

Abstract
p25α is a 219-residue proteinwhich stimulates aberrant tubulin polymerization and is implicated in a variety of other functions. The protein has unusual secondary structure involving significant amounts of random coil, and binding to microtubules is accompanied by a large structural change, suggesting a high degree of plasticity. p25α has been proposed to be natively unfolded, so that folding is coupled to interaction with its physiological partners. Here we show that recombinant human p25α is folded under physiological conditions, since it has a well structured and solvent-sequestered aromatic environment and considerable chemical shift dispersion of amide and aliphatic protons. With increasing urea concentrations, p25α undergoes clear spectral changes suggesting significant loss of structure. p25α unfolds cooperatively in urea according to a simple two-state transition with a stability in water of ~5 kcal/mol. The protein behaves as a monomer and refolds with a transient on-pathway folding intermediate. However, high sensitivity to proteolytic attack and abnormal gel filtration migration behavior suggests a relatively extended structure, possibly organized in distinct domains. A deletion mutant of p25α lacking residues 3–43 also unfolds cooperatively and with similar stability, suggesting that the N-terminal region is largely unstructured. Both proteins undergo significant loss of structure when bound to monomeric tubulin. The stoichiometry of binding is estimated to be 3–4 molecules of tubulin per p25α and is not significantly affected by the deletion of residues 3–43. In conclusion, we dismiss the proposal that p25α is natively unfolded, although the protein is relatively flexible. This flexibility may be linked to its tubulin-binding properties.
Keywords: natively unfolded protein, stability, folding kinetics, tubulin, flexibility, binding stoichiometry
Human p25α (also known as TPPP [tubulin polymerization promoting protein]) is a small basic 219-amino acid residue protein that is conserved among mammalian species (including cow and rodents) as well as having more distant homologs in insects and worms (Shiratsuchi et al. 1995; Seki et al. 1999; Tirian et al. 2003). The protein was originally identified as a protein copurified with tau protein kinases from bovine brain (Takahashi et al. 1991). Subsequently, it has been implicated in a number of different functions. Bovine p25α has submicromolar affinity for tubulin (Tirian et al. 2003) and causes aberrant microtubule assemblies at substoichiometric concentrations (Hlavanda et al. 2002; Tirian et al. 2003). Rat p25α is a potent inhibitor of glycogen synthase kinase 3 (GSK3) (Martin et al. 2002), and human p25α stimulates aggregation of α-synuclein, the major component of Lewy Bodies associated with Parkinson’s disease (Lindersson et al. 2005). The protein can be phosphorylated by GSK3 (Martin et al. 2002), tau kinase II (Takahashi et al. 1991; Martin et al. 2002), and protein kinase C isoforms (Yokozeki et al. 1998).
p25α is also interesting from a biophysical perspective. The protein appears to have an unusual nonglobular structure, since sequence analysis predicts 30%–43% α-helix content but only 4% is estimated from the far-UV spectrum (Hlavanda et al. 2002). Further, p25α may be plastic enough to undergo significant structural changes, since the spectrum of p25α incubated with tubulin was different from the spectrum predicted from the individual components (Hlavanda et al. 2002). The protein can be recovered from the supernatant of heat-treated tissue extract (Takahashi et al. 1991), indicating that it is either natively unfolded or does not aggregate upon unfolding. Very recently, Ovadi and coworkers proposed that p25α belonged to the class of natively unfolded proteins (Kovacs et al. 2004; Orosz et al. 2004). They based this on two experimental observations. Firstly, 1H-NMR spectroscopy showed a lack of signal dispersion, including low- and high-field resonances, as well as extremely broad amide proton chemical shifts (Kovacs et al. 2004). Secondly, the protein’s single Trp residue gave rise to an emission maximum around 350 nm, typical of high solvent-exposure (Orosz et al. 2004). They also showed that trifluoroethanol was able to induce α-helical structure in p25α, but this has also been shown to be the case for natively folded proteins (Buck et al. 1993; Chiti et al. 2000; Kumar et al. 2004). Finally, they used the neural network-based algorithm PONDR (Dunker et al. 2001) to predict that the first 52 or so residues of p25α, which are largely polar or charged, are intrinsically disordered. This observation is significant, as long disordered stretches (at least 40 residues) are predicted with good confidence; in contrast, the pattern of alternating ordered and disordered short regions may be less trustworthy.
In addition to p25α, two related proteins, subsequently named p25β and p25γ, have been identified at the DNA level (Zhang et al. 2002). The three proteins share a significant degree of sequence identity in the middle and C-terminal regions, including a highly conserved Rossmann fold known to be involved in nucleotide binding (Shiratsuchi et al. 1995; Zhang et al. 2002). Apart from this fold, no known structural motifs have been identified from the p25α sequence. Importantly, the putatively unstructured N-terminal region (residues 3–43) is missing in p25β and p25γ.
To shed more light on p25α’s unusual conformational properties, we investigated its biophysical properties by spectroscopic techniques. In contrast to the observations by Ovadi and coworkers, we found that p25α behaves like a conventional folded protein in nearly all aspects. It forms a compact structure with well defined tertiary interactions as judged from near-UV circular dichroism (CD), fluorescence, NMR, cross-linking, and acrylamide quenching experiments. Furthermore, the protein unfolds cooperatively under equilibrium conditions, and the unfolding and refolding kinetics suggest that the protein folds to the native state via a transiently populated partially folded intermediate. However, an unusual feature is the protein’s sensitivity to low concentrations of protease, which in conjunction with abnormal migration behavior on a gel filtration column and rapid refolding and unfolding rates suggests a dynamic and flexible structure. We have been able to titrate the binding of p25α to monomeric tubulin in solution using fluorescence spectroscopy, and we present evidence suggesting that the proteins form an oligomeric complex involving between 3 and 4 molecules of tubulin per p25α molecule. A similar set of biophysical and tubulin-binding studies have been carried out with p25αΔ3-43, a truncated version lacking the N-terminal region which is absent in p25β and p25γ. We found no significant differences between p25α and p25αΔ3-43, confirming that the N-terminal region is an unstructured region of the protein which does not contribute to its stability; furthermore, p25α’s ability to bind monomeric tubulin does not involve this region.
Results
p25α is folded under physiological conditions and unfolds cooperatively
p25α was purified to at least 95% purity using ion-exchange and gel-filtration chromatography (Fig. 1 ▶). We were able to reproduce the far-UVCD spectrum of bovine brain p25α reported by Ovadi and coworkers (Hlavanda et al. 2002) (Fig. 2A ▶). Secondary structure analysis of p25α’s structure using the k2d program (Andrade et al. 1993) predicts 15% α-helix, 30% β-helix, and 55% random coil, but the fit is rather poor (data not shown), suggesting unusual features in the protein structure. Nevertheless, p25α clearly has well defined secondary structure, since addition of 5 M urea (well above the midpoint of denaturation, see below) leads to a marked loss of spectral intensity (Fig. 2A ▶).
Figure 1.
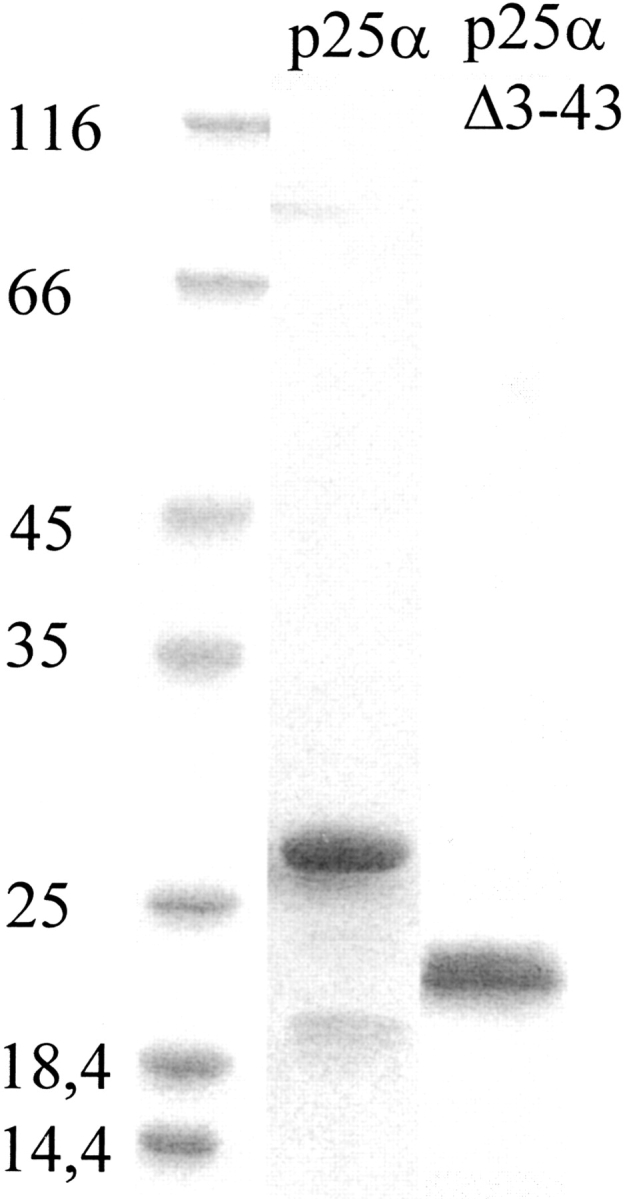
Purification of p25α (23.7 kDa) and p25αΔ3-43 (19.6 kDa) after ion exchange and gel filtration chromatography.
Figure 2.

Spectral properties of p25α. (A) Far-UV CD spectrum of native (solid line) and denatured (stippled line [5 M urea]) p25α. Native p25α is predicted to have 15% α-helix, 30% β-sheet, and 55% random coil, although the fit to the predicted spectrum is not good (data not shown). (B) Near-UV CD spectrum of native p25α. The peak at 280 nm indicates that the Trp residue experiences a well defined asymmetrical environment, typical of the native state. (C) Fluorescence emission spectra of native (solid line) and denatured (stippled line [5 M urea]) p25α. (D) Stern-Volmer plot of the quenching of native (•) and denatured (○ [5 M urea]) p25α. The slopes of the plots are 1.14 and 4.30 M−1, respectively, indicating that the Trp is much more accessible to acrylamide in the denatured rather than the native state.
p25α has one Trp (residue 76) and three Tyr residues, which provide convenient spectroscopic handles in the aromatic region. There is a distinct peak centered around 280 nm in the near-UV CD spectrum of p25α (Fig. 2B ▶). Since the near-UV CD spectrum reflects on the environment experienced by the aromatic side chains, we conclude that the aromatic chains experience a structured and asymmetric environment under physiological conditions. This is corroborated by the fluorescence emission spectrum, obtained by excitation at 295 nm, which is selective for Trp residues. Here the intensity peaks at 330 nm, typical of buried Trp residues (Fig. 2C ▶). In the presence of 5Murea, there is a dramatic red shift to a peak around 355 nm (Fig. 2C ▶). The fluorescence spectrum of p25α in 5 M urea, but not in buffer, bears a strong resemblance to the fluorescence spectrum of p25α in phosphate buffer (pH 7.0) reported by Ovadi and coworkers (Orosz et al. 2004).
A more direct test of the surface exposure of the Trp residue in p25α is to quench its intensity in the presence of increasing concentrations of acrylamide. The more surface-exposed the Trp residue is, the more accessible it is to acrylamide and the more its fluorescence will be quenched (Lakowicz 1999). The slope of the plot in Figure 2D ▶ (the Stern-Volmer constant kSV) is 1.32 ± 0.3 in PBS buffer alone and 5.21 ± 0.20 in 5 M urea. This makes it clear that the Trp is much more protected from quenching under physiological conditions than in 5 M urea (Fig. 2D ▶).
The NMR spectrum recorded in the absence of urea shows signs of a folded structure: the chemical shifts are dispersed, and 1H-signals are found between 0–0.7 ppm, 5–6 ppm, and 9–10 ppm (Fig. 3A ▶).
Figure 3.

1D NMR spectra of p25α in buffer (A) and buffer and 7 M urea (B). Note the loss of chemical shift dispersion in the region from 6–10 ppm (amide and aromatic protons) and the region from 0–0.7 ppm (the aliphatic region) in urea, suggesting unfolding.
Judging by the presence of the Hα signals with a shift greater than 4.7 ppm (which is indicative of β-sheet structure), the protein contains a very lowamount of β-structure.
Upon addition of 7 M urea, the chemical shift dispersion collapses completely, and no NMR signals indicative of secondary structure remain (Fig. 3B ▶). As was the case for fluorescence, the spectrum in urea, but not in buffer, resembles the 1H-NMR spectrum reported for p25α by Ovadi and coworkers (Kovacs et al. 2004).
To follow the nature of the conformational change at increasing urea concentrations, we incubated p25α at different concentrations of urea. This led to a marked spectral shift, as indicated in Figure 2C ▶, with an isosbestic point around 335 nm that is retained over a broad range of urea concentrations (data not shown). The presence of this point is in itself indicative of a two-state transition, i.e., only the native and denatured states are significantly populated at equilibrium. A denaturation curve is obtained with a denaturation midpoint at 3.71 ± 0.02 M (using the ratio of the intensities at 320 nm and 335 nm) and 3.82 ± 0.02 M (using the 354/335 nm ratio) (Fig. 4A ▶). This predicts a stability of 5.42 ± 0.62 kcal/mol, which is considered low for a conventional globular protein, whose ΔGD-N-values typically lie in the range 5–15 kcal/mol (Pace 1990). The protein does not appear to be involved in any stabilizing intermolecular interactions such as noncovalent dimerization, since a 20-fold change in protein concentration (from 1 to 20 μM) has no effect on the stability parameters (data not shown).
Figure 4.

Equilibrium and kinetic stability of p25α. (A) Equilibrium denaturation of p25α in urea, followed by the ratio of the emission intensities at 327, 335, and 355 nm.•, 327/335 nm; ○, 355/335 nm. (B) Time profiles of unfolding and refolding of p25α followed by stopped-flow. Both time profiles are fitted to a single exponential decay with offset. (C) Log of the observed rate constants of folding and unfolding of p25α vs. [urea]. The data are fitted to a three-state model (D ↔ I ↔ N) (Scheme 1). Results are given in Table 1. (D) Effect of [Na2SO4] on the log of the refolding rate in 0.5 M urea, where the intermediate accumulates transiently. The rise in refolding rates with [Na2SO4] is consistent with the scenario that the intermediate is on-pathway.
Another assay for a native fold is to cross-link the protein under different solvent conditions. The rationale is that the compact species should be more disposed toward internal cross-linking than more extended species. We used the primary amine-reactive cross-linking agent BS3 (spacer arm length ~11.4 Å). The experiment is performed between 0 and 6 M urea; over this concentration range, fluorescence data indicate that p25α will go from completely folded to completely unfolded. As controls we used lysozyme (which does not denature over this urea concentration range) and the natively unfolded α-synuclein. α-Synuclein remains unaffected by BS3, while a slightly faster-migrating lysozyme species is stabilized by the cross-linker, indicating the formation of a more compact (internally cross-linked) species in addition to the uncross-linked species (Fig. 5 ▶, left panel). The p25α band splits up into two bands, one migrating faster than the 25α kDa band seen in the absence of BS3, and the other migrating at ~25α kDa. The faster-migrating band gradually disappears above 3 M urea. As previously suggested, the lysozyme and α-synuclein data indicate that internal cross-linking by BS3 requires the presence of a native fold, which is present in p25α below 4 M urea. This is consistent with the urea-denaturation data, which indicate a transition occurring with a midpoint at ~3.7 M urea.
Figure 5.

(Left panel) Cross-linking of p25α, lysozyme, and α-synuclein with BS3 at different urea concentrations. BS3 cross-linking leads to the formation of a second faster-migrating band in the case of p25α and lysozyme (indicated by arrows), which for p25α disappears at higher urea concentrations. (Right panel) Digestion of p25α and lysozyme in the presence of various concentrations of protease. (A) p25α in PBS buffer with trypsin. (B) p25α in 0.5 M Na2SO4 with trypsin. (C) p25α in PBS buffer with chymotrypsin. (D) Lysozyme in PBS buffer with trypsin. The numbers on the gel indicate the fraction of protease relative to p25α or lysozyme (w/w). Note that the digestion pattern for p25α is the same when we stabilize the native state (in 0.5 M Na2SO4).
We also used analytical gel filtration to monitor the expansion of p25α at increasing denaturant concentrations. In the absence of buffer, p25α elutes with a Stokes radius of 30 Å (data not shown). For a globular protein, this would correspond to a molecular mass of ~49 kDa (Uversky et al. 1999). Given that the protein has a monomeric molecular weight of 23.7 kDa, this elution behavior suggests either dimerization (which is unlikely, given that p25α stability is independent of protein concentration, as mentioned above) or a relatively expanded conformation. Such an expanded monomer would not be completely unfolded, however. For example, the natively unfolded protein α-synuclein (molecular weight 14.5 kDa) elutes with a Stokes radius of ~32 Å, which for a globular protein corresponds to an apparent molecular weight of 60 kDa. In addition, the elution volume of p25α decreases by 10% in the presence of 6 M urea, where it is expected to be unfolded; in contrast, that of α-synuclein, which is only expected to undergo insignificant expansion under these conditions, is reduced by only 4.5%.
The above data strongly indicate that p25α folds to a well defined state under physiological conditions. A partially folded state, such as a “molten globule,” would not be expected to have a strong near-UV CD peak or to be protected so well against quenching. Another test for the existence of a partially folded state is to investigate the ability of p25α to bind the hydrophobic probe ANS1 (8-anilino-1-naphthalenesulfonic acid). ANS is typically used to test for the presence of hydrophobic patches in partially folded conformations (Goto and Fink 1989; Semisotnov et al. 1991), binding to which typically increases ANS fluorescence by more than an order of magnitude. However, p25α increases ANS fluorescence by <25% (data not shown), indicating no significant binding.
Folding of p25α occurs via a transient intermediate
To probe the stability of p25α in more detail, we measured the rates of the protein’s folding and unfolding. The kinetic profiles for both folding and unfolding yielded single exponential decays (Fig. 4B ▶). The log of the observed rate constant versus [urea] is shown in Figure 4C ▶. Under unfolding conditions (above ~4 M urea), the log of the rate constant increases linearly with [urea], indicating that unfolding occurs without an intermediate. However, under refolding conditions there is a marked “rollover” below ~2 M urea, which is the hall-mark of a folding intermediate (Tanford 1970; Baldwin 1996). The rollover can generally be attributed to a switch in the ground state from which folding occurs, namely from the intermediate state at low urea concentrations and the denatured state at higher concentrations (where the intermediate is destabilized and therefore does not accumulate).
Based on this plot alone, we cannot distinguish between an on-pathway intermediate I (D ↔ I ↔ N) and an off-pathway intermediate C (C ↔ D ↔ N) (Baldwin 1996). Both pathways can be fitted satisfactorily to the kinetic data and yield identical predictions of the stability of the native state. Complete resolution of the pathway requires us to measure the rate constants for formation and decay of the intermediate (Capaldi et al. 2001), which is beyond our technical scope. However, there is a simple assay, using sodium sulfate, which under favorable circumstances can distinguish between the two scenarios. Inorganic salts such as sodium sulfate favor compact protein conformations because they are preferentially excluded from the protein surface (Timasheff 2002). In addition to stabilizing the native state, these salts will also induce partially structured states to accumulate during the refolding process. Accumulation of a folding intermediate will have a profoundly different effect on folding rates, depending on whether it is on-pathway or off-pathway. In the off-pathway scenario, the folding rate is limited by the fraction of the protein which is in the denatured state D (fD=[D]/(D]+[C]), and the more C is stabilized by salts relative to D, the slower the observed folding rate. In contrast, the on-pathway scenario leads to the folding rate being limited by the fraction fI=[I]/([I]+[D]), and the more I is stabilized, the faster the rate should be. Under conditions where folding occurs directly from the denatured state and the intermediate does not accumulate, folding rates should in both scenarios be accelerated by salt. We showed previously that the intermediate, which accumulates when the ribosomal protein S6 folds in the presence of Na2SO4, by this criterion is off-pathway, since salt slows down folding (Otzen and Oliveberg 1999), and we obtained similar results for the folding intermediate of Bet v 1 (Mogensen et al. 2004). In the case of p25α, however, salt accelerates folding markedly in 0.5 M urea where the intermediate accumulates significantly (Fig. 4D ▶), suggesting that the intermediate is on the path between the denatured and native state.
The kinetic data are analyzed according to equation 4 as listed in Table 1. Due to the relative uncertainty of the refolding rate constants obtained at low denaturant concentrations, the slope of the chevron plot in the rollover region (mf) has been set to zero for simplicity. Our kinetic data predict that the native state is stabilized by 6.21 ± 0.23 kcal/mol relative to the denatured state, within error the same as the data obtained from equilibrium denaturation experiments. In addition, the sum of the kinetic m-values (1.27 ± 0.05 M−1) is close to the equilibrium m-value (1.06 ± 0.12 M−1). Taken together, these observations suggest that the folding behavior of p25α is described satisfactorily by a simple three-state model, and it is not necessary to invoke any additional states to describe our system (cf. Fersht 1999).
Table 1.
Biophysical parameters for p25α and p25αΔ3-43 obtained from equilibrium and kinetic experiments
| Parameter | p25α | p25αΔ3-43 |
| kSV (PBS buffer) (M−1)a | 1.32 ± 0.03 | 2.44 ± 0.11 |
| kSV (5M urea) (M−1)a | 5.21 ± 0.20 | 9.28 ± 0.20 |
| [urea]50% (equilibrium experiment) (M)b | 3.76 ± 0.03 | 3.84 ± 0.10 |
| [urea]50% (kinetic experiment) (M)c | 3.62 ± 0.25 | - |
| mD-Nequilibrium (M−1)d | 1.06 ± 0.12 | 0.95 ± 0.11 |
| mD-Nkinetic (M− 1)e | 1.27 ± 0.05 | - |
| kfwater (s−1) | 164.5 ± 1.4 | - |
| kuwater (s−1) | 2.01 ± 0.28 | - |
| mu (M−1) | 0.12 ± 0.01 | - |
| KI | 453 ± 28 | - |
| mI (M−1) | −1.15 ± 0.05 | - |
| ΔGD-Nequilibrium (kcal mol−1)f | 5.42 ± 0.62 | 4.96 ± 0.59 |
| ΔGD-Nkinetic (kcal mol−1)g | 6.21 ± 0.23 | - |
All experiments at 25°C in PBS pH 7.4 and 2 mM DTT.
a Data from a Stern-Volmer plot (Fig. 2D ▶; equation 1).
b The average of the two plots in Fig. 4A ▶. Data fitted to equation 2.
c The urea concentration at which [N]=[D], which means that [N]/[D]=[N]/[I] * [I]/[D]=KI * kf/ku=1 (cfr. equation 3).
d Obtained from equation 2.
emD-Nkinetic =mI + mf − mu. mf is set to zero because of the narrow denaturant concentration range over which this parameter can be determined.
f Calculated from the relationship ΔGD-N= − RT ln(10)*mD-N*[urea]50% (Pace 1986).
g Calculated from the relationship ΔGD-N= − RT ln(10)log(kfKI/Ku).
p25α is sensitive to proteolysis
While p25α by all spectroscopic approaches appears to behave as a normal cooperatively folded protein, the gel filtration analysis suggested that the protein is relatively expanded. We decided to probe this in more detail using proteolysis. Resistance to proteolysis is often used as a criterion for the integrity of a protein’s structure (Tsai et al. 2002). We therefore incubated the protein with decreasing concentrations of trypsin, which cleaves after the basic residues Lys and Arg. At [p25α]:[trypsin] ratios between 1:10−1 and 1:10−4, p25α was completely degraded by trypsin after 1 h at 37°C (Fig. 5A ▶). In contrast, hen egg white lysozyme, which has a compact folded structure, remains completely intact under the same conditions (Fig. 5D ▶). p25α contains an unusually large number of basic residues (26 Lys and 15 Arg out of a total of 219 residues), which might dispose the protein toward digestion despite a well consolidated fold. However, we obtained similar results with chymotrypsin, which cleaves after aromatic and large hydrophobic side-chains (Fig. 5C ▶). To test whether trypsin degraded the unfolded state of p25α or the native state, we shifted the conformational equilibrium toward the native state (in 0.5 M Na2SO4), but as shown in Figure 5B ▶, this did not appreciably alter the trypsin susceptibility (inhibition of trypsin activity in urea prevented us from drawing firm conclusions from similar experiments in 1 M urea). This suggests that trypsin is able to degrade the native state, which is the major species under all these conditions.
p25αΔ3-43 is just as stable as p25α
Amino acid residues 3–43 in p25α are absent in the close homologs p25β and p25γ (Zhang et al. 2002). The truncated protein p25αΔ3-43, lacking these residues, was produced (Fig. 1 ▶) and subjected to a biophysical analysis. Like p25α, the protein undergoes a marked change in its fluorescence spectrum upon transfer from zero to 5 M urea (Fig. 6A ▶), although the increase in intensity of the native state relative to the denatured state (compared to p25α ) shifts the isosbestic point from 335 to 340 nm. The midpoint of denaturation is 3.84 ± 0.10 M (Fig. 6B ▶), which translates to a stability of 4.59 ± 0.59 kcal/mol, only slightly less than that of p25α (Table 1). Quenching experiments provide a Stern-Volmer constant of 2.40 ± 0.03 M−1 in PBS and 8.56 ± 0.29 M−1 in 5 M urea (Fig. 6C ▶), again showing that the protein undergoes a significant structural change upon transfer from zero to 5 M urea, although the absolute values have increased for both the native and denatured states compared to p25α.
Figure 6.

Biophysical properties of p25αΔ3-43. Data summarized in Table 1. (A) Fluorescence emission spectra of native (solid line) and denatured (stippled line [5 M urea]) protein. (B) Equilibrium denaturation in urea, followed by the ratio of the emission intensities at 327 and 340 nm. (C) Stern-Volmer plot of the quenching of native (•) and denatured (○ [5 M urea]) protein.
Interaction between p25α/p25αΔ3-43 and tubulin
As previously mentioned, Ovadi showed that the spectrum of bovine p25α incubated with tubulin in a 2.8:1 ratio had a smaller intensity than the spectrum predicted from the sum of the individual components (Hlavanda et al. 2002). This suggests interactions between the two proteins, with complex formation leading to loss or alteration of structure. We are able to reproduce these results for both recombinant human p25α and p25αΔ3-43 (Fig. 7A,B ▶), indicating that the truncated version of p25α is still able to bind tubulin. Similar results are obtained by fluorescence; although there is no shift in peak maximum, the tubulin:p25α complex has a 20% reduced intensity compared to the sum of the individual proteins (Fig. 7C ▶), while the reduction is ~10% for the tubulin:p25αΔ3-43 complex (Fig. 7D ▶).
Figure 7.

Spectroscopic alterations induced in the p25α:tubulin (A,C) and p25αΔ3-43:tubulin (B,D) complexes as measured by far-UV CD (A,B) and Trp fluorescence (C,D). The actual spectrum of the complex is denoted by the stippled line with ○; the mathematical sum of the spectra of the two protein components is denoted by the joined line with •. The difference is indicated by the stippled line with x. For clarity, spectra of free p25α and tubulin are omitted.
This reduction in fluorescence induced by binding between the two components provides us with a sensitive method to monitor the binding of p25α and p25αΔ3-43 to tubulin. We have therefore titrated p25α and p25αΔ3-43 into a tubulin solution in small steps while measuring the fluorescence (cf. equation 4). The difference between the measured fluorescence and that expected from the contributions of the individual components (Fexp − Fobs) is an indication of the extent of binding between tubulin and p25α. In the absence of any interaction, the difference should remain zero throughout. However, a plot of Fexp − Fobs versus the ratio between p25α (or p25αΔ3-43) and tubulin reveals that Fexp − Fobs rises steeply at low [p25α]:[tubulin], before it reaches a constant level (Fig. 8 ▶) Remarkably, the data indicate that binding is saturated well below a ratio of one p25α per tubulin molecule. This makes it clear that the data cannot be analyzed with a simple 1:1 binding model.
Figure 8.

Titration of tubulin with p25α (•) and p25αΔ3-43 (○). The difference Fexp − Fobs (where Fexp is the mathematical sum of the fluorescence of the two protein components and Fobs is the measured fluorescence) is plotted vs. the ratio of the two protein components.
Binding isotherms involving simple 1:1 binding stoichiometry, in which excess ligand is titrated into the protein solution, often reach saturation at ligand:protein ratios well above 1 if the dissociation constant Kd is well above the protein concentration P. Let us designate p25α as the ligand and tubulin as the protein. If P is well above Kd, saturation by ligand binding would occur around a ligand:protein ratio ~1, since there is enough protein present to push all added ligand into complex. If, on the other hand, P is below Kd, saturation will now occur at protein:ligand ratios well above 1. Therefore saturation at ratios of [p25α]:[tubulin] well below 1 when p25α is being titrated into tubulin must reflect departure from simple 1:1 stoichiometry.
We can instead analyze the data as a simple titration plot (rather than a binding isotherm), in which it is assumed that all the added p25α binds to tubulin in the initial titration steps until all tubulin-binding sites are saturated. In this way we obtain an intersection between the linear titration part of the plot and the maximum value of the plot at a [p25α]:[tubulin] of 0.24–0.32, suggesting that up to 3–4 tubulin molecules can bind to each p25α molecule. Essentially the same results are obtained for p25αΔ3-43. Because of the difficulty of separating contributions from tubulin and p25α, however, we have not been able to estimate with sufficient precision the concentration of free tubulin at each [p25α]:[tubulin] value, and consequently a Scatchard analysis was not able to provide further insight (data not shown).
Structural changes are specific to tubulin and are not caused by polyanions, lipids, or nucleotides
p25α has a strongly basic domain in common with other tubulin-binding substances such as the binding domains of microtubule-binding proteins like tau and MAP2 as well as cationic substances such as DEAE-dextran, protamine, melittin, and spermidine (Wolff 1998). Nevertheless, the structural effect of tubulin on p25α cannot be mimicked by simple negative charge. We did not observe any effect of polymeric anions such as heparin, hyaluronic acid, carrageenan, xanthan, or alginate or negatively charged cyclodextrins such as sulfated β-cyclodextrin or carboxymethyl-cyclodextrin on the structure of p25α, as monitored by fluorescence or CD. Nor did zwitterionic (phosphocholine) or anionic (phosphoglycerol) phospholipids alter the structure or proteolytic sensitivity of p25α, although addition of phosphoglycerol led to extensive aggregation, probably due to unspecific electrostatic association (data not shown). Therefore, the basis of the specificity between p25α and tubulin must involve structural specificity, rather than simple electrostatics. Similarly, no structural changes were observed in the presence of ATP or GTP (data not shown), despite the existence of a nucleotide-binding Rossmann fold in the p25α sequence.
Discussion
p25α is folded but has a flexible structure
The first objective of this study was to establish whether p25α is natively folded. Prior to this study, the protein’s unusual far-UV CD spectrum indicated a low level of organized secondary structure (Hlavanda et al. 2002), much less than that predicted by sequence analysis. In addition, spectroscopic evidence had been presented suggesting that the protein is natively unfolded (Kovacs et al. 2004; Orosz et al. 2004).
Our work clearly shows by a number of mutually reinforcing methods that human recombinant p25α behaves like a conventional monomeric folded protein. It is folded according to far- and near-UV CD, fluorescence and NMR spectroscopy, unfolds cooperatively under equilibrium conditions, and can be internally cross-linked like other natively folded proteins. Furthermore, it appears to fold via an “on-pathway” partially folded intermediate, in common with many other proteins of the same size (Matouschek et al. 1990; Parker et al. 1995, 1998; Nölting et al. 1997), indicating a well organized accumulation of structure.
We find it difficult to explain the divergence between our data and those of Ovadi and coworkers (Kovacs et al. 2004; Orosz et al. 2004). There is no doubt that we are working with the same protein, since the far-UV CD spectra are essentially identical in buffer, and both protein samples undergo spectral changes upon binding tubulin. Since we have shown that p25α undergoes a dramatic change in its far-UV CD spectrum upon unfolding in urea, this suggests that the Ovadi p25α sample should also be folded. However, the fluorescence and NMR spectra recorded by Ovadi and coworkers (in 10–20 mM phosphate buffer [pH 7.0]) correspond to our spectra for p25α in 5–7 M urea. One difference is that we are working with recombinant p25α, while Ovadi and coworkers purify p25α from bovine brain in the presence of 5 mM EDTA. Thus there could be at least three reasons for the difference in behavior. Firstly, p25α could bind a metal ion which is removed in the presence of EDTA, leading to unfolding. Metal ions are known to bind and induce structure in natively unfolded proteins (Uversky and Narizhneva 1998). However, this option can be discounted for p25α since we find that p25α unfolds in an identical fashion in the presence and absence of 5 mM EDTA (data not shown). Secondly, a brain component copurifying with p25α could keep the protein unfolded, and this component would naturally be absent in E. coli. Nevertheless, such a component would presumably have to be present at stoichiometric concentrations and would therefore have been identified by SDS-PAGE analysis. Even if such a component existed, it does not resolve the discrepancy between the CD and fluorescence/NMR spectra presented by Ovadi and coworkers. Thirdly, p25α from bovine brain could be post-translationally modified, leading to an unfolded state. This possibility cannot be ruled out, although it would be without precedent.
Nevertheless, although we have shown that p25α is natively folded, it is unusually sensitive to proteolysis under physiological conditions. The sensitivity must be linked to the structure of the native state. It cannot be ascribed to the protein’s relatively low stability, first because even a stability of 5.4 kcal/mol predicted for p25α leads to only 0.01% denatured protein in the absence of denaturant, and secondly because it persists in the presence of stabilizing salts favoring the native state. This could indicate either that the protein is inherently dynamic and flexible, or that it contains a number of exposed loops that provide a point of attack for proteases (both trypsin and chymotrypsin). It is also possible that the protein contains several domains linked by flexible linker regions that would be the first points of attack by proteases, followed by proteolysis of the isolated domains. These domains could be arranged in an extended fashion rather than being tightly bound in a globular structure. That could explain the abnormal elution behavior of p25α on the gel filtration column, where the protein elutes with an apparent molecular weight of ~50 kDa rather than the expected 24 kDa, although there is no indication of dimerization. Although these domains may unfold as separate units during chemical denaturation, this will not necessarily be visible as independent transitions. Overlapping denaturation curves can merge to an apparent single denaturation transition. However, the appearance of a transient folding intermediate could be ascribed to the folding of one domain prior to another during the folding process. This has been observed for, e.g., barnase, where the intermediate mainly consists of contacts within the domain consisting of the major α-helix and the central strands of the β-sheet (Matouschek et al. 1992). Full elucidation of the modular structure of p25α requires more detailed structural information, e.g., by NMR or X-ray crystallography, although the flexibility suggested by our data will probably pose challenges for crystallization.
Some flexible regions of the protein can already be identified by indirect means. If a terminal part of a protein is unstructured, then it should not contribute to the protein’s stability, since it will not by itself contribute to the stabilization of the native state by, e.g., side-chain contacts and docking of secondary structure elements. The small difference in structural stability between p25α and p25αΔ3-43 suggests that the deleted N-terminal region (up to residue 43) is largely unstructured. This agrees well with the prediction by Orosz et al. (2004), where residues 1–47 have disorder values above 0.8 (a value of 1.0 indicates that the residue is fully disordered). Similar small effects on stability have been observed for other proteins in which unstructured regions have been deleted, e.g., CI2 (cf. the stabilities measured in Jackson and Fersht 1991 and Jackson et al. 1993). In contrast, deletion of even a small number of terminal residues which are integrated into a protein’s structure can have significant deleterious effects on stability (De Prat Gay et al. 1995; Hamill et al. 1998).
Biological implications of a flexible structure
While p25α is not unfolded under physiological conditions, its protease sensitivity is shared with another group of proteins, namely those which are natively unfolded. A growing number of such proteins have been reported (for reviews, see Dunker et al. 2002a; Dyson and Wright 2002; Tompa 2002; Uversky 2003) which in many cases only assume structure upon binding to specific protein partners (e.g., Uversky et al. 2000; Dunker et al. 2002b; Muro-Pastor et al. 2003; Lacy et al. 2004). Different reasons for their unfolded state have been proposed, including interactions with a larger number of proteins, reduced sensitivity to environmental perturbations (Lee et al. 2001), and a greater “capture radius” during the binding and folding process (Shoemaker et al. 2000). Of particular relevance for p25α may be the fact that structural flexibility and its accompanying protease sensitivity provides a useful additional level of control in cellular signaling processes, in which a response needs to be rapidly turned on and off. p25α’s suggested involvement in signaling cascades via the protein kinase C family (Yokozeki et al. 1998) and modulation of microtubule dynamics (Tirian et al. 2003) would require fine-tuning of its cellular population levels, which could be provided by protease-sensitivity. However, these speculations will have to be substantiated by further experiments, e.g., by detection of proteolytic fragments or by testing the physiological function of a p25α variant engineered to decrease its protease sensitivity.
p25α’s flexibility may also be relevant for its ability to bind tubulin both as monomers and polymers and assemble multiple monomers into microtubules (Tirian et al. 2003), although the partial loss of structure seen in the tubulin:p25α complex could in principle come from tubulin as well as p25α.
Nature of the complex formed between p25α and tubulin
Our titration data suggest that p25α is able to form oligomeric complexes with tubulin, in which 3–4 molecules of tubulin may engage each p25α molecule at very low [p25α]-[tubulin] ratios. This ties in well with the ability of substoichiometric concentrations of p25α to induce large alterations in microtubule behavior (Hlavanda et al. 2002). The extended state of p25α suggested by the gel filtration data would also provide a relatively large binding surface which might facilitate contact to several tubulin molecules. Our data are at odds with those of Ovadi and coworkers (Tirian et al. 2003), who presented elegant surface plasmon resonance (SPR) results consistent with the formation of a 1:1 complex between tubulin and p25α with an estimated Kd of 0.2 μM. A drawback of the SPR technique in this context may be that tubulin must be immobilized on a sensor chip, and this treatment is likely to hinder it from engaging in oligomeric contacts that may occur in solution and that may increase the affinity substantially. In solution, p25α appears to be tightly bound to tubulin at concentrations of 0.1 μM and less. However, it is inherently not feasible to estimate Kd accurately from a titration curve which assumes tight binding between the two components.
Formally we cannot rule out that the substoichiometric influence of p25α on microtubular morphology could occur by 1:1 complexation with a small fraction of the tubulin molecules which subsequently seed the formation of aberrant tubulin structures. There is probably a spectrum of different stoichiometries, leading to a displacement of the population of complexes toward smaller p25α:tubulin complexes as [p25α]:[tubulin] increases. This may also explain why the titration data in Figure 8 ▶ are not entirely linear at low p25α concentrations, but curve downward. Unfortunately, the narrow concentration range over which this shift occurs prevents us from distinguishing different populations of complexes, although future experiments with more sensitive equipment such as analytical centrifugation and a light scattering apparatus may shed more light on this issue.
Materials and methods
Materials
Bovine brain tubulin (>99% pure) was from Cytoskeleton. BS3 was from Pierce.
Purification of p25α and p25αΔ3-43
cDNA coding for human p25α cDNA was amplified by reverse transcription polymerase chain reaction (PCR) from a human fetal brain mRNA library (Clontech) using the primers p25α 5′: 5′-CACCCATGGCTGACAAGGCCAA-3′, p25α 3′:5′-CACG GATCCCTACTTGCCCCCTTGCAC-3′. For expression of native p25α, the PCR fragment was inserted in the pET-11d vector (Novagen). For expression of p25αΔ3-43, a deletion mutant lacking residues 3–43, a pET-11d vector was generated using the pET-11d p25α vector as template and the following primers (DNA Technology): 5′-CACGGATCCTACTTGCC CCCTTGCAC-3′and 5′-CACCCATGGCTGCATCCCCT GAGCTCAGT-3′. Correct insertion was verified by DNA sequencing (MWG-Biotech). For protein purification, E. coli BL21 (DE3) cells (Stratagene) were transformed, pelleted, and lysed by sonication on ice in buffer A (50 mM NaH2PO4 [pH 8.2]). The soluble proteins were heated to 100 °C for 10 min, and the heat-denatured proteins were removed by centrifugation. Heat-stable proteins were loaded onto a Poros HS50 cation column (PerSeptive Biosystems), which was eluted by a double linear gradient, first by buffer B (1 M NaCl, 50 mM NaH2PO4 [pH 8.2]) and subsequently by buffer C (1 M NaCl, 50 mM NaH2PO4 [pH 12]). p25α eluted shortly after the pH exceeded pH 8.2. The final purification and buffer exchange was done on a GF75 gel filtration column (Amersham Pharmacia) that had been pre-equilibrated with 7Murea, 120 mM NaCl, 20 mM Na-phosphate (pH 7.4) (PBS) supplemented with 1 mM DTE in order to avoid unspecific dimerization via the three free Cys residues. Mass spectroscopic analysis led to a mass deviating only 0.06% from the theoretical mass (23,693.7 Da). Furthermore, on 2D gels the recombinant p25α migrates in a fashion very similar to that of p25α purified from bovine brain (Lindersson et al. 2005). Thus chemical modification of p25α due to heating at 100 °C is likely to be insignificant.
Analytical gel filtration chromatography
This was performed on a Superdex 75 PC 3.2/30 column (Amersham Pharmacia) connected to the SMART system chromatographic unit (Amersham Pharmacia). Fifty μg human recombinant p25α was diluted in elution buffer (PBS [pH 7.2], 1 mM DTE, 0–6 M urea) to a final volume of 50 μL and loaded onto the column pre-equilibrated with the elution buffer. p25α was eluted using a constant flow rate of 50 μL/min, and elution was monitored at 280 nm. The elution volumes of the five molecular markers (12.5–158 kDa from Amersham Pharmacia) were compared to that of p25α to determine its molecular size in different concentrations of urea. To ensure that the size-exclusion properties of the column do not change with increasing urea concentrations, human recombinant p25α was included as a control. Fifty μg α-synuclein was loaded onto the column and run at identical conditions to those used for p25α.
Equilibrium fluorescence studies
Urea denaturation experiments were carried out at protein concentrations of ~2.5 μM in PBS buffer (pH 7.4) and 2 mM DTT at 25°C. DTT was included to avoid unspecific dimerization via the three free Cys residues, and 10 M urea stock solutions were prepared fresh on a daily basis. For equilibrium denaturation experiments, each protein sample was allowed to equilibrate ~2 h before measurement. Equilibrium fluorescence studies were carried out by excitation at 295 nm, measuring emission at 310–380 nm (slit widths 4 nm) on an LS-55 spectrofluorimeter (Perkin-Elmer). ANS binding studies were performed with 40 μM ANS and 2 μM p25α in PBS buffer (pH 7.4), and 2 mM DTT at 25°C. Excitation was at 360 nm, and emission was recorded between 400 and 600 nm. Quenching studies were performed by adding aliquots of acrylamide from a 1.5 M stock solution to p25α (initial concentration 4–7 μM protein) and recording the emission intensity at 330 nm (native state) or 354 nm (denatured state) upon excitation at 295 nm. The solution was continually stirred with a small magnet and was allowed to equilibrate for a few minutes between each reading.
Stopped flow studies
Kinetic fluorescence measurements were performed on an Applied Photophysics SX18MV stopped-flow apparatus with a 2-nm slit width and a 320-nm glass filter. Stopped-flow fluorescence folding and unfolding experiments were initiated by 10-fold dilution of the protein from 5.5 M and 0 M urea, respectively, to the appropriate final urea concentrations in PBS buffer (pH 7.4), and 2 mM DTT. This allowed us to measure over the range 0.5–9.1 M urea. Data were fitted to a single exponential with offset. It was not necessary to include drift or additional kinetic phases. All signal changes occurred within the first few seconds, and recording over longer time scales did not reveal any additional phases.
Circular dichroism
All circular dichroism (CD) studies were performed on a Jasco J-715 spectropolarimeter (Jasco Spectroscopic) with a Jasco PTC-348W temperature control unit. Spectra were recorded in a 0.1-cm path length cuvette with resolution 0.2 nm, bandwidth 1.0 nm, sensitivity 50 mdeg, response 2.0 sec, and speed 20 nm/min at 25°C. Three scans were averaged to yield the final spectrum. Protein concentrations were 20 μM (far-UV CD, 250–205 nm) and 200 μM (near-UV CD, 320–250 nm).
Cross-linking with BS3
Fifteen μM protein was incubated with 0–6 M urea and PBS plus 1 mM DTE for 30 min at room temperature while shaking. Bis(sulfosuccinimidyl)suberate (BS3) (Pierce) (1 mM for p25α and α-synuclein, 5 mM for lysozyme) was added to the sample, and after 1 min the reaction was quenched with SDS loading buffer containing 25 mM Tris (pH 6.8), 4% SDS, and 40% glycerol. Higher concentrations of BS3 than 1 mM gave fuzzy bands for p25α and α-synuclein.
Interactions between p25α and tubulin
Typically, 800 μL of a 3 μM solution of tubulin was added to a 1.7-mL quartz cuvette, and p25α or p25αΔ3-43 was added in small-volume steps from a 30–40 μM stock solution. The solution was continually stirred with a small magnet. After each aliquot had been added, the solution was allowed to equilibrate for a few minutes before the measuring of fluorescence intensity Fobs (excitation at 295 nm, emission at 337 nm, excitation and emission slit widths 5 nm). Intensities of the p25α or p25αΔ3-43 stock solutions were recorded separately in the same cuvette prior to the titration experiment. For p25α and p25αΔ3-43, protein concentrations in PBS buffer and 1 mM DTT were determined by a bicinchoninic acid assay using the BCA Protein Assay Kit (Pierce), which tolerates up to 1 mM DTE and DTT. The concentration determined in this way diverged by only ~20% from the less accurate approach of using the protein extinction coefficients at 280 nm, based on the content of Trp, Tyr, and Cys (Gill and von Hippel 1989). Tubulin concentrations were based on weighed aliquots provided by the manufacturer. This diverged by only 5% from the concentration estimated from the calculated extinction coefficient.
NMR spectroscopy
NMR spectra were acquired on a BRUKER DRX600 NMR spectrometer equipped with a xyz-gradient TXI (H/C/N) probe. Spectra were recorded at 298 K. The protein was concentrated to a final volume of 350 μL with a concentration of 5 mg/mL. The sample contained 1 mM DTT, 5% D2O, and had a pH of 6.5.
Data analysis
Quenching studies
The ratio Fo/F, where Fo is the fluorescence intensity in the absence of acrylamide and F the intensity at different acrylamide concentrations, was plotted versus acrylamide concentration. The data were fitted to the following equation:
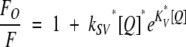 |
(1) |
where Fo is the fluorescence in the absence of quencher (acrylamide), kSV is the Stern-Volmer constant, [Q] is the concentration of quencher, and KV is a constant that takes into account static quenching (Lakowicz 1999). Static quenching is more pronounced in the denatured state, where the aromatic residues are more exposed.
Equilibrium denaturation
To take into account small variations in protein concentration in the different urea aliquots, we calculated the ratio Iλmaxnative/Iisosbestic, where Iλmaxnative is the wavelength of maximum emission in the native state (327 nm) and Iisosbestic is the wavelength at which the intensities of the native state and denatured state are the same (335 nm for p25α and 340 nm for p25αΔ3-43) and therefore can be taken as a measure of protein concentration. This ratio was plotted versus urea concentration, and the data were fitted to an equation assuming a linear dependence of the pre- and post-transition baselines on urea concentration (Pace 1986; Clarke and Fersht 1993):
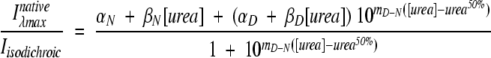 |
(2) |
where αN and αD denote the signal at 0 M urea for the native and denatured state, βN and βD are the slopes of the baselines of the native and denatured states, mD-N is the linear dependence of the log of the equilibrium denaturation constant KDN on urea, and urea50% is the urea concentration where 50% of the protein is denatured.
Kinetic analysis
The observed rate constant kobs was plotted versus urea concentration, and the data were analyzed according to a model involving an on-pathway folding intermediate (Scheme 1) (Baldwin 1996):
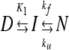 |
(Scheme 1) |
where KI=[I]/[D] whose log value depends linearly on [urea] with a slope of mI, while kf and ku are refolding and unfolding rate constants whose log-values depend linearly on [urea] with slopes of mf and mu, respectively. mf is set to zero because of the narrow denaturant concentration range over which this parameter can be determined.
Titration of tubulin with p25α and p25αΔ3-43
The fluorescence emission intensity Fexp expected from the titrated solution in the absence of interactions between tubulin and p25α or p25αΔ3-43 was calculated as follows (taking into account the effect of dilution of both tubulin and p25α or p25αΔ3-43 during the titration experiment):
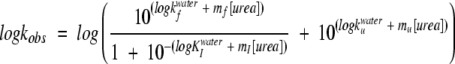 |
(3) |
where Iotubulin and Iop25 are the fluorescence intensities of the tubulin and p25α or p25αΔ3-43 stock solutions, while Vtubulin and Vp25α are the volumes of the tubulin solution (800 μL) and added p25α or p25αΔ3-43 (0–900 μL).
The difference Fexp − Fobs (where Fobs is the measured fluorescence) was plotted versus the ratio [p25α]:[tubulin] or [p25αΔ3-43]:[tubulin].
Acknowledgments
D.E.O. is supported by the Danish Technical Research Council. P.H.J. is supported by the Lundbeck Foundation and the Danish Medical Research Council (22–02–0140).
Abbreviations
ANS, 8-anilino-1-naphthalene-sulfonic acid
C, off-pathway intermediate
D, denatured state
I, on-pathway intermediate
N, native state
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041285605.
References
- Andrade, M.A., Chacón, P., Merelo, J.J., and Morán, F. 1993. Evaluation of secondary structure of proteins from UV circular dichroism using an unsupervised learning neural network. Protein Eng. 6 383–390. [DOI] [PubMed] [Google Scholar]
- Baldwin, R. 1996. On-pathway versus off-pathway folding intermediates. Fold. Des. 1 R1–R8. [DOI] [PubMed] [Google Scholar]
- Buck, M., Radford, S.E., and Dobson, C.M. 1993. A partially folded state of hen egg white lysozyme in trifluoroethanol: Structural characterization and implications for protein folding. Biochemistry 32 669–678. [DOI] [PubMed] [Google Scholar]
- Capaldi, A.P., Shastry, M.C., Kleanthous, C., Roder, H., and Radford, S.E. 2001. Ultrarapid mixing experiments reveal that Im7 folds via an on-pathway intermediate. Nat. Struct. Biol. 8 68–72. [DOI] [PubMed] [Google Scholar]
- Chiti, F., Taddei, N., Bucciantini, M., White, P., Ramponi, G., and Dobson, C.M. 2000. Mutational analysis of the propensity for amyloid formation by a globular protein. EMBO J. 19 1441–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, J. and Fersht, A.R. 1993. Engineered disulfide bonds as probes of the folding pathway of barnase: Increasing the stability of proteins against the rate of denaturation. Biochemistry 32 4322–4329. [DOI] [PubMed] [Google Scholar]
- De Prat Gay, G., Ruiz-Sanz, J., Neira, J.L., Corrales, F.J., Otzen, D.E., Ladurner, A.G., and Fersht, A.R. 1995. Conformational pathway of the polypeptide chain of CI2 growing from its N-terminus in vitro. Parallels with the protein folding pathway. J. Mol. Biol. 254 968–979. [DOI] [PubMed] [Google Scholar]
- Dunker, A.K., Lawson, J.D., Brown, C.J., Williams, R.M., Romero, P., Oh, J.S., Oldfield, C.J., Campen, A.M., Ratliff, C.M., and Hipps, K.W., et al. 2001. Intrinsically disordered protein. J. Mol. Graph. Model. 19 26–59. [DOI] [PubMed] [Google Scholar]
- Dunker, A.K., Brown, C.J., Lawson, J.D., Iakoucheva, L.M., and Obradovic, Z. 2002a. Intrinsic disorder and protein function. Biochemistry 41 6573–6580. [DOI] [PubMed] [Google Scholar]
- Dunker, A.K., Brown, C.J., and Obradovic, Z. 2002b. Identification and functions of usefully disordered proteins. Adv. Prot. Chem. 62 25–49. [DOI] [PubMed] [Google Scholar]
- Dyson, H.J. and Wright, P.E. 2002. Coupling of folding and binding for unstructured protein. Curr. Opin. Struct. Biol. 12 54–60. [DOI] [PubMed] [Google Scholar]
- Fersht, A.R. 1999. Structure and mechanism in protein science. A guide to enzyme catalysis and protein folding. Freeman & Co. New York.
- Gill, S.C. and von Hippel, P.H. 1989. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182 319–326. [DOI] [PubMed] [Google Scholar]
- Goto, Y. and Fink, A.L. 1989. Conformational states of β-lactamase molten-globule states at acidic and alkaline pH with high salt. Biochemistry 28 945–952. [DOI] [PubMed] [Google Scholar]
- Hamill, S.J., Meekhof, A.E., and Clarke, J. 1998. The effect of boundary selection on the stability and folding of the third fibronectin type III domain from human tenascin. Biochemistry 37 8071–8079. [DOI] [PubMed] [Google Scholar]
- Hlavanda, E., Kovacs, J., Olah, J., Orosz, F., Medzihradszky, K.F., and Ovadi, J. 2002. Brain-specific p25 protein binds to tubulin and microtubules and induces aberrant microtubule assemblies at substoichiometric concentrations. Biochemistry 41 8657–8664. [DOI] [PubMed] [Google Scholar]
- Jackson, S.E. and Fersht, A.R. 1991. Folding of chymotrypsin inhibitor 2. 1: Evidence for a two-state transition. Biochemistry 30 10428–10435. [DOI] [PubMed] [Google Scholar]
- Jackson, S.E., Moracci, M., elMasry, N., Johnson, C.M., and Fersht, A.R. 1993. Effect of cavity-creating mutations in the hydrophobic core of chymotrypsin inhibitor 2. Biochemistry 32 11259–11269. [DOI] [PubMed] [Google Scholar]
- Kovacs, G.G., Laszlo, L., Kovacs, J., Jensen, P.H., Lindersson, E., Boton, G., Molnar, T., Perczel, A., Hudecz, F., and Mezö, G. et al. 2004. Natively unfolded tubulin polymerization promoting protein TPPP/p25 is a common marker of α-synucleinopathies. Neurobiol. Dis. 17 155–162. [DOI] [PubMed] [Google Scholar]
- Kumar, Y., Tayyab, S., and Muzammil, S. 2004. Molten-globule like partially folded states of human serum albumin induced by fluoro and alkyl alcohols at low pH. Arch. Biochem. Biophys. 426 3–10. [DOI] [PubMed] [Google Scholar]
- Lacy, E.R., Filippov, I., Lewis, W.S., Otieno, S., Xiao, L., Weiss, S., Hengst, L., and Kriwacki, R.W. 2004. p27 binds cyclin-CDK complexes through a sequential mechanism involving binding-induced protein folding. Nat. Struct. Mol. Biol. 11 358–364. [DOI] [PubMed] [Google Scholar]
- Lakowicz, J.R. 1999. Principles of fluorescence spectroscopy, 2nd ed. Kluwer Academic/Plenum Publishers, New York.
- Lee, L.K., Stollar, E., Chang, J.G., Grossman, R., O’Brien, J., Ladbury, B., Carpenter, S., Roberts, S., and Luisi, B. 2001. Expression of the Oct-1 transcription factor and characterization of its interactions with the Bob1 coactivator. Biochemistry 40 6580–6588. [DOI] [PubMed] [Google Scholar]
- Lindersson, E., Lundvig, D., Petersen, C., Madsen, P., Højrup, P., Moos, T., Otzen, D.E., Gai, W.-P., and Jensen, P.H. 2005. P25a is co-expressed with α-synuclein in α-synucleinopathies and stimulates its aggregation. J. Biol. Chem. 280 5703–5715. [DOI] [PubMed] [Google Scholar]
- Martin, C.P., Vazquez, J., Avila, J., and Moreno, F.J. 2002. P24, a glycogen synthase kinase 3 (GSK 3) inhibitor. Biochim. Biophys. Acta 1586 113–122. [DOI] [PubMed] [Google Scholar]
- Matouschek, A., Kellis, J.T., Serrano, L., Bycroft, M., and Fersht, A.R. 1990. Transient folding intermediates characterized by protein engineering. Nature 346 440–445. [DOI] [PubMed] [Google Scholar]
- Matouschek, A., Serrano, L., and Fersht, A.R. 1992. The folding of an enzyme IV. Structure of the intermediate in the refolding of barnase analysed by a protein engineering procedure. J. Mol. Biol. 224 819–835. [DOI] [PubMed] [Google Scholar]
- Mogensen, J.E., Ibsen, H., Lund, J., and Otzen, D.E. 2004. Elimination of an off-pathway folding intermediate by a single point mutation. Biochemistry 43 3357–3367. [DOI] [PubMed] [Google Scholar]
- Muro-Pastor, M.I., Barrera, F.N., Reyes, J.C., Florencio, F.J., and Neira, J.L. 2003. The inactivating factor of glutamine synthetase, IF7, is a “natively unfolded” protein. Protein Sci. 12 1443–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nölting, B., Golbik, R., Neira, J.L., Soler-Gonzalez, A.S., Schreiber, G., and Fersht, A.R. 1997. The folding pathway of a protein at high resolution from microseconds to seconds. Proc. Natl. Acad. Sci. 94 826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orosz, F., Kovacs, G.G., Lehotzky, A., Olah, J., Vincze, O., and Ovadi, J. 2004. TPPP/p25: From unfolded protein to misfolding disease: Prediction and experiments. Biol. Cell 96 701–711. [DOI] [PubMed] [Google Scholar]
- Otzen, D.E. and Oliveberg, M. 1999. Salt-induced detour through compact regions of the protein folding landscape. Proc. Nat. Acad. Sci. 96 11746–11751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace, C.N. 1986. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 131 266–279. [DOI] [PubMed] [Google Scholar]
- ———. 1990. Conformational stability of globular proteins. Trends Biochem. Sci. 15 14–17. [DOI] [PubMed] [Google Scholar]
- Parker, M.J., Spencer, J., and Clarke, A.R. 1995. An integrated kinetic analysis of intermediates and transition states in protein folding reactions. J. Mol. Biol. 253 771–786. [DOI] [PubMed] [Google Scholar]
- Parker, M.J., Dempsey, C.E., Hosszu, L.L.P., Waltho, J.P., and Clarke, A.R. 1998. Topology, sequence evolution and folding dynamics of an immunoglobulin domain. Nat. Struct. Biol. 5 194–198. [DOI] [PubMed] [Google Scholar]
- Seki, N., Hattori, N., Sugano, S., Suzuki, Y., Nakagawara, A., Muramatsu, M., Hori, T., and Saito, T. 1999. A novel human gene whose product shares significant homology with the bovine brain-specific protein p25 on chromosome 5p15.3. J. Hum. Genet. 44 121–122. [DOI] [PubMed] [Google Scholar]
- Semisotnov, G.V., Rodinova, N.A., Razgulyaev, O.I., Uversky, V.N., Gripas, A.F., and Gilmanshin, R.I. 1991. Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers 31 119–128. [DOI] [PubMed] [Google Scholar]
- Shiratsuchi, A., Sato, S., Oomori, A., Ishiguro, K., Uchida, T., and Imahori, K. 1995. cDNA cloning of a novel brain-specific protein p25. Biochim. Biophys. Acta 1251 66–68. [DOI] [PubMed] [Google Scholar]
- Shoemaker, B.A., Portman, J.J., and Wolynes, P.G. 2000. Speeding molecular recognition by using the folding funnel: The fly-casting mechanism. Proc. Natl. Acad. Sci. 97 8868–8873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, M., Tomizawa, K., Ishiguro, K., Sato, K., Omori, A., Sato, S., Shiratsuchi, A., Uchida, T., and Imahori, K. 1991. A novel brain-specific 25 kDa protein (p25) is phosphorylated by a Ser-Thr-Pro kinase (Tpk-Ii) from tau protein-kinase fractions. FEBS Lett. 289 37–43. [DOI] [PubMed] [Google Scholar]
- Tanford, C. 1970. Protein denaturation. Part C. Theoretical models for the mechanism of denaturation. Adv. Prot. Chem. 24 1–95. [PubMed] [Google Scholar]
- Timasheff, S. 2002. Protein hydration, thermodynamic binding, and preferential hydration. Biochemistry 41 13473–13482. [DOI] [PubMed] [Google Scholar]
- Tirian, L., Hlavanda, E., Olah, J., Horvath, I., Orosz, F., Szabo, B., Kovacs, J., Szabad, J., and Ovadi, J. 2003. TPPP/p25 promotes tubulin assemblies and blocks mitotic spindle formation. Proc. Natl. Acad. Sci. 100 13976–13981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa, P. 2002. Intrinsically unstructured proteins. Trends Biochem. Sci. 27 527–533. [DOI] [PubMed] [Google Scholar]
- Tsai, C.-J., De Laureto, P.P., Fontana, A., and Nussinov, R. 2002. Comparison of protein fragments identified by limited proteolysis and by computational cutting of proteins. Protein Sci. 11 1753–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky, V.N. 2003. A protein-chameleon: Conformational plasticity of α-synuclein, a disordered protein involved in neurodegenerative disorders. J. Biomol. Struct. Dyn. 21 211–234. [DOI] [PubMed] [Google Scholar]
- Uversky, V.N. and Narizhneva, N.V. 1998. Effect of natural ligands on the structural properties and conformational stability of proteins. Biochemistry (Moscow) 63 420–433. [PubMed] [Google Scholar]
- Uversky, V.N., Abdullaev, Z.K., Arseniev, A.S., Bocharov, E.V., Dolgikh, D.A., Latypov, R.F., Melnik, T.N., Vassilenko, K.S., and Kirpichnikov, M.P. 1999. Structure and stability of recombinant protein depend on the extra N-terminal methionine residue: S6 permutein from direct and fusion expression systems. Biochim. Biophys. Acta 1432 324–332. [DOI] [PubMed] [Google Scholar]
- Uversky, V.N., Gillespie, J.R., and Fink, A.L. 2000. Whyare “natively unfolded” proteins unstructured under physiologic conditions? Proteins 41 415–427. [DOI] [PubMed] [Google Scholar]
- Wolff, J. 1998. Promotion of microtubule assembly by oligocations: Cooperativity between charged groups. Biochemistry 37 10722–10729. [DOI] [PubMed] [Google Scholar]
- Yokozeki, T., Homma, K., Kuroda, S., Kikkawa, U., Ohno, S., Takahashi, M., Imahori, K., and Kanaho, Y. 1998. Phosphatidic acid-dependent phosphorylation of a 29-kDa protein by protein kinase C α in bovine brain cytosol. J. Neurochem. 71 410–417. [DOI] [PubMed] [Google Scholar]
- Zhang, Z., Wu, C.C., Huang, W.-N., Wang, S., Zhao, E., Huang, Q., Xie, Y., and Mao, Y. 2002. A novel human gene whose product shares homology with bovine brain-specific protein p25 is expressed in fetal brain but not in adult brain. J. Hum. Genet. 47 266–268. [DOI] [PubMed] [Google Scholar]