

Abstract
The Escherichia coli chaperonin machinery, GroEL, assists the folding of a number of proteins. We describe a sequence-based approach to identify the natural substrate proteins (SPs) for GroEL. Our method is based on the hypothesis that natural SPs are those that contain patterns of residues similar to those found in either GroES mobile loop and/or strongly binding peptide in complex with GroEL. The method is validated by comparing the predicted results with experimentally determined natural SPs for GroEL. We have searched for such patterns in five genomes. In the E. coli genome, we identify 1422 (about one-third) sequences that are putative natural SPs. In Saccharomyces cerevisiae, 2885 (32%) of sequences can be natural substrates for Hsp60, which is the analog of GroEL. The precise number of natural SPs is shown to be a function of the number of contacts an SP makes with the apical domain (NC) and the number of binding sites (NB) in the oligomer with which it interacts. For known SPs for GroEL, we find ~4 < NC < 5 and 2 ≤ NB ≤ 4. A limited analysis of the predicted binding sequences shows that they do not adopt any preferred secondary structure. Our method also predicts the putative binding regions in the identified SPs. The results of our study show that a variety of SPs, associated with diverse functions, can interact with GroEL.
Keywords: chaperonins, protein recognition, E. coli, yeast genomes
Molecular chaperones are required to assist the folding of a subset of proteins in the cytosol of Escherichia coli (Horovitz et al. 2001; Thirumalai and Lorimer 2001; Fenton and Horwich 2003). Chaperonins GroEL and GroES from E. coli, which are biological nanomachines, utilize ATP in a coordinated manner to assist folding of a number of substrate proteins (SP) that are otherwise destined for aggregation. The roughly cylindrical shape of GroEL consists of two homo-heptameric rings stacked back-to-back. Each subunit is made up of apical, intermediate, and equatorial domains (Xu and Sigler 1998). The initial event in the GroEL function is the capture of the SP by the apical domain. GroEL can bind to a vast array of SPs that are unrelated in sequence as long as they are presented in a misfolded state (Viitanen et al. 1992). Misfolded proteins typically have exposed hydrophobic residues. The universal ability of GroEL to recognize a vast array of misfolded proteins suggests that the SP binding largely involves a favorable hydrophobic interaction with GroEL. Substrate proteins do not require a preferred secondary structure to bind to GroEL (Aoki et al. 2000). The crystal structure of peptides bound to the apical domain (“minichaperone”) (Chen and Sigler 1999), and the structure of the mobile group of GroES bound to GroEL (Xu et al. 1997) validate the earlier mutagenesis studies (Fenton et al. 1994) that identified the substrate protein-binding sites in the apical domain.
Although GroEL interacts with a vast array of proteins, including random polypeptide sequences, an important unresolved question is “What are the natural substrates in a genome that require the chaperonin machinery to reach their functionally competent states?” The answer to this question, under in vivo conditions depends on a number of factors, including the growth condition of E. coli. Based on reasonable assumptions about the amount of GroEL and GroES in E. coli and estimates of the rates of assisted in vitro folding, it has been argued (Lorimer 1996) that only ~2%–5% of the proteins can afford to recruit the GroEL machinery. Here, we identify the set of proteins in five genomes including E. coli and Saccharomyces cerevisiae that are capable of forming relatively stable complexes with chaperonins. Using the patterns of proteins that are known to form complexes with GroEL, we show that a large number of E. coli proteins can interact with GroEL in the same manner as GroES or the strongly binding peptide (SBP) (Chen and Sigler 1999). Our method also identifies putative stretches in the protein sequence that are responsible for this interaction. The binding patterns have diverse secondary structures even when found within the same protein.
Hypothesis
Nonnative polypeptides and GroES bind to GroEL at a mostly hydrophobic groove found on two helices from the apical domain of each subunit (Fig. 1A ▶). In the state of GroEL (T state), which has a high affinity for SP (Horovitz et al. 2001; Fenton and Horwich 2003), the binding sites form a near-continuous hydrophobic lining at the mouth of the cavity enclosed by each ring. NMR and X-ray structures of GroEL in complex with GroES (Xu et al. 1997), with peptides representing the GroES mobile loop (Landry et al. 1996), SBP (Chen and Sigler 1999), and the N terminus tag of the neighboring apical fragment (Buckle et al. 1997), suggest that significant ordering of the substrate takes place upon binding to GroEL. These structural studies also show that SPs must contain residues that can lock into the groove between helices H and I in the apical domain of GroEL. Based on the β-hairpin conformation adopted by the GroEL-bound GroES mobile-loop peptide and the SBP, as well as their sequence similarities, it has been suggested that the peptides that interact strongly with GroEL belong to the co-chaperonin class (Shewmaker et al. 2001).
Figure 1.

(A,B) Contacts between GroEL helices H and I (cyan) and (A) the GroES mobile loop (pink) and (B) the SBP (magenta). Side chains of the residues that form the closest contacts are shown in red (GroEL), green (GroES), and yellow (SBP). (C,D) Schematic representation of contacts between GroEL and (C) GroES and (D) SBP. A and B were produced using VMD (Humphrey et al. 1996) and PovRay (Persistence of Vision Raytracer Pty. Ltd.).
Building on these studies and noting the promiscuous nature of GroEL–SP interaction (Fayet et al. 1986; van Dyk et al. 1989; Gordon et al. 1994), we hypothesize that the natural substrates are likely to be those that have multiple (≥2) patterns of residues similar to that found in the mobile loop of GroES or the SBP. The presence of such patterns ensures that these proteins can be accommodated between the grooves of helices H and I. The requirement that natural SPs have at least two such patterns in a sequence guarantees that the SP can interact with multiple GroEL-binding sites simultaneously. Stringent substrates apparently interact with at least three consecutive subunits (Farr et al. 2000). The stability of the GroEL–SP complex is determined not only by binding to multiple sites, but also by specific sequence-dependent interactions between SP and the apical domain. Strong binding to a few binding sites can lead to more stable GroEL–SP than weak binding to several binding sites. Interaction with multiple subunits imparts stability to the GroEL–SP complex and ensures a dynamic role for GroEL-assisted folding. Using the structural complementarity to the residues in the binding pocket of GroEL, we determine protein sequences that exhibit the same complementarity to the GroEL-binding site as GroES and SBP. Among this set of putative GroEL substrates, we find several proteins that have been shown to interact strongly with GroEL in vitro (Houry et al. 1999), which indicates that other GroEL substrates might have the binding features of GroES.
The absence of structures for the SP–GroEL complex makes it difficult to estimate the typical number of contacts, NC, natural SPs make with the apical domain. As a result, we also searched for the number of potential SPs when NC and the number of distinct interaction sites, NB, of GroEL (see Materials and Methods for details) are varied. By varying NC between four and five, we identify most of the currently known SPs that recognize GroEL. The minimal hypothesis allows us to identify a vast number of potential natural SPs for chaperonins. The calculational details are given in Materials and Methods.
Putative E. coli substrate proteins
We find that 2754 of 4307 E. coli proteins (≅64%) contain at least one binding pattern that exactly matches with the one in GroES (Table 1). Only about half (1422) of these proteins, or 33% of the entire database, also contain multiple repeats of the GroES binding pattern. Of the 1422 proteins, only 332 (see Supplemental Material for a list) have four or more binding sites. The set of 1422 proteins should be considered natural GroEL SPs, given their ability to interact with several GroEL binding sites. The statistical significance of this result can be assessed by noting that a random arrangement of residues along the sequences in the E. coli database would result in only five pattern matches (equation 3)! The large number of predicted natural SPs in E. coli is consistent with the observation that many proteins are capable of interacting, at least transiently, with GroEL (Viitanen et al. 1992). Because of thermodynamic and kinetic constraints, it is unlikely that in the course of the cell cycle, GroEL can assist in the folding of all of these proteins (Lorimer 1996).
Table 1.
Number of proteins that contain the same sequence patterns as the binding motifs of GroES mobile loop peptide and the SBP
| GroES pattern matches | SBP pattern matches | ||||
| Genome | Database size | Single | Multiple | Single | Multiple |
| E. coli | 4307 | 2754 | 1422 | 942 | 188 |
| S. cerevisiae | 9626 | 5943 | 3064 | 2019 | 442 |
| U. urealyticum | 614 | 383 | 189 | 122 | 16 |
| T. acidophilum | 1481 | 870 | 415 | 274 | 65 |
| M. kandleri | 1687 | 838 | 323 | 222 | 23 |
Single matches refer to protein sequences containing at least one pattern, while multiple matches represent sequences containing two or more patterns with two successive patterns being separated by at least 23 residues.
The number of sequences that share SBP-like complementarity is small. Among the 22% of the proteins that have at least one SBP binding pattern, only ~4% have multiple patterns. A combined search for the GroES and SBP patterns separated by 23 amino acid positions along the sequence results in 153 putative substrate proteins. The reduced number of proteins containing the SBP binding pattern is because nature has counter-selected against SPs that form tight hyperstable complexes. Such strong interactions will produce complexes that may not easily dissociate, as is required for efficient functioning of the GroEL nanomachine.
Comparison between predictions and experiments
We compared the results of our database search with proteins that have been suggested by experiments to be natural SPs for GroEL. Using 2D gel analysis, Houry et al. (1999) have identified 52 proteins that interact with GroEL. Although this number is clearly a lower bound to the number of natural SPs, this data set allows us to test the validity of our method. As shown in Table 2, 34 (~66%) of these proteins are found to have the exact GroES-type complementarity and 13 (25%) have multiple putative binding regions. By reducing the number of contacts that an SP makes with helices H and I to five (see Materials and Methods and discussion below) the percentage of identified substrates exceeds 70 if the number of binding sites NB = 2. As a by-product, the proposed sequence-based approach predicts the binding regions also.
Table 2.
GroEL substrate proteins identified by Houry et al. (1999) that contain GroES-binding patterns with five and six contacts
| GroES contacts | |||||
| Protein | Swiss-Prot code | Lseq | Mw (kDa) | 6 | 5 |
| Phopspho-2-dehydro-3-deoxyheptonate aldolase, Phe-sensitive | AROG_ECOLI | 350 | 38.0 | 1 | 2 |
| Phopspho-2-dehydro-3-deoxyheptonate aldolase, Trp-sensitive | AROH_ECOLI | 348 | 38.7 | 0 | 2 |
| Tetrahydropteroyltriglutamate methyltransferase | METE_ECOLI | 753 | 84.7 | 1 | 2 |
| 5,10-Methylenetetrahydrofolate reductase | METF_ECOLI | 296 | 33.1 | 1 | 2 |
| S-adenosylmethionine synthetase | METK_ECOLI | 384 | 42.0 | 3 | 5 |
| Acetolactate synthase III large subunit | ILVI_ECOLI | 574 | 63.0 | 4 | 8 |
| 2-isopropylmalate synthase | LEU1_ECOLI | 523 | 57.3 | 4 | 5 |
| D-amino acid dehydrogenase small subunit | DADA_ECOLI | 432 | 47.6 | 2 | 3 |
| Glutamate decarboxylase-α | DCEA_ECOLI | 466 | 52.7 | 1 | 3 |
| Dihydrolipoamide dehydrogenase | DLDH_ECOLI | 474 | 50.7 | 1 | 5 |
| Threonine 3-dehydrogenase | TDH_ECOLI | 341 | 37.2 | 1 | 2 |
| Galactitol-1-phosphate 5-dehydrogenase | GATD_ECOLI | 346 | 37.4 | 1 | 5 |
| Tagatose-bisphosphate aldolase gatY | GATY_ECOLI | 286 | 31.1 | 2 | 3 |
| Enolase | ENO_ECOLI | 432 | 45.7 | 2 | 4 |
| Glyceraldehyde 3-phosphate dehydrogenase A | G3P1_ECOLI | 331 | 35.4 | 0 | 2 |
| NAGD protein | NAGD_ECOLI | 250 | 27.1 | 1 | 4 |
| Phosphate acetyltransferase | PTA_ECOLI | 714 | 77.1 | 4 | 9 |
| UDP-galactopyranose mutase | GLF_ECOLI | 367 | 43.0 | 1 | 1 |
| Capsular-synthesis regulator component B | RCSB_ECOLI | 216 | 23.7 | 1 | 3 |
| Aspartate carbamoyltransferase, catalytic chain | PYRB_ECOLI | 311 | 34.4 | 3 | 3 |
| Uracil phosphoribosyltransferase | UPP_ECOLI | 217 | 22.5 | 0 | 1 |
| Phosphomethylpyrimidine kinase | THID_ECOLI | 266 | 28.6 | 1 | 4 |
| THIG protein | THIG_ECOLI | 256 | 26.9 | 2 | 3 |
| THIH protein | THIH_ECOLI | 377 | 43.3 | 1 | 2 |
| 8-amino-7-oxononanoate synthase | BIOF_ECOLI | 384 | 41.6 | 1 | 4 |
| Lipoic acid synthetase | LIPA_ECOLI | 321 | 36.1 | 1 | 1 |
| DNA-directed RNA polymerase β chain fragment | RPOB_ECOLI | 1342 | 150.6 | 1 | 7 |
| 30S ribosomal protein S2 | RS2_ECOLI | 241 | 26.6 | 0 | 1 |
| Phenylalanyl-tRNA synthetase β chain | SYFB_ECOLI | 795 | 87.4 | 2 | 5 |
| Glycyl-tRNA synthetase β chain | SYGB_ECOLI | 689 | 76.8 | 2 | 4 |
| Threonyl-tRNA synthetase | SYT_ECOLI | 642 | 74.0 | 1 | 5 |
| NUSG protein | NUSG_ECOLI | 181 | 20.5 | 1 | 2 |
| Elongation factor Tu | EFTU_ECOLI | 394 | 43.2 | 0 | 4 |
| ATP-dependent Clp protease ATP-binding subunit ClpX | CLPX_ECOLI | 424 | 46.4 | 1 | 3 |
| DNA gyrase subunit A | GYRA_ECOLI | 875 | 97.0 | 1 | 7 |
| RecA protein | RECA_ECOLI | 353 | 38.0 | 2 | 3 |
| Cell division inhibitor minD | MIND_ECOLI | 270 | 29.6 | 1 | 2 |
| Phosphate transport ATP-binding protein PSTB | PSTB_ECOLI | 257 | 29.0 | 1 | 4 |
| Ferritin | FTN_ECOLI | 165 | 19.4 | 0 | 1 |
| Protein YCFV | YCFV_ECOLI | 233 | 24.9 | 0 | 2 |
| Protein in XERC-UVRD intergenic region | YIGB_ECOLI | 238 | 27.1 | 0 | 2 |
| Protein OXMY-DEOC intergenic region | YJJU_ECOLI | 357 | 40.0 | 2 | 6 |
There are several substrates that are known to interact with GroEL in vitro. We searched for exact GroES-like pattern (G_IVL_G), which has one amino acid less than that found in the GroEL–GroES-(ADP)7 complex. Using this pattern, we find that RuBisCo from pea, mitochondrial, and cytoplasmic malate dehydrogenase (mMDH and cMDH, respectively) have one pattern match. The method, while successful in rationalizing the interaction between these SPs and GroEL, is not capable of assessing the differences in the strength of interaction between mMDH and cMDH. The larger protein aconitase (Chaudhuri et al. 2001), which cannot be fully encapsulated in GroEL, has seven GroES-like patterns. This suggests that aconitase may also be recognized by the GroEL-apical domain.
The preceding comparisons have been made by searching for a precise GroES pattern, which was determined by noting that the mobile loop makes six contacts with H and I helices of the apical domain (see Materials and Methods). As explained in Materials and Methods, the typical number of contacts (NC) a polypeptide chain makes with GroEL during the capture process is not known. The value of NC is likely to be a function of the SP. Even for a given SP, NC is likely to fluctuate. To generalize our method, we made a search for the number of sequences as a function of NC and NB (see Materials and Methods). If NC is too small, then all SPs would be identified as natural SPs. However, small NC would lead to a highly unstable SP–GroEL complex. On the other hand, if NC is large as in the case for SBP, which makes eight contacts with the apical domain, then very few SPs would qualify as natural SPs (see Table 1). This is because unbinding of the SP in a hyperstable GroEL–SP complex is improbable. Thus, there should be a range of NCvalues that would give rise to a stable, but not hyperstable SP–GroEL complex. In terms of the variables (NC, NB), we find that as long as 4 < NC < 6, then interaction with about two to four binding sites suffices to identify the expected third of the E. coli proteins as natural SPs. From Figure 2 ▶, it follows that in excess of 80% of the 52 proteins identified by Houry et al. (1999) can be predicted using the two-dimensional map, as long as NC and NB are in the range given above.
Figure 2.

Contour plot of the percentage of E. coli proteins able to bind to a given number of GroEL sites, NB, as a function of the number of contacts per site, NC. The dotted line represents the boundary of (NC, NB) values below which in excess of 80% of the substrates in the experiments by Houry et al. (1999) are identified.
The results in Figure 2 ▶ can be profitably used in conjunction with experiments. For example, if NB for a SP is known experimentally, as in the study of Farr et al. (2000), then Figure 2 ▶ can be used to predict the number of contacts the SP makes with the apical domain. Given that these contacts involve predominantly hydrophobic residues, a pattern search can be used to obtain the binding regions in the sequence.
S. cerevisiae and E. coli genomes have similar percentages of substrate proteins that interact with chaperonins
The heat-shock protein 60 (Hsp60) from S. cerevisiae (Reading et al. 1989) and Rubisco subunit-binding protein (RsuBP) from pea (Hemmingsen et al. 1988) are analogs of GroEL. Both GroEL and Hsp60 enable refolding of dihydrofolate reductase, while GroEL and RsuBP assist in the refolding of Rubisco. A search for GroES- and SBP-like binding patterns among S. cerevisiae protein sequences is a way to identify the natural SPs for Hsp60 in the S. cerevisiae genome. The percentage of S. cerevisiae sequences that contain GroES patterns is similar to that in E. coli, with 62% having single patterns (vs. 12 expected for sequences with randomly arranged amino acids given by equation 3) and 32% multiple patterns (Table 1). We find that only 845 sequences have four or more binding sites. As in E. coli, the number of sequences with SBP-like patterns is considerably less (Table 1). Some SPs are found in both organisms, such as adenylate cyclase (two GroES repeats in E. coli and three in S. cerevisiae), enolase (two repeats in each), glutamine synthetase (three repeats in E. coli and two in S. cerevisiae).
Putative binding regions do not have a preferred secondary structure
Interaction of SPs with GroEL does not require the binding regions to adopt specific secondary structure (Aoki et al. 2000). For example, the SBP is a random coil in isolation (Chen and Sigler 1999; G. Stan, B.R. Brooks, and D. Thirumalai, unpubl.) as is the fragment of the mobile group. Nevertheless, both interact with the apical domain of GroEL. We have performed a limited search of the preferred secondary structures (using PSIPred; Jones 1999) adopted by the putative SPs identified using our method. For example, in aspartokinase I, with four sequence regions matching the GroES-binding pattern, segments 77–85, 130–138, and 557–565 are predicted to be mostly or exclusively α-helical, but 776–784 is a strand. Among the two threonine synthase binding regions, in the segment 27–35, a short strand is enclosed by two coiled regions, but segment 137–145 is predicted to form an α-helix. Similar variations occur among the four binding regions of the UDP-glucose lipid carrier transferase: 97–105 and 121–129 are α-helical, 151–159 includes a coil region and a helix, and 207–215 forms a β-strand and a coil region. These anecdotal examples support the experimental studies of Yoshida and coworkers (Aoki et al. 2000) who found that “random” sequences with no preference for any specific secondary structure can bind to GroEL.
The eubacterium Ureaplasma urealyticum contains putative GroEL substrates
The U. urealyticum genome (Glass et al. 2000) is unique in that it lacks the GroEL/S chaperonin system. Nevertheless, we find that even in this bacterium, the percentage of protein sequences containing the GroES and SBP-binding patterns is similar to that found in E. coli and S. cerevisiae. As shown in Table 1, among the 614 sequences in this genome, 383 (62%) contain the GroES-binding pattern (compared with one expected for random sequences according to equation 3), and 189 (31%) of these constitute putative GroEL substrates. The SBP binding pattern is present in 122 (20%) sequences (none expected in the random case), 16 (3%) of which contain it multiple times.
Thermophilic and hyperthermophilic bacteria contain different percentages of putative GroEL substrates
Thermophiles and hyperthermophiles are archeal organisms that grow at high (45°–80°C) and very high (above 80°C) temperatures. Thermoplasma acidophilum (Ruepp et al. 2000), which is found at 59°C, has a slightly smaller percentage of putative GroEL substrates (28%) than E. coli and S. cerevisiae (Table 1). For Methanopyrus kandleri (Slesarev et al. 2002), which exists at temperatures between 80° and 110°C, we predict that 323 (19%) proteins interact strongly with GroEL (Table 1). The smaller number of putative SPs in the M. kandleri genome may be related to the larger fraction (P_ = 0.16, compared with 0.11 in E. coli) of negatively charged residues in the M. kandleri genome. Because the percentage of negatively charged residues in M. kandleri is greater than in E. coli (and other genomes), it is less likely to find the largely hydrophobic GroES-like pattern in the protein sequences in M. kandleri.
A set of membrane proteins contains GroES-like binding patterns
Several membrane proteins form complexes with GroEL, resulting in solubilization of these substrates in the absence of detergents (Bochkareva et al. 1996; Deaton et al. 2004). The degree of solubilization of a set of membrane proteins depends on the ratio of GroEL to solute protein molecules and the detergent used to purify them. The membrane proteins BR, Tar, and MalGFK show GroEL-dependent solubilization, while Lacy and DAGK have transient solubilization (Deaton et al. 2004). We find that all of these membrane proteins contain a GroES-like binding pattern: BR has two matching sequence segments; Tar has four; MalGFK, two; LacY, six; and DAGK, one. This suggests that GroEL-solubilization might occur as a result of favorable hydrophobic interactions with regions containing patterns that are surrogates for GroES mobile loop peptide.
Assessing the sensitivity of results to variations in binding patterns
We have assumed that the sequence pattern found in the fragment of GroES mobile loop that interacts with GroEL or the SBP pattern (to a lesser extent) is representative of natural chaperonin SPs. To assess the sensitivity of the results to changes in the binding patterns, we have considered the effect of random substitutions in substrate residues or modified the length of the binding pattern. We replaced hydrophobic residues with hydrophilic ones, or vice versa, in the GroES pattern while preserving the gap positions and the overall pattern length. As shown in Table 3, these substitutions, which result in the patterns H_PPP_H_P, P_HHP_H_P, or P_PPH_H_P, do not affect the E. coli results significantly. However, introduction of charged residues in the GroES pattern (e.g., R_PPH_I_K) dramatically reduces the number of matches. The reduced number of sequences that contain charged patterns is indicative of functions requiring specific interactions. Indeed, a binding mechanism requiring specific interactions, namely, charged and polar, is found in the eukaryotic CCT (Gómez-Puertas et al. 2004). As a result, CCT has a high selectivity for actins and tubulins and it assists a limited number of proteins.
Table 3.
Number of E. coli proteins containing patterns resulting from substitutions in the GroES-binding patterns and shortening the SBP-binding pattern
| Substrate | Pattern | 4-type pattern | Single | Multiple |
| GroES | L_PPH_I_S | H_PPP_H_P | 2100 | 771 |
| S_IWH_L_S | P_HHP_H_P | 2609 | 1189 | |
| Q_GHV_I_S | P_PPH_H_P | 2143 | 819 | |
| R_PPH_I_K | +_PPP_H_− | 174 | 2 | |
| SBP | WM_ _ _WGFLH | HH_ _ _HPHHP | 1879 | 701 |
| WM_ _ _WGFL | HH_ _ _HPHH | 3105 | 1798 |
We also reduced the length of the SBP pattern obtained from the minichaperone-bound structure by removing one or two amino acids at the C terminus. This has the effect of significantly increasing the number of matches (Table 3). When the SBP pattern is shortened to nine residues, the number of protein matches in E. coli becomes similar to that for the GroES-binding patterns. This suggests that a less tightly bound SBP could resemble more closely the GroES binding. It also follows that the natural SPs may not interact as strongly with GroEL as SBP.
We also considered the consequence of varying the number of contiguous hydrophobic residues in our search pattern. This type of pattern could define an SP-binding pattern in view of the importance of the hydrophobic interactions for SP recognition by GroEL. The number of sequence matches as a function of the length of the hydrophobic pattern shows (Fig. 3 ▶) that short hydrophobic patterns (λ ≤ 4) are present in most protein sequences. The number of sequences that contain a continuous stretch of six hydrophobic residues is close to that containing the GroES-binding pattern is 2488. The similarity of the results for these two different patterns is also observed in the number of sequences that contain these patterns multiple times. It follows that polypeptide chains that contain a minimum number (between five and six) (Fig. 3 ▶) of contiguous stretch of hydrophobic residues can interact favorably with GroEL. To assess whether this conclusion depends on natural sequences, we generated a database of random sequences each with length L̄ = 314. The sequences are made from four types of amino acids with probability of occurrence corresponding to that in E. coli (Materials and Methods). Analysis of the database of random sequences shows that the number of sequences with five or six continuous stretches of hydrophobic residues is nearly the same as in E. coli (White and Jacobs 1990). From this, we conclude that GroEL can form complexes with random sequences as long as they possess a continuous stretch of at least five hydrophobic amino acid residues.
Figure 3.

The number of E. coli sequences, NHS, containing a contiguous set of hydrophobic residues of given length. For block length λ, NHS gives the total number of sequences with hydrophobic block length ≤λ. Circles represent the number of proteins containing single (filled symbols) and multiple matches (empty symbols).
Discussion
Based on the assumption that GroES mobile loop peptide and the SBP peptide sequence patterns are typical of substrate proteins that interact with GroEL, we have devised a sequence-based method to identify the natural substrates for chaperonins. We believe that putative substrate proteins are those that contain multiple repeats (≥2) of these patterns because they can form stable complexes (not hyperstable as in the SBP–apical domain complex) with GroEL through interactions at multiple binding sites. Several experimentally identified GroEL SPs are found to contain these patterns, suggesting that the GroES and SBP-like complementarity are features of a large class of substrates, including ones that are considered to be stringent SPs. The putative SPs have diverse functions and no common structural feature.
Our method is a physically based procedure for identifying natural SPs for GroEL and its analog in other organisms. The good agreement between the predictions and the experimentally identified GroEL substrates (provided NC and NB are allowed to vary), validates the methodology. However, it is clear that the sequence-based approach alone cannot identify all of the SPs that interact transiently with GroEL in a cellular environment. This could especially be the case for large SPs (those that cannot be fully encapsulated in the GroEL cavity) that form only marginally stable complexes with GroEL. Even such proteins like aconitase, which may not interact directly with the grooves of helices H and I could have binding patterns of the GroES mobile loop. Despite this limitation, the present method has, for the first time, provided a basis for identifying natural substrate proteins that require the chaperonin machinery. The predictions of a bioinformatics-based approach are amenable to experimental tests.
Materials and methods
Pattern identification
We determined the binding motifs for the co-chaperonin GroES (Xu et al. 1997) and the 12-mer SBP (Chen and Sigler 1999) by identifying their respective contacts with GroEL (Fig. 1 ▶). Inter-residue contacts form if the closest distance between heavy atoms is <4 Å and the contact surface area is at least 20 Å2. Using the Contacts of Structural Units program (Sobolev et al. 1999), the binding patterns for GroES and SBP are, respectively, G_IVL_G_A, and WM_ _ _WGFLHP, where “_” represents an arbitrary residue. Amino acids in GroEL that are closest to these substrates and the associated contact maps are displayed in Figure 1 ▶.
To perform a general search of the genome for putative chaperonin SPs, we divided the 20 amino acids into four classes, namely, hydrophobic (H), polar (P), positively (+), and negatively charged (−). The four classes are H (C, F, I, L, W, V, M, Y, and A), P (G, P, N, T, S, Q, and H), + (R and K), and − (D and E). Previously we showed (Stan et al. 2003) that, for GroEL and GroES functions, the chemical class, but not the identity of the residue, is conserved. Translating the binding sequences into the four residue types, the amino acid residues that are complementary to those in the strongly conserved residues in the GroEL-binding sites are HH_ _ _HPHHPP for SBP and P_HHH_P_H for GroES. Neither of these sequence patterns contain charged residues, which is consistent with the notion that predominantly hydrophobic attraction (however, see Buckle et al. 1997) helps the apical domain ensnare the SPs. The most likely substrates are those that contain at least two sequence patterns such as the ones found in the mobile loop of GroES or the SBP. Accordingly, we searched for these two patterns in five genomes: the Escherichia coli K12 (NCBI accession code NC_000913; Blattner et al. 1997), the Saccharomyces cerevisiae (the current version as of May 1, 2004 at the Saccharomyces Genome Database) (Goffeau et al. 1996), the Ureaplasma urealyticum (NCBI accession code NC_002162; Glass et al. 2000), the Thermoplasma acidophilum (NCBI accession code NC_002578; Ruepp et al. 2000), and the Methanopyrus kandleri (NCBI accession code NC_003551; Slesarev et al. 2002).
The patterns identified using GroES complementarity can vary to some extent. The extent of variation will depend on the strength of interaction between the binding sites and the apical domain. For efficient annealing, it has been argued (Orland and Thirumalai 1997) that the average stabilizing energy per residue between SP binding sites and GroEL be on the order of (1–2) kBT. The length of the GroES pattern, which is the natural complement to the helices H and I, can change to satisfy the stability criterion. However, it is crucial to have a set of core residues in the SP that serve as recognition sites by the hydrophobic residues in the GroEL apical domain. Our previous study (Stan et al. 2003) shows that the core residues in GroES are G_IVL.
Sequences containing multiple matches are more likely to be natural SPs, as each substrate protein can bind to up to seven GroEL-binding sites. Simultaneously binding to several (say, >4) sites is unlikely because the resulting SP–GroEL complex would have a very low dissociation constant. The successive matches must be separated by a minimal distance along the sequence corresponding to the spatial separation between adjacent binding sites. In the T state of GroEL, the binding sites from neighboring subunits are separated by 25 Å. To estimate the sequence length, 1, corresponding to this distance, we used the Flory formula R ≅ b 13/5, where R is the end-to-end distance and b ~3.8 Å, is the distance between Cα atoms. This leads to l ~23 residues between the end of one binding pattern and the beginning of the next along the sequence. The value of l ≅ 23 is approximate. It is likely that multiple binding sites that are separated by stiff loops (l < 23) can also serve as natural substrates. Most probable loops have l ≈ 10 (Camacho and Thirumalai 1995). Using 10 ≤ l ≤ 23 in the search for multiple binding patterns results in ~5% variation in the number of multiple pattern matches. Increasing l up to 60 results in only an ~10% change in the number of patterns.
The patterns that we search for are based on the number of contacts, NC, the mobile loop of GroES, or the SBP makes with the H and I helices of the apical domain. Because no SP–GroEL structure for a natural SP is available, we searched for the instances that these exact patterns occur in the various genomes. Until there are several SP–GroEL structures, it is uncertain whether the number of contacts that these peptides make is typical. Absent this information, we performed a general two-parameter search by varying the number of contacts, NC, and located a stretch of the polypeptide chain that can interact with the apical domain. The pattern changes as NC is changed. For a fixed pattern, we also searched the genomes for the number of binding sites (NB) in each sequence. A typical range of NB is likely to be 2 ≤ NB ≤ 4.
Statistical significance of sequence matches
To establish the statistical significance of our results, it is necessary to ascertain whether the observed number of patterns can arise randomly. The probability that a stretch of L randomly arranged residues matches a given pattern is (Karlin 1995)
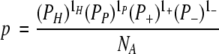 |
(1) |
where Pi, i = H,P,+, − is the probability of a residue being of type i, and li is the number of residues of type i in the pattern. The number of random arrangements of the four residue types and gaps (g) of length lg in the given pattern is
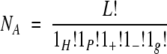 |
(2) |
where L = 1H + 1P + 1+ + 1− + 1g. The expected number of sequences in a database that contain at least one pattern is
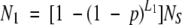 |
(3) |
where L1 = L̄S − L + 1, and NS is the number of sequences. We assumed that the average sequence length, L̄S, satisfies the condition L̄S > L. The frequencies of the four chemical types in the E. coli genome are PH = 0.46, PP = 0.38, P+ = 0.10, and P− = 0.11. Excluding sequences of <20 residues, L̄S for E. coli is 314. For S. cerevisiae, the corresponding values are PH = 0.40, PP = 0.37, P+ = 0.12, P− = 0.12, and L̄S = 458, while for U. urealyticum PH = 0.43, PP = 0.32, P+ = 0.13, P− = 0.12, and L̄S = 372.
Acknowledgments
We thank the reviewers for several thoughtful comments. This work is supported by a grant from the NIH to G.H.L. and D.T. through grant number 1R01GM067851-01.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04933205.
Supplemental material: see www.proteinscience.org
References
- Aoki, K., Motojima, F., Taguchi, H., Yomo, T., and Yoshida, M. 2000. GroEL binds artificial proteins with random sequences. J. Biol. Chem. 275 13755–13758. [DOI] [PubMed] [Google Scholar]
- Blattner, F.A., Plunkett III, G., Bloch, C.A., Perna, N.T., Burland, V., Riley, M., Collado-Vides, J., Glasner, J.D., Rode, C.K., Mayhew, G.F., et al. 1997. The complete genome sequence of Escherichia coli K-12. Science 277 1453–1462. [DOI] [PubMed] [Google Scholar]
- Bochkareva, E., Seluanov, A., Bibi, E., and Girshovich, A. 1996. Chaperonin-promoted post-translational membrane insertion of a multispanning membrane protein lactose permease. J. Biol. Chem. 271 22256–22261. [DOI] [PubMed] [Google Scholar]
- Buckle, A.M., Zahn, R., and Fersht, A.R. 1997. A structural model for GroEL - polypeptide recognition. Proc. Natl. Acad. Sci. 94 3571–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho, C. and Thirumalai, D. 1995. Theoretical predictions of folding pathways by using the proximity rule, with applications to bovine pancreatic trypsin inhibitor. Proc. Natl. Acad. Sci. 92 1277–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri, T.K., Farr, G.W., Fenton, W.A., Rospert, S., and Horwich, A.L. 2001. GroEL/GroES-mediated folding of a protein too large to be encapsulated. Cell 107 223–233. [DOI] [PubMed] [Google Scholar]
- Chen, L.L. and Sigler, P.B. 1999. The crystal structure of a GroEL/peptide complex: Plasticity as a basis for substrate diversity. Cell 99 757–768. [DOI] [PubMed] [Google Scholar]
- Deaton, J., Sun, J., Holzenburg, A., Struck, D.K., Berry, J., and Young, R. 2004. Functional bacteriorhodopsin is efficiently solubilized and delivered to membranes by the chaperonin GroEL. Proc. Natl. Acad. Sci. 101 2281–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr, G.W., Furtak, K., Rowland, M.R., Ranson, N.A., Saibil, H.R., Kirchhausen, T., and Horwich, A.L. 2000. Multivalent binding of nonnative substrate proteins by the chaperonin GroEL. Cell 100 561–573. [DOI] [PubMed] [Google Scholar]
- Fayet, O., Louarn, J.M., and Georgopoulos, C. 1986. Suppression of Escherichia coli DNA46 mutation by amplification of the groes and groel genes. Mol. Gen. Genet. 202 434–445. [DOI] [PubMed] [Google Scholar]
- Fenton, W.A. and Horwich, A.L. 2003. Chaperonin-mediated protein folding: Fate of substrate polypeptide. Q. Rev. Biophys. 36 229–256. [DOI] [PubMed] [Google Scholar]
- Fenton, W.A., Kashi, Y., Furtak, K., and Horwich, A.L. 1994. Residues in chaperonin GroEL required for polypeptide binding and release. Nature 371 614–619. [DOI] [PubMed] [Google Scholar]
- Glass, J.I., Lefkowitz, E.J., Glass, J.S., Heiner, C.R., Chen, E.Y., and Cassell, G.H. 2000. The complete sequence of the mucosal pathogen Ureaplasma urealyticum. Nature 407 757–762. [DOI] [PubMed] [Google Scholar]
- Goffeau, A., Barrell, B.G., Bussey, H., Davis, R.W., Dujon, B., Feldmann, H., Galibert, F., Hoheisel, J.D., Jacq, C., Johnston, M., et al. 1996. Life with 6000 genes. Science 274 546–567. [DOI] [PubMed] [Google Scholar]
- Gómez-Puertas, P., Martín-Benito, J., Carrascosa, J.L., Willison, K.R., and Valpuesta, J.M. 2004. The substrate recognition mechanisms in chaperonins. J. Mol. Recognit. 17 85–94. [DOI] [PubMed] [Google Scholar]
- Gordon, C.L., Sather, S.K., Casjens, S., and King, J. 1994. Selective in vivo rescue by GroEL/ES of thermolabile folding intermediates to phage P22 structural proteins. J. Biol. Chem. 269 27941–27951. [PubMed] [Google Scholar]
- Hemmingsen, S.M., Woolford, V., van der Vies, S.M., Tilly, K., Dennis, D.T., Georgopoulos, C.P., Hendrix, R.W., and Ellis, R.J. 1988. Homologous plant and bacterial proteins chaperone oligomeric protein assembly. Nature 333 330–334. [DOI] [PubMed] [Google Scholar]
- Horovitz, A., Fridmann, Y., Kafri, G., and Yifrach, O. 2001. Allostery in chaperonins. J. Struct. Biol. 135 104–114. [DOI] [PubMed] [Google Scholar]
- Houry, W.A., Frishman, D., Eckerskorn, C., Lottspeich, F., and Hartl, F.U. 1999. Identification of in vivo substrates of the chaperonin GroEL. Nature 402 147–154. [DOI] [PubMed] [Google Scholar]
- Humphrey, W., Dalke, A., and Schulten, K. 1996. VMD—visual molecular dynamics. J. Mol. Graphics 14 33–38. [DOI] [PubMed] [Google Scholar]
- Jones, D.T. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292 195–202. [DOI] [PubMed] [Google Scholar]
- Karlin, S. 1995. Statistical significance of sequence patterns in proteins. Curr. Opin. Struct. Biol. 5 360–371. [DOI] [PubMed] [Google Scholar]
- Landry, S.J., Taher, A., Georgopoulos, C., and van der Vies, S.M. 1996. Interplay of structure and disorder in cochaperonin mobile loops. Proc. Natl. Acad. Sci. 93 11622–11627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorimer, G.H. 1996. A quantitative assessment of the role of the chaperonin proteins in protein folding in vivo. FASEB J. 10 5–9. [DOI] [PubMed] [Google Scholar]
- Orland, H. and Thirumalai, D. 1997. A kinetic model for chaperonin assisted protein folding. J. Phys. 7 533–560. [Google Scholar]
- Reading, D.S., Hallberg, R.L., and Myers, A.M. 1989. Characterization of the yeast HSP60 gene coding for a mitochondrial assembly factor. Nature 337 655–659. [DOI] [PubMed] [Google Scholar]
- Ruepp, A., Graml, W., Santos-Martinez, M.L., Koretke, K.K., Volker, C., Mewes, H.W., Frishman, D., Stocker, S., Lupas, AN., and Baumeister, W. 2000. The genome sequence of the thermoacidophilic scavenger Thermoplasma acidophilum. Nature 407 508–513. [DOI] [PubMed] [Google Scholar]
- Shewmaker, F., Maskos, K., Simmerling, C., and Landry, S.J. 2001. The disordered mobile loop of GroES folds into a defined β-hairpin upon binding GroEL. J. Biol. Chem. 276 31257–31264. [DOI] [PubMed] [Google Scholar]
- Slesarev, A.I., Mezhevaya, K.V., Makarova, K.S., Polushin, N.N., Shcherbinina, O.V., Shakhova, V.V., Belova, G.I., Natale, L.A.D.A., Rogozin, I.B., Tatusov, R.L., et al. 2002. The complete genome of hyperthermophile Methanopyrus kandleri AV19 and monophyly of archaeal methanogens. Proc. Natl. Acad. Sci. 99 4644–4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolev, V., Sorokine, A., Prilusky, J., Abola, E.E., and Edelman, M. 1999. Automated analysis of interatomic contacts in proteins. Bioinformatics 15 327–332. [DOI] [PubMed] [Google Scholar]
- Stan, G., Thirumalai, D., Lorimer, G.H., and Brooks, B.R. 2003. Annealing function of GroEL: Structural and bioninformatic analysis. Biophys. Chem. 100 453–467. [DOI] [PubMed] [Google Scholar]
- Thirumalai, D. and Lorimer, G.H. 2001. Chaperonin-mediated protein folding. Annu. Rev. Biophys. Biomol. Struct. 30 245–269. [DOI] [PubMed] [Google Scholar]
- van Dyk, T.K., Gatenby, A.A., and LaRossa, R.A. 1989. Demonstration by genetic suppression of interaction of GroE products with many proteins. Nature 342 451–453. [DOI] [PubMed] [Google Scholar]
- Viitanen, P.V., Gatenby, A.A., and Lorimer, G.H. 1992. Purified chaperonin 60 (GroEL) interacts with the non-native states of a multitude of Escherichia coli proteins. Protein Sci. 1 363–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, S.H. and Jacobs, R.E. 1990. Statistical distribution of hydrophobic residues along the length of protein chains: Implications for protein folding and evolution. Biophys. J. 57 911–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Z. and Sigler, P.B. 1998. GroEL/GroES: Structure and function of a two-stroke folding machine. J. Struct. Biol. 124 129–141. [DOI] [PubMed] [Google Scholar]
- Xu, Z., Horwich, A.L., and Sigler, P.B. 1997. The crystal structure of the assymetric GroEL-GroES-(ADP)7 chaperonin complex. Nature 388 741–750. [DOI] [PubMed] [Google Scholar]