

Abstract
Bothrojaracin (BJC) is a 27-kD snake venom protein from Bothrops jararaca that has been characterized as a potent thrombin inhibitor. BJC binds to exosites I and II, with a dissociation constant of 0.7 nM, and influences but does not block the proteinase catalytic site. BJC also binds prothrombin through an interaction that has not been characterized. In the present work we characterize the interaction of BJC with prothrombin quantitatively for the first time, and identify the BJC binding site on human prothrombin. Gel filtration chromatography demonstrated calcium-independent, 1:1 complex formation between fluorescein-labeled BJC ([5F]BJC) and prothrombin, whereas no interactions were observed with activation fragments 1 or 2 of prothrombin. Isothermal titration calorimetry showed that binding of BJC to prothrombin is endothermic, with a dissociation constant of 76 ± 32 nM. The exosite I-specific ligand, hirudin54–65 (Hir54–65 (SO3−), displaced competitively [5F]BJC from prothrombin. Titration of the fluorescent hirudin54–65 derivative, [5F]Hir54–65(SO3−), with human prothrombin showed a dissociation constant of 7.0 ± 0.2 μM, indicating a ∼100-fold lower binding affinity than that exhibited by BJC. Both ligands, however, displayed a similar, ∼100-fold increase in affinity for exosite I when prothrombin was activated to thrombin. BJC efficiently displaced [5F]Hir54–65(SO3−) from complexes formed with thrombin or prothrombin with dissociation constants of 0.7 ± 0.9 nM and 11 ± 80 nM, respectively, indicating that BJC and Hir54–65(SO3−) compete for the same exosite on these molecules. The results indicate that BJC is a potent and specific probe of the partially exposed anion-binding exosite (proexosite I) of human prothrombin.
Keywords: Bothrojaracin, prothrombin, proexosite I, α-thrombin, exosite I, hirudin54-65, titration calorimetry, fluorescence polarization
The blood-clotting serine proteinase, thrombin, is formed by cleavage of two peptide bonds in prothrombin by blood coagulation factor Xa. Thrombin generation is accompanied by separation of the prothrombin activation fragment 1 and 2 domains and the protease domain (thrombin). The prothrombin fragment 1 domain functions in prothrombin activation by binding calcium and negatively charged phospholipid surfaces (Gitel et al. 1973; Dombrose et al. 1979). Binding of prothrombin to the cofactor protein, factor Va, which regulates the activation of prothrombin by factor Xa, is thought to involve fragment 2 and/or an exosite in the proteinase domain of prothrombin (Kotkow et al. 1995; Krishnaswamy and Walker 1997; Anderson et al. 2000b). During the conversion of prothrombin into thrombin, the catalytic site is activated and additional allosteric regulatory sites become expressed on the enzyme (Liu et al. 1991b; Wu et al. 1994). Anion-binding exosite I is a positively charged site on thrombin that interacts with fibrinogen (Naski et al. 1990), the platelet thrombin receptor (Liu et al. 1991a), thrombomodulin (Ye et al. 1992), heparin cofactor II (Hortin et al. 1989), factor V and Va (Dharmawardana et al. 1999), and with C-terminal hirudin peptides (Krstenansky and Mao 1987). Through these interactions, exosite I plays key roles in the specificity of thrombin towards macromolecular substrates, cofactors, and some inhibitors (Stubbs and Bode 1993; Guillin et al. 1995). Recently, exosite I has been shown to be partially exposed on prothrombin in a precursor state (proexosite I), and is fully expressed on thrombin, with a ∼100-fold increase in affinity for hirudin peptides (Anderson et al. 2000a). Another positively charged site on thrombin is anion-binding exosite II, which binds heparin (Sheehan and Sadler 1994) and fragment 2 (Walker and Esmon 1979). The crystal structure of the thrombin–fragment 2, domain–domain complex indicates that exosite II is not accessible on prothrombin (Arni et al. 1993).
Bothrojaracin (BJC) is a potent and highly specific thrombin inhibitor isolated from the venom of the jararaca snake common to Brazil (Bothrops jararaca) (Zingali et al. 1993). BJC is a 27-kD disulfide-linked heterodimer that belongs to the C-type (Ca2+-dependent) lectin-like related protein family (Arocas et al. 1997). The mechanism of thrombin inhibition is unusual in that BJC interacts with both exosites I and II on thrombin (Arocas et al. 1996), forming a 1:1 noncovalent complex and inhibiting thrombin activity toward fibrinogen, platelets, protein C, and factor V (Zingali et al. 1993; Arocas et al. 1998), while the activity towards tripeptide chromogenic substrates is modulated but not impaired (Monteiro et al. 1999). In addition, BJC has been found to form a noncovalent complex with prothrombin (Arocas et al. 1996), and this was associated with inhibition by BJC of prothrombin activation by Oxyuranus scutellatus venom (Monteiro and Zingali 2000). However, the mechanism by which BJC binds to prothrombin is not known, and the binding site on prothrombin has not been identified.
In the present work we quantitatively characterize the binding of BJC to prothrombin for the first time, and identify the binding site on prothrombin as proexosite I in the catalytic domain of prothrombin. Competitive binding studies employing fluorescence probes showed that specific binding of sulfated hirudin54–65 (Hir54–65[SO3−]) to proexosite I displaced BJC from the complex formed with prothrombin. BJC similarly disrupted the prothrombin complex with Hir54–65(SO3−), indicating that BJC and Hir54–65(SO3−) bind competitively to proexosite I on human prothrombin. The results for BJC represent the highest affinity interaction with proexosite I described thus far, and may therefore aid the development of novel anticoagulants based on exosite inhibitors.
Results
Formation of the [5F]BJC–prothrombin complex and the absence of interaction with prothrombin fragments 1 and 2
Binding of BJC to prothrombin was examined first by Superose 12 gel filtration, measured by employing a fluorescein-labeled derivative of BJC ([5F]BJC) to monitor elution (Fig. 1A ▶). In a typical experiment, the retention time of [5F]BJC was ∼25 min, while coinjection with a threefold molar excess of prothrombin displaced the peak to ∼22 min (Fig. 1B ▶), corresponding to a ∼100-kD molecular weight protein complex. These results were consistent with formation of a 1:1 complex between [5F]BJC (27 kD) and prothrombin (72 kD). Thus, as seen previously with thrombin (Zingali et al. 1993), BJC bound to a single prothrombin molecule. The elution pattern in the presence of BJC and prothrombin was not altered when the equilibration buffer contained 10 mM CaCl2 or 10 mM EGTA (data not shown), indicating that the BJC–prothrombin interaction did not depend on calcium binding. Mixtures of [5F]BJC and fragment 1 or fragment 2, even at 100-fold molar excesses (Fig. 1 ▶, C and D, respectively), had an elution pattern similar to that found with [5F]BJC alone. Again, the presence of Ca2+ or EGTA produced no effect. Thus, [5F]BJC did not interact with prothrombin fragment 1 or fragment 2, indicating that the BJC binding site on human prothrombin was localized to the catalytic domain.
Fig. 1.

Formation of [5F]BJC–prothrombin complex and the absence of interaction of [5F]BJC with prothrombin fragments investigated by gel filtration chromatography. Superose 12 was equilibrated in TBS, and elution of [5F]BJC was monitored by the elution volume of the fluorescein fluorescence in the absence and presence of other proteins. The following samples (20-μL loop) were applied onto the column: (A) 0.33 μM [5F]BJC (0.18 μg); (B) 0.33 μM [5F]BJC (0.18 μg) + 1 μM human prothrombin (1.4 μg); (C) 0.3 μM [5F]BJC (0.18 μg) + 30 μM prothrombin fragment 1 (14 μg); (D) 0.3 μM [5F]BJC (0.18 μg) + 30 μM prothrombin fragment 2 (11 μg).
Binding of BJC to prothrombin was measured by Isothermal Titration Calorimetry, with the results shown in Figure 2 ▶. Analysis of the results according to a single set of identical binding sites gave a dissociation constant of 76 ± 32 nM for native BJC binding to prothrombin. Binding was entropically driven, with an unfavorable enthalpy (ΔH) of 13.5 ± 1.8 kcal/mol. These data allowed the calculation of the free energy (ΔG = −9.7 kcal/mol) and entropic term (− TΔS = −3.8 kcal/mol) for binding of prothrombin to BJC. In agreement with the results obtained by gel filtration chromatography, the stoichiometry of the binding (n = 0.9 ± 0.1) confirms that one prothrombin binds to one BJC.
Fig. 2.

Calorimetric titration of BJC with human prothrombin. The binding isotherm at 25°C for titration of prothrombin (Pro) in the calorimetric cell containing 1 μM BJC was analyzed according to the independent single-site model as described in Materials and Methods. The solid line represents the least-squares fit of the experimental data with the parameters given in the text.
Titration of [5F]BJC with prothrombin produced a saturable increase of 15 ± 1 mP units (Fig. 3 ▶, inset), thought to reflect the decrease in the molecular rotation of the [5F]BJC–prothrombin complex in relation to free [5F]BJC. Analysis of these results indicated a tighter dissociation constant of 7 ± 10 nM for labeled BJC compared to native BJC, suggesting an enhanced affinity for prothrombin due to the presence of the fluorescent label. Direct titration of [5F]BJC with prothrombin fragments 1 and 2 up to 8 μM, by contrast, showed no changes in the fluorescence polarization, confirming the absence of evidence for binding to these domains. The ability of prothrombin fragment 1 or fragment 2 to dissociate the [5F]BJC–prothrombin complex was evaluated further in competitive titrations. As shown in Figure 3 ▶, addition of prothrombin fragments 1 or 2 did not displace [5F]BJC from the prothrombin complex. These data indicate that the complex formed between BJC and prothrombin did not occur through interaction with the γ-carboxyglutamic acid domain or the fragment 1 or 2 domains of the zymogen.
Fig. 3.

Fluorescence polarization titration of [5F]BJC–prothrombin complex with prothrombin fragments. Titration with prothrombin (Pro) fragment 1 (open circles) or fragment 2 (closed circles) was performed in TBS containing 100 nM [5F]BJC and 250 nM prothrombin. Experiments were performed at 25°C and fluorescence polarization measured as described in Materials and Methods. The arrow indicates the polarization value obtained for free [5F]BJC (closed diamonds). Inset: Direct titration of 100 nM [5F]BJC with human prothrombin (Pro) along with the fitted curve calculated with the parameters given in the text.
Effect of Hir54–65(SO3−) on dissociation of the [5F]BJC-prothrombin complex
Anion-binding exosite I is present on the prothrombin surface in a low-affinity, precursor state (Ni et al. 1993; Van de Locht et al. 1996; Anderson et al. 2000a). The recent observation that hirudin54–65 peptides interact with prothrombin with affinities ∼100-fold lower than that found for thrombin, suggested that exosite I has a distinct conformation in the zymogen (proexosite I) (Anderson et al. 2000a). Fluorescence polarization titrations employing the fluorescein-labeled peptide, [5F]Hir54–65(SO), confirmed the formation of a specific proexosite I complex between the peptide and prothrombin (Fig. 4 ▶). By contrast to [5F]BJC, saturation of [5F]Hir54–65 (SO3−) occurred at higher prothrombin concentrations, giving a dissociation constant of 7.0 ± 0.2 μM for the peptide (Fig. 4 ▶), which was in good agreement with the value of 3.2 ± 0.3 μM determined previously by fluorescence intensity changes of [5F]Hir54–65 (SO3−) (Anderson et al. 2000a).
Fig. 4.

Direct titration of [5F]Hir54–65(SO3−) with human prothrombin. The change in fluorescence polarization of a solution containing 100 nM [5F]Hir54–65(SO3−) titrated with prothrombin (Pro) was performed in TBS at 25°C as described in Materials and Methods. Inset: Scatchard plot for the [5F]Hir54–65(SO3−) interaction with prothrombin.
The ability of Hir54–65(SO3−) to interfere in the formation of the [5F]BJC–prothrombin complex was studied by fluorescence polarization titration of a mixture of [5F]BJC and prothrombin with Hir54–65(SO3−) (Fig. 5 ▶). Hir54–65(SO3−) gradually decreased the polarization toward the initial value, and the results were well described by a competitive binding model with indistinguishable dissociation constants of 7 ± 5 μM and 7 ± 0.2 μM for binding of the unlabeled and fluorescein-labeled hirudin peptides to prothrombin, respectively. In agreement with crystallographic studies demonstrating that the heparin binding site (exosite II) is not available on prothrombin (Arni et al. 1993), titration of the [5F]BJC–prothrombin complex with heparin showed no dissociation up to 15 U/mL heparin (data not shown).
Fig. 5.

Competitive titration of the [5F]BJC–prothrombin complex with Hir54–65(SO3−). Titration with Hir54–65(SO3−) was performed in TBS containing 100 nM [5F]BJC and 250 nM prothrombin. Experiments were performed at 25°C, and polarization was measured as described in Materials and Methods. The solid line represents the nonlinear least-squares fit by the model for competitive binding of [5F]BJC and the unlabeled hirudin peptide to prothrombin, with the fitted parameters given in the text.
Effect of BJC on dissociation of [5F]Hir54–65(SO3−) from complexes with thrombin or prothrombin
The relationships between the binding sites for BJC and [5F]Hir54–65(SO3−) on native thrombin and prothrombin were investigated in additional competitive binding titrations. Titration with BJC of a solution of 40 nM [5F]Hir54–65 (SO3−) containing 160 nM α-thrombin, gradually decreased the polarization of the labeled complex toward the initial value (Fig. 6A ▶). Analysis of the titration gave a dissociation constant of 0.7 ± 0.9 nM for native BJC binding to thrombin, in agreement with the previously estimated affinity (Arocas et al. 1996). This result was also in good agreement with a previous finding in which BJC and Hir54–65 (SO3−) interfered with binding to immobilized thrombin (Arocas et al. 1996). BJC was similarly able to induce a progressive decrease in the initial polarization value of a mixture of 40 nM [5F]Hir54–65(SO3−) and 2.1 μM human prothrombin. Thus, at 3.5 μM BJC, the enhanced polarization values returned to those obtained for free [5F]Hir54–65 (SO3−). Analysis of these results gave a dissociation constant of 111 ± 80 nM for binding of native BJC to prothrombin, in very good agreement with the calorimetric result of 76 ± 32 nM (Fig. 6B ▶). These results indicate that BJC and [5F]Hir54–65(SO3−) compete for binding to (pro)exosite I on thrombin and prothrombin.
Fig. 6.

Competitive titration of the [5F]Hir54–65(SO3−)–thrombin and [5F]Hir54–65(SO3−)–prothrombin complexes with BJC. Titration with BJC of solutions containing: (A) 160 nM thrombin and 40 nM [5F]Hir54–65 (SO3−). (B) Titration of 2.1 μM prothrombin plus 40 nM [5F]Hir54–65 (SO3−) with BJC. Experiments were performed and analyzed as described in Materials and Methods and the text. The solid lines represent the best fits with the parameters given in the text. For the analysis of the thrombin titration, the dissociation constant for the hirudin peptide binding to thrombin was fixed at the determined value of 25 nM (Anderson et al. 2000a). Similarly, for the prothrombin titration, the dissociation constant for hirudin peptide binding to prothrombin was fixed at the determined value of 7 μM.
Discussion
Quantitative characterization of the interaction of BJC with prothrombin and thrombin in the present work supports the conclusion that BJC binds specifically to proexosite I of prothrombin with a dissociation constant 76–111 nM and to thrombin with the ∼100-fold higher affinity of 0.7 ± 0.9 nM. The high affinity is thought to reflect the dependence on simultaneous interactions of BJC with both exosite I and II on thrombin (Arocas et al. 1996). These results confirmed the previous observation that BJC binding to thrombin was displaced by occupation of exosite I with Hir54–65(SO3−) in solid-phase assays (Arocas et al. 1996). Investigation of the effect of Hir54–65(SO3−) on dissociation of the BJC–prothrombin complex showed that the hirudin peptide bound competitively with BJC to prothrombin. In addition, BJC was highly efficient in displacing [5F]Hir54–65(SO3−) from complexes formed with either thrombin or prothrombin, with dissociation constants of 0.7 ± 0.9 nM and 111 ± 80 nM, respectively. This demonstrated that BJC and hirudin54–65 compete for one of the same binding sites on both the zymogen and the active enzyme. Comparison of the affinities of BJC for the zymogen and thrombin shows that BJC has about a ∼100-fold higher affinity for thrombin compared to prothrombin. This was consistent with the ∼100-fold enhancement in affinity observed with prothrombin and thrombin for the binding of hirudin54–65 peptides (Anderson et al. 2000a). Thus, the results indicate that similar large changes in exosite affinity for both the hirudin peptide and BJC accompany the activation of prothrombin to thrombin.
BJC belongs to the snake venom C-type lectin-like related family of proteins (Arocas et al. 1997), which contain many structural similarities (Atoda et al. 1991). One of them is the IX/X-bp, a protein with anticoagulant effect that has been isolated from Bothrops jararaca (Sekiya et al. 1993) and Trimeresurus flavoridis venoms (Atoda and Morita 1989). Although this protein does not bind to prothrombin, it does interact with the γ-carboxyglutamic domain of factors IX and X in a Ca2+-dependent fashion (Atoda et al. 1994). As shown here, the mechanism of interaction of BJC with prothrombin is clearly distinct, not being modulated by calcium ions. This is supported by the absence of detectable interaction between BJC and fragment 1, which contains the prothrombin γ-carboxyglutamic acid domain. Fragments 1 and 2 each also contain a kringle domain that is a common motif in blood coagulation proteins involved in protein–protein interactions (Tulinsky 1991). The absence of interaction of BJC with fragments 1 and 2 ruled out the involvement of the kringles in BJC–prothrombin complex formation.
The positive enthalpy value found for BJC–prothrombin complex formation indicated that the binding of prothrombin to BJC was entropically driven. A favorable entropy is usually observed in hydrophobic interactions due to the release of structured water from the interacting surfaces into the bulk solvent. On the other hand, when the interaction of the biomolecule leads to conformational changes where a portion of the molecule could become more mobile, a favorable positive entropy can also be observed (Tame et al. 1998). Whether BJC interaction with exosite I is driven by electrostatic interactions, as seen with the hirudin peptides (Karshikov et al. 1992), or by a hydrophobic interaction, as in the case of the inhibitor, triabin (Fuentes-Prior et al. 1997), remains unclear. On the other hand, several studies have demonstrated conformational changes in thrombin induced by ligand binding to exosite I (Hortin and Trimpe 1991; Liu et al. 1991a; Ye et al. 1992; Fredenburgh et al. 1997) and exosite II (Bock 1992; Fredenburgh et al. 1997). This is also observed for BJC, which interacts with exosites I and II to induce distinct alterations in the thrombin catalytic site environment (Monteiro et al. 1999). On this basis, modulation of other prothrombin domain interactions by ligand binding to proexosite I would not be surprising. BJC binding to proexosite I may induce conformational changes in the zymogen that account for the positive enthalpy value observed upon complex formation.
Together, our studies establish that BJC is the highest affinity, specific probe of proexosite I on human prothrombin described to date. This finding is of particular interest because of the implication of proexosite I in the mechanism of the regulatory interactions of prothrombin with factor Va during prothrombin activation. Proexosite I may mediate binding of prothrombin to factor Va directly, and thereby assist the formation of productive complexes (Anderson et al. 2000b). On this basis, BJC may be used as a sensitive probe of the function of the newly characterized proexosite on prothrombin and its role in regulation of prothrombin activation.
Materials and methods
Chemicals and proteins
Fluorescein 5-isothiocyanate was purchased from Molecular Probes Inc.; [Tyr(SO3H)63]-hirudin fragment (54–65) (Hir54–65 (SO3−) was from Sigma Co. Fluorescein-labeled [Tyr(SO3H)63]-hirudin fragment (54–65) ([5F]Hir54–65(SO3−) was prepared as described previously (Bock et al. 1997). Bothrojaracin (BJC) was purified from lyophilized crude Bothrops jararaca venom purchased from Instituto Butantan as described elsewhere (Arocas et al. 1996). Protein concentration was determined from the 280-nm absorbance with an absorption coefficient of 2.00 (mg/mL)−1 cm−1 and molecular weight of 27,000. Purity was assessed by SDS-PAGE. Fluorescein-labeled bothrojaracin ([5F]BJC) was prepared by incubating 18 μM BJC with a 10-fold molar excess of fluorescein 5–isothiocyanate in 20 mM Na2HPO4, 150 mM NaCl, pH 7.5 for 1 h at room temperature. The sample was extensively dialyzed against 20 mM Tris-HCl, 150 mM NaCl, pH 7.5 (TBS). A typical [5F]BJC preparation contained about 0.2 mol of fluorescein/mol of BJC, and had the same specific inhibitory activity in thrombin-induced platelet aggregation as native BJC.
Human thrombin and prothrombin were purified from frozen human plasma following previously described procedures (Bezeaud et al. 1985; Ngai and Chang 1991). Prothrombin fragment 1 and fragment 2 were subproducts of thrombin purification: nonretained proteins from heparin–agarose were chromatographed on Mono-Q 10/10 HR (Pharmacia). The peak eluted at 0.4 M NaCl showed a single 22-kD band on SDS-PAGE corresponding to fragment 1. The peak eluting at 0.3 M NaCl was further subjected to gel filtration on Superdex G-75 HR, yielding a peak corresponding to a single 14-kD band on SDS-PAGE. N-terminal sequencing of the 22- and 14-kD proteins, performed on a Porton Integrated Microsequencer system (model PI 2090), confirmed their identification as human prothrombin fragments 1 and 2, respectively. Concentration of proteins was estimated by the 280-nm absorbance with the following molecular weights and absorption coefficients ([mg/mL]−1 cm−1), respectively: human prothrombin (71,600), 1.47 (Mann et al. 1981); prothrombin fragment 1 (21,700), 1.19 (Mann et al. 1981); prothrombin fragment 2 (14,000), 1.15 (Church et al. 1991); and human thrombin (36,600), 1.83 (Fenton et al. 1977).
Gel filtration
Gel filtration chromatography was performed using a Superose 12 HR column (Pharmacia) attached to a Shimadzu high-pressure liquid chromatography system. The column was equilibrated with TBS at a flow rate of 0.5 mL/min. Protein samples were preincubated at room temperature for 2 min prior to 20-μL injections. Elution was followed using two detectors: one for absorbance at 280 nm, and another for fluorescein fluorescence excited at 490 nm and observed at 510 nm.
Isothermal titration calorimetry
Heats of prothrombin binding to BJC were obtained with a MCS titration microcalorimeter from Microcal Inc. at 25°C. Titration experiments were performed by making successive injections of 2–4 μL each of prothrombin into the 1.34-mL sample cell containing 1 μM BJC until near saturation was achieved. The solution of BJC in 50 mM Tris-HCl, pH 7.5, was titrated with prothrombin prepared in the same buffer, by the use of a 100-μL syringe rotating at 200 rpm. The calorimetric enthalpy (ΔHcal) for each injection was calculated after correction for the heat of prothrombin dilution obtained in control experiments performed by titrating prothrombin into buffer.
The binding isotherms were fitted according to a model for a single set of identical binding sites by nonlinear squares analysis using the Scientist software (MicroMath) or Origin 5.0 software provided by Microcal. Dissociation constants (KD) were determined from the fitting with Scientist software. The enthalpy change (ΔH), and stoichiometry (n) were determined from the fitting with Origin software, according to Equation (1), fixing the association constant obtained from KD:
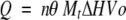 |
1 |
where Q is the total heat content of the solution contained in the cell volume (Vo), at fractional saturation θ, ΔH is the molar heat of ligand binding, n is the number of sites, and Mt is the bulk concentration of macromolecule in Vo. The binding constant, Ka, is described as:
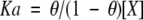 |
2 |
where [X] is the free concentration of ligand.
The free energy (ΔG) and entropy term (TΔS) of association were calculated according to:
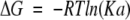 |
3 |
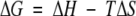 |
4 |
Fluorescence polarization studies
Fluorescence polarization assays were performed on a ISS-PC1 spectrofluorimeter (ISS Inc.). Samples were excited at 485 nm and observed through a 3–69 cutoff filter in the emission path. Polarization was calculated from:
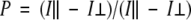 |
5 |
where I| and I⊥ were the fluorescence intensities detected through a polarizer oriented parallel (I|) or perpendicular (I⊥) to the direction of the polarization plane of the excitation beam. Results were expressed as millipolarization units (mP). The error for the polarization measurements did not exceed ±1 mP. All titrations were performed with measurements made after 10 min incubation at 25°C in 20 mM Tris-HCl, 150 mM NaCl, pH 7.5. Direct titrations were analyzed by nonlinear least-squares fitting of the quadratic binding equation, and competitive titrations were analyzed with the cubic equation for tight competitive binding using Scientist software (Anderson et al. 2000a).
Acknowledgments
We thank Dr. C. Nogueira (Serviço de Hemoterapia/HUCFF/UFRJ) for providing the human plasma samples used for prothrombin and α-thrombin purifications; Instituto Butantan (São Paulo, Brazil) for providing Bothrops jararaca venom. We thank Dr. J.L. Silva and D. Foguel for the use of the fluorescence equipment, and Dr. L. de Meis for calorimetric facilities; Drs. C. Barja-Fidalgo and Patricia J. Anderson for help with the analysis and careful revision of the manuscript; Ana Lúcia O. Carvalho and Denis L.S. Dutra for their technical assistance; the Laboratory of Microsequencing of Proteins and Peptides (LMPP) from the Medical Biochemistry Department (UFRJ); and Prof. L.M.T. Lima for helpful discussions. This work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Financiadora de Estudos e Projetos do Brasil (FINEP), Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), European Economic Community (EEC) contract Cl1*-CT94-0073 BR, and in part supported by National Institutes of Health Grant HL-38779 to P.E.B.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
BJC, bothrojaracin
[5F]BJC, bothrojaracin labeled with fluorescein 5-isothiocyanate
Hir54-65 (SO3−), Gly-Asp-Phe-Glu-Glu-Ile-Pro-Glu-Glu-Tyr(SO3−)-Leu-Gln
[5F]Hir54-65 (SO3−)
Hir54-65 (SO3−) labeled at the amino terminus with 5-carboxy(fluorescein)
TBS, Tris-buffered saline (20 mM Tris-HCl, 150 mM NaCl, pH 7.5)
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/
References
- Anderson, P.J., Nesset, A., Dharmawardana, K.R., and Bock P.E. 2000a. Characterization of proexosite I on prothrombin. J. Biol. Chem. 275 16428–16434. [DOI] [PubMed] [Google Scholar]
- ———. 2000b. Role of proexosite I in factor Va-dependent substrate interactions of prothrombin activation. J. Biol. Chem. 275 16435–16442. [DOI] [PubMed] [Google Scholar]
- Arni, R.K., Padmanabhan, K., Padmanabhan, K.P., Wu, T.P., and Tulinsky, A. 1993. Structures of the noncovalent complexes of human and bovine prothrombin fragment 2 with human PPACK-thrombin. Biochemistry 32 4727–4737. [DOI] [PubMed] [Google Scholar]
- Arocas, V., Zingali, R.B., Guillin, M.C., Bon, C., and Jandrot-Perrus, M. 1996. Bothrojaracin, a potent two-site-directed thrombin inhibitor. Biochemistry 35 9083–9089. [DOI] [PubMed] [Google Scholar]
- Arocas, V., Castro, H.C., Zingali, R.B., Guillin, M.C., Jandrot-Perrus, M., Bon, C., and Wisner, A. 1997. Molecular cloning and expression of bothrojaracin, a potent thrombin inhibitor from snake venom. Eur. J. Biochem. 248 550–557. [DOI] [PubMed] [Google Scholar]
- Arocas, V., Lemaire, C., Bouton, M.C., Bezeaud, A., Bon, C., Guillin, M.C., and Jandrot-Perrus, M. 1998. Inhibition of thrombin-catalyzed factor V activation by bothrojaracin. Thromb. Haemost. 79 1157–1161. [PubMed] [Google Scholar]
- Atoda, H. and Morita, T. 1989. A novel blood coagulation factor IX/factor X-binding protein with anticoagulant activity from the venom of Trimeresurus flavoviridis (Habu snake): Isolation and characterization. J. Biochem. (Tokyo) 106 808–813. [DOI] [PubMed] [Google Scholar]
- Atoda, H., Hyuga, M., and Morita, T. 1991. The primary structure of coagulation factor IX/factor X-binding protein isolated from the venom of Trimeresurus flavoviridis. Homology with asialoglycoprotein receptors, proteoglycan core protein, tetranectin, and lymphocyte Fc epsilon receptor for immunoglobulin E. J. Biol. Chem. 266 14903–14911. [PubMed] [Google Scholar]
- Atoda, H., Yoshida, N., Ishikawa, M., and Morita, T. 1994. Binding properties of the coagulation factor IX/factor X-binding protein isolated from the venom of Trimeresurus flavoviridis. Eur. J. Biochem. 224 703–708. [DOI] [PubMed] [Google Scholar]
- Bezeaud, A., Denninger, M.H., and Guillin, M.C. 1985. Interaction of human alpha-thrombin and gamma-thrombin with antithrombin III, protein C and thrombomodulin. Eur. J. Biochem. 153 491–496. [DOI] [PubMed] [Google Scholar]
- Bock, P.E. 1992. Active-site-selective labeling of blood coagulation proteinases with fluorescence probes by the use of thioester peptide chloromethyl ketones. II. Properties of thrombin derivatives as reporters of prothrombin fragment 2 binding and specificity of the labeling approach for other proteinases. J. Biol. Chem. 267 14974–14981. [PubMed] [Google Scholar]
- Bock, P.E., Olson, S.T., and Björk, I. 1997. Inactivation of thrombin by antithrombin is accompanied by inactivation of regulatory exosite I. J. Biol. Chem. 272 19837–19845. [DOI] [PubMed] [Google Scholar]
- Church, W.R., Ouellette, L.A., and Messier, T.L. 1991. Modulation of human prothrombin activation on phospholipid vesicles and platelets using monoclonal antibodies to prothrombin fragment 2. J. Biol. Chem. 266 8384–8391. [PubMed] [Google Scholar]
- Dharmawardana, K.R., Olson, S.T., and Bock, P.E. 1999. Role of regulatory exosite I in binding of thrombin to human factor V, factor Va, factor Va subunits, and activation fragments. J. Biol. Chem. 274 18635–18643. [DOI] [PubMed] [Google Scholar]
- Dombrose, F.A., Gitel, S.N., Zawalich, K., and Jackson, C.M. 1979. The association of bovine prothrombin fragment 1 with phospholipid. J. Biol. Chem. 254 5027–5040. [PubMed] [Google Scholar]
- Fenton II, J.W., Fasco, M.J., Stackrow A.B., Aronson, D.L., Young, A.M., and Finlayson, J.S. 1977. Human thrombins. Production, evaluation, and properties of alpha-thrombin. J. Biol. Chem. 252 3587–3598. [PubMed] [Google Scholar]
- Fredenburgh, J.C., Stafford, A.R., and Weitz, J.I. 1997. Evidence for allosteric linkage between exosites 1 and 2 of thrombin. J. Biol. Chem. 272 25493–25499. [DOI] [PubMed] [Google Scholar]
- Fuentes-Prior, P., Noeske-Jungblut, C., Donner, P., Schleuning, W.D., Huber, R., and Bode, W. 1997. Structure of the thrombin complex with triabin, a lipocalin-like exosite-binding inhibitor derived from a triatomine bug. Proc. Natl. Acad. Sci. 94 11845–11850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitel, S.N., Owen, W.G., Esmon, C.T., and Jackson, C.M. 1973. A polypeptide region of bovine prothrombin specific for binding to phospholipids. Proc. Natl. Acad. Sci. 70 1344–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillin, M.C., Bezeaud, A., Bouton, M.C., and Jandrot-Perrus, M. 1995. Thrombin specificity. Thromb. Haemost. 74 129–133. [PubMed] [Google Scholar]
- Hortin, G.L. and Trimpe, B.L. 1991. Allosteric changes in thrombin's activity produced by peptides corresponding to segments of natural inhibitors and substrates. J. Biol. Chem. 266 6866–6871. [PubMed] [Google Scholar]
- Hortin, G.L., Tollefsen, D.M., and Benutto, B.M. 1989. Antithrombin activity of a peptide corresponding to residues 54–75 of heparin cofactor II. J. Biol. Chem. 264 13979–13982. [PubMed] [Google Scholar]
- Karshikov, A., Bode, W., Tulinsky, A., and Stone, S.R. 1992. Electrostatic interactions in the association of proteins: An analysis of the thrombin–hirudin complex. Protein Sci. 1 727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotkow, K.J., Deitcher, S.R., Furie, B., and Furie, B. 1995. The second kringle domain of prothrombin promotes factor Va-mediated prothrombin activation by prothrombinase. J. Biol. Chem. 270 4551–4557. [DOI] [PubMed] [Google Scholar]
- Krishnaswamy, S. and Walker, R.K. 1997. Contribution of the prothrombin fragment 2 domain to the function of factor Va in the prothrombinase complex. Biochemistry 36 3319–3330. [DOI] [PubMed] [Google Scholar]
- Krstenansky, J.L. and Mao, S.J.T. 1987. Antithrombin properties of C-terminus of hirudin using synthetic unsulfated N-alpha-acetyl-hirudin45–65. FEBS Lett. 211 10–16. [DOI] [PubMed] [Google Scholar]
- Liu, L.W., Vu, T.K.H., Esmon, C.T., and Coughlin, S.R. 1991a. The region of the thrombin receptor resembling hirudin binds to thrombin and alters enzyme specificity. J. Biol. Chem. 266 16977–16980. [PubMed] [Google Scholar]
- Liu, L.W., Ye, J., Johnson, A.E., and Esmon, C.T. 1991b. Proteolytic formation of either of the two prothrombin activation intermediates results in formation of a hirugen-binding site. J. Biol. Chem. 266 23632–23636. [PubMed] [Google Scholar]
- Mann, K.G., Elion, J., Butkowski, R.J., Downing, M., and Nesheim, M.E. 1981. Prothrombin. Methods Enzymol. 80 286–302. [DOI] [PubMed] [Google Scholar]
- Monteiro, R.Q. and Zingali, R.B. 2000. Inhibition of prothrombin activation by bothrojaracin, a C-type lectin from Bothrops jararaca venom. Arch. Biochem. Biophys. 382 123–128. [DOI] [PubMed] [Google Scholar]
- Monteiro, R.Q., Rapôso, J.G., Wisner, A., Guimarães, J.A., Bon, C., and Zingali, R.B. 1999. Allosteric changes of thrombin catalytic site induced by interaction of bothrojaracin with anion-binding exosites I and II. Biochem. Biophys. Res. Commun. 262 819–822. [DOI] [PubMed] [Google Scholar]
- Naski, M.C., Fenton II, J.W., Maraganore, J.M., Olson, S.T., and Shafer, J.A. 1990. The COOH-terminal domain of hirudin. An exosite-directed competitive inhibitor of the action of alpha-thrombin on fibrinogen. J. Biol. Chem. 265 13484–13489. [PubMed] [Google Scholar]
- Ngai, P.K. and Chang, J.Y. 1991. A novel one-step purification of human α-thrombin after direct activation of crude prothrombin enriched from plasma. Biochem. J. 280 805–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni, F., Ning, Q., Jackson, C.M., and Fenton II, J.W. 1993. Thrombin exosite for fibrinogen recognition is partially accessible in prothrombin. J. Biol. Chem. 268 16899–16902. [PubMed] [Google Scholar]
- Sekiya, F., Atoda, H., and Morita, T. 1993. Isolation and characterization of an anticoagulant protein homologous to botrocetin from the venom of Bothrops jararaca. Biochemistry 32 6892–6897. [DOI] [PubMed] [Google Scholar]
- Sheehan, J.P. and Sadler, J.E. 1994. Molecular mapping of the heparin-binding exosite of thrombin. Proc. Natl. Acad. Sci. 91 5518–5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs, M.T. and Bode, W. 1993. A player of many parts: The spotlight falls on thrombin's structure. Thromb. Res. 69 1–58. [DOI] [PubMed] [Google Scholar]
- Tame, J.R., O'Brian, R., and Ladbury, J.E. 1998. Isothermal titration calorimetry of biomolecules. In Biocalorimetry: Applications of Calorimetry in the Biological Sciences (eds. J.E. Ladbury and B.Z. Chowdhry), pp. 27–38. John Wiley & Sons Ltd., Chichester, England.
- Tulinsky, A. 1991. The structures of domains of blood proteins. Thromb. Haemost. 66 16–31. [PubMed] [Google Scholar]
- Van de Locht, A., Stubbs, M.T., Bauer, M., and Bode, W. 1996. Crystallographic evidence that F2 kringle catalytic domain linker of prothrombin does not cover the fibrinogen recognition exosite. J. Biol. Chem. 271 3413–3416. [DOI] [PubMed] [Google Scholar]
- Walker, F.J. and Esmon, C.T. 1979. The effect of prothrombin fragment 2 on the inhibition of thrombin by antithrombin III. J. Biol. Chem. 254 5618–5622. [PubMed] [Google Scholar]
- Wu, Q., Picard, V., Aiach, M., and Sadler, J.E. 1994. Activation-induced exposure of the thrombin anion-binding exosite. J. Biol. Chem. 269 3725–3730. [PubMed] [Google Scholar]
- Ye, J., Liu, L.W., Esmon, C.T., and Johnson, A.E. 1992. The fifth and sixth growth factor-like domains of thrombomodulin bind to the anion-binding exosite of thrombin and alter its specificity. J. Biol. Chem. 267 11023–11028. [PubMed] [Google Scholar]
- Zingali, R.B., Jandrot-Perrus, M., Guillin, M.C., and Bon, C. 1993. Bothrojaracin, a new thrombin inhibitor isolated from Bothrops jararaca venom: Characterization and mechanism of thrombin inhibition. Biochemistry 32 10794–10802. [DOI] [PubMed] [Google Scholar]