor conformation, the conformation of the FMN part of the FAD molecule is nearly identical in all family members. In particular, the isoalloxazine ring is always inserted into the protein at the FAD- and NAD(P)H-domain boundaries, whereas the adenine and ribose moieties are mostly exposed (Fig. 4B ▶). As a result, the binding of the isoalloxazine, ribityl, and pyrophosphate moieties are well preserved in the FR family, whereas the adenosine displays large conformational differences and a small number of contacts with protein residues. This observation implies that the adenine and ribose moieties are more flexible, contributing to the variability in cofactor flexibility observed in the FR family. Furthermore, the loop between β5 and the α-helix involved in the adenine binding differs among the various family members. Its length varies, and in some family members it is altogether missing. This variation in loop length possibly contributes to the varied cofactor conformations.
Conserved sequence motifs
Despite their significant structural similarities, the FR sequences vary greatly, showing conserved residues at only a few crucial positions (Correll et al. 1993; Lu et al. 1994; Ingelman et al. 1997) (Fig. 6 ▶ and Table 1). One of the most conserved sequence motifs is RxYS(T) (Fig. 6 ▶, red box), which is part of β4 (Fig. 3B ▶), where the invariant positively charged Arg residue forms hydrogen bonds to the negative pyrophosphate oxygen atom (OP1 or OP2) in all family members (Ingelman et al. 1997). Another side chain involved in cofactor binding is Tyr, which makes extensive van der Waals contacts with the flavin and hydrogen bonds to the ribityl 4′ hydroxyl (O4*). An additional highly conserved sequence motif is GxxS(T)xxL(x)5G(x)7PxG (Fig. 6 ▶, green box), which is part of α1β6 and is known as the phosphate-binding motif (Fig. 3B ▶) (Sridhar Prasad et al. 1998). In all family members, the Ser or Thr residues are part of the loop connecting β5 to α1, and are within hydrogen bonding distance to one of the pyrophosphate oxygen atoms (OP1 or OP2). The absence of side chain at the first Gly allows a close approach of the main chain to the pyrophosphate moiety. The remaining conserved residues cluster in the helix and β6 of the FAD-binding domain.
Fig. 6.

Sequence alignment of all FAD-containing proteins resembling the ferredoxin reductase structure based on structure-sequence alignment as obtained using the CE program (Shindyalov and Bourne 1998). Conserved sequence motifs are depicted in boxes: RxYS(T) in red, GxxS(T)xxL(x)5G(x)7PxG in green, and MxxxGT(S)G(A)IxP in blue. In yellow are conserved residues not interacting with the cofactor. The data bank accession code and position numbers according to the sequence and according to the PDB are given. Proteins: 1FDR, flavodoxin reductase from Escherichia coli; 1A8P, ferredoxin oxidoreductase from Azotobacter vinelandii; 1NDH, cytochrome b5 reductase from Sus scrofa; 2CND, nitrate reductase from Zea mays; 1AMO, NADPH-cytochrome P450 reductase from Rattus norvegicus; 1CQX, flavohemoprotein from Alcaligenes eutrophus.
Another conserved sequence motif, MxxxGT(S)G(A)IxP (Fig. 6 ▶, blue box), is part of the NAD(P)H-binding domain and comprises the βαβ structure motif. The conserved Gly residue is located near the N-terminus of the sequence involved in pyrophosphate binding of the NAD(P)H cofactor, and is known as the phosphate-binding loop (Karplus et al. 1991). In addition, the Thr residue interacts with the isoalloxazine ring of the FAD cofactor in selected family members, and in the other family members it is in the vicinity of the cofactor. The remaining residues cluster in the interface between the NAD(P)H- and FAD-binding domains and around the NAD(P)H binding site. There is similarity between the GxGxxG motif observed in dehydrogenases and the central part of the phosphate-binding motif, GT(S)G(A)IxP, present in the NAD(P)H-binding domain of FR members. Both of these motifs are rich in Gly, but the specific residues interacting with the NAD(P)H cofactor are different in the two types of domains (Ingelman et al. 1997).
The p-cresol methylhydroxylase (PCMH) structural family
Structural conservation
The third structural family of FAD-binding proteins is exemplified by p-cresol methylhydroxylase (PCMH); all family members are listed in Table 1 (Benson et al. 1997; Dobbek et al. 1999; Cunane et al. 2000; Dym et al. 2000; Fraaije et al. 2000). These enzymes catalyze diverse redox reactions. The FAD-binding domain consists of two α + β subdomains: one is composed of three parallel β-strands (β1–3) surrounded by α-helices, and is packed against the second subdomain containing five antiparallel β-strands (β4–8) surrounded by α-helices (Fig. 4C ▶). A schematic diagram of the topological arrangement of the secondary structure of a PCMH family representative is shown in Figure 3C ▶.
FAD binding and conformation
The binding pocket for the FAD cofactor is essentially identical for all family members. The two subdomains accommodate the FAD cofactor between them (Fig. 4C ▶). They envelope most of the FAD cofactor and interact with it by hydrogen bonds and van der Waals contacts. In fact, the extended opening between the two subdomains in PCMH family members allows the FAD to penetrate in between them and adopt an elongated conformation (Table 1). In all family members, the adenine ring points towards the FAD-binding domain (Fig. 4C ▶). Although the conformation of FAD is conserved, it may be either covalently or noncovalently bound to the protein (Table 1).
Conserved sequence motifs
Although the sequence conservation of the PCMH family along the entire alignment is very small, members share some conserved residues in the N-terminal and C-terminal regions comprising the FAD-binding domain. Sequence-structure alignment is shown in Figure 7 ▶, and conserved sequence motifs are listed in Table 1. One of the most conserved and well studied residues is an Arg residue situated at the C terminus which is within hydrogen bonding distance of both the O2 atom of the isoalloxazine ring and O3* of the ribitol, to provide charge compensation (Fig. 7 ▶, pink box). An additional most conserved sequence motif is P(x)6G(A)xN (Fig. 7 ▶, blue box) in which the middle x residue is within hydrogen bonding distance of OP1 or OP2 and forms a coil segment connecting β2 to α1 (Fraaije et al. 1998) (Fig. 3C ▶). This loop binds to the adenine ring and compensates for the negative charge of the FAD molecule through the formation of hydrogen bonds between backbone nitrogen atoms of Gly and some other residues belonging to the sequence motif. Another well-studied conserved sequence is G(x)8GY (Fig. 6 ▶, green box) in which the x residues are part of α2 and are within hydrogen bonding distance of the pyrophosphate and the isoalloxazine moieties (Fraaije et al. 1998) (Fig. 3c ▶). Similar to the FR and PO family members, the negatively charged pyrophosphate group of the FAD is compensated by a hydrogen bond to a positive residue (Arg if it is present) or by the backbone to an amide group (in this sequence motif, a Gly residue). In contrast to some other flavoproteins (e.g., Mande et al. 1996; Mizutani et al. 1996; Sridhar Prasad et al. 1998; Yue et al. 1999) however, there is no helix dipole oriented toward the pyrophosphate moiety to help compensate for the negative charge.
Fig. 7.
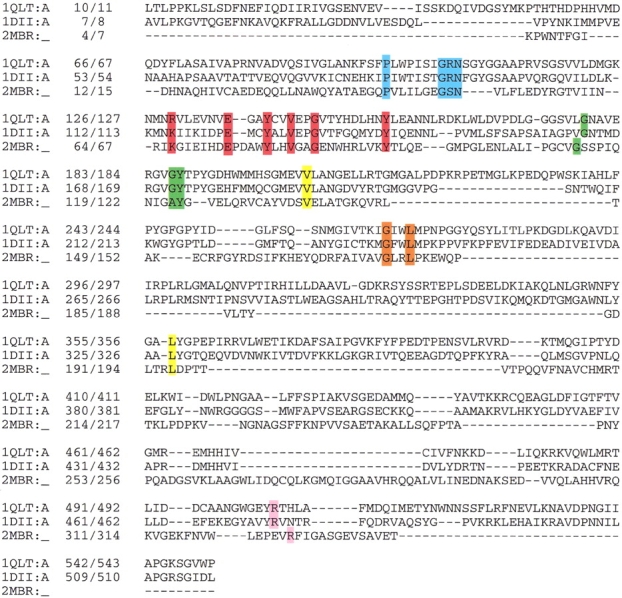
The sequence alignment of FAD-containing proteins resembles the p-cresol methylhydroxylase structure. Conserved sequence motifs are depicted in boxes: R(x)6ExxYxxVxxG(x)8Y in red, P(x)6G(A)xN in blue, GxxL in orange, R in pink, and G(x)8GY in green. The data bank accession code and position numbers according to the sequence and according to the PDB are given. Proteins: 1DII, p-cresol methylhydroxylase from Pseudomonas putida; 1QLT, vanillyl-alcohol oxidase from Penicillium simplicissimum; 2MBR, uridine diphospho-N-acetylenolpyruvylglucosamine reductase from Escherichia coli.
Based on our sequence-structure analysis, we identified two partially conserved sequence motifs. The first is GxxL, part of β8, which interacts with the adenine ring (Fig. 7 ▶, orange box). The second motif we found is R(x)6ExxYxxVxxG(x)8Y (Fig. 7 ▶, red box), part of β4 and β5 with the Glu, Tyr, and Gly residues conserved. The central part of this motif, VxxG, is located near the adenine ring and is in close proximity to the GxxL and P(x)6G(A)xN sequence motifs.
The pyruvate oxidase (PO) structural family
Structural conservation
Pyruvate oxidase (PO) was previously considered a single-member family (Muller et al. 1994). We found that electron transfer flavoprotein (Roberts et al. 1996) has a similar fold and shares several conserved sequence motifs with PO (Table 1). In both proteins, the FAD-binding domain fold is similar to the Rossmann fold of the dehydrogenases that binds NADH (Rossmann et al. 1974) (Fig. 3D ▶ and Fig. 1 ▶, respectively). The basic structure consists of five instead of six parallel β-strands (β1–5) interspersed by α-helices that lie on both sides of the five-stranded β-sheet.
FAD binding and conformation motifs
The FAD cofactor adopts an elongated conformation and lies roughly perpendicular to the core β-strands (Fig. 4D ▶, Table 1). In contrast to NADH in dehydrogenases, the FAD cofactor in the PO structural family is bound in the opposite direction with the adenosine moiety at the second βαβαβ unit rather than at the first such unit. In addition, the conserved motif GxGxxG found in both the FAD-binding domain in the GR family and in NADH-binding protein in dehydrogenases is absent in the PO family. A characteristic of the FAD-binding site in the PO family members involves internal hydrogen bonding in the FAD molecule between the ribityl 4′-hydroxyl group and N1 of the isoalloxazine ring (Roberts et al. 1999).
Conserved sequence
Although the determination of the consensus sequence motif must await the identification of the structures of similar proteins, possible conserved motifs, based on the sequence-structure alignment, are proposed in Figure 8 ▶ and Table 1. The more conserved sequence motif KxLxxLxxxL(x)6S(T)(x)6GxV (Fig. 7 ▶, red box), is located in α1 and β2, which are part of the βαβαβ Rossmann fold (Fig. 3D ▶). The Thr or Ser are within hydrogen bonding distance of the OP1 or OP2 atoms of the pyrophosphate moiety. An additional conserved sequence motif is RxxxxxGxG (Fig. 8 ▶, blue box), in which the backbone nitrogen atom from the last x residue, Arg in the case of electron transfer flavoprotein and Ile in pyruvate oxidase, interacts with the pyrophosphate moiety for charge compensation. These two conserved sequence motifs are potentially phosphate-binding motifs in the PO protein family. Another conserved motif is VGxS(N) (Fig. 8 ▶, green box), which is part of the β3 of the second Rossmann fold, where Ser in electron transfer flavoprotein and Asn in pyruvate oxidase interact with the pyrophosphate group (Fig. 3D ▶). It is noteworthy that although the residue hydrogen bonded to OP1 or OP2 atoms is different in the two proteins, they both align well in the sequence-structure alignment (Fig. 8 ▶). Moreover, residues in the electron transfer flavoprotein family are highly conserved (Roberts et al. 1999). These include Gly 223 and Gly 225, part of RxxxxxGxG; Ser 249, part of KxLxxLxxxL(x)6S(T)(x)6GxV; and Ser 282, part of VGxS(N). An additional conserved motif, K(x)7IxxDP(x)9 D(x)4AD(x)2K (Fig. 8 ▶, pink box), is part of α3β4 of the second Rossmann fold, in which the first Asp residue interacts with the adenine ring (Fig. 3D ▶).
Fig. 8.

Structure-sequence alignment for the two members of the PO family fold as derived from the program CE (Shindyalov and Bourne 1998). Potential conserved sequence motifs are shown in boxes: KxLxxLxxxL(x)6S(T)(x)6GxV in red, RxxxxxGxG in blue, VGxS(N) in green, and K(X)7IXXDP(x)9D(X)4AD(X)2K in pink. The data bank accession code and position numbers according to the sequence (starting from one) and according to the PDB are given. Proteins: 1POW, pyruvate oxidase from Lactobacillus plantarum; 1EFV, electron transfer flavoprotein from Homo sapiens.
Single-member FAD folds
There are four FAD-containing proteins (the last four proteins listed in Table 1) with different folds, all distinct from the above four family folds. Our amino acid sequence analysis revealed that none of them contains the conserved FAD-binding sequence motifs identified in the FAD families.
The FAD-binding domain fold of Acyl coenzyme A dehydrogenase (MCAD) is composed of three domains. Domains 1 and 3 are mostly α-helical and are closely associated. Domain 2 consists of a four-stranded parallel β-sheet sandwich between three-stranded antiparallel β-sheets (Tiffany et al. 1997). The FAD in the enzyme has an elongated conformation.
The structure of methylenetetrahydrofolate reductase is a β8α8-barrel, which resembles the structure of flavin mononucleotide (FMN)-binding proteins rather than FAD-binding proteins. However, the FAD cofactor binds to a different part of the FAD-binding domain compared to the barrels that bind FMN (Guenther et al. 1999).
In DNA photolyase, the FAD-binding domain is composed of 14 α-helices grouped into two clusters with the FAD-binding site in between (Park et al. 1995). Cluster I comprises α7 to α11 and contains all residues that interact with the phosphate oxygen of the FAD. Cluster II consists of α12 to α20, and residues within this bundle have polar interactions with the riboflavin part of the FAD. The catalytic cofactor FAD is bound to photolyase in a U-shaped conformation with the isoalloxazine and adenine rings in close proximity.
The topology of the FAD-binding domain in quinone oxidoreductase is composed of a twisted central parallel β-sheet surrounded on both sides by connecting helices (Foster et al. 1999). Although at first glance it seems that this fold is reminiscent of the dehydrogenase fold or the PO family fold, the exact topology differs. The most prominent difference is the absence of the conserved FAD-binding sequence motifs identified in dehydrogenases and in the PO family (Smith et al. 1977).
Identifying phosphate-binding sites
In attempts to detect phosphate analog-binding sites, we used conserved phosphate-binding sequence motifs identified in the FAD-family folds as a query in searching the Protein Data Bank. This search was carried on using the option FINDPATTERNS in the GCG server (Williams and Gibbs 1990). Specifically, there are nine frequently occurring conserved phosphate-binding sequence motifs: GxGxxG(x)17–19E(D) and T(S)(x)5F(Y)hhGD(E) from the GR protein family, RxYS(T) and GxxS(T)xxL(x)5G found in the FR family, P(x)6G(A)xN and R(x)6ExxYxxV from the PCMH family, and K(R)xLxxLxxxL, R(x)5GxG, and K(R)(x)7IxxDP(x)9D from the PO family.
Forty nonFAD-containing protein structures were identified from the above query sequence motifs, each containing a phosphate-containing ligand. Approximately half of these protein structures contain a ligand closely related to the FAD cofactor (e.g., NAD(P)H, ATP, and FMN), and most sequences contain the dinucleotide-binding motif GxGxxG(x)17–19E of the GR family, known to bind these cofactors (Schulz 1992). Interestingly, two of these structures were solved in the presence of NAD(P)H and contain one of our newly derived phosphate-binding sequence motifs from the new PO family, adopting a fold similar to the Rossmann fold of dehydrogenases that bind NAD(P)H (Table 2). The structures of the remaining half (∼20) of the phosphate-containing proteins were solved in the presence of ligands distantly related to FAD cofactor. Nevertheless, in only half of them is the phosphate-containing ligand within hydrogen bonding or van der Waals distance of the phosphate-binding sequence motif (Table 2). In all examples, the pyrophosphate moiety of the cofactor forms hydrogen bonds to residues from the phosphate-binding motifs. In addition, in phospholipase A2 and basic fibroblast growth factor, the phosphate-binding motif interacts with more than one phosphate-containing ligand (Table 2).
Table 2.
Results of a search of the Protein Data Bank for phosphate containing ligands binding to proteins, with known three-dimensional structures, containing one of the pyrophosphate-binding consensus motifs identified in the FAD-family folds of Table 1
| Sequence Motif | FAD fold | PDB Code | Protein name | Phosphate-containing ligands |
| GxGxxG(x)17–19E | GR | 1agr | Guanine nucleotide-binding protein | guanosine-5′-diphosphate |
| GR | 1vip | Phospholipase A2 | SO4 | |
| GR | 1pbo | Phospholipase A2 | 1-o-octyl-2-heptylphosphonyl-sn-glycerole-3-3-phosphoethanolamin | |
| GR | 1gia | Gi α1 | 5′-guanosine-diphosphate monothiophosphate | |
| GR | 1csn | Casein Kinase-1 | ATP, SO4 | |
| GR | 1pgn | 6-phosphogluconate dehydrogenase | pyrophosphate 2 | |
| GR | 2cae | Catalase | S-dioxyneyhionine | |
| RxxxxxGxG | PO | 1kvr | Udp-galactose 4-epimerase | uridine-5′-diphosphate, NADH |
| K(x)7IxxD(x)10D | PO | 1bfb | Basic Fibroblast Growth Factor | N,O6-disulfoglucosamine, 1,4-dideoxy-o2-sulfo-glucuronic acid, 1,4-dideoxy-5-dehydro-o2-sulfo-glucuronic acid, SO4 |
| PO | 1gts | Glutaminyl-tRNA synthetase | AMP, ATP | |
| PO | 1ad3 | Aldehyde dehydrogenase | NADH | |
| KxLxxLxxxL | PO | 1tpu | Triosephosphate isomerase | Phosphoglycolohydroxamic acid |
| PO | 1cwn | Aldehyde reductase | NADPH | |
| GxSxxL(x)5G | FD | 1ajr | Aspartate transaminase | 2-lysine(3-hydroxy-2-2-methyl-5 phosphonooxymethyl-pyridin-4 ylmethane) |
Discussion
FAD-family folds
Protein motif identification has become an essential part of sequence analysis in general and genome research in particular. FAD-containing proteins offer a useful case study, as the three-dimensional structures of 32 different flavoproteins are known to atomic resolution and their folding pattern appears much more similar than would be suggested by comparison of their sequences. Our comparative sequence-structure study of these proteins revealed four different FAD-folds as well as various conserved sequence motifs distinguished from one fold to another. Three of the family folds, GR, FR, and PCMH, were previously defined, while PO is newly derived. Among all of the conserved sequence motifs listed in Table 1, D(x)6GxxP in the GR family, R(x)6ExxYxxVxxG(x)8Y and GxxL in the PCMH family, and all four in the PO family are newly derived. In addition, there are four proteins that exhibit FAD-binding domains with folds different from the ones displayed by the four FAD families. Conserved sequence motifs identified in the FAD families are absent in these proteins.
The FAD-binding domain folds adopted by the FAD families vary. The largest family is represented by GR enzymes, and all family members adopt the βαβαβ fold known as the Rossmann fold. A related fold is found in the newly derived PO family members, adopting a fold similar to the double (βαβαβ and βαβαβ) Rossmann fold found in dehydrogenases. Nevertheless, sequence-structure homology revealed that their sequences diverge greatly and in fact share no conserved sequence motifs. This could potentially be explained by the finding that in contrast to NADH in dehydrogenases, the FAD cofactor in the PO structural family is bound in the opposite direction with the adenosine moiety at the second βαβαβ unit rather than at the first. Although NADH and FAD share moieties, they are not identical and would therefore be expected to interact with different parts of the polypeptide chain. Therefore, despite the topological and functional similarities of FAD-binding domains found in PO family members to NAD(P)H-binding domains found in dehydrogenases, a divergent evolutionary relationship between these structures cannot be assumed. The FAD-binding fold adopted by the FR and PCMH family members is composed of a cylindrical β-domain and two α + β subdomains respectively. Both folds are distinct from the Rossmann fold.
Atomic interactions of cofactor with proteins
For each FAD family we analyzed the correlation between the fold and various cofactor properties. Although some of the FAD properties are conserved among proteins belonging to the same FAD-family fold, others are not. One of the least conserved properties is the bond type (covalent versus noncovalent). When there is covalent interaction, FAD is bound to protein residues through atoms from the isoalloxazine ring. This is in agreement with the observation that the isoalloxazine ring interacts with residues located in the poorly conserved regions along the polypeptide sequence.
Another less conserved cofactor property is the number of protein residues–cofactor atoms interactions. Whereas all proteins studied have at least one contact with the pyrophosphate moiety (OP1 or OP2 atoms), the number of contacts varies from one protein to another and there is no strong preference for a particular number of contacts in the different families. This observation is reinforced by the variety of conserved sequence motifs present in the different family folds. Moreover, some members of a FAD-fold family contain all conserved sequence motifs whereas others contain only some.
A partially conserved property is the geometry of the FAD, which is controlled by the surrounding protein as either an elongated or bent-butterfly conformation. The cofactor conformation in most of the FAD-family folds is elongated with slight flexibility that varies from one protein to another. In the FR family, the cofactor adopts either an elongated or bent conformation (Table 1). This variation can be explained by the observation that although the general fold of the individual FAD- and NAD(P)H-binding domains in the FR family is invariant, their relative orientation differs among the family members. As a result the FAD, situated at the junction between the two domains, can adopt either an elongated or bent conformation depending on the proximity of the two domains. In contrast, in the PCMH family, the FAD-binding domain is composed of two subdomains distant from each other, which accommodate the FAD in between them. In fact the FAD holds the two subdomains together and therefore can adopt only an elongated conformation. Similarly in the PO family, the FAD molecule is situated on top of the core β-strands and adopts an elongated conformation for maximal interaction with the FAD-binding domain. Therefore, it seems that the fold of the FAD-binding domain and the cofactor conformation are not independent, because the fold determines the position and shape of the pocket on which the cofactor conformation depends. This variability in the cofactor conformation and shape of cofactor pocket in the FAD-binding domain of the different FAD-family folds implies that there is no single protein `pharmacophore' for binding FAD.
One of the most conserved properties of the FAD is the direction in which it approaches the FAD-binding domain. In all FR family members, the isoalloxazine ring points toward the FAD-binding domain, whereas in the rest of the FAD-family folds it is the adenosine moiety that does so.
Cofactor atoms interacting with conserved sequence motifs
Another characteristic of the cofactor is evident from our analysis of the correlation between FAD atoms interacting with the conserved sequence motifs identified for each of the families. These conserved sequence motifs are distinctive in the four FAD families. Nevertheless, residues from the most conserved sequence motifs in a specific family fold make hydrophobic and/or hydrophilic contacts with cofactor atoms. In contrast, residues from partially conserved sequence motifs are distant (>4.0Å) from cofactor atoms and therefore probably form at best weak interactions with the cofactor. Residues involved in binding oxygen atoms (OP1 and OP2) of the pyrophosphate moiety are more highly conserved relative to the alignment as a whole, and in fact are part of the most conserved sequence motifs. In contrast, residues involved in binding the adenine, ribose, ribitol, and isoalloxazine moieties are part of less conserved regions. Because the pyrophosphate moiety is negatively charged, it is expected to bind to regions in the protein with positive charges. This can be achieved by interacting with positively charged amino acids, peptide groups, or α-helix dipoles. Notably, when there is no helix to stabilize the negative charge of the pyrophosphate group, Arg is the most common residue and is capable of forming multiple hydrogen bonds with the pyrophosphate moiety. When, however, there is an α-helix dipole, as observed in most of the GR family members, Gly is the most common amino acid, and its main chain is within hydrogen bonding distance of the pyrophosphate group.
In contrast to the conserved pyrophosphate binding, the residues involved in binding the isoalloxazine and adenosine moieties are usually part of the partially conserved sequence motifs. This is consistent with the fact that the catalytic function of FAD is restricted to the isoalloxazine ring, and with the variety of family members involved in diverse redox reactions. Consequently, diverse residues from the different proteins are involved in catalytic function and contact the isoalloxazine ring. Similarly, the function of the adenosine moiety is mainly to stabilize cofactor binding through interaction with the protein. This stability can be achieved by interaction with different residues in the different proteins belonging to the same FAD-family fold.
The variety of redox functions may also explain the variability in cofactor conformation observed in the FAD families. As mentioned above, the flexibility is achieved by movement of the adenine ring toward or away from the isoalloxazine ring. As a result, the two rings can interact with different parts of the protein, usually in less conserved regions. We therefore propose that the pyrophosphate moiety anchors the FAD-binding domain by binding to the most conserved sequence motif, with less specific binding of the isoalloxazine and adenine rings to less conserved sequence motifs. This is consistent with the observation that some of the most conserved sequence motifs described above are known to bind ATP, FMN, and GTP as well. They all share a common pyrophosphate group. This observation further implies that it is possible to predict the pyrophosphate-binding site by identifying the most conserved sequence motif in a multiple alignment of other prosthetic groups containing pyrophosphate moieties such as AMP, FMN, and GTP. It is conceivable to assume that inspection of the residues interacting with the OP1 or OP2 atoms can be used as a tool for identifying the most conserved sequence motif in the FAD-containing proteins adopting a unique fold, as for the four single-member (SM) protein families of Table 1.
These conserved phosphate-binding sequence motifs have a predictive use: they can be applied to searches for phosphate analog-containing proteins. In fact, we identified a dozen nonFAD-containing proteins that bind phosphate-containing ligands as cofactors (Table 2). Each contains one of the phosphate-binding motifs assigned for the FAD families. These motifs are within hydrogen bond distance of the phosphate moiety of the phosphate-containing ligand. Interestingly, three of the phosphate analog ligands were actually sulphate ions. This implies that conserved sulfate- and sulphate analog-binding sequence motifs could be considered potential binding sites for phosphate ions.
Conclusion
The FAD-dependent enzymes illustrate how similar cofactors are utilized by nature in a wide variety of protein families. The availability of the three-dimensional structures of a large collection of FAD-containing proteins provides an attractive opportunity for sequence-function-structure analysis. Four different FAD-family folds, each with distinctive conserved sequence motifs were identified. Furthermore, the FAD pocket shape can be distinguished from one fold to another as well, implying that there is no unique `pharmacophore' for binding FAD. In contrast to the diversity of the FAD fold and its sequence motifs, the pyrophosphate moiety makes hydrogen bonds with residues from the most conserved sequence motifs in each fold family. Therefore, we propose that the pyrophosphate moiety is crucial for molecular recognition, whereas the isoalloxazine ring is involved in catalytic function, and the adenine ring stabilizes cofactor binding, both rings interacting with protein residues in partially conserved sequence motifs. As a result, the isoalloxazine and adenine rings interact with different residues in the different proteins belonging to the same FAD-family fold. This is consistent with the variability in bond type (covalent versus noncovalent) and cofactor conformation (elongated versus bent) observed in the FAD-family folds. Nonetheless, in each FAD-family fold there exists a clear correlation between the fold, the shape and position of the pocket, and the cofactor conformation. This is in agreement with the finding that the cofactor exhibits directionality, which is highly conserved within a family fold.
An unexpected outcome of our analysis is that the conserved pyrophosphate-binding sequence motifs that were identified in FAD-dependent proteins may be applied to discover phosphate-binding motifs in sequences of nonFAD-containing proteins. Where the structures of these proteins are known, the identified motifs are found to bind to phosphate- or sulfate-containing ligands. Building on this result, the sequence motifs found to bind to pyrophosphate in four single-membered FAD protein families (last four entries in Table 1) may serve to identify new members of these families.
Materials and methods
Sequences and three-dimensional structures of all flavoproteins were retrieved from the Protein Data Bank (www.rcsb.org/pdb). A total of 150 flavoprotein X-ray structures solved in the presence of FAD were available from the October 2000 release, and consist of 32 nonredundant proteins. Table 1 lists the proteins studied, data bank accession codes, and references to one of the structural papers on each protein. Homology searching for identifying three-dimensional similarities in protein structure was performed using the program Combinatorial Extension (CE) (http://cl.sdsc.edu/ce.html). The program aligns two or more polypeptide chains using characteristics of their local geometry as defined by vectors between Cα positions (Shindyalov and Bourne 1998). Each of the 32 proteins was used as a query in the sequence-structure search, resulting in the identification of four FAD-family folds. Based on this sequence-structure alignment, conserved sequence motifs unique for each of the FAD families were identified (Table 1). Next, protein residues forming van der Waals interactions and hydrogen bond contacts with the cofactor were analyzed using the program LigPlot (Wallace et al. 1995). We then correlated these protein residues with residues from the most conserved sequence motif. Finally, each of the conserved sequence motifs was used as query in a search of the Protein Data Bank for nonFAD-containing proteins solved in the presence of phosphate-containing ligands. This search was aided by the option FINDPATTERNS on the Genetic Computer Group (GCG) server (Williams and Gibbs 1990).
Acknowledgments
We thank Dr. I. Xenorios for discussion and NIH and DOE for support.
Abbreviations
FAD, flavin adenine dinucleotide
FMN, flavin mononucleotide
ATP, adenosine triphosphate
NADH, nicotinamide adenine dinucleotide
NADPH, nicotinamide adenine dinucleotide phosphate
GR, glutathione reductase
FR, ferredoxin reductase
PO, pyruvate oxidase
PCMH, p-cresol methylhydroxylase
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.12801.
References
- Bacon, D.J. and Anderson, W.F. 1988. A fast algorithm for rendering space-filling molecule pictures. J. Mol. Graph. 6 219–220. [Google Scholar]
- Benson, T.E., Walsh, C.T., and Hogle, J.M. 1997. X-ray crystal structures of the S229A mutant and wild-type MurB in the presence of the substrate enolpyruvyl-UDP-N-acetylglucosamine at 1.8-Å resolution. Biochemistry 36 806–811. [DOI] [PubMed] [Google Scholar]
- Binda, C., Coda, A., Angelini, R., Federico, R., Ascenzi, P., and Mattevi, A. 1999. A 30-angstrom-long U-shaped catalytic tunnel in the crystal structure of polyamine oxidase. Structure Fold. Des. 7 265–276. [DOI] [PubMed] [Google Scholar]
- Bond, C.S., Zhang, Y., Berriman, M., Cunningham, M.L., Fairlamb, A.H., and Hunter, W.N. 1999. Crystal structure of Trypanosoma cruzi trypanothione reductase in complex with trypanothione, and the structure-based discovery of new natural product inhibitors. Structure Fold. Des. 7 81–89. [DOI] [PubMed] [Google Scholar]
- Chen, Z.W., Koh, M., Van Driessche, G., Van Beeumen, J.J., Bartsch, R.G., Meyer, T.E., Cusanovich, M.A., and Mathews, F.S. 1994. The structure of flavocytochrome c sulfide dehydrogenase from a purple phototrophic bacterium. Science 266 430–432. [DOI] [PubMed] [Google Scholar]
- Correll, C.C., Ludwig, M.L., Bruns, C.M., and Karplus, P.A. 1993. Structural prototypes for an extended family of flavoprotein reductases: Comparison of phthalate dioxygenase reductase with ferredoxin reductase and ferredoxin. Protein Sci. 2 2112–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunane, L.M., Chen, Z.W., Shamala, N., Mathews, F.S., Cronin, C.N., and McIntire, W.S. 2000. Structures of the flavocytochrome p-cresol methylhydroxylase and its enzyme-substrate complex: Gated substrate entry and proton relays support the proposed catalytic mechanism. J. Mol. Biol. 295 357–374. [DOI] [PubMed] [Google Scholar]
- Dobbek, H., Gremer, L., Meyer, O., and Huber, R. 1999. Crystal structure and mechanism of CO dehydrogenase, a molybdo iron-sulfur flavoprotein containing S-selanylcysteine. Proc. Natl. Acad. Sci. 96 8884–8889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle, R.F. 1986. Of URFs and ORFs: A primer on how to analyse derived amino acid sequences, University Science Books, Mill Valley, CA.
- Dym, O., Pratt, E.A., Ho, C., and Eisenberg, D. 2000. The crystal structure of D-lactate dehydrogenase, a peripheral membrane respiratory enzyme. Proc. Natl. Acad. Sci. 97 9413–9418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggink, G., Engel, H., Vriend, G., Terpstra, P., and Witholt, B. 1990. Rubredoxin reductase of Pseudomonas oleovorans. Structural relationship to other flavoprotein oxidoreductases based on one NAD and two FAD fingerprints. J. Mol. Biol. 212 135–142. [DOI] [PubMed] [Google Scholar]
- Enroth, C., Neujahr, H., Schneider, G., and Lindqvist, Y. 1998. The crystal structure of phenol hydroxylase in complex with FAD and phenol provides evidence for a concerted conformational change in the enzyme and its cofactor during catalysis. Structure 6 605–617. [DOI] [PubMed] [Google Scholar]
- Eppink, M.H., Schreuder, H.A., and van Berkel, W.J. 1998. Lys42 and Ser42 variants of p-hydroxybenzoate hydroxylase from Pseudomonas fluorescens reveal that Arg42 is essential for NADPH binding. Eur. J. Biochem. 253 194–201. [DOI] [PubMed] [Google Scholar]
- Ermler, U., Siddiqui, R.A., Cramm, R., and Friedrich, B. 1995. Crystal structure of the flavohemoglobin from Alcaligenes eutrophus at 1.75 Å resolution. EMBO J. 14 6067–6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster, C.E., Bianchet, M.A., Talalay, P., Zhao, Q., and Amzel, L.M. 1999. Crystal structure of human quinone reductase type 2, a metalloflavoprotein. Biochemistry 38 9881–9886. [DOI] [PubMed] [Google Scholar]
- Fraaije, M.W., Van Berkel, W.J., Benen, J.A., Visser, J., and Mattevi, A. 1998. A novel oxidoreductase family sharing a conserved FAD-binding domain [published erratum appears in Trends Biochem. Sci. 1998 Jul;23(7):271]. Trends Biochem. Sci. 23 206–207. [DOI] [PubMed] [Google Scholar]
- Fraaije, M.W., van Den Heuvel, R.H., van Berkel, W.J., and Mattevi, A. 2000. Structural analysis of flavinylation in vanillyl-alcohol oxidase. J. Biol. Chem. 275 38654–38658. [DOI] [PubMed] [Google Scholar]
- Guenther, B.D., Sheppard, C.A., Tran, P., Rozen, R., Matthews, R.G., and Ludwig, M.L. 1999. The structure and properties of methylenetetrahydrofolate reductase from Escherichia coli suggest how folate ameliorates human hyperhomocysteinemia. Nat. Struct. Biol. 6 359–365. [DOI] [PubMed] [Google Scholar]
- Ingelman, M., Bianchi, V., and Eklund, H. 1997. The three-dimensional structure of flavodoxin reductase from Escherichia coli at 1.7 A resolution. J. Mol. Biol. 268 147–157. [DOI] [PubMed] [Google Scholar]
- Karplus, P.A., Daniels, M.J., and Herriott, J.R. 1991. Atomic structure of ferredoxin-NADP+ reductase: Prototype for a structurally novel flavoenzyme family. Science 251 60–66. [PubMed] [Google Scholar]
- Karplus, P.A. and Schulz, G.E. 1987. Refined structure of glutathione reductase at 1.54 Å resolution. J. Mol. Biol. 195 701–729. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Cryst. 24 946–950. [Google Scholar]
- Lennon, B.W., Williams, C.H., Jr., and Ludwig, M.L. 1999. Crystal structure of reduced thioredoxin reductase from Escherichia coli: Structural flexibility in the isoalloxazine ring of the flavin adenine dinucleotide cofactor. Protein Sci. 8 2366–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leys, D., Tsapin, A.S., Nealson, K.H., Meyer, T.E., Cusanovich, M.A., and Van Beeumen, J.J. 1999. Structure and mechanism of the flavocytochrome c fumarate reductase of Shewanella putrefaciens MR-1. Nat. Struct. Biol. 6 1113–1117. [DOI] [PubMed] [Google Scholar]
- Lu, G., Campbell, W.H., Schneider, G., and Lindqvist, Y. 1994. Crystal structure of the FAD-containing fragment of corn nitrate reductase at 2.5 Å resolution: Relationship to other flavoprotein reductases. Structure 2 809–821. [DOI] [PubMed] [Google Scholar]
- Mande, S.S., Sarfaty, S., Allen, M.D., Perham, R.N., and Hol, W.G. 1996. Protein–protein interactions in the pyruvate dehydrogenase multienzyme complex: Dihydrolipoamide dehydrogenase complexed with the binding domain of dihydrolipoamide acetyltransferase. Structure 4 277–286. [DOI] [PubMed] [Google Scholar]
- Mathews, F.S. 1991. New flavoenzymes. Curr. Opin. Struct. Biol. 1 954–967. [Google Scholar]
- Merritt, E.A. and Murphy, M.E.P. 1994. Raster3D Version 2.0 — A program for photorealistic molecular graphics. Acta. Crystallogr. D. Biol. Crystallogr. 50 869–873. [DOI] [PubMed] [Google Scholar]
- Mizutani, H., Miyahara, I., Hirotsu, K., Nishina, Y., Shiga, K., Setoyama, C., and Miura, R. 1996. Three-dimensional structure of porcine kidney D-amino acid oxidase at 3.0 Å resolution. J. Biochem. (Tokyo) 120 14–17. [DOI] [PubMed] [Google Scholar]
- Moller, W. and Amons, R. 1985. Phosphate-binding sequences in nucleotide-binding proteins. FEBS Lett. 186 1–7. [DOI] [PubMed] [Google Scholar]
- Muller, Y.A., Schumacher, G., Rudolph, R., and Schulz, G.E. 1994. The refined structures of a stabilized mutant and of wild-type pyruvate oxidase from Lactobacillus plantarum. J. Mol. Biol. 237 315–335. [DOI] [PubMed] [Google Scholar]
- Nishida, H., Inaka, K., Yamanaka, M., Kaida, S., Kobayashi, K., and Miki, K. 1995. Crystal structure of NADH-cytochrome b5 reductase from pig liver at 2.4 Å resolution. Biochemistry 34 2763–2767. [DOI] [PubMed] [Google Scholar]
- Park, H.W., Kim, S.T., Sancar, A., and Deisenhofer, J. 1995. Crystal structure of DNA photolyase from Escherichia coli [see comments]. Science 268 1866–1872. [DOI] [PubMed] [Google Scholar]
- Rao, S.T., and Rossmann, M.G. 1973. Comparison of super-secondary structures in proteins. J. Mol. Biol. 76 241–256. [DOI] [PubMed] [Google Scholar]
- Roberts, D.L., Frerman, F.E., and Kim, J.J. 1996. Three-dimensional structure of human electron transfer flavoprotein to 2.1-Å resolution. Proc. Natl. Acad. Sci. 93 14355–14360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, D.L., Salazar, D., Fulmer, J.P., Frerman, F.E., and Kim, J.J. 1999. Crystal structure of Paracoccus denitrificans electron transfer flavoprotein: Structural and electrostatic analysis of a conserved flavin binding domain. Biochemistry 38 1977–1989. [DOI] [PubMed] [Google Scholar]
- Rossmann, M.G., Moras, D., and Olsen, K.W. 1974. Chemical and biological evolution of nucleotide-binding protein. Nature 250 194–199. [DOI] [PubMed] [Google Scholar]
- Schulz, G.E. 1992. Binding of nucleotides by proteins.Curr. Opin. Struct Biol. 2 61–67. [Google Scholar]
- Schulz, G.E. and Schirmer, R.H. 1974. Topological comparison of adenyl kinase with other proteins. Nature 250 142–144. [DOI] [PubMed] [Google Scholar]
- Schulz, G.E., Schirmer, R.H., and Pai, E.F. 1982. FAD-binding site of glutathione reductase. J. Mol. Biol. 160 287–308. [DOI] [PubMed] [Google Scholar]
- Shindyalov, I.N. and Bourne, P.E. 1998. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 11 739–747. [DOI] [PubMed] [Google Scholar]
- Smith, W.W., Burnett, R.M., Darling, G.D., and Ludwig, M.L. 1977. Structure of the semiquinone form of flavodoxin from Clostridum MP. Extension of 1.8 Å resolution and some comparisons with the oxidized state. J. Mol. Biol. 117 195–225. [DOI] [PubMed] [Google Scholar]
- Sridhar Prasad, G., Kresge, N., Muhlberg, A.B., Shaw, A., Jung, Y.S., Burgess, B.K., and Stout, C.D. 1998. The crystal structure of NADPH: Ferredoxin reductase from Azotobacter vinelandii. Protein Sci. 7 2541–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiffany, K.A., Roberts, D.L., Wang, M., Paschke, R., Mohsen, A.W., Vockley, J., and Kim, J.J. 1997. Structure of human isovaleryl-CoA dehydrogenase at 2.6 Å resolution: Structural basis for substrate specificity. Biochemistry 36 8455–8464. [DOI] [PubMed] [Google Scholar]
- Trickey, P., Wagner, M.A., Jorns, M.S., and Mathews, F.S. 1999. Monomeric sarcosine oxidase: Structure of a covalently flavinylated amine oxidizing enzyme. Structure Fold. Des. 7 331–345. [DOI] [PubMed] [Google Scholar]
- Vallon, O. 2000. New sequence motifs in flavoproteins: Evidence for common ancestry and tools to predict structure. Proteins 38 95–114. [DOI] [PubMed] [Google Scholar]
- Van Driessche, G., Koh, M., Chen, Z.W., Mathews, F.S., Meyer, T.E., Bartsch, R.G., Cusanovich, M.A., and Van Beeumen, J.J. 1996. Covalent structure of the flavoprotein subunit of the flavocytochrome c: Sulfide dehydrogenase from the purple phototrophic bacterium Chromatium vinosum. Protein Sci. 52753–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace, A.C., Laskowski, R.A., and Thornton, J.M. 1995. LIGPLOT: A program to generate schematic diagrams of protein–ligand interactions. Protein Eng. 8 127–134. [DOI] [PubMed] [Google Scholar]
- Wang, M., Roberts, D.L., Paschke, R., Shea, T.M., Masters, B.S., and Kim, J.J. 1997. Three-dimensional structure of NADPH-cytochrome P450 reductase: Prototype for FMN- and FAD-containing enzymes. Proc. Natl. Acad. Sci. 94 8411–8416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga, R.K., Drenth, J., and Schulz, G.E. 1983. Comparison of the three-dimensional protein and nucleotide structure of the FAD-binding domain of p-hydroxybenzoate hydroxylase with the FAD- as well as NADPH-binding domains of glutathione reductase. J. Mol. Biol. 167 725–739. [DOI] [PubMed] [Google Scholar]
- Wierenga, R.K., Terpstra, P., and Hol, W.G. 1986. Prediction of the occurrence of the ADP-binding beta alpha beta-fold in proteins, using an amino acid sequence fingerprint. J. Mol. Biol. 187 101–107. [DOI] [PubMed] [Google Scholar]
- Williams, G.W., and Gibbs, G.P. 1990. Automatic updating of the EMBL database via EMBNet. Comput. Appl. Biosci. 6 122–123. [DOI] [PubMed] [Google Scholar]
- Wohlfahrt, G., Witt, S., Hendle, J., Schomburg, D., Kalisz, H.M., and Hecht, H.J. 1999. 1.8 and 1.9 A resolution structures of the Penicillium amagasakiense and Aspergillus niger glucose oxidases as a basis for modelling substrate complexes. Acta Crystallogr. D. Biol. Crystallogr. 55 969–977. [DOI] [PubMed] [Google Scholar]
- Yeh, J.I., Claiborne, A., and Hol, W.G. 1996. Structure of the native cysteine-sulfenic acid redox center of enterococcal NADH peroxidase refined at 2.8 Å resolution. Biochemistry 35 9951–9957. [DOI] [PubMed] [Google Scholar]
- Yue, Q.K., Kass, I.J., Sampson, N.S., and Vrielink, A. 1999. Crystal structure determination of cholesterol oxidase from Streptomyces and structural characterization of key active site mutants. Biochemistry 38 4277–4286. [DOI] [PubMed] [Google Scholar]
- Ziegler, G.A., Vonrhein, C., Hanukoglu, I., and Schulz, G.E. 1999. The structure of adrenodoxin reductase of mitochondrial P450 systems: Electron transfer for steroid biosynthesis. J. Mol. Biol. 289 981–990. [DOI] [PubMed] [Google Scholar]