

Abstract
Fusion to cationic peptides, such as nonaarginine (R9), provides a means to deliver molecular cargo into mammalian cells. Here, we provide a thorough analysis of the effect of an R9 tag on the attributes of a model protein: bovine pancreatic ribonuclease (RNase A). The R9 tag diminishes the conformational stability of RNase A (ΔTm=−8°C in phosphate-buffered saline). This effect is nearly mitigated by the addition of salt. The tag does not compromise the enzymatic activity of RNase A. An R9 tag facilitates the purification of RNase A by cation-exchange chromatography and enables the adsorption of RNase A on glass slides and silica resin with the retention of enzymatic activity. The tag can be removed precisely and completely by treatment with carboxypeptidase B. Finally, the R9 tag increases both the cellular uptake of RNase A and the cytotoxicity of G88R RNase A, a variant that evades the cytosolic ribonuclease inhibitor protein. Thus, we conclude that polyarginine is a versatile protein fusion tag.
Keywords: affinity tag, conformational stability, enzymatic activity, protein adsorption, protein transduction domain, ribonuclease A
Numerous cationic peptides have been shown to permeate mammalian cells (Vives et al. 1997; Suzuki et al. 2002; Coeytaux et al. 2003). These peptides can serve as protein transduction domains (PTDs) to carry both small-molecule drugs and macromolecules such as proteins into cells (Schwarze et al. 1999; Rothbard et al. 2002). Although details of the internalization mechanism are unclear, the positive charge of these peptides is known to be critical for internalization (Mitchell et al. 2000). This requirement is probably due to favorable Coulombic interactions between the peptide and anionic cell-surface molecules, especially heparin sulfate (Fuchs and Raines 2004; Gonçalves et al. 2005). The length and composition of the cationic peptide is important for internalization, as seven to nine cationic charges are optimal and arginine residues are preferred over lysine (Wender et al. 2000).
Despite the substantial interest among protein scientists in using PTDs, we are not aware of a detailed analysis of the effect of a PTD on the intrinsic properties of a protein cargo. Unlike small molecules, proteins have fragile conformations that are essential for their function. The addition of a nonaarginine (R9) fusion tag could disrupt that conformation, and hence, compromise function.
While much work has focused on the utility of an R9 fusion tag in mediating cellular internalization, there is evidence that similar tags confer other useful attributes. Over 20 years ago, conjugation to short oligomers of arginine was used to facilitate protein purification (Sassenfeld and Brewer 1984). More recently, green fluorescent protein (GFP) containing a hexaarginine tag was adsorbed reversibly onto mica surfaces (Nock et al. 1997). Cationic peptides have also been shown to interact strongly with both plastic and glass (Chico et al. 2003).
Here, we report on the impact of an R9 fusion tag on the conformational stability and biological function of its protein cargo. We also describe numerous applications for an R9 tag that are unrelated to cellular internalization, as well as a means to remove the tag precisely and completely. As a model protein, we use bovine pancreatic ribonuclease (RNase A; EC 3.1.27.5), which was perhaps the most studied enzyme of the 20th century (Raines 1998) and is now the basis for a new class of cytotoxins (Leland and Raines 2001). The intrinsic enzymatic and cytotoxic activities of RNase A provide a sensitive measure of the effect of an R9 tag on both physical and biological attributes of a protein cargo. We find that R9 has an ensemble of virtues that is unique among known fusion tags.
Results
Purification of an R9-tagged protein and proteolytic degradation of the R9 tag
One impetus for the development of novel peptide tags is to facilitate the purification of proteins produced by recombinant DNA (rDNA) technology (Nilsson et al. 1997; Stevens 2000; Hearn and Acosta 2001). Tags that can be utilized for purification and then removed are especially desirable. The addition of R9 to RNase A increased the positive charge of the protein and thus strengthened its interaction with inexpensive, cation-exchange resins (Fig. 1A ▶). A concentration of NaCl exceeding 1 M was required for the elution of RNase A-R9 from a HiTrap SP column. The resulting RNase-R9 was homogeneous according to SDS-PAGE (Fig. 1B ▶), and had MS (MALDI) m/z 15,404 (expected for Met-RNase A-Gly3-Arg9: 15,390).
Figure 1.
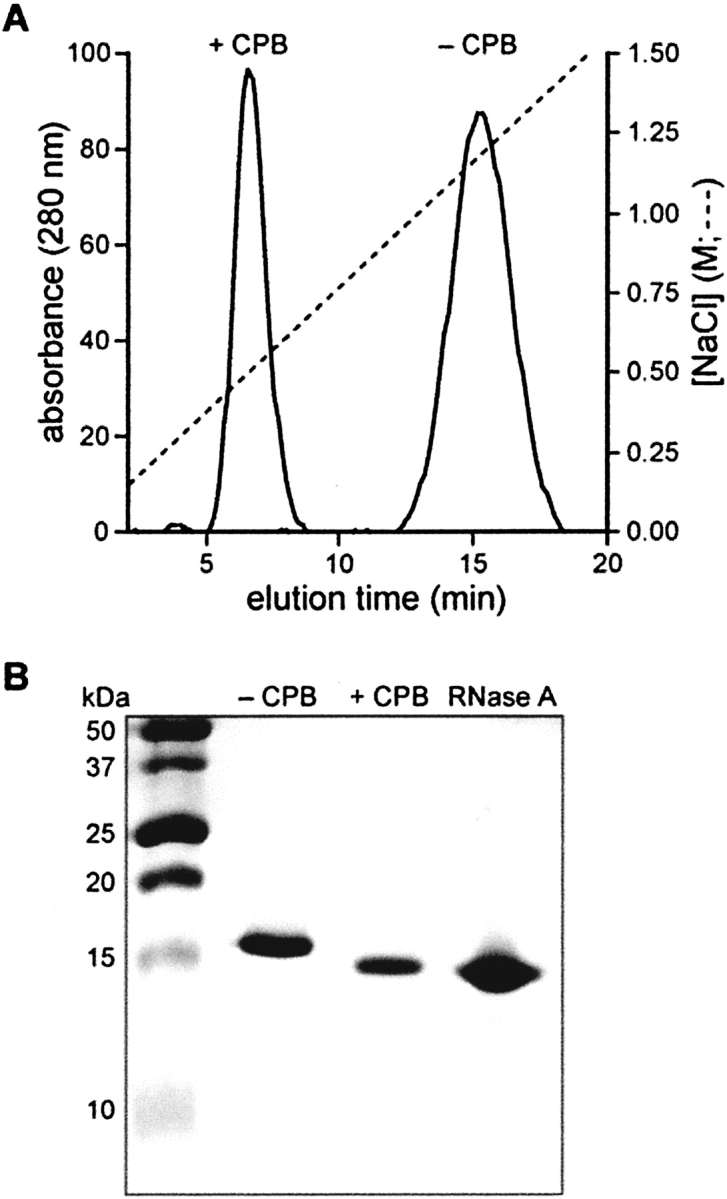
Effect of an R9 tag on the purification of a protein by cation-exchange chromatography. (A) RNase A-R9 was purified by cation-exchange chromatography before (−CPB) and after (+CPB) the addition of carboxypeptidase B. (B) SDS-PAGE gel of RNase A-R9 before (−CPB) and after (+CPB) the addition of carboxypeptidase B. Purified RNase A is a standard.
The C-terminal R9-tag was removable by incubation with the exoprotease carboxypeptidase B (CPB), which catalyzes the sequential hydrolysis of peptide bonds of C-terminal basic residues. Removal of the tag was complete in <1 h. The resulting protein eluted at a lower salt concentration from SP Sepharose (Fig. 1A ▶), had greater mobility during SDS-PAGE (Fig. 1B ▶), and had MS (MALDI) m/z 14,000 (expected for Met-RNase A-Gly3: 13,985), such that Δ(m/z)=−1404 (expected for the loss of Arg9: −1406).
Effect of R9 tag on conformational stability and enzymatic activity
To be useful, a tag cannot greatly diminish the stability or activity of a protein. We measured the effect of an R9 tag on the conformational stability of RNase A. The value of Tm for RNase A decreased from 63.7 ± 1.0 to 54.0 ± 1.0°C upon addition of the R9 tag (Fig. 2A ▶). RNase A is a cationic protein with a measured pI=9.3 (Ui 1971). Adding additional cationic residues is likely to decrease the conformational stability of RNase A by increasing unfavorable Coulombic interactions within the protein (Shaw et al. 2001; Ramos and Baldwin 2002). To test this hypothesis, we measured the Tm of the two proteins at increasing concentrations of NaCl, which can ameliorate unfavorable Coulombic interactions. The value of Tm for RNase A increased gradually as a function of salt concentration (Fig. 2B ▶). In contrast, the value of Tm for RNase A-R9 increased dramatically between 0.10 and 0.50 M NaCl, approaching that of RNase A. This result indicates that the destabilization does indeed arise from unfavorable Coulombic interactions.
Figure 2.
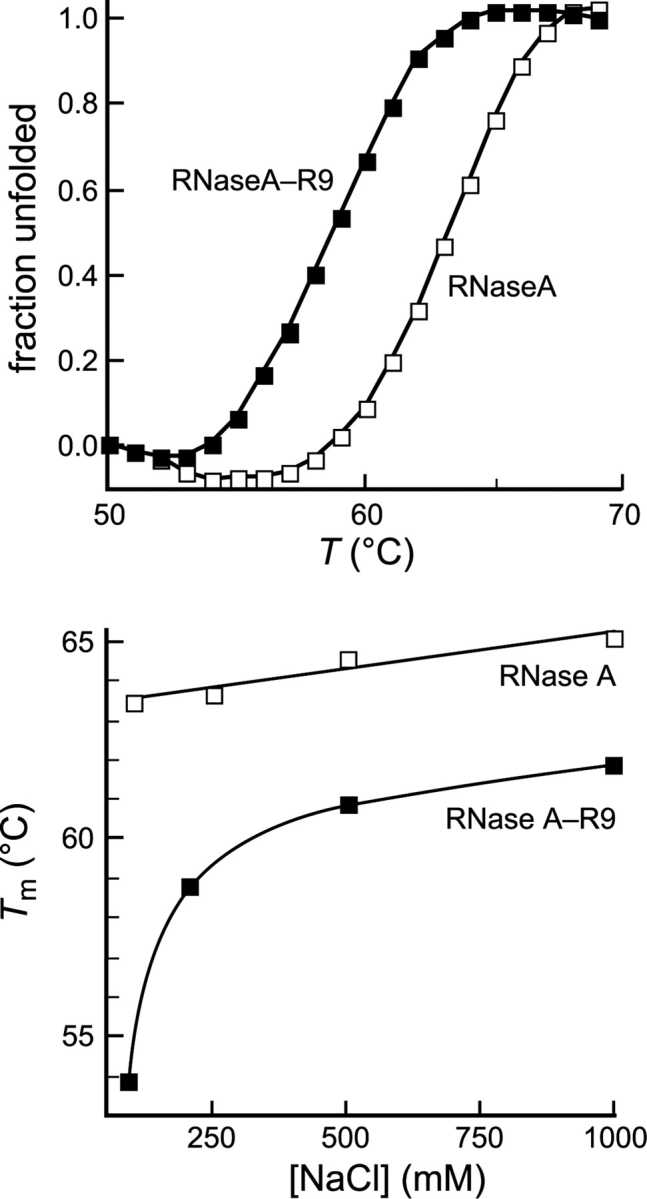
Effect of an R9 tag on the conformational stability of a protein. (Top) Thermal denaturation of RNase A (□) (Tm=63.7 ± 1.0°C) and RNase A-R9 (▪) (Tm=54.0 ± 1.0°C) in PBS. (Bottom) Conformational stability of RNase A (□) and RNase A-R9 (▪) in 50 mM sodium phosphate buffer (pH 7.2), containing NaCl (0–1.00 M).
The enzymatic activity of RNase A and RNase A-R9 was determined by using a continuous assay with a fluorogenic substrate. The addition of R9 had no noticeable effect on the activity of RNase A (Table 1). It is noteworthy, however, that these assays were performed in a buffer containing 0.50 M NaCl to minimize adsorption to the quartz cuvette.
Table 1.
Biochemical parameters of ribonuclease A and its variants
| Ribonuclease | pIa | Tmb (°C) | kcat/KMc (106M−1s−1) | IC50d (μM) |
| RNase A | 8.6 | 63.7 | 1.8 ± 0.2 | >25 |
| RNase A-R9 | 9.6 | 56 | 1.0 ± 0.2 | >25 |
| G88RNase A | 8.8 | 60 | 12.6 ± 0.4 | 5.8 ± 1.0 |
| G88R RNase A-R9 | 9.8 | 54.0 | 9.6 ± 0.2 | 2.0 ± 0.4 |
a Values of pI were estimated from amino acid composition (Bjellqvist et al. 1993, 1994).
b Values of Tm (±1°C) were determined in PBS by UV spectroscopy.
c Values of kcat/KM(±SE) are for the catalysis of 6-FAM-dArU(dA)2–6-TAMRA cleavage at 25°C in 10 mM Bis-Tris-HCl buffer, pH 6.0, containing NaCl (0.50 M).
d Values of IC50 (±SE) are for the incorporation of [methyl-3H]thymidine into the DNA of K-562 cells.
Cellular uptake of an R9-tagged protein
The most often noted function of cationic peptides such as R9 is as a protein transduction domain. The uptake of fluorescein-labeled RNase A and RNase A-R9 into CHO-K1 cells was visualized by confocal microscopy. The uptake of RNase A-R9 was evident after 30 min at 37°C (Fig. 3A ▶). In contrast, the uptake of RNase A was not apparent under the same conditions (Fig. 3B ▶).
Figure 3.

Effect of an R9 tag on the uptake of a protein by living mammalian cells. CHO-K1 cells were incubated with fluorescein-labeled RNase A-R9 (10 μM, A) or fluorescein-labeled RNase A (10 μM, B) for 15 min at 37°C before visualization by fluorescence microscopy. Scale bar: 10 μm.
Cytotoxicity of R9-tagged variants of ribonuclease A
In addition to being well-studied model proteins, ribonucleases are potentially useful as cancer chemotherapeutics (Leland and Raines 2001). Although RNase A itself is not cytotoxic, its G88R variant is known to be toxic toward a wide variety of cancer cell types by virtue of its ability to evade ribonuclease inhibitor (RI) (Leland et al. 1998; Antignani et al. 2001; Haigis et al. 2003). RI is a cytosolic protein that binds to wild-type RNase A with nearly fM affinity, but has 104-fold less affinity for G88R RNase A. Adding the R9 tag to G88R RNase A resulted in a three-fold increase in its cytotoxic activity (Fig. 4 ▶; Table 1). This result suggests that cellular uptake limits the cytotoxicity of G88R RNase A. Interestingly, adding the R9 tag did not endow wild-type RNase A with cytotoxic activity (Fig. 4 ▶), despite an evident increase in cellular uptake (Fig. 3 ▶). This result suggests that the concentration of RNase A-R9 in the cytosol does not reach 4 μM, which is the cytosolic concentration of RI (Haigis et al. 2003).
Figure 4.

Effect of an R9 tag on the cytotoxicity of wild-type ribonuclease A, its G88R variant, and Onconase. Cell proliferation was determined by incorporation of [methyl-3H]thymidine into cellular DNA after a 44-h incubation with a ribonuclease. Each data point is expressed as a percentage of a PBS control. Variants with R9 tags are denoted with filled symbols: RNase A-R9,•; G88R RNase A-R9, ▪ Variants without R9 tags are denoted with open symbols: RNase A, ○; G88R RNase A,□; ONC, ▵. IC50 values are listed in Table 1.
Adsorption of an R9-tagged protein
The immobilization of proteins on surfaces is enabling the creation of protein microarrays for probing protein–ligand and protein–protein interactions (Kabsch 1988; Kodadek 2001; Zhu and Snyder 2003). We used an R9 tag to adsorb RNase A to glass slides and silica resin (Fig. 5 ▶). The R9 tag enhances the binding of fluorescein-labeled RNase A by >30-fold (Fig. 5A ▶). The tagged protein remains associated with a glass slide, even in the presence of 1 M NaCl (data not shown). Adsorption of RNase A-R9 to silica resin affords active enzyme on a solid support (Fig. 5B ▶). All detectable enzymatic activity is associated with the silica resin, rather than the supernatant. Virtually no wild-type RNase A adsorbed to silica resin under the same conditions.
Figure 5.

Effect of an R9 tag on the adsorption of a protein to a glass slide and silica resin. (A) Fluorescent images of fluorescein-labeled RNase A-R9 and RNase A (10–0.01 μM) adsorbed on to a glass slide. (B) Ribonucleolytic activity in a solution containing silica resin with adsorbed RNase A-R9 or RNase A, and in the supernatant upon removal of the silica resin with adsorbed RNase A-R9.
Discussion
A great benefit of rDNA technology is the ability to fuse peptide sequences with useful attributes to a protein of interest. Until recently, such peptide tags have been used solely to facilitate purification or detection (Nilsson et al. 1997; Stevens 2000; Hearn and Acosta 2001). A few tags, such as the S-peptide of RNase A (Kim and Raines 1993), can play both roles. Recently, new tags have emerged that endow additional functions, such as cellular internalization or surface immobilization (Elia et al. 2002; Kabouridis 2003). We have shown that a polyarginine (R9) tag can provide all of these attributes.
We fused R9 to the C terminus of RNase A by using rDNA technology. There, the R9 tag greatly increased the affinity of RNase A for a cation-exchange resin (Fig. 1 ▶). Further, the tag was removable precisely and completely by the addition of carboxyprotease B. In additional experiments, we fused the R9 tag to the N terminus of RNase A. The resulting fusion protein had a degraded tag (data not shown). This result has precedent, as the endogenous ompT protease of Escherichia coli has been shown to cleave proteins between adjacent basic residues (Ford et al. 1991). Because protein biosynthesis begins at the N terminus, a C-terminal R9 tag on RNase A (which forms inclusion bodies in E. coli; delCardayré et al. 1995) is less vulnerable to degradation by the ompT protease than is an N-terminal tag.
Enzymatic catalysis provides an extremely sensitive measure of native protein structure (Knowles 1987). We conclude that the addition of an R9 tag appears to have little effect on the three-dimensional structure of wild-type RNase A or its G88R variant, based on a less than twofold change in values of kcat/KM for RNA cleavage (Table 1). The R9 tag does, however, decrease the conformational stability of RNase A at physiological salt concentration (Fig. 2 ▶). This effect is mitigated by the addition of salt, indicating that it originates from unfavorable Coulombic interactions between the cationic tag and the cationic protein. Because RNase A is an especially cationic protein, the destabilizing effect of an R9 tag is likely to be less severe in most proteins.
One major hurdle in the development of protein chemotherapeutics is the inability to deliver them into cells. Some success has been achieved by fusing proteins to molecules that have cell surface receptors, such as folate or transferrin (Leamon and Low 1991; Rybak et al. 1991; Suzuki et al. 1999). A more recent approach has been to fuse proteins to peptide tags that are capable of entering cells (Beerens et al. 2003; Green et al. 2003). Indeed, we observed that RNase A-R9 is internalized more efficiently than is RNase A (Fig. 3 ▶). More rapid uptake is consistent with the three-fold increase in cytotoxic activity conferred by an R9 tag to G88R RNase A (Fig. 4 ▶). This increase is noteworthy, given that approximately 99% of RNase A suffers lysosomal degradation before reaching the cytoplasm (Kothandaraman et al. 1998).
Finally, we find that RNase A-R9 but not RNase A is adsorbed strongly on glass or silica surfaces (Fig. 5 ▶). The RNase A-R9 remains adsorbed in the presence of 1 M NaCl, and is thus irreversibly immobilized for many purposes. Most significantly, the adsorbed RNase A retains enzymatic activity, as had been observed previously upon covalent immobilization of RNase A at a specific site (Sweeney et al. 2000; Soellner et al. 2003).
Summary
We provide a thorough analysis of the effect of an R9 tag on the attributes of a model protein: RNase A. We find that the R9 tag diminishes the conformational stability of the protein (ΔTm=−8°C in PBS), but that this effect is ameliorated by the addition of salt. The tag has a negligible effect on the structure of the protein, as evidenced by the retention of full enzymatic activity. We find that an R9 tag facilitates protein purification by cation-exchange chromatography and enables the adsorption of functional protein on glass slides and silica resin. The tag can be removed precisely and completely by treatment with carboxypeptidase B. Finally, the R9 tag increases both the cellular uptake of the protein and the cytotoxicity of a protein variant (which is manifested in the cytosol). We conclude that polyarginine is a versatile protein fusion tag.
Materials and methods
Cells and chemicals
Escherichia coli strain BL21 (DE3) PlysS and the pET22b(+) expression vector were from Novagen. Human erythroleukemia cells (line K-562) and Chinese hamster ovary (line CHO-K1) were from the American Type Culture Collection. [Methyl-3H]Thymidine (6.7 Ci/mmol) was from NEN Life Science Products. All other chemicals and reagents were of commercial reagent grade or better, and were used without further purification.
Terrific Broth (TB) liquid medium contained (in 1 L) tryptone (12 g), yeast extract (24 g), glycerol (4 mL), KH2PO4 (2.31 g), and K2HPO4 (12.54 g). Phosphate-buffered saline (PBS) was 10 mM sodium phosphate buffer (pH 7.4), containing NaCl (138 mM) and KCl (2.7 mM).
Instruments
The mass of ribonuclease variants was confirmed by matrix-assisted laser desorption ionization-time-of-flight (MALDI-TOF) mass spectrometry using a Voyager-DE-PRO Biospectrometry Workstation (Applied Biosystems) using 3,5-dimethoxy-4-hydroxycinnamic acid as a matrix. Fluorescence measurements were performed with a QuantaMaster 1 photon counting fluorometer equipped with sample stirring (Photon Technology International). Radioactivity was quantified with a Microbeta TriLux liquid scintillation counter (Perkin-Elmer MA).
Site-directed mutagenesis
Oligonucleotides were obtained from Integrated DNA Technology. cDNA encoding variants of RNase A were created in plasmid pBXR, which directs the production of RNase A in E. coli (delCardayré et al. 1995) by using the QuikChange mutagenesis kit from Stratagene. All variants of RNase A possessed an N-terminal methionine residue, which has been reported to have no effect on ribonucleolytic activity (Arnold et al. 2002). An R9 tag was separated from the remainder of a protein by a triglycine linker.
Production and purification of protein variants
Untagged variants of RNase A and Onconase (which is the most cytotoxic known homolog of RNase A) (Matousek et al. 2003) were produced in E. coli and purified as described previously (Leland et al. 1998). Variants of RNase A containing a C-terminal R9 tag were prepared as follows: BL21(DE3)PlysS cells containing a plasmid that encodes an RNase A variant were grown at 37°C with shaking (250 rpm) in TB containing ampicillin (200 μg/mL) and chloramphenicol (35 μg/mL) to an OD=1.6 at 600 nm. cDNA expression was induced by adding isopropyl β-D-thioglucopyranoside (IPTG) (to 1 mM). Cells were grown for an additional 4 h before harvesting. Cell pellets were resuspended in lysis buffer, which was 10 mM Tris-HCl buffer (pH 8.0), containing ethylenediaminetetraacetic acid (EDTA) (1.0 mM), NaCl (0.10 M), and phenylmethylsulfonyl fluoride (1.0 mM), and lysed by sonication. Inclusion bodies were isolated by centrifugation at 11,000g for 45 min and solubilized in denaturing solution, which was 20 mM Tris-HCl buffer (pH 8.0), containing guanidine hydrochloride (7.0 M) and EDTA (10 mM), for 4 h at room temperature. Solubilized inclusion bodies were diluted 10-fold with acetic acid (20mM) and clarified by centrifugation. The supernatant was dialyzed overnight against the same buffer. The resulting protein was then folded overnight at 4°C in a redox buffer, which was 100 mM Tris-HCl buffer (pH 8.0), containing EDTA (10 mM), L-arginine (0.5 M), reduced glutathione (1 mM), and oxidized glutathione (0.2 mM). Refolded protein was purified by cation-exchange chromatography on a 5-mL HiTrap SP-sepharose FF column (Amersham Biosciences) in 50 mM sodium acetate buffer (pH 5.0), with a linear gradient (50+50 mL) of NaCl (0–1.5 M). The identity of each variant was verified by MALDI-TOF mass spectrometry.
Assays of enzymatic activity
Ribonucleolytic activity was measured by monitoring the increase in the fluorescence of 6-FAM-dArU(dA)2–6-TAMRA (Integrated DNA Technologies) upon enzyme-catalyzed cleavage, as described previously (Kelemen et al. 1999) with minor modifications. Polyarginine-containing peptides are known to bind to glass surfaces (Chico et al. 2003). We observed this phenomenon (data not shown), and so performed all assays in 10 mM Bis-Tris-HCl buffer (pH 6.0), containing NaCl (0.50 M). In this high-salt buffer, the binding of protein to a quartz cuvette was found to be insignificant.
Assays of cytotoxicity
The effect of ribonucleases on cell proliferation was determined by measuring the incorporation of [methyl-3H]thymidine into cellular DNA. K-562 cells were grown in RPMI 1640 medium containing fetal bovine serum (10% v/v), penicillin (100 units/mL), and streptomycin (100 μg/mL). Cytotoxicity assays were performed using asynchronous log-phase cultures grown at 37°C in a humidified incubator containing CO2(g) (5% v/v). To assay toxicity, cells (95 μL of a solution of 5×104 cells/mL) were incubated with PBS containing a ribonuclease (5 μL) in a 96-well plate. The cells were grown for 44 h and then pulsed for 4 h with radiolabeled thymidine (0.25 μCi/well), which is only incorporated into the DNA of living cells. DNA was harvested onto glass fiber filters using a PHD cell harvester (Cambridge Technology). Filters were washed with water and dried with methanol, and their 3H content was quantified with liquid scintillation counting.
Semisynthesis of fluorescent proteins
RNase A was labeled with fluorescein at one specific residue in a surface loop by using variants in which Ala19 was replaced with a cysteine residue (Haigis and Raines 2003). A 19C RNase A-R9 or A 19 C RNase A (100 μM) were incubated in PBS containing a 20-fold molar excess of 5-iodoacetamidofluorescein (Molecular Probes) and a threefold molar excess of tris[2- carboxyethylphosphine] hydrochloride (TCEP) for 4 h at room temperature. The resulting solution was dialyzed overnight against 50mM sodium acetate buffer (pH 5.0), and then purified by cation-exchange chromatography using a HiTrap CMSepharose Fast Flow column (5 mL) with a linear gradient (50+50 mL) of NaCl (0–1.00 M for A19C RNase A; 0–2.00 M for A19C RNase A-R9). Conjugation to the fluorophore was confirmed by MALDI-TOF mass spectrometry.
Fluorescence microscopy
Fluorescence microscopy was used to follow the internalization of fluorescent proteins by mammalian cells. CHO-K1 cells were maintained in Ham’s F-12 Medium containing fetal bovine serum (5% v/v), penicillin (100 units/mL), and streptomycin (100 μg/mL). Cells were seeded onto glass-bottom culture dishes and grown overnight in the same medium. The medium was replaced with fresh medium (100 μL) immediately before the addition of fluorescein-labeled proteins. Cells were allowed to incubate with fluorescein-labeled RNase A-R9 or RNase A for 15 min, and were then washed three times with the same medium containing n-propyl gallate (1 mM) to protect against photobleaching. Images were obtained with a Nikon C1 laser-scanning confocal microscope equipped with 60× and 100× lenses.
Thermal denaturation
As RNase A is denatured, its six tyrosine residues become exposed to solvent and its molar absorptivity at 287 nm decreases significantly (Hermans and Scheraga 1961). RNase A-R9 or RNase A (25 μM) was placed in PBS or 50 mM sodium phosphate buffer (pH 7.2), containing NaCl (0–1.00 M). Unfolding was monitored by the change in absorbance at 287 nm as the temperature was raised at a rate of 0.15°C/min. Data were fitted to a two-state model to calculate the value of Tm, which is the temperature at the midpoint of the transition between the folded and unfolded states (Pace and Scholtz 1997).
Adsorption of proteins to glass slides
Fluorescein-labeled RNase A-R9 and RNase A were adsorbed on glass microscope slides by adding 5 μL of a solution of protein in PBS to wells formed by secure-seal hybridization chambers (Schleicher and Schuell Bioscience). After 15 min, the wells were washed three times with PBS. The adsorption of proteins was visualized with a Typhoon 9410 variable mode fluorimager (Amersham Biosciences). Similarly, slides with adsorbed fluorescein-labeled RNase A-R9 were washed with 50 mM sodium phosphate buffer (pH 7.2), containing NaCl (1.00 M) in an attempt to desorb the protein.
Adsorption of proteins to silica resin
A solution of RNase A-R9 or RNase A (10 μM) in PBS was allowed to adsorb to acid-washed silica resin (10 mg; Sigma Chemical) for 1 h. The resulting resin was washed extensively with 50 mM sodium phosphate buffer (pH 7.2), containing NaCl (1.00 M). The activity of the adsorbed enzyme was measured by adding resin (1 μL of a 1 mg/mL suspension) to a cuvette containing assay buffer, which was 50 mM MES-NaOH buffer (pH 6.0), containing NaCl (0.10 M) and the fluorogenic substrate 6-FAM-dArU(dA)2–6-TAMRA. The MES in the assay buffer was purified prior to use by anion-exchange chromatography to remove the potent ribonuclease inhibitor oligo(vinylsulfonic acid) that contaminates buffers derived from ethanesulfonic acid (Smith et al. 2003). Substrate cleavage was monitored by the change in fluorescence (excitation, 495 nm; emission, 515 nm).
Acknowledgments
We are grateful to Prof. P.A. Frey, B.D. Smith, and K.A. Dickson for contributive discussions, and Prof. T.F.J. Martin for the use of his confocal microscope. This work was supported by grants GM44783 and CA73808 (NIH). S.M.F. was supported by Biotechnology Training Grant 08349 (NIH).
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051393805.
References
- Antignani, A., Naddeo, M., Cubellis, M.V., Russo, A., and D’Alessio, G. 2001. Antitumor action of seminal ribonuclease, its dimeric structure, and its resistance to the cytosolic ribonuclease inhibitor. Biochemistry 40 3492–3496. [DOI] [PubMed] [Google Scholar]
- Arnold, U., Hinderaker, M.P., and Raines, R.T. 2002. Semisynthesis of ribonuclease A using intein-mediated protein ligation. Sci. World J. 2 1823–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerens, A.M., Al Hadithy, A.F., Rots, M.G., and Haisma, H.J. 2003. Protein transduction domains and their utility in gene therapy. Curr. Gene Ther. 3 486–494. [DOI] [PubMed] [Google Scholar]
- Bjellqvist, B., Hughes, G.J., Pasquali, C., Paquet, N., Ravier, F., Sanchez, J.C., Frutiger, S., and Hochstrasser, D. 1993. The focusing positions of polypeptides in immobilized pH gradients can be predicted from their amino acid sequences. Electrophoresis 14 1023–1031. [DOI] [PubMed] [Google Scholar]
- Bjellqvist, B., Basse, B., Olsen, E., and Celis, J.E. 1994. Reference points for comparisons of two-dimensional maps of proteins from different human cell types defined in a pH scale where isoelectric points correlate with polypeptide compositions. Electrophoresis 15 529–539. [DOI] [PubMed] [Google Scholar]
- Chico, D.E., Given, R.L., and Miller, B.T. 2003. Binding of cationic cell-permeable peptides to plastic and glass. Peptides 24 3–9. [DOI] [PubMed] [Google Scholar]
- Coeytaux, E., Coulaud, D., Le Cam, E., Danos, O., and Kichler, A. 2003. The cationic amphipathic α-helix of HIV-1 viral protein R (Vpr) binds to nucleic acids, permeablizes membranes, and efficiently transfects cells. J. Biol. Chem. 278 18110–18116. [DOI] [PubMed] [Google Scholar]
- delCardayré, S.B., Ribó, M., Yokel, E.M., Quirk, D.J., Rutter, W.J., and Raines, R.T. 1995. Engineering ribonuclease A: Production, purification, and characterization of wild-type enzyme and mutants at Gln11. Protein Eng. 8 261–273. [DOI] [PubMed] [Google Scholar]
- Elia, G., Silacci, M., Scheurer, S., Scheuermann, J., and Neri, D. 2002. Affinity-capture reagents for protein arrays. Trends Biotechnol. 20 S19–S22. [DOI] [PubMed] [Google Scholar]
- Ford, C.F., Suominen, I., and Glatz, C.E. 1991. Fusion tails for the recovery and purification of recombinant proteins. Protein Expr. Purif. 2 95–107. [DOI] [PubMed] [Google Scholar]
- Fuchs, S.M. and Raines, R.T. 2004. Pathway for polyarginine entry into mammalian cells. Biochemistry 43 2438–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves, E., Kitas, E., and Seelig, J. 2005. Binding of oligoarginine to membrane lipids and heparan sulfate: Structural and thermodynamic characterization of a cell-penetrating peptide. Biochemistry 44 2692–2702. [DOI] [PubMed] [Google Scholar]
- Green, I., Christison, R., Voyce, C.J., Bundell, K.R., and Lindsay, M.A. 2003. Protein transduction domains: Are they delivering? Trends Pharmacol. Sci. 24 213–215. [DOI] [PubMed] [Google Scholar]
- Haigis, M.C. and Raines, R.T. 2003. Secretory ribonucleases are internalized by a dynamin-independent endocytic pathway. J. Cell Sci. 116 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis, M.C., Kurten, E.L., and Raines, R.T. 2003. Ribonuclease inhibitor as an intracellular sentry. Nucleic Acids Res. 31 1024–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hearn, M.T. and Acosta, D. 2001. Applications of novel affinity cassette methods: Use of peptide fusion handles for the purification of recombinant proteins. J. Mol. Recognit. 14 323–369. [DOI] [PubMed] [Google Scholar]
- Hermans Jr., J. and Scheraga, H.A. 1961. Structural studies of ribonuclease. V. Reversible change of configuration. J. Am. Chem. Soc. 83 3283–3292. [Google Scholar]
- Kabouridis, P.S. 2003. Biological applications of protein transduction technology. Trends Biotechnol. 21 498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch, W. 1988. Evaluation of single-crystal x-ray diffraction data from a position-sensitive detector. J. Appl. Crystallogr. 21 916–924. [Google Scholar]
- Kelemen, B.R., Klink, T.A., Behlke, M.A., Eubanks, S.R., Leland, P.A., and Raines, R.T. 1999. Hypersensitive substrate for ribonucleases. Nucleic Acids Res. 27 3696–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J.-S. and Raines, R.T. 1993. Ribonuclease S-peptide as a carrier in fusion proteins. Protein Sci. 2 348–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles, J.R. 1987. Tinkering with enzymes: What are we learning? Science 236 1252–1258. [DOI] [PubMed] [Google Scholar]
- Kodadek, T. 2001. Protein microarrays: Prospects and problems. Chem. Biol. 8 105–115. [DOI] [PubMed] [Google Scholar]
- Kothandaraman, S., Hebert, M.C., Raines, R.T., and Nibert, M.L. 1998. No role for pepstatin-A-sensitive acidic proteinase in reovirus infections of L or MDCK cells. Virology 251 264–272. [DOI] [PubMed] [Google Scholar]
- Leamon, C.P. and Low, P.S. 1991. Delivery of macromolecules into living cells: A method that exploits folate receptor endocytosis. Proc. Natl. Acad. Sci. 88 5572–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leland, P.A. and Raines, R.T. 2001. Cancer chemotherapy—ribonucleases to the rescue. Chem. Biol. 8 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leland, P.A., Schultz, L.W., Kim, B.-M., and Raines, R.T. 1998. Ribonuclease A variants with potent cytotoxic activity. Proc. Natl. Acad. Sci. 98 10407–10412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matousek, J., Soucek, J., Slavík, T., Tomá nek, M., Lee, J.E., and Raines, R.T. 2003. Comprehensive comparison of the cytotoxic activities of onconase and bovine seminal ribonuclease. Comp. Biochem. Physiol. C136 343–356. [DOI] [PubMed] [Google Scholar]
- Mitchell, D.J., Kim, D.T., Steinman, L., Fathman, C.G., and Rothbard, J.B. 2000. Polyarginine enters cells more efficiently than other polycationic homopolymers. J. Pept. Res. 56 318–325. [DOI] [PubMed] [Google Scholar]
- Nilsson, J., Stahl, S., Lundeberg, J., Uhlen, M., and Nygren, P.A. 1997. Affinity fusion strategies for detection, purification, and immobilization of recombinant proteins. Protein Expr. Purif. 11 1–16. [DOI] [PubMed] [Google Scholar]
- Nock, S., Spudich, J.A., and Wagner, P. 1997. Reversible, site-specific immobilization of polyarginine-tagged fusion proteins on mica surfaces. FEBS Lett. 414 233–238. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. and Scholtz, J.M. 1997. Measuring the conformational stability of a protein. In Protein structure (ed. Creighton, T.E.), pp. 299–321. Oxford University Press, New York.
- Raines, R.T. 1998. Ribonuclease A. Chem. Rev. 98 1045–1065. [DOI] [PubMed] [Google Scholar]
- Ramos, C. and Baldwin, R. 2002. Sulfate anion stabilization of native ribonuclease A both by anion binding and by the Hofmeister effect. Protein Sci. 11 1771–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbard, J.B., Kreider, E., VanDeusen, C.L., Wright, L., Wylie, B.L., and Wender, P.A. 2002. Arginine-rich molecular transporters for drug delivery: Role of backbone spacing in cellular uptake. J. Med. Chem. 45 3612–3618. [DOI] [PubMed] [Google Scholar]
- Rybak, S.M., Saxena, S.K., Ackerman, E.J., and Youle, R.J. 1991. Cytotoxic potential of ribonuclease and ribonuclease hybrid proteins. J. Biol. Chem. 266 21202–21207. [PubMed] [Google Scholar]
- Sassenfeld, H.M. and Brewer, S.J. 1984. A polypeptide fusion designed for the purification of recombinant proteins. BioTechnology 2 76–81. [Google Scholar]
- Schwarze, S.R., Ho, A., Vocero-Akbani, A., and Dowdy, S.F. 1999. In vivo protein transduction: Delivery of a biologically active protein into a mouse. Science 285 1569–1572. [DOI] [PubMed] [Google Scholar]
- Shaw, K.L., Grimsley, G.R., Yakovlev, G.I., Makarov, A.A., and Pace, C.N. 2001. The effect of net charge on the solubility, activity, and stability of ribonuclease Sa. Protein Sci. 10 1206–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, B.D., Soellner, M.B., and Raines, R.T. 2003. Potent inhibition of ribonuclease A by oligo (vinylsulfonic acid). J. Biol. Chem. 278 20934–20938. [DOI] [PubMed] [Google Scholar]
- Soellner, M.B., Dickson, K.A., Nilsson, B.L., and Raines, R.T. 2003. Sitespecific protein immobilization by Staudinger ligation. J. Am. Chem. Soc. 125 11790–11791. [DOI] [PubMed] [Google Scholar]
- Stevens, R.C. 2000. Design of high-throughput methods of protein production for structural biology. Struct. Fold. Des. 15 R177–R185. [DOI] [PubMed] [Google Scholar]
- Suzuki, M., Saxena, S.K., Boix, E., Prill, R.J., Vasandani, V.M., Ladner, J.E., Sung, C., and Youle, R.J. 1999. Engineering receptor-mediated cytotoxicity into human ribonucleases by steric blockage of inhibitor interaction. Nat. Biotechnol. 17 265–270. [DOI] [PubMed] [Google Scholar]
- Suzuki, T., Futaki, S., Niwa, M., Tanaka, S., Ueda, K., and Sugiura, Y. 2002. Possible existence of common internalization mechanisms among arginine-rich peptides. J. Biol. Chem. 277 2437–2443. [DOI] [PubMed] [Google Scholar]
- Sweeney, R.Y., Kelemen, B.R., Woycechowsky, K.J., and Raines, R.T. 2000. A highly active immobilized ribonuclease. Anal. Biochem. 286 312–314. [DOI] [PubMed] [Google Scholar]
- Ui, N. 1971. Isolectric points and conformation of proteins. I. Effect of urea on the behavior of some proteins in isoelectric focusing. Biochim. Biophys. Acta 229 567–581. [PubMed] [Google Scholar]
- Vives, E., Brodin, P., and Lebleu, B. 1997. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 272 16010–16017. [DOI] [PubMed] [Google Scholar]
- Wender, P.A., Mitchell, D.J., Pattabiraman, K., Pelkey, E.T., Steinman, L., and Rothbard, J.B. 2000. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc. Natl. Acad. Sci. 97 13003–13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, H. and Snyder, M. 2003. Protein chip technology. Curr. Opin. Chem. Biol. 7 55–63. [DOI] [PubMed] [Google Scholar]