

Abstract
The aim of this work is to shed more light on the effect of domain–domain interactions on the kinetics and the pathway of protein folding. A model protein system consisting of several single-tryptophan variants of the two-domain yeast phosphoglycerate kinase (PGK) and its individual domains was studied. Refolding was initiated from the guanidine-unfolded state by stopped-flow or manual mixing and monitored by tryptophan fluorescence from 1 msec to 1000 sec. Denaturant titrations of both individual domains showed apparent two-state unfolding transitions. Refolding kinetics of the individual domains from different denaturant concentrations, however, revealed the presence of intermediate structures during titration for both domains. Refolding of the same domains within the complete protein showed that domain–domain interactions direct the folding of both domains, but in an asymmetric way. Folding of the N domain was already altered within 1 msec, while detectable changes in the folding of the C domain occurred only 60–100 msec after initiating refolding. All mutants showed a hyperfluorescent kinetic intermediate. Both the disappearance of this intermediate and the completion of the folding were significantly faster in the individual N domain than in the complete protein. On the contrary, folding of the individual C domain was slower than in the complete protein. The presence of the C domain directs the refolding of the N domain along a completely different pathway than that of the individual N domain, while folding of the individual C domain follows the same path as within the complete protein.
Keywords: protein folding, domain interactions, phosphoglycerate kinase, two-state folding, tryptophan fluorescence
The general mechanism of protein folding, i.e., how the amino acid sequence directs the organization of the polypeptide chain towards the native structure, is still an unresolved problem. Despite the growing interest, the gap between the sizes of the sequence and structural databases seems to be widening. Due to the obvious advantage of simplicity, the experimental and theoretical work leading to our present understanding of protein folding was mostly performed on small single domain proteins and on model peptides (Snow et al. 2002; Gillespie and Plaxco 2004). Although these studies contributed significantly to our knowledge about the folding mechanisms, it is not trivial how, and to what extent, this knowledge can be translated to larger proteins comprised of several structural domains.
The definition of a domain is not rigorous. Several definitions can be found in the protein folding literature. One can define it as a folding unit, i.e., as part of the protein that can fold to a stable native-like structure even when isolated from the other parts of the protein. Another classification often used in the literature defines the domain as a compact dominion of the native protein structure within which elements interact more extensively, than with elements outside of it. Relatively few studies address the folding properties of multidomain proteins (Adams et al. 1996; Hosszu et al. 1997; Dinner et al. 2000; Wenk et al. 2000; Jäger et al. 2001). These studies revealed that individual structural domains are often able to fold independently; thus, the structure-based and the folding-based domain definitions often coincide. It was also shown that domain interactions could result in both productive and unproductive folding intermediates (Dinner et al. 2000; Jäger and Plückthun 2000). Folding of the domains within a multidomain protein can differ significantly from the folding of the individual domains, since folding paths depend sensitively on the relative stability of the domains and the strength of the domain interactions (Brandts et al. 1989; Semisotnov et al. 1991; Freire et al. 1992; Szilágyi and Vas 1998).
Because of its two-domain structure, phosphoglycerate kinase (PGK) from various organisms has proven a good model to analyze domain interactions (Missiakas et al. 1990; Vas et al. 1990; Ritco-Vonsovici et al. 1995a; Reed et al. 2003). It has been used in many unfolding and refolding studies, and different denatured states of PGK have been well characterized (Chardot et al. 1988; Griko et al. 1989; Damaschun et al. 1993, 1998; Gast et al. 1993).
Yeast PGK is a 415 residue monomeric protein that is built up of two structurally similar domains, linked by a helical hinge. The two domains also interact through the C-terminal helix, which folds back to the N domain, and proved to be crucial for enzyme activity (Ritco-Vonsovici et al. 1995b). In the folded structure the contacts between the two domains are formed through hydrophobic interactions and hydrogen bonds. There are two tryptophan residues in the wild-type yeast PGK that undergo fluorescence changes upon the folding/unfolding transition (Damaschun et al. 1998).
Folding of fragments containing less than one structural domain is incomplete and not cooperative, but both individual full-length domains fold into native-like structures (Pecorari et al. 1996; Hosszu et al. 1997). The isolated C domain retains the ability to bind ATP. The isolated N domain loses the substrate binding ability. For the PGK from Bacillus stearothermophilus it has been shown that the domain retains its native structure. The lack of the contacts with the C-terminal helix, however, leads to an increased flexibility of the 3-phosphoglycerate binding region resulting in the loss of the substrate binding ability (Hosszu et al. 1997).
Förster energy transfer between covalently bound fluorophores was used to determine intramolecular distances measuring the unfolding kinetics and equilibrium titrations of yeast PGK (Lillo et al. 1997a,b). Fluorescence techniques based on specific tryptophan mutants of multidomain proteins are capable of characterizing the behavior of the single domains along the folding process. Several enzymatically active single tryptophan mutants of yeast PGK have been constructed in order to monitor the folding and functional properties of its domains (Szpikowska et al. 1994; Beechem et al. 1995; Cheung and Mas 1996; Szpikowska and Mas 1996).
Most folding studies performed on multidomain proteins address the effect of the domain interactions on the enzyme activity and on the stability of the folded structure. In this work we intend to clarify the effect of the domain interactions on the kinetics and pathway of the folding reaction.
Results and Discussion
Thermodynamic characterization of the studied protein model system
A model system based on the two-domain protein, yeast PGK, was constructed to study the role of domain interactions in protein folding. The model consists of tryptophan mutants that allow the selective detection of the kinetics of conformational changes in the domains of PGK, both in the presence and in the absence of domain interactions.
Figure 1 ▶ illustrates the structure of the four components of the studied model system, based on X-ray crystallography data (Watson et al. 1982). The positions where the single tryptophan fluorophores were placed are shown in red. Sequence differences between the wild type and the mutants are summarized in the table included in the figure
Figure 1.

Structure of the proteins of the model system. 3D representation of the PGK variants used in our model protein system (Watson et al. 1982). The location where the single tryptophan reporters were introduced is indicated in red. Constructing the model system involved the mutation of the residues Trp 308, Trp 333, and Tyr 122; these are shown as “spacefill.” Alpha helices are colored blue, and beta sheets green. Sequence differences between the wild-type yeast PGK and the mutants are listed in italics in the table.
A detailed fluorescence and circular dichroism characterization of the used mutants was presented earlier (Osváth et al. 2003). In that work, a fit based on a three-state model (Folded [F] Intermediate [I] Unfolded [U]) yielded the free-enthalpy differences between the different states, and their sensitivities to GuHCl concentration. Based on the thermodynamic data published there, here we calculated the concentration of the folded, intermediate, and unfolded states during the GuHCl titration for the two mutants of the complete protein (Fig. 2 ▶). Two types of refolding experiments were completed: diluting GuHCl to conditions favoring the folded state from the unfolded and from the intermediate state, as indicated in Figure 2 ▶.
Figure 2.
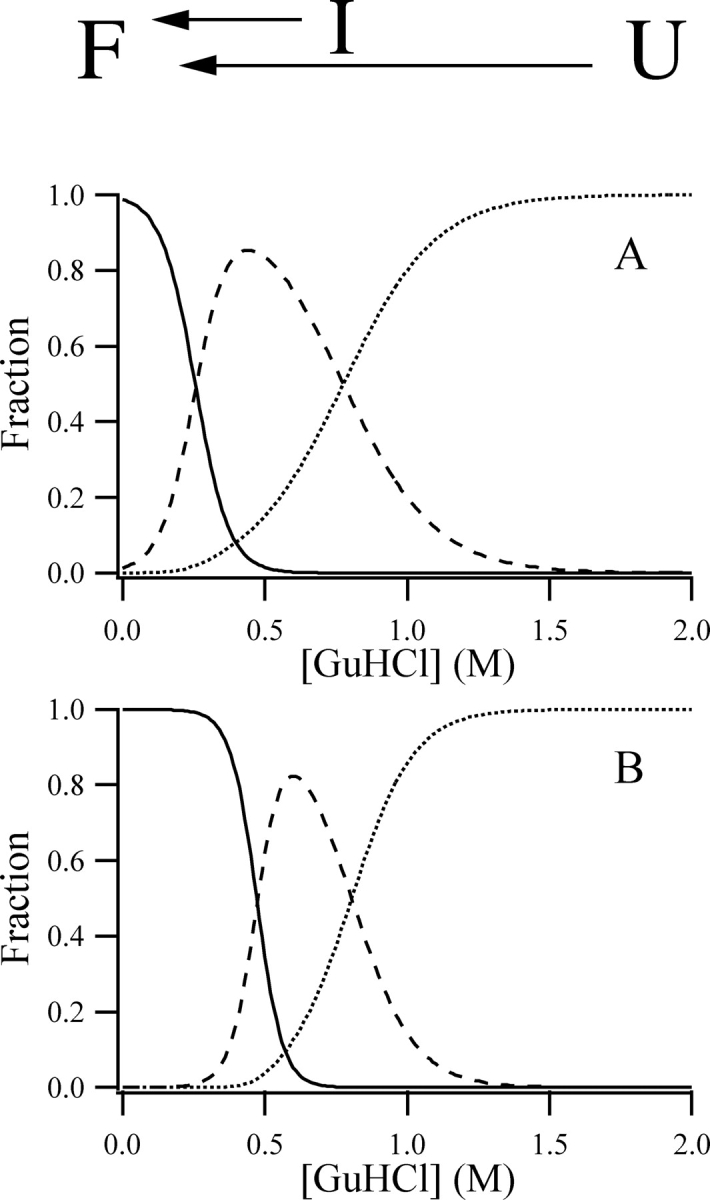
Changes in the population of different states during GuHCl titrations. Variations in the populations of the folded (continuous line), intermediate (dashed line), and unfolded (dotted line) states of the hisPGK W122 (A) and hisPGK W333 (B). The populations were calculated based on the free-enthalpy (also called Gibbs free energy) differences and denaturant sensitivities published earlier (Osváth et al. 2003). The different GuHCl concentration jumps performed in the refolding experiments are indicated.
No intermediate was detected in the circular dichroism measured during the equilibrium GuHCl titrations of the individual N and C domains. A two-state model yielded a perfect fit of the data. This was in agreement with published cold and heat denaturation experiments (Gast et al. 1995; Damaschun et al. 1998). The tryptophan fluorescence spectra measured at intermediate denaturant concentrations, however, could not be generated as the average of the emission spectra of a folded and an unfolded population measured at 0 M and 1.7 M GuHCl, respectively (Fig. 3 ▶). Nonnative fluorescence spectra do not clearly prove the presence of an intermediate; it could indicate a flexibility change of the native state as well (Ervin et al. 2002). Refolding experiments with these mutants were done from the fully unfolded state and also from intermediate denaturant concentrations. The kinetic data shown in this paper unequivocally prove the presence of equilibrium intermediates in the GuHCl titration of the individual domains.
Figure 3.

Fluorescence emission spectra. Fluorescence emission spectra of the hisN W122 (A) and hisC W333 (B) mutants excited at 290 nm in the native state (0 M GuHCl; continuous line), unfolded state (1.7 M GuHCl; dotted line), and at intermediate denaturant concentrations (0.65 M and 0.55 GuHCl for the hisN W122 and hisC W333, respectively; dashed line). The peak emission intensity of the protein in 1.7 M GuHCl was taken as unity. Samples contained 40–60 μM protein, 50 mM potassium phosphate, 1 mM EDTA, 1 mM DTT (pH 6.2).
Recording the refolding kinetics from 1 msec to 1000 sec
Refolding of the GuHCl denatured protein was monitored from 1 msec to 1000 sec after an 11-fold dilution by stopped-flow (1 msec to 50 sec) or manual mixing (30 sec to 1000 sec). Conformational changes were followed by tryptophan fluorescence. The stopped-flow and the manual mixing traces were recorded separately, but at the same time and on the same sample. The two kinetic curves were fit together by normalizing the two fluorescence traces to overlap between 30 sec and 50 sec.
The above procedure resulted in smooth curves, which confirmed the validity of the method. No other corrections were needed, with the only exception of the refolding from the partially unfolded state of the hisN W122 shown in Figure 5A ▶ (below). For this trace the slopes of the two curves did not match after the above procedure. The stopped-flow and the manual mixing experiments were done with different detection spectral width. In the partial refolding of hisN W122 the very small fluorescence quantum yield change of the last folding step is combined with a spectral shift. This situation made necessary an additional stretching of the manual mixing trace to match the slopes of the two measurements between 30 sec and 50 sec. The trace shown in Figure 5A ▶ (below) is the result of this procedure.
Figure 5.

Refolding from the partially denatured state. Refolding kinetics of the hisN W122 (A; dotted line in B), hisPGK W122 (continuous line in B), hisC W333 (C), and hisPGK W333 (D) mutants from a partially unfolded state. Samples contained 40–60 μM protein, 50 mM potassium phosphate (pH 6.2), 1 mM EDTA, 1 mM DTT, 0.58 M, 0.63 M, 0.56 M, and 0.65 M GuHCl for the hisN W122, the hisPGK W122, the hisC W333, and the hisPGK W333, respectively. Kinetics were recorded after an 11-fold dilution with a similar buffer containing no GuHCl. The fluorescence of the unfolded state in 1.7 M GuCHl was taken as unity.
In the unfolded protein there are no long-range interactions and the tryptophans are solvent-exposed; thus, their fluorescence should be sequence-independent. The fluorescence yield of the tryptophan residues could depend only on possible effects of their immediate neighbors in the amino acid sequence. Such nearest-neighbor effects would be identical for the mutant pairs anyway, but we found that the fluorescence spectra measured in 1.7 M GuHCl are identical within experimental error for all four single-tryptophan mutants between 320 nm and 400 nm. A small difference was observed between these spectra in the 300-nm and 320-nm region, which can be explained by a weak contribution from the tyrosine residues. The contribution of tyrosines was eliminated in the kinetic measurements using a 320-nm cutoff filter.
Taking advantage of the above findings, all fluorescence intensities were normalized to the fluorescence of the tryptophan in the unfolded protein in 1.7 M GuHCl. The final fluorescence measured at 1000 sec was scaled to this normalized value based on equilibrium GuHCl titration curves from which the fluorescence at the GuHCl concentration after the stopped flow mixing could be determined. We supported that this was the value that the kinetic experiments were approaching.
Kinetics of refolding from the fully unfolded state
Figure 4 ▶ compares refolding from the fully unfolded state of four single-tryptophan mutants (hisN W122, hisPGK W122, hisC W333, and hisPGK W333), as detected by fluorescence excited at 295 nm. Refolding was initiated by an 11-fold dilution of the protein sample solubilized in 1.7 M GuHCl. Table 1 summarizes the fluorescence levels for the studied mutants in the folded state, and 1 msec after the stopped-flow mixing. All mutants showed tryptophan fluorescence quenching upon folding. The extent of the quenching was different for the individual domains and mutants of the complete protein, the fluorescence of the individual domains being stronger. This is probably caused by a change in the flexibility of the native structure due to the lack of the interdomain contacts (Hosszu et al. 1997; Ervin et al. 2002).
Figure 4.

Refolding from the unfolded state. Refolding kinetics of the hisN W122 (A; dotted line in B), hisPGK W122 (continuous line in B), hisC W333 (C; dotted line in D), and hisPGK W333 (continuous line in D) mutants. Samples of 40–60 μM protein unfolded in 1.7 M GuHCl, 50 mM potassium phosphate (pH 6.2), 1 mM EDTA, 1 mM DTT aqueous solution, were diluted 11-fold with a similar solution containing no protein and no GuHCl. The fluorescence of the unfolded state in 1.7 M GuCHl was taken as unity.
Table 1.
Fluorescence levels of the studied mutants
| hisN W122 | hisPGK W122 | hisC W333 | hisPGK W333 | |
| Fluorescence 1 msec after mixing | 1.17 ± 0.1 | 2.58 ± 0.1 | 1.18 ± 0.1 | 1.15 ± 0.1 |
| Fluorescence of the native state in 0 M GuHCl | 0.89 ± 0.1 | 0.51 ± 0.05 | 0.73 ± 0.1 | 0.36 ± 0.05 |
Relative fluorescence intensities of the hisN W122, the hisPGK W122, the hisC W33, and the hisPGK W333 mutants in the folded state, and 1 msec after initiating refolding. The fluorescence of the unfolded state in 1.7 M GuHCl was taken as unity.
The fluorescence of the hisPGK W122 mutant is substantially higher after the burst phase than the fluorescence of the protein unfolded in 1.7 M GuHCl, but the fluorescence of the other three mutants is close to unity. An increase in fluorescence was observed in all four mutants up to the seconds timescale, except for the hisN W122, where a hyperfluorescent intermediate is formed in <100 msec. This is followed by a fluorescence decrease and, in the case of the hisN W122, the formation of a state with lower fluorescence than that of the native state. By the end of the 1000-sec time window all mutants practically reach folding equilibrium. The estimation of how far the system is from equilibrium after 1000 sec was made doing an exponential fit to the kinetics between 200 sec and 1000 sec. The equilibrium fluorescence estimated from the baseline of the fit differed from the fluorescence level measured at 1000 sec by <1% for the hisN W122, 7% for the hisPGK W122, 3% for the hisC W333, and 5% for the hisPGK W333. During the normalization of the kinetic data no correction was made for these small differences since they are smaller than the error of the relative fluorescence levels. Taking into account the size of the fluorescence changes during refolding, we estimate that at 1000 sec the folding reaction is <3%–4% away from equilibrium in all the mutants of our model system. This is in agreement with earlier findings (Betton et al. 1992; Missiakas et al. 1992; Otto et al. 1994; Gast et al. 1997).
The fluorescence changes accompanying refolding were different for the individual domains and the corresponding complete proteins. The folding of the individual N domain differs from the trace obtained for the corresponding complete protein already at 1 msec. The two kinetics are compared on the same scale in Figure 4B ▶. Contrary to this, the folding kinetics of the individual C domain is different from the folding of the same domain within the complete protein only 60–100 msec after mixing (Fig. 4D ▶). This is in agreement with earlier results that found no influence of the domain interactions on the folding of the C-terminal up to 10 msec (Osváth et al. 2003).
The large difference in the fluorescence level of the hyperfluorescent intermediates observed indicates that the transient structure formed in the vicinity of the residue 122 is different in the individual N domain and the corresponding complete protein. Apparently the domain interactions influence the submillisecond collapse of the N terminus of the yeast PGK. This differs from earlier findings of Parker and coworkers (Parker et al. 1996b), who concluded that the two domains of B. stearothermophilus phosphoglycerate kinase do not influence each other’s early collapse. The same group working on the same protein found a hierarchical folding, with the long-range contacts influencing only the slow steps of the folding (Parker et al. 1996a). In contrast, our results indicate that long-range contacts are important in both the fast and slow steps of protein folding; thus, folding is not hierarchical in our case. It has been shown that the folding pathway in a two-domain protein depends sensitively on the stability of the domains relative to the energy of the domain–domain interaction (Brandts et al. 1989; Freire et al. 1992). The above differences between yeast and B. stearothermophilus PGK folding probably come from the difference in the stability of the domains. Since the B. stearothermophilus is a thermostable bacterium, its proteins have evolutionary been designed to be more stable than the proteins of organisms living at lower temperatures, e.g., yeast (Závodszky et al. 1998; Szilágyi and Závodszky 2000). Also, our measurements were done at pH 6.2, where the protein is less stable than at pH 7.2, used in the experiments done on the B. stearothermophilus PGK. The stronger effect of the domain interactions and the lack of hierarchical folding in the yeast enzyme can probably be accounted for the smaller stability of the individual domains of the yeast protein compared to the B. stearothermophilus.
Our results show that the tryptophan fluorescence detected folding of the individual N domain is qualitatively different from the folding of the N domain within the complete protein indicating that the N domain follows different folding pathways in the two cases. Contrary to the behavior of the N domain, the overall fluorescence course for the individual C domain and the corresponding complete protein were analogous. The individual N-terminal folds faster than the individual C domain, but due to the domain interactions the folding rates change, and the C domain acquires its native structure faster than the N domain in the complete protein.
Kinetics of refolding from intermediate denaturant concentrations
Figure 5 ▶ shows the fluorescence-detected stopped-flow refolding from partially unfolded states of the four single tryptophan mutants of our model system. Similar to the refolding from the fully unfolded state, folding of the N domain is altered radically due to the domain interactions, while the changes in the folding of the C domain are less fundamental.
The fluorescence changes accompanying the refolding from a completely unfolded state (Fig. 4A,C ▶) and the refolding from the partially unfolded state (Fig. 5A,C ▶) are strikingly different for both individual domains. In a two-state folder only the unfolded and the folded states are populated significantly during the equilibrium titrations; thus, the kinetics of the refolding should be independent of the initial state. This is not the case for either of the individual domains, indicating that intermediate structures accumulate during the GuHCl titrations of both individual domains. We believe that the best way to confirm the two-state character of a folding reaction is based on a kinetic check. For a two-state folder the folding kinetics should depend solely on the final state and not on the initial extent of denaturation.
Conclusions
Domain–domain interactions alter the folding of yeast PGK considerably. The effect of the domain interactions is asymmetric.
Domain interactions were found to exert a measurable effect on the refolding of the N domain within <1 msec, while the effect of these interactions on the C domain became detectable only after 60–100 msec.
The individual N domain folds faster than the individual C domain, but folding of the N domain is slowed down, and folding of the C domain is accelerated due to the domain interactions. As a result, within the complete protein the C domain acquires its native structure faster than the N domain.
Our data suggest that domain–domain interactions direct the folding of the N-terminal domain on a completely different pathway within the full-length protein compared to the individual N domain. Folding of the C domain is influenced by the interdomain contacts, but the folding pathway remains the same.
Materials and methods
Mutants of hisPGK, hisN, and hisC were constructed and expressed using T7 promoter in Rosetta cells, a codon plus variant of Escherichia coli BL-21, as described earlier (Osváth and Gruebele 2003). By inserting the gene into the pET28 plasmid, these proteins were automatically fused to a His-tag on their N-terminal. His-affinity purification was done on Ni-NTA column (Qiagen), the purified protein was flash frozen and stored at −80°C as described previously (Osváth et al. 2003). Experiments on PGK mutants with the affinity tag cleaved proved that the His-tag does not change the folding stability of the mutants.
Refolding was initiated by a rapid dilution of the protein unfolded by different amounts of GuHCl using a stopped-flow apparatus (Applied Photophysics π*-180) and followed by time-resolved changes of tryptophan fluorescence excited at 295 nm, 5 nm bandwidth, and detected through a 320-nm cutoff filter. Fifty to 100 shots were averaged to obtain the traces shown in the figures. Fluorescence spectra were measured on an Edinburgh Analytical Instruments C-900 luminometer. Manual mixing experiments were recorded on the same instrument with 5-nm bandwidth excitation at 295 nm, and 10 nm-wide detection at 340 nm. The curves shown are the average of three to five individual measurements.
A comparison of the manual mixing and stopped-flow refolding kinetics showed that after 2–3 min artifacts occur in the stopped-flow measurements, probably due to the diffusion of the two mixed solutions into the observed sample compartment. To avoid this problem, the stopped-flow measurements were limited to 50 sec. Another artifact that needed special attention during the stopped-flow measurements was light scattering. Using the filters described above the contribution of the scattered light to the signal was reduced to <3%.
Acknowledgments
This work was supported by Hungarian grants OTKA D-38480 (S.O.), OTKA TS-044730/2002 (J.F.), and OTKA T-046412 (P.Z.); ETT S01/2003 (J.F.); and the Austrian–Hungarian exchange grant TéT-ö AD-A-17/2002 (J.F., G.K.). Expression system for some of the mutants were made while S.O. was working in the group of Dr. Martin Gruebele. We thank Dr. Mária Vas for useful discussions and critical reading of the paper.
Abbreviations
GuHCl, Guanidine hydrochloride
DTT, 1,4-Dithio-L-threitol
EDTA, Ethylenediamine-tetraacetic acid disodium salt
PGK, phosphoglycerate kinase
hisPGK, histidine-tagged variant of yeast PGK
hisN, histidine-tagged variant of the N-terminal domain (1–186) of yeast PGK
hisC, histidine-tagged variant of the C-terminal domain (187–412) of yeast PGK
hisPGK W333, W308F mutant of hisPGK
hisPGK W122, W308F, W333F, Y122W triple mutant of hisPGK
hisN W122, Y122W mutant of hisN
hisC W333, W308F mutant of hisC
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051359905.
References
- Adams, B., Fowler, R., Hudson, M., and Pain, R.H. 1996. The role of the C-terminal lysine in the hinge bending mechanism of yeast phosphoglycerate kinase. FEBS Lett. 385 101–104. [DOI] [PubMed] [Google Scholar]
- Beechem, J.M., Sherman, M.A., and Mas, M.T. 1995. Sequential domain unfolding in phosphoglycerate kinase: Fluorescence intensity and anisotropy stopped-flow kinetics of several tryptophan mutants. Biochemistry 34 13943–13948. [DOI] [PubMed] [Google Scholar]
- Betton, J.-M., Missiakas, D., and Yon, J.M. 1992. The slow-refolding step of phosphoglycerate kinase as monitored by pulse proteolysis. Arch. Biochem. Biophys. 296 95–101. [DOI] [PubMed] [Google Scholar]
- Brandts, J.F., Hu, C.Q., and Lin, L.-N. 1989. A simple model for proteins with interacting domains. Applications to scanning calorimetry data. Biochemistry 28 8588–8596. [DOI] [PubMed] [Google Scholar]
- Chardot, T., Mitraki, A., Amigues, Y., Desmadril, M., Betton, J.M., and Yon, J.M. 1988. The effect of phosphate on the unfolding-refolding of phosphoglycerate kinase induced by guanidine hydrochloride. FEBS Lett. 228 65–68. [DOI] [PubMed] [Google Scholar]
- Cheung, C.-W. and Mas, M.T. 1996. Substrate-induced conformational changes in yeast 3-phosphoglycerate kinase monitored by fluorescence of single tryptophan probes. Protein Sci. 5 1144–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damaschun, G., Damaschun, H., Gast, K., Misselwitz, R., Müller, J.J., Pfeil, W., and Zirwer, D. 1993. Cold denaturation-induced conformational changes in phosphoglycerate kinase from yeast. Biochemistry 32 7739–7746. [DOI] [PubMed] [Google Scholar]
- Damaschun, G., Damaschun, H., Gast, K., and Zirwer, D. 1998. Denatured states of yeast phosphoglycerate kinase. Biochemistry (Moscow) 63 259–275. [PubMed] [Google Scholar]
- Dinner, A.R., Šali, A., Smith, L.J., Dobson, C.M., and Karplus, M. 2000. Understanding protein folding via free-energy surfaces from theory and experiment. Trends Biochem. Sci. 25 331–339. [DOI] [PubMed] [Google Scholar]
- Ervin, J., Larios, E., Osváth, S., Schulten, K., and Gruebele, M. 2002. What causes hyperfluorescence: Folding intermediates or conformationally flexible native states? Biophys. J. 83 473–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire, E., Murphy, K.P., Sanchez-Ruiz, J.M., Galisteo, M.L., and Privalov, P.L. 1992. The molecular basis of cooperativity in protein folding. Thermodynamic dissection of interdomain interactions in phosphoglycerate kinase. Biochemistry 31 250–256. [DOI] [PubMed] [Google Scholar]
- Gast, K., Damaschun, G., Damaschun, H., Misselqitz, R., and Zirwer, D. 1993. Cold denaturation of yeast phophoglycerate kinase: Kinetics of changes in secondary structure and compactness on unfolding and refolding. Biochemistry 32 7747–7752. [DOI] [PubMed] [Google Scholar]
- Gast, K., Damaschun, G., Desmadril, M., Minard, P., Müller-Frohne, M., Pfeil, W., and Zirwer, D. 1995. Cold denaturation of yeast phophoglycerate kinase: Which domain is more stable? FEBS Lett. 358 247–250. [DOI] [PubMed] [Google Scholar]
- Gast, K., Noppert, A., Mullerfrohne, M., Zirwer, D., and Damaschun, G. 1997. Stopped flow dynamic light scattering as a method to monitor compaction during protein folding. Eur. Biophys. J. 25 211–219. [Google Scholar]
- Gillespie, B. and Plaxco, K.W. 2004. Using protein folding rates to test protein folding theories. Annu. Rev. Biochem. 73 837–859. [DOI] [PubMed] [Google Scholar]
- Griko, Y.V., Venyaminov, S.Y., and Privalov, P.L. 1989. Heat and cold denaturation of phosphoglycerate kinase (interaction of domains). FEBS Lett. 244 276–278. [DOI] [PubMed] [Google Scholar]
- Hosszu, L.L.P., Craven, C.J., Spencer, J., Parker, M.J., Clarke, A.R., Kelly, M., and Waltho, J.P. 1997. Is the structure of the n-domain of phosphoglycerate kinase affected by isolation from the intact molecule? Biochemistry 36 333–340. [DOI] [PubMed] [Google Scholar]
- Jäger, M. and Plückthun, A. 2000. Direct evidence by H0D exchange and ESI-MS for transient unproductive domain interaction in the refolding of an antibody scFv fragment. Protein Sci. 9 552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger, M., Gehrig, P., and Plückthun, A. 2001. The scFv fragment of the antibody hu4D5–8: Evidence for early premature domain interaction in refolding. J. Mol. Biol. 305 1111–1129. [DOI] [PubMed] [Google Scholar]
- Lillo, M.P., Beechem, J.M., Szpikowska, B.K., Sherman, M.A., and Mas, M.T. 1997a. Design and characterization of a multisite fluorescence energy-transfer system for protein folding studies: A steady-state and time-resolved study of yeast phosphoglycerate kinase. Biochemistry 36 11261–11272. [DOI] [PubMed] [Google Scholar]
- Lillo, M.P., Szpikowska, B.K., Mas, M.T., Sutin, J.D., and Beechem, J.M. 1997b. Real-time measurement of multiple intramolecular distances during protein folding reactions: A multisite stopped-flow fluorescence energy-transfer study of yeast phosphoglycerate kinase. Biochemistry 36 11273–11281. [DOI] [PubMed] [Google Scholar]
- Missiakas, D., Betton, J.-M., Minard, P., and Yon, J.M. 1990. Unfolding-refolding of the domains in yeast phosphoglycerate kinase: Comparison with the isolated engineered domains. Biochemistry 29 8683–8689. [DOI] [PubMed] [Google Scholar]
- Missiakas, D., Betton, J.-M., Chaffotte, A., Minard, P., and Yon, J.M. 1992. Kinetic studies of the refolding of yeast phosphoglycerate kinase: Comparison with the isolated engineered domains. Protein Sci. 1 1485–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osváth, S. and Gruebele, M. 2003. Proline can have opposite effects on fast and slow protein folding phases. Biophys. J. 85 1215–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osváth, S., Sabelko, J.J., and Gruebele, M. 2003. Tuning the heterogeneous early folding dynamics of phosphoglycerate kinase. J. Mol. Biol. 333 187–199. [DOI] [PubMed] [Google Scholar]
- Otto, M.R., Lillo, M.P., and Beechem, J.M. 1994. Resolution of multi-phasic reactions by the combination of fluorescence total-intensity and anisotropy stopped-flow kinetic experiments. Biophys. J. 67 2511–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker, M.J., Sessions, R.B., Badcoe, I.G., and Clarke, A.R. 1996a. The development of tertiary interactions during the folding of a large protein. Fold. Des. 1 145–156. [DOI] [PubMed] [Google Scholar]
- Parker, M.J., Spencer, J., Jackson, G.S., Burston, S.G., Hosszu, L.L.P., Craven, C.J., Waltho, J.P., and Clarke, A.R. 1996b. Domain behavior during the folding of a thermostable phosphoglycerate kinase. Biochemistry 35 15740–15752. [DOI] [PubMed] [Google Scholar]
- Pecorari, F., Guilbert, C., Minard, P., Desmadril, M., and Yon, J.M. 1996. Folding and functional complementation of engineered fragments from yeast phosphoglycerate kinase. Biochemistry 35 3465–3476. [DOI] [PubMed] [Google Scholar]
- Reed, M.A.C., Hounslow, A.M., Sze, K.H., Barsukov, I.G., Hosszu, L.L.P., Clarke, A.R., Craven, C.J., and Waltho, J.P. 2003. Effects of domain dissection on the folding and stability of the 43 kDa protein PGK probed by NMR. J. Mol. Biol. 330 1189–1201. [DOI] [PubMed] [Google Scholar]
- Ritco-Vonsovici, M., Minard, P., Desmadril, M., and Yon, J.M. 1995a. Is the continuity of the domains required for the correct folding of a two-domain protein? Biochemistry 34 16543–16551. [DOI] [PubMed] [Google Scholar]
- Ritco-Vonsovici, M., Mouratou, B., Minard, P., Desmadril, M., Yon, J.M., Andrieux, M., Leroy, E., and Guittet, E. 1995b. Role of the C-terminal helix in the folding and stability of yeast phosphoglycerate kinase. Biochemistry 34 833–841. [DOI] [PubMed] [Google Scholar]
- Semisotnov, G.V., Vas, M., Chemeris, V.V., Kashparova, N.J., Ktotova, N.V., Razgulyaev, O.I., and Sienv, M.A. 1991. Refolding kinetics of pig muscle and yeast 3-phosphoglycerate kinase and of their proteolytic fragments. Eur. J. Biochem. 202 1083–1090. [DOI] [PubMed] [Google Scholar]
- Snow, C.D., Nguyen, H., Pande, V.S., and Gruebele, M. 2002. Absolute comparison of simulated and experimental protein-folding dynamics. Nature 420 102–106. [DOI] [PubMed] [Google Scholar]
- Szilágyi, A.N. and Vas, M. 1998. Sequential domain refolding of pig muscle 3-phosphoglycerate kinase: Kinetic analysis of reactivation. Fold. Des. 3 565–575. [DOI] [PubMed] [Google Scholar]
- Szilágyi, A. and Závodszky, P. 2000. Structural differences between mesophilic, moderately thermophilic and extremely thermophilic protien subunits: Results of a comprehensive survey. Structure 8 493–504. [DOI] [PubMed] [Google Scholar]
- Szpikowska, B.K. and Mas, M.T. 1996. Urea-induced equilibrium unfolding of single tryptophan mutants of yeast phosphoglycerate kinase: Evidence for stable intermediate. Arch. Biochem. Biophys. 335 173–182. [DOI] [PubMed] [Google Scholar]
- Szpikowska, B.K., Beechem, J.M., Sherman, M.A., and Mas, M.T. 1994. Equilibrium unfolding of yeast phosphoglycerate kinase and its mutants lacking one or both tryptophans: A circular dichroism and steady-state and time resolved fluorescence study. Biochemistry 33 2217–2225. [DOI] [PubMed] [Google Scholar]
- Vas, M., Sinev, M.A., Kotova, N., and Semisotnov, G.V. 1990. Reactivation of 3-phosphoglycerate kinase from its proteolytic fragments. Eur. J. Biochem. 189 575–579. [DOI] [PubMed] [Google Scholar]
- Watson, H.C., Walker, N.P.C., Shaw, P.J., Bryant, T.N., Wendell, P.L., Fothergill, L.A., Perkins, R.E., Conroy, S.C., Dobson, M., Tuite, M.F., et al. 1982. Sequence and structure of yeast phosphoglycerate kinase. EMBO J. 1 1635–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenk, M., Herbst, R., Hoeger, D., Kretschmar, M., Lubsen, N.H., and Jaenicke, R. 2000. γ S-crystallin of bovine and human eye lens: Solution structure, stability and folding of the intact two-domain protein and its separate domains. Biophys. Chem. 86 95–108. [DOI] [PubMed] [Google Scholar]
- Závodszky, P., Kardos, J., Svingor, Á ., and Petsko, G. 1998. Adjustment of conformational flexibility is a key event in the thermal adaptation of proteins. Proc. Natl. Acad. Sci. 95 7406–7411. [DOI] [PMC free article] [PubMed] [Google Scholar]