

Abstract
RNase III enzymes are a highly conserved family of proteins that specifically cleave double-stranded (ds)RNA. These proteins are involved in a diverse group of functions, including ribosomal RNA processing, mRNA maturation and decay, snRNA and snoRNA processing, and RNA interference. Here we report the crystal structure of the nuclease domain of RNase III from the pathogen Mycobacterium tuberculosis. Although globally similar to other RNase III folds, this structure has some features not observed in previously reported models. These include the presence of an additional metal ion near the catalytic site, as well as conserved secondary structural elements that are proposed to have functional roles in the recognition of dsRNAs.
Keywords: RNaseIII, dsRNA, RNA processing, RNA interference, enzymes, protein–nucleic acid interactions, crystallography, genomics—structural
The ribonuclease III (RNase III) family of enzymes are endoribonucleases specific for double-stranded RNA (dsRNA). Cleavage of dsRNA by RNase III is an essential step in both the maturation and decay of many coding and noncoding RNAs (Court 1993; Nicholson 1999, 2003). RNase III activity is required in prokaryotes for the processing of pre-rRNA (Srivastava and Schlessinger 1990), and the maturation and degradation of specific mRNAs (Regnier and Grunberg-Manago 1990). In eukaryotes, additional RNase III functions include the processing of snoRNAs and snRNAs, and of the microRNAs and siRNAs that are essential components of RNAi and RNA silencing (Court 1993; Nicholson 1999, 2003; Conrad and Rauhut 2002). RNase III is found in all bacterial and eukaryotic species. Although not essential in Escherichia coli, strains deficient in RNase III are attenuated (Talkad et al. 1978), and rnc appears to be essential in both Bacillus subtilis and Mycobacterium tuberculosis (Wang and Bechhofer 1997; Herskovitz and Bechhofer 2000; Sassetti et al. 2003).
Bacterial RNase III proteins are the simplest members of the RNase III family, comprising a single N-terminal nuclease domain fused to a C-terminal dsRNA binding domain (dsRBD). The functional unit of RNase III is a dimer, formed in the bacterial proteins as a homooligomer of equivalent subunits. Cleavage of dsRNA by RNase III is dependent on divalent cations. Metal binding is coordinated by four strictly conserved acidic residues: a glutamate and aspartate (E. coli residues Glu41Ec and Asp45Ec) from within the RNase III signature box (NERLEFLGDS), and two additionally conserved residues, Asp114Ec and Glu117Ec (Blaszczyk et al. 2001). Of these, Asp45Ec and Glu117Ec are essential for catalysis in vitro (Sun et al. 2004; Zhang et al. 2004).
Despite several existing structures, the mechanisms by which RNase III recognizes and cleaves dsRNA are not fully understood. To better understand its function, and as a part of the M. tuberculosis structural genomics initiative, we determined the crystal structure of the nuclease domain of RNase III from the bacterium M. tuberculosis (TB) to 2.1 Å. The TB RNase III structure reveals a previously unseen metal binding site adjacent to the known active site metal. Modeling of dsRNA onto this structure shows potential implications for the second metal and for additional conserved elements in the recognition of dsRNA substrate.
Results and Discussion
Overall structure
Full-length TB RNase III crystallized as a monomer in the asymmetric unit, and forms a homodimer via crystallographic twofold symmetry operators. The structure of the RNase III nuclease domain displays the all α-helical fold seen previously for the A. aeolicus (AA) RNase III and T. maritima (TM) RNase III (PDB entries 1JFZ and 1O0W) nuclease domains (Blaszczyk et al. 2001, 2004; Fig. 1 ▶). Two additional helices, α2′ and α5′, as well as a second 310 helix, that are not observed in the AARNase III structure also are seen in the TM model. The overall dimer structure resembles a shallow oblong bowl with helices α2′, α5′, and α4 forming the rim and α3 forming the base (Fig. 1 ▶). Two symmetry-related active sites, defined by conserved catalytic residues, are located at either end of the central cleft.
Figure 1.
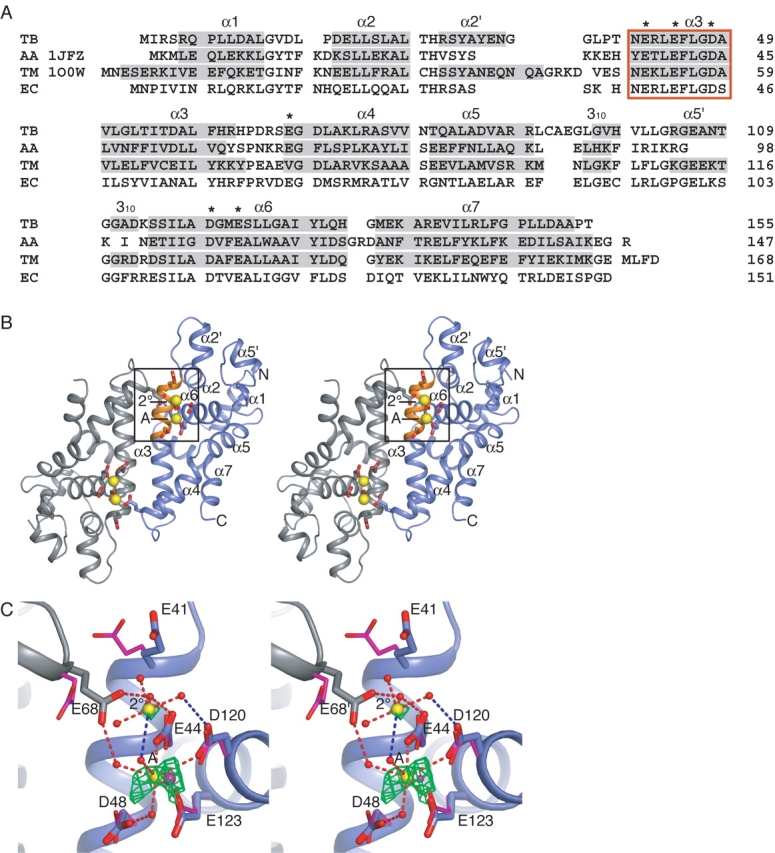
(A) Sequence and secondary structure of the nuclease domains from M. tuberculosis (TB), A. aeolicus (AA), T. maritima (TM), and E. coli (EC) RNase III. Secondary structure assignments (shaded gray) were assigned using the STRIDE server (Frishman and Argos 1995). The RNase III signature sequence is enclosed in the orange box. Residues marked with an asterisk (*) are referred to in the text. (B) Stereo diagram of RNase III dimer. The active site residues coordinate the A- and 2°-site metal ions (yellow spheres). The signature sequence (orange), which includes three of the active site residues, is contained within helix α4. The box encloses one of the two symmetry-related active sites shown in C. (C) Stereo diagram showing coordination of metal ions in the active site. Interactions indicated by red dashes are 3.2 Å, and shorter interactions indicated in blue are 3.2–3.5 Å. The A-site ion is directly coordinated by residues Glu44, Asp120, and Glu123, and to Asp48 and Glu68′ through water molecules. The 2°-site ion is coordinated through its hydration shell to residues Glu44 and Asp120 and to Glu68′ from the adjacent monomer. Fo-Fc positive difference electron density map (green) contoured at 3σ was calculated from a model in which the Ca2+ ions were replaced with water molecules, and the resultant structure was subjected to CNS simulated annealing and positional and B-factor refinement. In magenta are the active site residues and A-site metal ion from the A. aeolicus Mn2+-bound structure (PDB ID 1JFZ; Blaszczyk et al. 2001).
Based on experimental and refined maps, we located two calcium ions within each active site. The first of these (the A-site) has been seen in prior nuclease structures and is thought to participate directly in catalysis. The second ion (2°-site) occupies a new region of the dimer where metal binding has not been observed previously. Although the experimental electron density for the second ion is somewhat weak, the position of the ion, as well as the first, was confirmed by replacement of the modeled ions with water, followed by simulated annealing, positional and B-factor refinement; the resultant Fo-Fc difference density maps had significant positive difference density peaks at the ion sites, consistent with the presence of atoms heavier than water (Fig. 1C ▶). The geometric arrangement of the groups liganding the ions likewise is typical of that seen in other metal-bound structures.
In the initial solution of the TB RNase III structure, only two of the four expected Selenium sites were found by SOLVE, both of which reside in the nuclease domain. No credible electron density was evident for the C-terminal dsRNA-binding domain in initial or refined maps, and the dsRBD’s two resident Se sites were never located, even after refinement of the structure. SDS-PAGE showed that full-length RNase III is the only species present in the crystals (data not shown). As a consequence, the dsRNA binding domain appears to be highly mobile with respect to the nuclease domain. Since the dsRBD is ~40% of the protein mass in the crystal, this may explain why the Rwork and Rfree values are relatively high given the quality of the data.
The dimer interface is principally constructed from contacts between the α3 helices of each subunit. A total of 1188 Å2 is buried in each monomer. Over a third of this surface area is accounted for by just three residues: Arg42, Phe45, and Phe60. Residues Phe45 and Phe60 form “ball-and-socket” interactions across the dimer interface at either end of α3 (Fig. 2A ▶). The importance of Phe45 was demonstrated previously, as nonconservative mutations of this residue inactivate the nuclease (Blaszczyk et al. 2001). Arg42 bridges the dimer interface, making a number of contacts with backbone carbonyl oxygens while ordering the loop connecting helices α3 and α4 (Fig. 2B ▶). Additionally, Arg42 forms a large part of the socket that accommodates Phe60 of the adjacent subunit. Two additional nonhydrophobic dimer interactions are observed in the TB structure (Fig. 2A ▶). The first is near the active site, in which Glu68 bridges the dimer interface to coordinate the 2°-site ion in conjunction with residues from the neighboring protomer. The other interaction connects Tyr130 with Asp57′ and Arg139′ (where “′” signifies residues from the adjoining monomer).
Figure 2.

(A) Stereo diagram of dimer interface showing the surface of one monomer and the interacting region from the adjacent monomer. The surface regions corresponding to the polar side-chain atoms which bridge the interface are colored: Glu68 OE1 and OE2, red; Tyr130 OH, red; and Arg42 NH1 and NH2, blue. The box outlines the region shown in B. (B) 2Fo-Fc refined electron density map contoured at 1.5σ detailing the interaction between Arg42 and backbone carbonyls of residues Phe60′ through Ser67′ of the dimermate. (C) A-form dsRNA modeled on the TB nuclease domain so that the scissile phosphates (green spheres) lie adjacent to the A-site metal ions (yellow spheres). The 2°-site ions are shown in magenta. The bases forming the two-nucleotide overhang product are indicated with orange bars. This arrangement places the minor groove corresponding to the distal box “anti-determinant” bases (cyan) (Zhang and Nicholson 1997) in close proximity to helices α2′ and α5′ (red). Surface electrostatics calculation using APBS (D) (Baker et al. 2001) and surface conservation (E) (Glaser et al. 2003) show negatively charged and conserved surface residues which align with the proposed dsRNA binding region.
Signature sequence and active site
The TB RNase III signature sequence comprises residues 40–49 (NERLEFLGDA) (Fig. 1A ▶). Residues Glu44 and Asp48 in combination with Asp120 and Glu123 coordinate the A-site metal ion, as seen in earlier structures (Blaszczyk et al. 2001, 2004; Fig. 1C ▶). These four residues are strictly conserved, and the E. coli equivalents, Glu45Ec and Glu117Ec, are essential for function both in vivo and in vitro. These findings have led to the conclusion that the A-site is catalytic (Sun and Nicholson 2001; Sun et al. 2005). Overall, the position and the orientation of the four A-site coordinating residues align closely with those in previously solved RNase III structures (Blaszczyk et al. 2001, 2004; Fig. 1D ▶). However, we also see concerted rotamer differences for residues Glu41 and Glu68 (Glu38Ec and Glu67Ec), with respect to the AA RNase III model (Fig. 1D ▶). Both of these residues lie adjacent to the 2°-site, and their inward rotations likely would be energetically disfavored in the absence of the positive charge provided by the 2°-site ion. Indeed, the inward rotation of Glu68 allows this residue to coordinate both the A-site and 2°-site metals through their respective hydration shells. Glu41 does not directly contact the 2°-site metal, and at 4.0 Å the distance between Glu41 and the closest inner-shell water is longer than would be expected for a direct interaction. However, given the conserved nature of Glu41 it seems probable that it may have a role in the positioning of the 2°-site ion.
Implications for dsRNA interactions
The spacing between A-site ions across the dimer symmetry axis is ~21 Å. When considered in the context of a dsRNA duplex, this distance approaches the spacing between a pair of backbone phosphates that are positioned two base pairs (bp) apart across the minor groove of A-form dsRNA (17 Å) (Fig. 2C ▶, green spheres). RNase III enzymes leave a 2-bp overhang following cleavage, and simple modeling of a dsRNA, such that two scissile phosphates lie proximal to the A-site metals, orients the minor groove across the longest width of the bowl-shaped dimer. This model, first proposed by Zhang et al. (2004) for AA RNase III, is based on the observation that each active site is responsible for one strand cleavage reaction and that this orientation would lay the dsRNA along a broad stripe of negatively charged, conserved residues that form the metal binding sites (Fig. 2D,E ▶).
Although recognition of dsRNA by RNase III is generally sequence-independent, two regions have been identified in which specific base pair combinations inhibit dsRNA cleavage by the enzyme (Zhang and Nicholson 1997). These regions, termed “anti-determinants,” consist of the proximal box, which lies at positions −4 to −6 in relation to the cut site, and the distal box, which covers positions −11 and −12. Modeling of dsRNA as outlined above places the distal box (Fig. 2C ▶, cyan bars) adjacent to helices α2′ and α5′ (Fig. 2C ▶, red). Mutation to alanine of the E. coli residue Glu100Ec, equivalent to Glu106 within helix α5′, increases Km by about eightfold with a concomitant increase in Kcat of about fivefold (Sun et al. 2004). Since Glu100Ec is not near the active site, and because the rate-limiting step in catalysis appears to occur subsequent to hydrolysis (Campbell et al. 2002), these results can be rationalized by a model in which decreased affinity for dsRNA leads to a faster rate of product release. Our model suggests that this effect may be a result of Glu100Ec directly interacting with dsRNA, and that this interaction may occur within the distal box anti-determinant region.
Implications for the 2°-site ion
The role of the 2°-site metal is less clear. One possibility is that the 2°-site is equivalent to the B-site in a two-metal ion catalyzed reaction. Measurement of the Hill coefficient for RNase III, along with inhibitor studies using 2-hydroxy-4H-isoquinoline-1,3-dione, a compound specific for two-metal ion nucleases, implies that the hydrolysis reaction catalyzed by RNase III proceeds through a two-metal ion mechanism (Sun et al. 2005). However, in the absence of large distortion of the dsRNA helix, the spacing of the 2°-sites would seem to be too far for direct interaction with the scissile phosphates (Fig. 2C ▶). Additionally, at 5.4 Å, the distance between the A-site and the 2°-site is greater than the canonical spacing of ~3.9 Å between ions in a two-metal ion hydrolysis active site (Beese and Steitz 1991; Steitz and Steitz 1993). In general, the inner liganding shell of the B-site ion in a two-metal mechanism is directly filled by at least one acidic side chain. In contrast, the 2°-site ion coordination we observe is exclusively through outer-shell contacts. Taken together, the 2°-site is unlikely to correspond to the B-site used in two-metal phosphodiester hydrolysis reactions.
What purpose then might Glu41, Glu68, and the 2°-site ion serve? It is worth noting that Glu41 and Glu68 are highly conserved, although not as strictly as the four catalytic residues, and that mutation of the equivalent residues in E. coli RNase III results in an increase of fivefold and threefold, respectively, in the Km for dsRNA, implicating these residues in dsRNA contacts (Zhang et al. 2004). Since the active site of RNase III has a highly conserved and negatively charged surface (Fig. 2D,E ▶) it has been proposed that dsRNA contacts may be mediated through ionic interactions. In support of this hypothesis, a model in which dsRNA interacts with RNase III across the width of the dimer would position the 2°-site proximal to the dsRNA backbone (Fig. 2C ▶). We note that the association of the 2°-site ion with Glu41 and Glu68 is likely a direct result of the high metal ion concentration used in the crystallization conditions for these studies. Given the high Ca2+ concentration and the high B-factor (80 Å2) for metal modeled at the 2°-site, this locus appears to interact only weakly with divalent cations in the absence of dsRNA substrate. As nucleic acids often have a shell of metal ions associated with their phosphodiester backbone, it is possible that the 2°-site might serve as a repository for cations brought in with dsRNA upon binding.
The model proposed here for dsRNA binding to RNase III agrees well with that suggested by Zhang et al. (2004). By taking advantage of the symmetry of both RNase III and dsRNA, as well as the spacing of the likely A-site metal ions, the scissile phosphates of a dsRNA can be readily positioned near the active sites in a manner that would be expected to produce the appropriate two-base overhang products. This arrangement is supported by the conservation of residues along the dsRNA binding region, and suggests that helices α2′ and α5′ may contact the distal box anti-determinant region of dsRNA. The model also provides a role for the conserved but noncatalytic, residues Glu41 and Glu68 in dsRNA recognition. More extensive definition of the mechanisms by which RNase III recognizes and cleaves RNA awaits future co-crystal studies.
Materials and methods
Cloning and protein expression and purification
The gene for M. tuberculosis RNase III (Rv2925c) was amplified from genomic M. tuberculosis strain H37Rv DNA by PCR. The PCR product was ligated into the KasI and HindIIII sites of the pSV271 vector (gift from N. Pokola and T. Handel, U.C. Berkeley), which encodes for a hexa-histadine tag followed by a TEV protease site. The coding sequence was verified by DNA sequencing.
His-tagged RNase IIII was expressed at OD600 of 0.4–0.6 in E. coli BL21(DE3)pLysS at 37°C and induced with 1 mM IPTG for 3–5 h. Harvested cells were resuspended and frozen at −80°C in lysis buffer (50 mM Tris [pH 7.5], 300 mM NaCl, 10% glycerol) with protease inhibitors PMSF and pepstatin A. Expression of selenomethionyl (Se-Met) protein was performed using the protocol of Van Duyne et al. (1993).
For purification, thawed cells were sonicated and centrifuged, and soluble His-tagged RNase III was purified by nickel affinity chromatography. Fractions containing RNase III were pooled and subjected to cleavage of the His-tag by tobacco etch virus (TEV) protease, followed by a second nickel affinity step. Protein eluting in the flow-through was concentrated and run over a gel-filtration column. TB RNase III eluted as a dimer in solution from this step. Protein purity was assessed by SDS-PAGE, and identity was confirmed by ESI mass spectroscopy.
Crystallization
Protein was dialyzed against 10 mM Tris-HCl (pH 7.5), 50 mM NaCl, and 1 mM DTT. Initial crystals were identified with the use of microfluidic protein crystallization chips (Hansen et al. 2002) set with a protein concentration of 8.5 mg/mL. Crystals were observed in conditions containing 20% PEG 8000, 100 mM MES (pH 6.5), and 200 mM Ca-acetate. Crystals, of the space group P43212 with a = b = 72.6 Å and c = 90.1 Å, were grown at 18°C in 15%–20% PEG 3000 with 100 mM Ca-acetate and 100 mM Tris-HCl (pH 7.5), and transferred to an equivalent solution containing 12%–18% xylitol in steps of 2% xylitol before freezing. Crystal trays were moved to 4° before transfer to cryoprotectant and freezing.
Data collection and structure solution
X-ray diffraction data were collected at beamline 8.3.1 at the Advanced Light Source, Lawrence Berkeley National Laboratory. Data were processed using Denzo and Scalepack (Otwinowski and Minor 1997). Collection and processing statistics are given in Table 1. SOLVE and RESOLVE (Terwilliger and Berendzen 1999; Terwilliger 2000) were used to find the Selenium sites, calculate experimental phases, and generate and refine initial electron density maps. Model building was carried out with O, using the A. aeolicus RNase III structure (Blaszczyk et al. 2001) as a starting model. Upon availability of a higher-resolution (2.1 Å) native data set, phases were extended from 2.5 Å using DM (Collaborative Computational Project No. 4 1994). Refinement was carried out using CNS (Brunger et al. 1998).
Table 1.
Data collection and processing statistics
| Data collection | Se-Met peak | Se-Met remote | Native |
| Wavelength (Å) | 0.9793 | 1.0000 | 1.1000 |
| Resolution (Å) | 2.50 (2.59–2.50) | 2.50 (2.59–2.50) | 2.10 (2.18–2.10) |
| Rsyma | 0.048 (0.138) | 0.047 (0.133) | 0.049 (0.252) |
| I/σ | 17.1 (6.4) | 16.6 (6.5) | 28.0 (5.0) |
| Redundancy | 6.6 | 6.6 | 4.6 (4.7) |
| Completeness (%) | 97.3 (88.7) | 96.0 (81.9) | 99.1 (99.6) |
| Wilson B factor | 51.4 | 51.7 | 43.3 |
| Mean figure of merit | 0.51 | ||
| Se found/Expected | 2/4 | ||
| Refinement | |||
| Rworkb | 0.249 | ||
| Rfreeb | 0.286 | ||
| RMSD bond (Å) | 0.008 | ||
| RMSD angle (8) | 1.40 | ||
| Residues built | 155 | ||
| Water atoms | 254 | ||
| Calcium atoms | 2 | ||
| Ramachandran space | |||
| Residues in generously allowed regions | 1 | ||
| Residues in disallowed regions | 0 | ||
aRsym = ∑∑J|(IJ−<I>)|/∑IJ, where I is the observed intensity.
bRwork (Rfree ) = ∑||Fobs|−|Fcalc||/∑|Fobs|, where Fobs and Fcalc are observed and calculated structure factors, respectively. Five percent of reflections were excluded from refinement and used to calculate Rfree.
Acknowledgments
We thank Emmanuel Skordalakes and members of the Berger lab for helpful discussions, insights, and technical assistance. We are grateful to James Holton and the staff at ALS beamline 8.3.1 for support and assistance in data acquisition, and David King for mass spectroscopy analysis. This work was supported by NIH P50 GM62410 (M. tuberculosis structural genomics consortium grant). Coordinates have been deposited in the RSCB PDB (ID 2A11).
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051665905.
References
- Baker, N.A., Sept, D., Joseph, S., Holst, M.J., and McCammon, J.A. 2001. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. 98 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beese, L.S. and Steitz, T.A. 1991. Structural basis for the 3′-5′ exonuclease activity of Escherichia coli DNA polymerase I: A two metal ion mechanism. EMBO J. 10 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaszczyk, J., Tropea, J.E., Bubunenko, M., Routzahn, K.M., Waugh, D.S., Court, D.L., and Ji, X. 2001. Crystallographic and modeling studies of RNase III suggest a mechanism for double-stranded RNA cleavage. Structure (Camb) 9 1225–1236. [DOI] [PubMed] [Google Scholar]
- Blaszczyk, J., Gan, J., Tropea, J.E., Court, D.L., Waugh, D.S., and Ji, X. 2004. Noncatalytic assembly of ribonuclease III with double-stranded RNA. Structure (Camb) 12 457–466. [DOI] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54 (Pt. 5) 905–921. [DOI] [PubMed] [Google Scholar]
- Campbell Jr., F.E., Cassano, A. G., Anderson, V.E., and Harris, M.E. 2002. Pre-steady-state and stopped-flow fluorescence analysis of Escherichiacoli ribo-nuclease III: Insights into mechanism and conformational changes associated with binding and catalysis. J.Mol. Biol. 317 21–40. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project No. 4. 1994. The CCP4 Suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50 (Pt. 5) 760–763. [DOI] [PubMed] [Google Scholar]
- Conrad, C. and Rauhut, R. 2002. Ribonuclease III: New sense from nuisance. Int. J. Biochem. Cell Biol. 34 116–129. [DOI] [PubMed] [Google Scholar]
- Court, D.L. 1993. RNA processing and degradation by RNase III. In Control of messenger RNA stability (eds. J.G. Belasco and G. Brawerman), pp. 71–116. Academic Press, New York.
- Frishman, D. and Argos, P. 1995. Knowledge-based protein secondary structure assignment. Proteins 23 566–579. [DOI] [PubMed] [Google Scholar]
- Glaser, F., Pupko, T., Paz, I., Bell, R.E., Bechor-Shental, D., Martz, E., and Ben-Tal, N. 2003. ConSurf: Identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 19 163–164. [DOI] [PubMed] [Google Scholar]
- Hansen, C.L., Skordalakes, E., Berger, J.M., and Quake, S.R. 2002. A robust and scalable microfluidic metering method that allows protein crystal growth by free interface diffusion. Proc. Natl. Acad. Sci. 99 16531–16536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskovitz, M.A. and Bechhofer, D.H. 2000. Endoribonuclease RNase III is essential in Bacillus subtilis. Mol. Microbiol 38 1027–1033. [DOI] [PubMed] [Google Scholar]
- Nicholson, A.W. 1999. Function, mechanism and regulation of bacterial ribonucleases. FEMS Microbiol. Rev. 23 371–390. [DOI] [PubMed] [Google Scholar]
- ———. 2003. The ribonuclease superfamily: Forms and functions in RNA maturation, decay, and gene silencing. In RNAi: A guide to gene silencing (ed. G.J. Hannon), pp. 149–174. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Otwinowski, Z. and Minor, V. 1997. Processing of X-ray diffraction data collected in oscillation mode. In Macromolecular crystallography, Part A, pp. 307–326. Academic Press, New York. [DOI] [PubMed]
- Regnier, P. and Grunberg-Manago, M. 1990. RNase III cleavages in non-coding leaders of Escherichia coli transcripts control mRNA stability and genetic expression. Biochimie 72 825–834. [DOI] [PubMed] [Google Scholar]
- Sassetti, C.M., Boyd, D.H., and Rubin, E.J. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48 77–84. [DOI] [PubMed] [Google Scholar]
- Srivastava, A.K. and Schlessinger, D. 1990. Mechanism and regulation of bacterial ribosomal RNA processing. Annu. Rev. Microbiol. 44 105–129. [DOI] [PubMed] [Google Scholar]
- Steitz, T.A. and Steitz, J.A. 1993. A general two-metal-ion mechanism for catalytic RNA. Proc. Natl. Acad. Sci. 90 6498–6502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, W. and Nicholson, A.W. 2001. Mechanism of action of Escherichia coli ribonuclease III. Stringent chemical requirement for the glutamic acid 117 side chain and Mn2+ rescue of the Glu117Asp mutant. Biochemistry 40 5102–5110. [DOI] [PubMed] [Google Scholar]
- Sun, W., Li, G., and Nicholson, A.W. 2004. Mutational analysis of the nuclease domain of Escherichia coli ribonuclease III. Identification of conserved acidic residues that are important for catalytic function in vitro. Biochemistry 43 13054–13062. [DOI] [PubMed] [Google Scholar]
- Sun, W., Pertzev, A., and Nicholson, A.W. 2005. Catalytic mechanism of Escherichia coli ribonuclease III: Kinetic and inhibitor evidence for the involvement of two magnesium ions in RNA phosphodiester hydrolysis. Nucleic Acids Res. 33 807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkad, V., Achord, D., and Kennell, D. 1978. Altered mRNA metabolism in ribonuclease III-deficient strains of Escherichia coli. J. Bacteriol. 135 528–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger, T.C. 2000. Maximum-likelihood density modification. Acta Crystallogr. D Biol. Crystallogr. 56 (Pt. 8) 965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger, T.C. and Berendzen, J. 1999. Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 55 (Pt. 4) 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Duyne, G.D., Standaert, R.F., Karplus, P.A., Schreiber, S.L. and Clardy, J. 1993. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol. 229 105–124. [DOI] [PubMed] [Google Scholar]
- Wang, W. and Bechhofer, D.H. 1997. Bacillus subtilis RNase III gene: Cloning, function of the gene in Escherichia coli, and construction of Bacillus subtilis strains with altered rnc loci. J. Bacteriol. 179 7379–7385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, K. and Nicholson, A.W. 1997. Regulation of ribonuclease III processing by double-helical sequence antideterminants. Proc. Natl. Acad. Sci. 94 13437–13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H., Kolb, F.A., Jaskiewicz, L., Westhof, E., and Filipowicz, W. 2004. Single processing center models for human Dicer and bacterial RNase III. Cell 118 57–68. [DOI] [PubMed] [Google Scholar]