

Abstract
The thermodynamic and kinetic stabilities of the eye lens family of βγ-crystallins are important factors in the etiology of senile cataract. They control the chance of proteins unfolding, which can lead to aggregation and loss of transparency. βB2-Crystallin orthologs are of low stability and comprise two typical βγ-crystallin domains, although, uniquely, the N-terminal domain has a cysteine in one of the conserved folded β-hairpins. Using high-temperature (500 K) molecular dynamics simulations with explicit solvent on the N-terminal domain of rodent βB2-crystallin, we have identified in silico local flexibility in this folded β-hairpin. We have shown in vitro using two-domain human βB2-crystallin that replacement of this cysteine with a more usual aromatic residue (phenylalanine) results in a gain in conformational stability and a reduction in the rate of unfolding. We have used principal components analysis to visualize and cluster the coordinates from eight separate simulated unfolding trajectories of both the wild-type and the C50F mutant N-terminal domains. These data, representing fluctuations around the native well, show that although the mutant and wild-type appear to behave similarly over the early time period, the wild type appears to explore a different region of conformational space. It is proposed that the advantage of having this low-stability cysteine may be correlated with a subunit-exchange mechanism that allows βB2-crystallin to interact with a range of other β-crystallin subunits.
Keywords: age-related cataract, βγ-crystallin fold, essential dynamics, principal components analysis, protein molecular dynamics simulation, protein stability, protein unfolding rates
For the eye lens to refract light it needs to remain transparent for the lifetime of an organism. High protein concentration within lens fiber cells provides both refraction and transparency (Delaye and Tardieu 1983). However, as there is little protein turnover in the lens, long-term stability is required to maintain solubility and avoid aggregation (Harding and Crabbe 1984). There are three main classes of proteins in the eye lenses of vertebrates: α-, β-, and γ-crystallins (Land and Fernald 1992). The α-crystallins belong to the small heat-shock proteins family (sHsps) (de Jong et al. 1993), while βγ-crystallins share distant relationships with microbial stress proteins (Wistow et al. 1985; Wistow 1990) of high stability (Jaenicke and Slingsby 2001). It is hypothesized that the role of α-crystallins in the lens is to bind lens proteins in the early stage of unfolding and so prevent formation of light-scattering aggregates (Horwitz 1992). In age-related cataracts, the level of unfolding/unfolded protein is thought to exceed the chaperoning capacity of the α-crystallins (Clark and Muchowski 2000; Bloemendal et al. 2004).
All vertebrate lens βγ-crystallins are two-domain proteins. A single βγ-crystallin domain consists of two Greek key motifs each containing four β-strands, and these form two β-sheets, with one strand, the c-strand, swapped between sheets (Fig. 1 ▶). Two domains are paired about a hydrophobic interface showing pseudo-twofold symmetry that is similar in all lens β - and γ-crystallins. β-Crystallins form oligomers and have sequence extensions not present in monomeric γ-crystallins, with the basic β-crystallins, such as βB1- and βB2-crystallin, having both N- and C-terminal extensions (Werten et al. 1999). It has been shown that βB2-crystallin helps to solubilize other β-crystallins by heteromer formation (Bateman and Slingsby 1992; Werten et al. 1999). The most significant difference between dimeric βB2-crystallin and the monomeric γ-crystallins observed in the solved crystal structures is domain swapping leading to intermolecular domain pairing in the case of the β-crystallin, whereas the γ-crystallins have intramolecular domain pairing (Bax et al. 1990). However, in human dimeric βB1-crystallin with truncated extensions the domain pairing is intramolecular (Van Montfort et al. 2003), showing that even in basic β-crystallin homodimers, different dimerization mechanisms are used.
Figure 1.

Cartoon of the N-terminal domain of βB2-crystallin. The first motif is colored blue and the second motif is colored red, with key residues mentioned in the text appended in stick representation. Cys50 is the location of the mutation. Tyr45 normally stacks with either a Phe or a Tyr at position 50 in other βγ-crystallin domains. Cys22 is the c-1strand β-bridge residue. Cys50 and Tyr45 are in motif 2, and Cys22 is from motif 1.
βB2-Crystallin, containing two cysteine residues per polypeptide chain, is the least thermodynamically stable members of the βγ-crystallin family as measured by equilibrium unfolding, with a C½ urea of ~2 M (Wieligmann et al. 1999), although it is less prone to heat-induced precipitation, particularly in the bovine protein (Bateman et al. 2003). Mass spectroscopic analyses from human lenses from the ages of 2 to 84 years old show it has undergone, paradoxically, less chemical modification than any other crystallin (Zhang et al. 2001). In contrast, bovine γB-crystallin and human γD-crystallin are extremely stable and can only be denatured by the combined action of chaotropes with either low pH (Rudolph et al. 1990) or higher temperature (Kosinski-Collins and King 2003). Interestingly, βB1-crystallin also shows much greater thermodynamic stability than its close relative βB2-crystallin with a C½ urea of 5.9 M (Kim et al. 2002). There are few folding/unfolding kinetic rate measurements of βγ-crystallins, and thermodynamic measurements are complicated by the fact that denaturant unfolding is often irreversible and thermal unfolding often results in aggregation (Kosinski-Collins and King 2003; Bloemendal et al. 2004).
With respect to the important question of the long-term stability of crystallins, of interest is the unfolding rate, which depends on both the height of the transition state and the depth of the native state energy well. A study of the folding/unfolding rates of cold-shock proteins (a set of orthologous bacterial proteins that have all β structure and widely differing energetic stabilities) has shown that there was no link between folding rate and conformational stability, whereas the unfolding rate was correlated to stability (Perl et al. 1998). A similar correlation is possible in βγ-crystallins, which also have a wide range of thermodynamic stabilities. It is known that in the case of γD-crystallin, one extremely thermodynamically stable member of the family for which data are available, the refolding rate is slow (Kosinski-Collins and King 2003). The unfolding rates were measured as t1/2 = 55 sec for the first detectable intermediate and t1/2 = 2700 sec for the final transition in 5.5 M GuHCl. Kinetic unfolding data are available for the even more stable microbial βγ-crystallin homolog, spherulin 3a, which differs from the crystallins by binding calcium (Kretschmar et al. 1999). This study shows that spherulin unfolded with a half time of 498 sec in 2.5 M GdmCl in the absence of Ca2+ but a half time in excess of 9 h in the presence of Ca2+ in 7.5 M GdmCl.
The slow unfolding rates of members of the βγ-crystallin fold family may be related to their complex topology (Fig. 1 ▶). Folding rates of single domain proteins can be correlated to the topology of the protein using a measure of topology called relative contact order, which calculates the average distance in sequence between contacting residues divided by total polypeptide length (Plaxco et al. 1998). It is postulated that proteins with high contact order take longer to find the transition state for entropic reasons. The intrinsic (long-term) stability of βγ-crystallin domains may be provided by their complex topology, which has a high relative contact order of ~0.2. This is likely to be the major contributor to a high-energy transition state and would provide an additional barrier to unfolding.
A prime objective in crystallin research is to identify molecular details of the age-related unfolding process that leads to light scattering aggregation and cataract. Molecular simulations provide atomistic detail of unfolding intermediates not obtainable experimentally, providing clues as to the kind of states that may be bound by the sHsp α-crystallin, as well as states that may lead to aggregation (Sathish et al. 2004). A previous molecular simulations study on the high-temperature unfolding of bovine γB-crystallin, a very stable member of the family, shows little change to tertiary contacts over 1 nsec at 500 K, although there is an increase in some hydrophobic residue exposure (Purkiss et al. 2000). Another molecular dynamics study of βγ-crystallins looked at the effect of adducts covalently bonded to key residues on bovine γB-crystallin with 300 K simulations of 100 psec (Crabbe et al. 2003). Over this timescale, disruption to the surface charge network is observed and this is speculated to destabilize the protein. Due to the computational limitations of in silico unfolding of stable proteins, most studies target fast folding/unfolding proteins such as barnase (Bond et al. 1997; Purkiss et al. 2004). However, rapid increases in computational power allow similar studies into the unfolding pathways of more kinetically stable proteins such as the βγ-crystallins, with the aim of identifying early unfolding states rather than the complete unfolding pathway.
Here we present a molecular simulations study of the N-terminal domain of rodent βB2-crystallin, chosen as it is a low stability member of the βγ-crystallin fold family for which coordinates are available, on the premise that it may unfold more rapidly than the previously studied γB-crystallin. When the simulation is carried out at high temperature to induce unfolding, a distinctive feature is the identification of changes early in the unfolding pathway in the region of the folded hairpin around Cysteine 50 (as numbered by alignment to γB-crystallin).
The second aim of this paper is to assess the contribution that residue 50 makes to stability by undertaking experiments in solution on both wild-type and a C50F mutant form of human βB2-crystallin dimer. In addition, the effect of this mutation on the unfolding pathways is investigated by performing molecular simulations on modelled C50F mutant coordinates of the N-terminal domain of rodent βB2-crystallin.
Results
Molecular simulations
Four 500 K trajectories (of 5 nsec each) and one 300 K control trajectory (5 nsec) were carried out for both the wild-type and mutant proteins. The backbone root mean square deviation (RMSD) from the crystal structure was calculated for all of the snapshots from each of the trajectories to give an approximate idea of the degree of structural perturbation during the course of the simulation.
It can be seen that the 300 K control trajectories have little variation from the initial wild-type crystal structure (or modelled mutant structure), with the RMSD remaining between 1 and 1.5 Å for the majority of the trajectory (Fig. 2A ▶). This indicates that the simulated wild-type and mutant protein are essentially stable during the simulation at room temperature.
Figure 2.
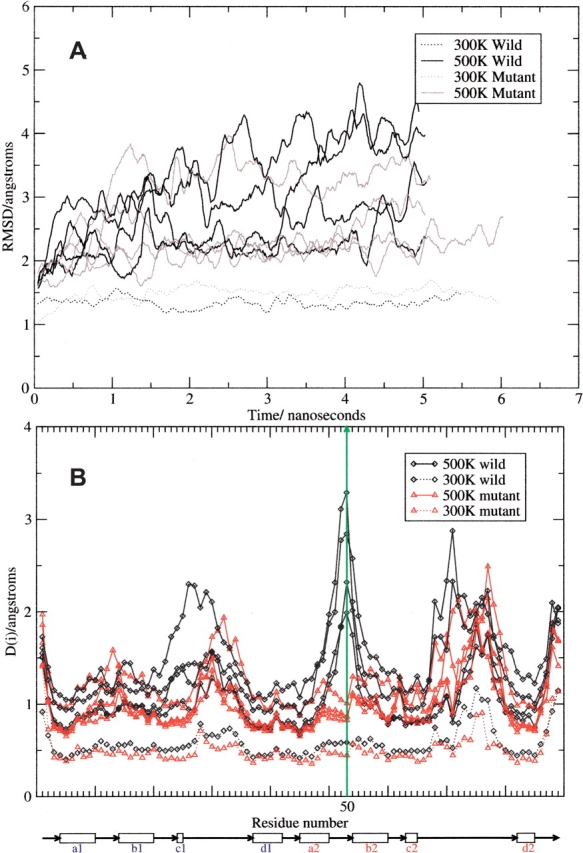
(A) Backbone RMSD. (B) Per Residue Structural Variation. Underneath the figure a diagram is shown indicating the position of the β-strands in the native structure. The vertical green line shows the position of the mutated residue, Cys50 (numbered by alignment to γB as in the PDB entry 1E7N).
Although the mutant and wild-type 500 K trajectories do behave somewhat differently, there is some overlap. For example, all but one of the 500 K mutant trajectories lose structure to a maximum RMSD of less than 3.0 Å; the other mutant trajectory loses structure to a maximum RMSD of around 4.0 Å. Two of the 500 K wild-type trajectories unfold to a maximum around 4 Å, one to around 3.5 Å (albeit only at the very end), and one remains at around 2.5 Å. With this small sample of independent trajectories, it looks as if the wild-type domain is less stable than the mutant, as expected.
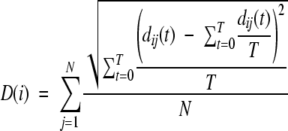
A new method was developed for measuring local structural change. The per residue fluctuation, D(i), during each trajectory is calculated by taking the standard deviation in distance between residue pairs over time, and taking the mean of these standard deviations between each residue and all other residues. So that
 |
(1) |
where dij(t) is the distance between residue i and residue j at time t, N is the number of residues, and T is the length of the trajectory.
The distances, dij, are between closest non-hydrogen atoms in each residue. This gives a measurement of the fluctuations of each residue over the course of the trajectory. The position of the mutation (Cys50 to Phe when numbered according to alignment with γB) is indicated by the vertical green line in Figure 2B ▶ with the horizontal axis showing the position of the β-strands in the native structure (see Fig. 1 ▶). It can be seen that in the wild type, Cys50 is one of the least stable parts of the protein and appears to be fluctuating greatly. However, in the mutant form, this position is significantly stabilized such that it is no longer even a peak but a minimum in the plot. The region around Cys22 (topologically equivalent to Cys22 in γB-crystallin and highly conserved in βγ-crystallins) also appears to have been stabilized, and indeed Phe50 does interact with residues in this region. The side chain of Phe50 (motif 2) is involved in interactions with Pro21 and Pro23, which could contribute extra energetic stability by increased van der Waals forces (Fig. 1 ▶). There are two small peaks that have appeared in the mutant trajectories that do not appear in the wild type at Gln12 (on the loop connecting the a1- and b1-strands) and Lys59 (on the loop connecting b2- and c2-strands). The reason for this is not apparent, as these residues are distant from the mutation site both in sequence and in folded structure. Other than these differences, the wild-type and mutant proteins follow a similar pattern of structural variation.
Principal components analysis
Principal components analysis (PCA) is used to observe if the motions of the proteins’ atoms observed during each simulation trajectory can be described by a small set of variables to gain insight into “paths” each trajectory is following. PCA is a mathematical method that takes a number of correlated variables and transforms them into the same number of uncorrelated variables (the principal components) ordered by the degree of variance in the data it describes. In molecular dynamics this is applied to the atom coordinates, and the resulting eigenvectors can indicate correlated motions during the trajectory. It is sometimes referred to as “essential dynamics” in this context (Amadei et al. 1993). PCA is carried out on only the coordinates of Cα-atoms from the structures to avoid problems due to the wild-type and mutant proteins having different numbers of atoms. The individual structure coordinates from the trajectories can be projected onto the principal components to represent the same data within a different coordinate system. The new coordinate system can be called principal components space (PC-space).
The individual 300 K control trajectories are analyzed using PCA (data not shown). It was observed that the initial wild-type structure is not equilibrated as it does not remain in one well-defined cluster of points but moves over time to another part of PC-space. As is perhaps expected, the initial in silico model of the C50F mutant structure is not populated by many structures in the 300 K simulation and the trajectory quickly proceeds to cluster at a different point in PC space. This result appears to show that the initial structures are not at equilibrium under the explicit solvent conditions of the molecular dynamics (MD) simulation. The native state appears to be in a different region of PC-space, although overall the structural changes are minor. This is likely to be due to a combination of the protein relaxing from a structure packed in the crystal lattice to one in solution and the differences in the force fields used for the crystal structure determination and the simulation. In this study it is not essential that 500 K production runs are taken from an equilibrated structure as equilibrium properties are not being measured.
Clustering
The wild-type and mutant 500 K trajectories are analyzed together using PCA on the Cα atom coordinates to ascertain the degree to which the motions in the trajectories can be described using the same eigenvectors and thus indicate if they are unfolding down different pathways. This PCA analysis is used to cluster structures from the trajectories using the first few principal components. This method of clustering trajectories guarantees the input data, on which clustering is undertaken, are orthogonal.
The first three principal components are shown in Figure 3A ▶. The first two eigenvectors describe 57.1% of variance in the data; the first three, 63.9%; and the first four, 69.0%. The first four principal components are used to cluster the structures using Euclidian distances and hierarchical group-average linkage. The results of this are represented on Figure 3A ▶ by showing the positions of the average structures of each cluster with a green sphere numbered from 1 to 8, with 1 being the largest cluster and 8 being the smallest. To give an idea of the structural changes that correspond to each cluster, the average structures are shown in Figure 3B ▶. They are organized so that the middle column shows average structures from clusters that contain both wild-type and mutant members, the left-hand column shows structures from clusters that contain predominantly wild-type members, and the right-hand column shows structures from clusters that contain predominantly mutant members. The columns are ordered with reference to Figure 3A ▶ and distance from the native state in PC-space.
Figure 3.

(A) The first three principal components of the wild-type and mutant 500 K trajectories. The first component is horizontal, the second is vertical, and the third is represented by depth. The four wild-type 500 K trajectories are in blue and the four mutant 500 K trajectories are red. The initial eight structures are the medium-sized red and blue spheres. The average structures from cluster analysis are the large green numbered spheres. Numbering is by size of the cluster (where 1 is the largest and 8 is the smallest). (B) Average structures from mutant and wild-type cluster analysis using the principal components, numbered as in A.
Early in the simulated unfolding process, mutant and wild-type trajectories share the same clusters. Later clusters contain predominantly either wild-type or mutant structures. The obvious difference between the pathways is that the wild-type structures have lost the β-hairpin loop between strands a2 and b2; clusters 3 and 7 also appear to have had their second β-sheet (strands a2, b2, d2, and c1) disrupted, although the residues are still in β-strand conformations. This can be observed in the greater loss of native contacts in the wild-type clusters than in the mutant clusters (Fig. 3B ▶; Table 1). The mutant clusters have none of these characteristics and their differences are due to different conformations of the flexible loop regions between c and d strands. With the exception of cluster 2, no clear separation between the clusters can be observed in Figure 3A ▶, from which it may be concluded that none of the trajectories have escaped the native well. Cluster 2, which consists of mutant structures only, is characterized by a movement apart of the two β-sheets and shortening of the β-strands and is the only cluster with a clear separation in PC-space from all the other clusters. This observation in a single mutant trajectory could be an aberration, an unfolding intermediate, or a structure off the unfolding pathway. (An analysis was also performed whereby the different mutant trajectory was excluded, along with a random wild-type trajectory in order not to bias the analysis toward the wild-type protein. In this reanalysis, cluster 2 still contains mainly mutant structures but is not such an outlier and contains the highest number of retained native contacts of all the clusters.) All the trajectories retain the majority of their native contacts (Fig. 3B ▶; Table 1) and there are no dramatic changes to the domain tertiary structure.
Table 1.
Retained native contacts and relative solvent accessibilities of the side chains of Trp42, Trp65, and Trp68

Expression and protein purification
Both mutant and wild-type proteins are highly expressed and are present in the supernatant after the cells are disrupted. The purification protocol of the C50F mutant is the same as for the wild type (Bateman et al. 2001), eluting at 20% NaCl on the Q-Sepharose column and 6% NaCl on the Mono-Q column. No unusual behavior is observed and the protein does not precipitate. Confirmation that the mutant protein has been correctly expressed is by mass spectrometry (measured MW = 23,294 Da; calculated MW = 23,293 Da). Far UV circular dichroism spectra of wild-type and mutant protein confirm that the mutant protein has maintained the same secondary structure (Fig. 4A ▶).
Figure 4.

(A) SRCD spectra of the wild-type (black line) and mutant (gray line) protein collected at 2 mg/mL in 10 mM sodium phosphate buffer at 4°C. (B) Equilibrium unfolding of the wild-type (triangle) and mutant (inverted triangle) protein at 0.02 mg/mL in 10 mM sodium phosphate after 24 h at 21°C. Kinetic unfolding [4M urea (C); 6M urea (D)] at 0.02 mg/mL in 10 mM sodium phosphate at 21°C.
Unfolding under equilibrium conditions
On excitation at 280 nm, the fluorescence emission spectra of the folded wild-type and mutant proteins are the same, as are the fully denatured spectra at 6 M urea. This indicates that the proteins have similar environments around the fluorophores in both states. Having predicted, from molecular dynamics simulation, that the mutation C50F should stabilize the intrinsic stability of the N-terminal domain, the prediction is tested by unfolding 0.02 mg/mL of each protein in varying amounts of urea at pH 7 and 21°C and probing the degree of unfolding by tryptophan fluorescence at 317 nm. As expected, there is a red shift in the fluorescent emission spectra, with increased urea concentration as the tryptophans go into a more polar environment on unfolding. In common with many proteins, the fluorescence intensity of β B2-crystallin decreases on unfolding, indicating a greater degree of quenching of the fluorophores in the solvent than in the protein. This is in contrast to human γD-crystallin, where quenching is greater in the folded state (Evans et al. 2004; Kosinski-Collins et al. 2004). Mutational studies suggest that the topologically equivalent N-terminal domain Trp68 and C-terminal domain Trp157 surrounded by tyrosines and a histidine are responsible for the anomalous quenching in γD-crystallin (Kosinski-Collins et al. 2004). βB2-crystallin does not contain a tryptophan at position 157 and this could explain the difference.
Wild-type protein is found to unfold between ~1 M and ~3.5 M urea, while the mutant protein is found to unfold between ~2 M and ~3.5 M urea (Fig. 4B ▶). Both proteins are thus continuing to unfold up to the same urea concentration, but the wild-type is starting to unfold at ~1 M lower urea concentration. The mutant unfolding curve can be fitted to a two-state unfolding model, but the wild-type unfolding curve cannot fit such a model and therefore must have at least one intermediate. Attempts to fit the curve to various three-state models do not produce a stable solution. The mutation can be seen to have significantly increased the conformational stability of parts of the protein, confirming the prediction from molecular dynamics simulation that Cys50 is a region of instability of the wild-type protein.
Kinetic unfolding
It is possible that having increased the conformational stability of the protein, the mutant will take longer to unfold as is found with the cold-shock proteins (Perl et al. 1998). Protein buffer solution containing no denaturant is rapidly increased to 6 M urea (and 4 M) by addition of 8 M urea stock buffer solution taking the protein concentration to 0.01 mg/mL. The sample is excited at 280 nm and the fluorescence emission measured at 317 nm with time. The wild-type protein takes approximately 25–30 min to fully unfold, whereas the mutant takes more than 50 min at both 4 M and 6 M urea (Fig. 4C,D ▶). The wild-type protein shows a rapid initial transition between 0 and 120 sec of adding urea, followed by a slower unfolding phase. The mutant protein does not show such a rapid initial transition. After the initial unfolding phase, both wild-type and mutant then appear to unfold at more similar rates, thus the mutation has a greater effect on the initial phase but less on the later phase.
Tryptophan accessibilities
To give some comparison between the tryptophan fluorescence solution experiments and the molecular simulations, the mean side chain relative solvent accessibility (RSA) of the three tryptophans in the N-terminal domain, for each cluster derived from PCA analysis, is calculated (Table 1). It can be seen that the exposure of the tryptophans does not vary greatly between the clusters. The core Trp42 has almost no exposure to solvent in any cluster, showing the hydrophobic core is still intact. The side chain of Trp65 becomes slightly more buried relative to the native state in all clusters, while Trp68 has a slightly increased exposure to solvent. Interestingly, Trp68 is more exposed to solvent in the predominantly mutant clusters than in the wild-type clusters. Overall, no dramatic changes are observed in these measures, suggesting that the simulations are at a non-spectroscopically observable stage in the unfolding process.
Discussion
A distinctive feature of the high-temperature molecular simulations of the wild-type N-terminal domain of βB2-crystallin is the identification, using a local distance criterion, of an early unfolding intermediate centered around Cys50 (as numbered by alignment to γB) embedded within a folded β-hairpin, a structural feature characteristic of all Greek key motifs of the βγ-crystallin fold family. A curiosity of βB2-crystallin orthologs is the presence of Cys at residue 50 within the second Greek key motif, which is either a phenylalanine or a tyrosine in all other lens βγ-crystallins (Fig. 1 ▶). It is considered that this aromatic residue along with a conserved aromatic residue at position 45 stabilizes the packing of the folded β-hairpin against the β-sheet in all motifs in all βγ-crystallins and their homologs (Blundell et al. 1981; Wistow et al. 1983). However, position 50 in motif 2 of βB2-crystallin and topologically equivalent residues in the other three motifs in all other βγ-crystallins have φ- and ψ-angles corresponding to the left-handed α-helical region of the Ramachandran plot. Thus, these residues are a source of conformational strain in the structure, which an aromatic side chain can balance by providing stabilizing interactions. Simulations were therefore performed on coordinates in which this cysteine is replaced with a phenylalanine, and it is found that the region is indeed stabilized. It is likely then that absence of a stabilizing aromatic side chain at position 50 in βB2-crystallin sequences might contribute to its lower stability.
It has been shown by solution experiments that mutating Cys50 to Phe increases the conformational stability of two-domain human βB2-crystallin, confirming the hypothesis derived from molecular dynamics simulation on the rodent N-terminal domain. A significant difference was observed in equilibrium unfolding conditions with the mutant starting to unfold at higher urea concentrations. It has previously been shown that the rodent N-terminal domain is less stable than the C-terminal domain by equilibrium unfolding experiments on intact and the isolated N- and C-terminal domains of rat βB2-crystallin (Wieligmann et al. 1999). Those authors showed that the equilibrium unfolding curve of the intact βB2-crystallin is not just a superposition of the unfolding curves of the two isolated domains, thus indicating a degree of cooperativity in the folding of the two domains of the protein. They concluded that the N-terminal domain appears to be significantly stabilized by interaction with the C-terminal domain, whereas the C-terminal domain is slightly destabilized relative to the isolated C-terminal domain.
Differences between kinetic unfolding of the wild-type human βB2-crystallin and C50F mutant protein are also observed. The kinetic unfolding curves of the wild-type protein appear to have two phases—an initial fast phase and then a second slower phase. The initial faster phase does not appear to exist in the mutant unfolding curve, while the second phase appears more similar.
The in vitro unfolding experiments described here also support the conclusion that the human N-terminal domain is less stable than the C-terminal domain, as there is a clear stabilization of the initial unfolding transition in the mutant in both equilibrium and kinetic unfolding experiments. The wild-type protein continues to show spectroscopic changes up to the same urea concentration in equilibrium measurements (~3.5 M urea) as the stabilized mutant. This shows that while the mutation has stabilized the N-terminal domain of the protein, it has not increased the thermodynamic stability of the C-terminal domain of the protein significantly, but it does appear to have dramatically increased the kinetic stability of the whole protein. The mutant protein is still unfolding long after the wild-type protein has reached equilibrium. One explanation as to why the equilibrium unfolding curve appears to go from an apparent multistate transition in the wild-type to an apparent two-state transition with the mutation is that the two domains are now of a similar thermodynamic stability and so unfold together.
A PCA clustering method is used to look at the conformational space being explored by the two sets of four 500 K unfolding trajectories of both wild-type and mutant. This appears to show that at this early stage in the unfolding process the wild-type and mutant trajectories have yet to escape the native-state well (with the possible exception of cluster 2) (Fig. 3B ▶) and are exploring different regions of conformational space. Whether this means the mutant is unfolding down a very different unfolding pathway or just altering specific details of a generic unfolding pathway consistent with the topological considerations of protein folding (Plaxco et al. 1998; Makarov and Plaxco 2003) is not known. Further simulations are required to answer this question. This can be achieved by extending the length of the existing simulations or by using an implicit solvent technique such as Generalized Born/Surface Area (GB/SA) to increase the length of simulated time available. Unfolding of barnase by GB/SA has been shown to give similar results to the explicit solvent method used here (Purkiss et al. 2004). The unfolding of barnase, under explicit solvent conditions, is much more rapid than that seen for the N-terminal domain of βB2-crystallin, with the protein having completely unfolded in 5 nsec. The complex topology of the crystallin domain is the likely reason for the much slower rate.
A question that naturally leads from this result is what is the biological purpose of βB2-crystallin having a thermodynamically destabilizing cysteine at a position occupied by a phenylalanine or tyrosine in other members of the βγ-crystallin family. The answer may lie in the consequences of having an expanded β-crystallin gene family that results in the synthesis of several polypeptides with the ability to subunit exchange. The ensuing array of β-crystallin hetero-oligomers contributes to the heterogeneity of the highly concentrated crystallins in the lens cytoplasm (Bloemendal et al. 2004). It is noteworthy that Cys50 and Cys22 in βB2-crystallin are located on the β-sheet involved at the domain-pairing interface, although they are not themselves involved in close interactions (Fig. 1 ▶). The domain-pairing interface plays a crucial role in dimeric assembly in this domain-swapped dimer. Furthermore βB2-crystallin dimer is in rapid equilibrium with its (unstable) monomeric form (Slingsby and Bateman 1990). The dynamic behavior of the βB2-crystallin dimer interface may facilitate subunit-exchange among homodimers and heterodimers and explain βB2-crystallin’s ability to solubilize other β-crystallins (Bateman and Slingsby 1992; Bateman et al. 2003). This would be particularly advantageous for ensuring solubility of those β-crystallins that have endured covalent modifications during their long life in the lens.
Materials and methods
Molecular coordinates
The initial single domain structure used for the wild-type βB2-crystallin simulations was taken from the 2.35 Å resolution crystal structure of the N-terminal domain of rat βB2-crystallin (Clout et al. 2000; PDB code 1E7N). The initial structure of the C50F mutant was derived from the crystal structure using the program Swiss-PdbViewer (Guex and Peitsch 1997) with the cysteine mutated to a phenylalanine side chain. Missing side-chain hydrogens were added using the TLEAP program, but terminal residues not observed in the crystal structure were ignored.
Molecular simulations
The PMEMD (R.E. Duke and L.G. Pedersen, University of North Carolina-Chapel Hill) program developed from the AMBER 7 software suite was used for the simulations with parameters from the parm99 force field (without use of “extra-points”) (D.A. Case et al., University of California, San Francisco.). The simulations were undertaken in the isothermal-isobaric ensemble, with the conditions of constant number of particles, pressure, and temperature (NPT). SHAKE was used to constrain bonds containing hydrogen and the Particle Mesh Ewald summation method was used to calculate electrostatic forces. A non-bonded cutoff of 10 Å was used. The proteins were simulated in a box of TIP3P waters with a minimum of 9 Å from the protein to the box edge with two IB ions (large cations) to neutralize the negative charge of the system. The simulation protocol involved two stages of minimization: first the added hydrogens and waters, and then all atoms. This was followed by four stages of equilibration: (1) 5 psec of warming just the water and counter-ions (the protein coordinates being restrained to their initial values) from 50 K–300 K at constant volume (initial velocities from assigning random velocities from the Maxwell-Boltzmann distribution); (2) 5 psec of MD of just the water and counter-ions at 300 K under constant pressure; (3) 10 psec of warming the whole system to 300 K at constant volume using new, random velocities; and (4) 20 psec MD of the whole system at a constant temperature of 300 K and constant pressure of 1 atmosphere. The generation of a control trajectory at 300 K continued from this point. Production high-temperature simulations were started at different points along the control trajectory by warming to 500 K over 20 psec, under constant volume, to generate independent trajectories at this temperature. Snapshots of the structures were saved every 10 psec during the trajectories.
Four 500 K trajectories (5 nsec) and one 300 K control trajectory (5 nsec) were undertaken on both the wild-type and mutant domains after the start-up protocol described above. The simulations were undertaken on a Beowulf cluster using four or eight 1.3-GHz Athlon processors configured in dual processor nodes. The resulting coordinates were analyzed using PCA, using the program G_COVAR and G_ANAEIG from the GROMACS software suite. Residue distances and native contacts were calculated using the program CONTAX (A.G. Purkiss, http://people.cryst.bbk.ac.uk/~bpurk01/contax/index.html). Root mean squared differences were calculated using the program CARNAL. Graphs were produced using XMGRACE, and molecular pictures were produced using VMD (Humphrey et al. 1996) and PyMOL (DeLano Scientific) and then rendered using POVRAY version 3.5. Solvent accessibilities were calculated using NACCESS (S.J. Hubbard and J.M. Thornton, http://wolf.bms.umist.ac.uk/naccess/).
Expression and purification of proteins
A pET3a (Novagen) plasmid containing the human βB2-crystallin coding sequence, prepared as described previously (Bateman et al. 2001), was transformed into XL Gold cloning cells. Colonies were selected and incubated overnight in LB with 100 μg/mL ampicillin at 37°C, and shaking at 200 rpm. DNA was extracted using the Qiagen miniprep kit and transformed into Escherichia coli strain BL21 (DE3) (Novagen) for expression. An overnight culture (10 mL) was used to inoculate 1 L of 2yt medium supplemented with 100 μg/mL ampicillin, incubated at 37°C, and shaken at 200 rpm. Recombinant human βB2-crystallin overexpression was induced by adding IPTG to 500 μM at an optical density corresponding to an absorbance at 550 nm (A550) of 0.5. Four hours after induction, the cells were harvested by centrifugation at 5000g for 15 min and the pellet was resuspended in 10 mL of bugbuster (Novagen) and frozen at −20°C. The thawed pellet was sonicated on ice for 5 min with 5-sec pulses, the soluble and insoluble fractions of the cell lysate were separated by centrifugation at 4°C for 30 min at 18,000g, and the soluble fraction was dialyzed overnight against 25 mM Tris-HCl (pH 8.5), 1 mM DTT at 4°C. Ion exchange chromatography using a HiLoad 16/10 Q-Sepharose anion exchange column (Amersham Biosciences) equilibrated with 25 mM Tris-HCl (pH 8.5), 1 mM DTT buffer, and a 1 M NaCl gradient was the initial purification step. The peak fractions containing βB2-crystallin were combined and desalted by filtration under pressure using an Amicon cell fitted with a 10-kDa molecular weight cut-off membrane. The final purification step was by Mono-Q HR 10/10 (Pharmacia) anion exchange column in 25 mM BTP (pH 7.5), 1 mM DTT buffer, eluted with a 1 M NaCl gradient. The protein solution was then buffer-exchanged into 10 mM sodium phosphate using a HiPrep 26/10 desalting column (Amersham Biosciences). The protein was homogeneous as judged by SDS-PAGE, and the dimeric nature of the protein was assessed by size exclusion chromatography on a Superose 12 HR 10/30 (Pharmacia) equilibrated with 25 mM BTP (pH 7), 200 mM NaCl. The purified protein solution was then buffer exchanged into 10 mM sodium phosphate using a HiPrep 26/10 desalting column (Amersham Biosciences).
The C50F mutant of human βB2-crystallin was produced by site-directed mutagenesis. The plasmid was purified for mutagenesis and sequencing using the Qiagen miniprep kit. Primers were designed for the C50F mutation and the human βB2-crystallin plasmid used as the template for mutagenesis using the QuikChange kit (Stratagene). Colonies were screened by restriction digest, and verified by DNA sequencing (The DNA Sequencing Facility, Cambridge, UK). Expression and protein purification was performed following the same protocols as for the wild-type protein. Protein identification was by electrospray mass spectrometry (Micromass).
Urea-induced unfolding
Stock solutions of 10 mM sodium phosphate buffer at pH 7, containing between no urea and 8 M urea were prepared. The concentration of the 8 M urea stock was checked by measurement of the refractive index on an ABBE mark II refractometer (Reichert). For the equilibrium unfolding experiments, 0.02 mg/mL protein was added to varying ratios of the two stock solutions and left to equilibrate for 24 h at 21°C. Each sample was excited at 280 nm and fluorescence emission was measured at 317 nm (the wavelength of maximum difference between the folded and unfolded fluorescence spectra) using a Hitachi F-2500 spectrometer. The excitation and emission slits were 10 nm, and the samples were placed in 1-cm path length cell (Helma Ltd.).
The kinetic unfolding measurements were taken using the same stock solutions, excited at the same wavelength, and emission measured at the same wavelength using a SPEX FluoroMax-3 (Jobin Yvon Ltd.)spectrofluorometer. However, due to signal loss caused by bleaching of the fluorophores in the protein, an emission slit width of 1 nm was used. This produced a stable signal in the presence of protein without denaturant. Appropriate amounts of buffer and protein were added to the cell, followed by the rapid addition of the appropriate volume of 8 M urea stock solution. The fluorescence emission signal was monitored over time with an integration time of 1 sec.
CD spectroscopy
Synchrotron Radiation Circular Dichroism (SRCD) spectra were collected on Station UV1 at the Institute for Storage Ring Facilities, University of Aarhus, Denmark. Spectra were collected at a concentration of 2 mg/mL, in a 10 mM sodium phosphate buffer (pH 7.0). Cuvettes with a path length of 50 microns were used, and temperature was maintained at 4°C. Spectra were averaged over three scans and baseline spectra subtracted. The SRCD instrument was calibrated using camphor sulphonic acid (Miles et al. 2003). Spectra were converted to mean residue ellipticity units using a mean residue weight value of 113.9. Data processing was performed using the CDTool software suite (Lees et al. 2004).
Acknowledgments
We thank Drs. Dave Houldershaw and Richard Westlake for computation support, Dr. Orval Bateman for guidance and mass spectrometry measurements, Dr. Paul Rothwell for help with the fluorescence measurements, and the group of Professor B.A. Wallace for help with the SRCD. We acknowledge the BBSRC for funding the Beowulf cluster. C.S. and A.G.P. are grateful for the financial support of the Medical Research Council, and J.T.M. is the recipient of a Medical Research Council studentship.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041227805.
References
- Amadei, A., Linssen, A., and Berendsen, H.J.C. 1993. Essential dynamics of proteins. Proteins 17 412–425. [DOI] [PubMed] [Google Scholar]
- Bateman, O.A. and Slingsby, C. 1992. Structural studies on β H-crystallin from bovine eye lens. Exp. Eye Res. 55 127–133. [DOI] [PubMed] [Google Scholar]
- Bateman, O.A., Lubsen, N.H., and Slingsby, C. 2001. Association behaviour of human βB1-crystallin and its truncated forms. Exp. Eye Res. 73 321–331. [DOI] [PubMed] [Google Scholar]
- Bateman, O.A., Sarra, R., van Genesen, S.T., Kappe, G., Lubsen, N.H., and Slingsby, C. 2003. The stability of human acidic β-crystallin oligomers and hetero-oligomers. Exp. Eye Res. 77 409–422. [DOI] [PubMed] [Google Scholar]
- Bax, B., Lapatto, R., Nalini, V., Driessen, H., Lindley, P.F., Mahadevan, D., Blundell, T.L., and Slingsby, C. 1990. X-ray analysis of βB2-crystallin and evolution of oligomeric lens proteins. Nature 347 776–780. [DOI] [PubMed] [Google Scholar]
- Bloemendal, H., de Jong, W., Jaenicke, R., Lubsen, N.H., Slingsby, C., and Tardieu, A. 2004. Ageing and vision: Structure, stability and function of lens crystallins. Prog. Biophys. Molec. Biol. 86 407–485. [DOI] [PubMed] [Google Scholar]
- Blundell, T., Lindley, P., Miller, L., Moss, D., Slingsby, C., Tickle, I., Turnell, B., and Wistow, G. 1981. The molecular structure and stability of the eye lens: X-ray analysis of γ-crystallin II. Nature 289 771–777. [DOI] [PubMed] [Google Scholar]
- Bond, C.J., Wong, K.B., Clarke, J., Fersht, A.R., and Daggett, V. 1997. Characterization of residual structure in the thermally denatured state of barnase by simulation and experiment: Description of the folding pathway. Proc. Natl. Acad. Sci. 94 13409–13413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, J.I. and Muchowski, P.J. 2000. Small heat-shock proteins and their potential role in human disease. Curr. Opin. Struct. Biol. 10 52–59. [DOI] [PubMed] [Google Scholar]
- Clout, N.J., Basak, A., Wieligmann, K., Bateman, O.A., Jaenicke, R., and Slingsby, C. 2000. The N-terminal domain of βB2-crystallin resembles the putative ancestral homodimer. J. Mol. Biol. 304 253–257. [DOI] [PubMed] [Google Scholar]
- Crabbe, M.J., Cooper, L.R., and Corne, D.W. 2003. Use of essential and molecular dynamics to study γB-crystallin unfolding after non-enzymic post-translational modifications. Comput. Biol. Chem. 27 507–510. [DOI] [PubMed] [Google Scholar]
- de Jong, W.W., Leunissen, J.A., and Voorter, C.E. 1993. Evolution of the α-crystallin/small heat-shock protein family. Mol. Biol. Evol. 10 103–126. [DOI] [PubMed] [Google Scholar]
- Delaye, M. and Tardieu, A. 1983. Short-range order of crystallin proteins accounts for eye lens transparency. Nature 302 415–417. [DOI] [PubMed] [Google Scholar]
- Evans, P., Wyatt, K., Wistow, G.J., Bateman, O.A., Wallace, B.A., and Slingsby, C. 2004. The P23T cataract mutation causes loss of solubility of folded γD-crystallin. J. Mol. Biol. 343 435–444. [DOI] [PubMed] [Google Scholar]
- Guex, N. and Peitsch, M.C. 1997. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modelling. Electrophoresis 18 2714–2723. [DOI] [PubMed] [Google Scholar]
- Harding, J.J. and Crabbe, M.J.C. 1984. The lens: Development, proteins, metabolism and cataract. In The eye, 3rd ed. (ed. H. Davson), pp. 207–492. Academic Press, New York.
- Horwitz, J. 1992. α-Crystallin can function as a molecular chaperone. Proc. Natl. Acad. Sci. 89 10449–10453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey, W., Dalke, A., and Schulten, K. 1996. VMD—Visual Molecular Dynamics. J. Mol. Graph. 14 33–38. [DOI] [PubMed] [Google Scholar]
- Jaenicke, R. and Slingsby, C. 2001. Lens crystallins and their microbial homologs: Structure, stability, and function. Crit. Rev. Biochem. Mol. Biol. 36 435–499. [DOI] [PubMed] [Google Scholar]
- Kim, Y.H., Kapfer, D.M., Boekhorst, J., Lubsen, N.H., Bachinger, H.P., Shearer, T.R., David, L.L., Feix, J.B., and Lampi, K.J. 2002. Deamidation, but not truncation, decreases the urea stability of a lens structural protein, βB1-crystallin. Biochemistry 41 14076–14084. [DOI] [PubMed] [Google Scholar]
- Kosinski-Collins, M.S. and King, J. 2003. In vitro unfolding, refolding, and polymerization of human γD crystallin, a protein involved in cataract formation. Protein Sci. 12 480–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosinski-Collins, M.S., Flaugh, S.L., and King, J. 2004. Probing folding and fluorescence quenching in human γD crystallin Greek key domains using triple tryptophan mutant proteins. Protein Sci. 13 2223–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretschmar, M., Mayr, E.M., and Jaenicke, R. 1999. Kinetic and thermodynamic stabilization of the βγ-crystallin homolog spherulin 3a from Physarum polycephalum by calcium binding. J. Mol. Biol. 289 701–705. [DOI] [PubMed] [Google Scholar]
- Land, M.F. and Fernald, R.D. 1992. The evolution of eyes. Annu. Rev. Neurosci. 15 1–29. [DOI] [PubMed] [Google Scholar]
- Lees, J.G., Smith, B.R., Wien, F., Miles, A.J., and Wallace, B.A. 2004. CDTool—An integrated software package for circular dichroism spectroscopic data processing, analysis, and archiving. Anal. Biochem. 332 285–289. [DOI] [PubMed] [Google Scholar]
- Makarov, D.E. and Plaxco, K.W. 2003. The topomer search model: A simple, quantitative theory of two-state protein folding kinetics. Protein Sci. 12 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles, A.J., Wien, F., Lees, J.G., Rodger, A., Janes, R.W., and Wallace, B.A. 2003. Calibration and standardization of synchrotron radiation circular dichroism and conventional circular dichroism spectrophotometers. Spectroscopy 17 653–661. [Google Scholar]
- Perl, D., Welker, C., Schindler, T., Schroder, K., Marahiel, M.A., Jaenicke, R., and Schmid, F.X. 1998. Conservation of rapid two-state folding in mesophilic, thermophilic and hyperthermophilic cold shock proteins. Nat. Struct. Biol. 5 229–235. [DOI] [PubMed] [Google Scholar]
- Plaxco, K.W., Simons, K.T., and Baker, D. 1998. Contact order, transition state placement and the refolding rates of single domain proteins. J. Mol. Biol. 277 985–994. [DOI] [PubMed] [Google Scholar]
- Purkiss, A.G., Slingsby, C., and Goodfellow, J.M. 2000. Simulation of the highly stable protein: Bovine γB-crystallin at room and high temperature. Protein Pept. Lett. 7 211–217. [Google Scholar]
- Purkiss, A.G., MacDonald, J.T., Goodfellow, J.M., and Slingsby, C. 2004. Comparison of Generalised Born/Surface Area with periodic boundary simulations to study protein unfolding. Mol. Simul. 30 335–342. [Google Scholar]
- Rudolph, R., Siebendritt, R., Nesslauer, G., Sharma, A.K., and Jaenicke, R. 1990. Folding of an all-β protein: Independent domain folding in γ II-crystallin from calf eye lens. Proc. Natl. Acad. Sci. 87 4625–4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathish, H.A., Koteiche, H.A., and McHaourab, H.S. 2004. Binding of destabilized βB2-crystallin mutants to α-crystallin: The role of a folding intermediate. J. Biol. Chem. 279 16425–16432. [DOI] [PubMed] [Google Scholar]
- Slingsby, C. and Bateman, O.A. 1990. Quaternary interactions in eye lens β-crystallins: Basic and acidic subunits of β-crystallins favor heterologous association. Biochemistry 29 6592–6599. [DOI] [PubMed] [Google Scholar]
- Van Montfort, R.L., Bateman, O.A., Lubsen, N.H., and Slingsby, C. 2003. Crystal structure of truncated human βB1-crystallin. Protein Sci. 12 2606–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werten, P.J., Lindner, R.A., Carver, J.A., and de Jong, W.W. 1999. Formation of βA3/βB2-crystallin mixed complexes: Involvement of N- and C-terminal extensions. Biochim. Biophys. Acta 1432 286–292. [DOI] [PubMed] [Google Scholar]
- Wieligmann, K., Mayr, E.M., and Jaenicke, R. 1999. Folding and self-assembly of the domains of βB2-crystallin from rat eye lens. J. Mol. Biol. 286 989–994. [DOI] [PubMed] [Google Scholar]
- Wistow, G. 1990. Evolution of a protein superfamily: Relationships between vertebrate lens crystallins and microorganism dormancy proteins. J. Mol. Evol. 30 140–145. [DOI] [PubMed] [Google Scholar]
- Wistow, G., Turnell, B., Summers, L., Slingsby, C., Moss, D., Miller, L., Lindley, P., and Blundell, T. 1983. X-ray analysis of the eye lens protein γ-II crystallin at 1.9 Å resolution. J. Mol. Biol. 170 175–202. [DOI] [PubMed] [Google Scholar]
- Wistow, G., Summers, L., and Blundell, T. 1985. Myxococcus xanthus spore coat protein S may have a similar structure to vertebrate lens β γ-crystallins. Nature 315 771–773. [DOI] [PubMed] [Google Scholar]
- Zhang, Z., David, L.L., Smith, D.L., and Smith, J.B. 2001. Resistance of human βB2-crystallin to in vivo modification. Exp. Eye Res. 73 203–211. [DOI] [PubMed] [Google Scholar]