

Abstract
Chemokines, like stromal cell-derived factor-1 (SDF1/CXCL12), are small secreted proteins that signal cells to migrate. Because SDF1 and its receptor CXCR4 play important roles in embryonic development, cancer metastasis, and HIV/AIDS, this chemokine signaling system is the subject of intense study. However, it is not known whether the monomeric or dimeric structure of SDF1 is responsible for signaling in vivo. Previous structural studies portrayed the SDF1 structure as either strictly monomeric in solution or dimeric when crystallized. Here, we report two-dimensional NMR, pulsed-field gradient diffusion and fluorescence polarization measurements at various SDF1 concentrations, solution conditions, and pH. These results demonstrate that SDF1 can form a dimeric structure in solution, but only at nonacidic pH when stabilizing counterions are present. Thus, while the previous NMR structural studies were performed under acidic conditions that strongly promote the monomeric state, crystallographic studies used nonacidic buffer conditions that included divalent anions shown here to promote dimerization. This pH-sensitive aggregation behavior is explained by a dense cluster of positively charged residues at the SDF1 dimer interface that includes a histidine side chain at its center. A heparin disaccharide shifts the SDF1 monomer–dimer equilibrium in the same manner as other stabilizing anions, suggesting that glycosaminoglycan binding may be coupled to SDF1 dimerization in vivo.
Keywords: chemokines, heparin, NMR, fluorescence polarization, monomer–dimer equilibrium
Chemokines are small, secreted proteins that induce cell migration through activation of G protein-coupled receptors (GPCR), and also bind extracellular matrix glycosaminoglycans (GAG) in order to direct chemotaxis along a gradient of increasing chemokine concentration. Chemokines are categorized into four families, C, CC, CXC, and CX3C, based on the arrangement of conserved cysteine residues near the amino terminus. The well-characterized chemokine tertiary fold consists of a flexible amino terminus, followed by a three-stranded anti-parallel β-sheet and a carboxyl terminal α-helix. Chemokine quaternary structure is more varied; some chemokines are constitutively monomeric, whereas others self-associate to form homodimers mediated by residues of the amino terminus (CC chemokines) or the first β-strand (CXC chemokines). Dimerization was initially thought to be an artifact of the high concentrations necessary for structural studies, since chemokines are fully functional in chemotaxis and calcium flux assays at low nanomolar concentrations where the monomeric species should predominate (Rajarathnam et al. 1994, 1995; Paavola et al. 1998). Moreover, disruption of the IL-8 dimer through mutagenesis does not alter its properties as a receptor agonist in vitro (Rajarathnam et al. 1994, 1995). In contrast, a recent study showed that dimerization is critical for the in vivo leukocyte recruitment activity of three dimeric chemokines: MCP-1, RANTES, and MIP-1β (Proudfoot et al. 2003a). Those results suggest that GAG binding, GPCR activation, and self-association are all essential functional interactions for at least a subset of the ~50 known chemokines.
The CXC chemokine stromal cell-derived factor-1 (SDF1/CXCL12) and its cognate receptor CXCR4 normally function in leukocyte trafficking, hematopoiesis, and proper vertebrate fetal development (Nagasawa et al. 1996; Tachibana et al. 1998; Zou et al. 1998; Moepps et al. 2000; Murdoch 2000; Rossi and Zlotnik 2000; Doitsidou et al. 2002; Ara et al. 2003; Knaut et al. 2003a; Kunwar and Lehmann 2003; Molyneaux et al. 2003; Proudfoot et al. 2003b). While most chemokine and chemokine receptor gene knockouts result in no apparent phenotype, SDF1−/−and CXCR4−/− mice display an embryonic lethal phenotype with defects involving B-cell lymphopoiesis, bone marrow myelopoiesis, vascularization of the gastrointestinal tract, cardiac ventral septum formation, and cerebellar development (Nagasawa et al. 1996; Tachibana et al. 1998; Zou et al. 1998). SDF1–CXCR4 signaling in zebrafish and mice is also critical for the colocalization of primordial germ cells into gonads (Doitsidou et al. 2002; Ara et al. 2003; Knaut et al. 2003b; Kunwar and Lehmann 2003; Molyneaux et al. 2003). In addition to their roles in normal development and homeostasis, SDF1 and its receptor participate in the pathology of cancer and HIV/AIDS. SDF1 and CXCR4 direct the migration of metastatic breast cancer cells to specific tissues (Muller et al. 2001; Helbig et al. 2003), and this homing mechanism has been implicated in other cancers as well. CXCR4 serves as a coreceptor for entry of X4 HIV-1 strains into T cells, and SDF1 can inhibit this process by blocking gp120 binding to CXCR4, thus preventing the subsequent gp41-mediated membrane fusion event (Bleul et al. 1996; D’Souza et al. 2000).
SDF1 adopts the conserved chemokine tertiary fold, but it is unclear whether its functional form is monomeric or dimeric. The quaternary structure of SDF1 has been described in previous studies by a monomeric NMR structure determined at pH 4.9 (Crump et al. 1997), two dimeric crystal structures obtained in the presence of 1.6 or 1.9 M ammonium sulfate at pH 7.0 and 8.5 (Dealwis et al. 1998; Ohnishi et al. 2000), and an analytical ultracentrifugation study in a phosphate buffer at pH 7.4 revealing a monomer–dimer equilibrium (Holmes et al. 2001). Taken together, these results suggest that solution conditions may affect the oligomeric state of SDF1, similar perhaps to the pH- and salt-dependent aggregation and conformational properties observed for other chemokines (Skelton et al. 1995; Lowman et al. 1997; Laurence et al. 1998; Kuloglu et al. 2002).
Because SDF1 and CXCR4 play important roles in normal physiology and human disease states, we investigated the effect of solution conditions on the quaternary structure of SDF1. Our results show that SDF1 exists in a monomer–dimer equilibrium only under certain conditions. In particular, we found that acidic pH promotes the monomeric state by destabilizing the dimeric structure, while physiological pH and anions, including phosphate, sulfate, and citrate, shift this equilibrium toward the dimeric state. Basic residues of the dimer interface that control SDF1 oligomerization have previously been implicated in GAG binding (Amara et al. 1999; Mbemba et al. 2000; Sadir et al. 2001). We therefore examined the influence of sulfated GAGs on the SDF1 monomer–dimer equilibrium using a heparin disaccharide. Our results suggest that, at physiological pH, heparin binding promotes SDF1 dimer formation. As demonstrated for the chemokines RANTES, MIP-1β, and MCP-1 (Proudfoot et al. 2003a), SDF1α self-association may be essential to its ability to chemoattract cells in vivo.
Results
Production of recombinant SDF1α
Multiple SDF1 isoforms are produced in vivo through alternative pre-mRNA splicing, including the α and β variants, which differ only in the carboxyl-terminal extension of the β isoform by four amino acids. We produced both SDF1α andβ, but our experiments revealed no distinguishing structural or biochemical features. Because no functional differences have been reported for the two species, we describe only results obtained for the SDF1α isoform.
Previous structural studies of SDF1α have typically relied on chemically synthesized protein, precluding the efficient incorporation of stable isotopes for multinuclear NMR studies. Recombinant SDF1α was isolated from the insoluble fraction of bacterial cell cultures, purified by affinity chromatography, and refolded, yielding biologically active chemokine in a transwell chemotaxis assay (data not shown). With 15N/13C-labeled SDF1α produced in the same manner, we assigned its 1H, 13C, and 15N chemical shifts using standard triple-resonance methods (Fig. 1A ▶).
Figure 1.

SDF1α exists in a monomer–dimer equilibrium. (A) 2D 15N-1H HSQC spectrum of SDF1α in 20 mM sodium phosphate at pH 6.0, with residue assignments indicated. (B) A subset of signals in the 2D 15N-1H HSQC spectrum shift as the protein concentration is varied from 1.2 mM (green) to 10 μM (red). (C) Combined 1H/15N chemical shift perturbations for each SDF1α residue upon dilution from 1.2 mM to 10 μM. No values are plotted for residues 2, 10, 32, and 53 (prolines) or Leu 26 (not observed). (D) Residues with the largest 1H/15N chemical shift perturbations (>0.4) are highlighted in red on the surface of one monomer from the dimeric crystal structure (PDB code 1QG7). (E) Residues participating in the SDF1αintermonomer interface are highlighted on the structure as in D.
SDF1α monomer–dimer equilibrium
SDF1α has been previously described as a monomer (Crump et al. 1997), as a dimer (Dealwis et al. 1998; Ohnishi et al. 2000), and in a monomer–dimer equilibrium (Holmes et al. 2001). During the chemical shift assignment process, we noticed unusual line broadening for residues K24, H25, L26, K27, and L66 in the 15N-1H HSQC spectrum acquired in 20 mM sodium phosphate buffer at pH 6.0. To further investigate the oligomeric state of SDF1α we used 15N-1H HSQC spectra to observe the incremental dilution of SDF1α from 1.2 mM to 0.010 mM in 20 mM sodium phosphate at pH 6.0. As seen in Figure 1B ▶, the signals for some residues shift upon dilution while others remain constant. The combined 15N and 1H chemical shift perturbations shown in Figure 1C ▶ provide a sensitive means of monitoring changes in the chemical environment for each amino acid in SDF1α. Residues that shift significantly as a function of protein concentration cluster in the β1-strand and the carboxyl terminal end of the α-helix (Fig. 1D ▶), both of which contribute to the dimer interface observed in the SDF1α crystal structures (Fig. 1E ▶) (Dealwis et al. 1998; Ohnishi et al. 2000). The strong correlation between concentration-dependent chemical shift perturbations and residues of the dimer interface suggests that SDF1α exists in a monomer–dimer equilibrium. Interestingly, a very similar pattern of 1H/15N shift perturbations accompanied the titration of pH from 8.0 to 6.0, leading us to consider the effect of pH on the SDF1α monomer–dimer equilibrium, as discussed below.
In conjunction with HSQC dilution and titration experiments, we also monitored SDF1α aggregation state by pulse field gradient (PFG) self-diffusion measurements. PFG diffusion experiments work by effectively recording the average distance traveled by solute molecules in the NMR sample during a fixed diffusion period. An exponential decrease in signal intensities is observed for spectra acquired with increasingly intense gradient pulses bracketing the diffusion delay. More rapidly decaying signals reflect increased values of Ds, the self-diffusion coefficient, corresponding to lower apparent molecular weight. We observed that the signal intensities for samples of 250 μM SDF1α at pH 7.4 decayed at different rates depending on whether 20 mM phosphate or 20 mM MES was the buffer, corresponding to Ds values of 1.16 × 10−6 cm2sec−1 or 1.49 × 10−6 cm2sec−1, respectively (Fig. 2A ▶). From Ds values measured over a range of SDF1α concentrations, we obtained Kd values of 120 ± 80 μM in sodium phosphate and 1 ± 1 mM in MES for a monomer–dimer equilibrium (Fig. 2B ▶). While the Kd determination in MES buffer is imprecise, three-parameter fits to the MES and phosphate data yielded the same endpoint Ds values for pure monomer (1.66 × 10−6 cm2sec−1) and pure dimer (1.0 × 10−6 cm2sec−1). Thus, we conclude that both experiments monitored the same concentration-dependent equilibrium, though with apparently different dissociation constants.
Figure 2.

SDF1α dimer formation requires phosphate or sulfate. (A) Differences in translational self-diffusion coefficients measured for SDF1α (250 μM) at pH 7.4 suggest that SDF1α oligomerization is favored in the presence of 20 mM phosphate relative to 20 mM MES. Peak intensities from a series of 1D 1H spectra acquired in phosphate (♦, upper curve) or MES (○, lower curve) buffer using a longitudinal encode-decode pulsed field gradient diffusion pulse scheme (diffusion delay Δ = 80 msec; gradient pulse Δ = 5 msec) were analyzed by nonlinear fitting to Equation 1 (solid lines) to obtain values for the self-diffusion coefficient, Ds (Altieri et al. 1995). Every fifth data point has been enlarged for clarity. (B) Variations in Ds as a function of protein concentration were fit to a function describing a monomer–dimer equilibrium, revealing a ~10-fold change in the Kd for dimer dissociation depending on the presence (•) (Kd = 120 ± 80 μM) or absence (○) (Kd = 1000 ± 1000 μM) of phosphate. (C) Fluorescence polarization (FP) measurements over a range of SDF1α concentrations at pH 7.4 in the presence (▪) or absence (□) of 100 mM sodium phosphate were analyzed by nonlinear fitting to Equation 5. An equilibrium dissociation constant for the SDF1α dimer of 140 ± 19 μM was obtained in 100 mM sodium phosphate. In contrast, FP values for SDF1α measured in 100 mM HEPES at pH 7.4 vary only slightly, consistent with a dimer Kd > 10,000 μM.
Negative counterions promote SDF1α dimer formation
To more precisely determine the effects of pH and buffer components on the SDF1α dimerization, we measured Kd values under various solution conditions using fluorescence polarization (FP) of the single tryptophan residue. FP values recorded at SDF1α concentrations ranging from 0.01 to 1 mM were analyzed by nonlinear fitting to a model for monomer–dimer equilibrium (Equation 5). In samples containing only HEPES buffer at pH 7.4, minimal variation in FP values was observed over this SDF1α concentration range, as shown in Figure 2C ▶, consistent with a Kd value estimated at >10 mM. However, the Kd in 100 mM sodium phosphate at pH 7.4 from FP measurements was 140 × 19 μM (Fig. 2C ▶), similar to the result from PFG diffusion (120 μM) and previously published Kd values of 180 and 130 μM derived from analytical ultracentrifugation and dynamic light scattering measurements, respectively (Holmes et al. 2001). Additional FP measurements summarized in Table 1 reveal a similar stabilizing effect on the SDF1α dimer by the multivalent anions sulfate and citrate. Since the monomer–dimer equilibrium is virtually undetectable in a solution containing only HEPES buffer, multivalent anions such as phosphate, sulfate, or citrate therefore seem to be essential for SDF1 dimer formation.
Table 1.
SDF1 dimer dissociation Kdvalues (μM) determined by fluorescence polarization
| SDF1α variant | pH 7.4 100 mM PO4 | pH 7.4HEPES | pH 7.4 HEPES 100 mM SO4 | pH 5.5 MES 100 mM SO4 | pH 7.4 HEPES 100 mM citrate | pH 7.4 HEPES 5 mM I-S heparin |
| WT | 140 ± 19 | >10,000 | 261 ± 65 | 1638 ± 387 | 103 ± 29 | 172 ± 29 |
| H25R | 1522 ± 226 | 1793 ± 492 | 2063 ± 440 | |||
| H25L | 268 ± 44 | 617 ± 170 | 740 ± 160 | |||
| H25A | 820 ± 248 | 2295 ± 857 | 2661 ± 628 |
The SDF1α monomer–dimer equilibrium is sensitive to pH
Based on the similar patterns of HSQC shift perturbations observed as a function of protein concentration and pH, we used FP to determine SDF1α dimer Kd values in 20 mM HEPES buffer at pH 7.4 and in 20 mM MES buffer at pH 5.5. Since the monomer–dimer equilibrium is apparent only when a negative counterion is present, sodium sulfate (100 mM) was included in each sample instead of phosphate, since the sulfate anion remains divalent from pH 5.5 to 7.4. An obvious difference in the monomer–dimer equilibrium is apparent from the FP values (Fig. 3A ▶), with a change in SDF1α dimer Kd from 261 ± 65 μM at pH 7.4 to 1.6 ± 0.4 mM at pH 5.5. These results confirmed that, like PO42− and other suitable counterions, pH also affects the SDF1α monomer–dimer equilibrium.
Figure 3.
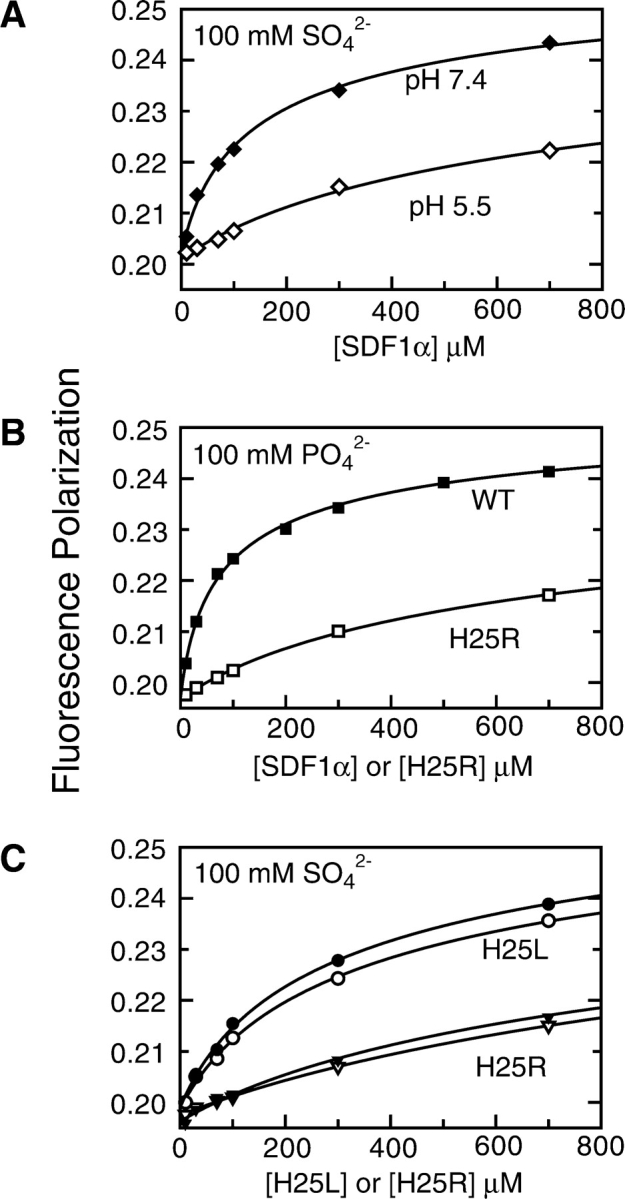
Titration of His 25 gives rise to the pH sensitivity of the SDF1α monomer–dimer equilibrium. Dimer dissociation Kd values were obtained by measuring fluorescence polarization (FP) as a function of wild type, H25R, and H25L SDF1α protein concentration and nonlinear fitting to Equation 5. (A) The Kd for SDF1α dimer dissociation rises from 261 ± 65 μM (♦) (100 mM HEPES [pH 7.4], 100 mM sodium sulfate) to 1.6 ± 0.4 mM (⋄) (100 mM MES [pH 5.5], 100 mM sodium sulfate), demonstrating that the monomer–dimer equilibrium is pH sensitive. Sulfate was used instead of phosphate because its ionization state is unchanged from pH 5.5–7.4. (B) Substitution of His 25 with Arg, which remains positively charged at pH 7.4, destabilizes the SDF1α dimer. In 100 mM sodium phosphate (pH 7.4), the Kd for H25R (□) is 1.5 ± 0.2 mM, compared to 140 ± 19 μM for wild-type SDF1α (▪). (C) Substitution of His 25 with Leu, an uncharged side chain, disrupts SDF1α dimerization only slightly, and eliminates the pH sensitivity. In 100 mM sodium sulfate the dimer Kd for H25L is 617 ± 170 μM at pH 7.4 (•) and 740 ± 160 μM at pH 5.5 (○). The Kd value for H25R in 100 mM sodium sulfate is 1.7 ± 0.5 mM at pH 7.4 (▾) and 2.0 ± 0.4 mM at pH 5.5 (▿).
Protonation state of His25 governs the SDF1α monomer–dimer equilibrium
As in other dimeric CXC chemokines, SDF1α self-associates by joining the β1-strands of two monomers to form a single six-stranded anti-parallel sheet. The β1-strand of SDF1α contains a number of basic amino acids (V23KHLKIL29). These positively charged amino acids are clustered in the SDF1α structure (Dealwis et al. 1998; Ohnishi et al. 2000), with the His25 side chain from one subunit positioned within 3.8 Å of the Lys27 side chain of the opposing subunit, as illustrated in Figure 4 ▶. We hypothesized that, at pH values below the pKa of His25, electrostatic repulsion between the positively charged histidine and lysine side chains would disfavor SDF1α dimerization. Higher pH would diminish this destabilizing effect by reducing the number of positively charged side chains at the intermolecular interface from six to four.
Figure 4.

Electrostatic disruption and stabilization of the SDF1α dimer. Crystal structure of SDF1α with basic residues at the dimer interface highlighted (PDB 1QG7). The side chains of K24 (green), H25 (red), and K27 (blue) are shown. The dashed line highlights the dimer interface, and the close proximity (3.8 Å) of positively charged H25 and K27 side chains from different monomers.
To test this hypothesis, we replaced His25 with a series of different amino acids by site-directed mutagenesis. We verified that each SDF1α variant was correctly folded by 2D 15N-1H HSQC or 1D 1H NMR (data not shown), and measured the dimer Kd under various solution conditions and pH values, as summarized in Table 1. As reflected in Figure 3B ▶, the dimer Kd measured at pH 7.4 with 100 mM phosphate for the H25R variant (1.5 ± 0.2 mM) is an order of magnitude higher than the wild-type SDF1α Kd in the same buffer conditions (140 ± 19 μM). As expected, the monomer–dimer equilibrium of the H25R variant, which remains fully charged at pH 5.5–7.4, displays essentially no pH dependence (Fig. 3C ▶). Moreover, the Kd values measured in the presence of 100 mM sulfate for H25R at pH 7.4 (1.8 ± 0.5 mM) and pH 5.5 (2.0 ± 0.4 mM) are roughly equal to the Kd of wild-type SDF1α at pH 5.5 (1.6 ± 0.4 mM). Thus, the H25R variant mimics the dimerization behavior of wild-type SDF1α with a protonated His25.
In a similar fashion, we determined the dimer dissociation Kd values for the H25L and H25A variants of SDF1α (Table 1). In the case of the H25L variant, the Kd measured at pH 7.4 in 100 mM phosphate is less than a factor of 2, different from that of the wild-type protein, suggesting that the dimer interface remains essentially intact. The H25A substitution perturbs the dimer Kd more dramatically and likely disrupts important contact surfaces. However, as in the case of H25R, substitution of the titratable His side chain with either Leu or Ala eliminates the pH dependence of dimerization (Fig. 3C ▶). On this basis, we conclude that titration of H25 in wild-type SDF1α gives rise to the pH sensitivity of the monomer–dimer equilibrium.
I-S heparin promotes SDF1α dimer formation
Like most chemokines, SDF1α binds glycosaminoglycans, highly sulfated oligosaccharide components of the extracellular matrix (Amara et al. 1999; Mbemba et al. 2000; Sadir et al. 2001), and these interactions are essential to the in vivo function of some inflammatory chemokines (Proudfoot et al. 2003a). Recent studies by McCornack et al. (2003) evaluated the binding of heparin disaccharides to the CC chemokine MIP-1β and showed that these small glycosaminoglycan fragments alter the monomer–dimer equilibrium of MIP-1β by stabilizing the dimer species (McCornack et al. 2004). Based on these and other reports linking GAG binding and chemokine dimerization, as well as our results demonstrating the impact of inorganic sulfate on SDF1α oligomerization, we tested the ability of a commercially available heparin fragment to shift the monomer–dimer equilibrium. We used FP to measure the SDF1α dimer Kd at pH 7.4 in HEPES buffer in the presence and absence of 5 mM I-S heparin disaccharide as shown in Figure 5 ▶. In these buffer conditions there is no measurable SDF1α dimerization in the absence of a stabilizing counterion (Table 1), but when I-S heparin disaccharide is added, the apparent Kd for dimer dissociation is 172 ± 29 μM. Binding of I-S heparin shifts the SDF1α monomer–dimer equilibrium toward dimer in a manner very similar to that observed for phosphate, sulfate, and citrate but at much lower concentration of heparin (Table 1). This suggests that extracellular matrix GAGs may lower the dimer dissociation Kd and promote SDF1α oligomerization in vivo.
Figure 5.

(Top) I-S heparin disaccharide stabilizes the SDF1α dimer. (Bottom) SDF1 self-association monitored by FP in the presence of 5 mM I-S heparin disaccharide (♦) revealed a dimer Kd of 172 ± 29 μM. In the absence of heparin (⋄), SDF1α is essentially monomeric in these buffer conditions (HEPES pH 7.4).
To better understand the functional link between dimerization and GAG binding, we had hoped to identify residues of SDF1α that interact with phosphate, sulfate, or heparin by chemical shift mapping. However, we were unable to define specific counterion binding sites on the SDF1α dimer, due to the complexity of 15N-1H HSQC spectra acquired in the presence of these stabilizing ligands. Since the monomer–dimer equilibrium is also altered, changes in the SDF1α NMR spectrum upon the addition of phosphate or sulfate may be due to either protein–protein or protein–ligand interactions. Another factor complicating the analysis of the NMR spectra is the potential for exchange between degenerate ligand binding sites, which may generate multiple signals for the same residue or cause significant line broadening. Despite these limitations, changes in the NMR spectra of SDF1α acquired with increasing amounts of either phosphate, sulfate, or heparin disaccharide showed clear evidence of the altered monomer–dimer equilibrium predicted from Kd values determined by FP.
For example, the HSQC spectrum of 250 μM SDF1α in pH 6.8 MES buffer shown in Figure 6A ▶ is consistent with the presence of a homogeneous monomeric species as predicted by the FP results (dimer Kd > 10 mM). With the addition of 1 mM I-S heparin disaccharide, a large number of new, broader peaks appeared in the HSQC spectrum (Fig. 6B ▶). The SDF1α dimer Kd is ~350 μM under these conditions based on FP analysis (data not shown), so the protein should be nearly equally distributed between the monomer and dimer species, consistent with the intensity ratios of old and new HSQC peaks. As the protein concentration was reduced to 10 μM in the presence of 1 mM I-S heparin, the HSQC spectra simplified to a pattern consistent with a monomeric state (Fig. 6C ▶). Thus, we concluded that the addition of heparin promoted SDF1α dimerization, with new signals in Figure 6B ▶ corresponding to the dimeric SDF1α species in complex with heparin, and as the protein concentration was reduced substantially below the measured dimer Kd, the dimeric SDF1α–heparin complex dissociated, returning to a homogeneous monomeric state.
Figure 6.

Heparin binds preferentially to the SDF1α dimer. (A) 15N-1H HSQC spectrum of 250 μM SDF1α in 25 mM MES (pH 6.8). (B) 15N-1H HSQC spectrum of 250 μM SDF1α in 25 mM MES (pH 6.8) with 1 mM I-S heparin disaccharide. (C) 15N-1H HSQC spectrum of 10 μM SDF1α in 25 mM MES (pH 6.8) with 1 mM I-S heparin disaccharide.
Discussion
Our overall goal is a structural understanding of all the intermolecular interactions required for SDF1α activity in vivo. High-affinity chemokine binding and activation of a specific GPCR is obviously essential to induce a chemotactic response in target cells. In addition, most chemokines bind cell-surface GAG, and many CC and CXC chemokines adopt conserved homodimeric arrangements. However, until the recent work by Proudfoot et al. (2003a) showing that GAG binding and dimerization are necessary for in vivo leukocyte recruitment by MCP-1, RANTES, and MIP-1β, the functional relevance of these common chemokine binding interactions had been uncertain. In this study, we focused initially on the question of whether the CXC chemokine SDF1α is dimeric in solution, since previous reports appeared to disagree on this aspect of its structure (Crump et al. 1997; Dealwis et al. 1998; Ohnishi et al. 2000; Holmes et al. 2001).
Factors controlling the SDF1α oligomeric state
We showed that SDF1α exists in a monomer–dimer equilibrium, but that the dimer dissociation Kd is highly dependent on both the solution pH and the presence of stabilizing counterions. Specifically, for SDF1α dimerization to occur, multivalent anions like phosphate, sulfate, citrate, or heparin must be present and the pH must be above the presumed pKa of His25, a residue positioned at the dimer interface. Importantly, our results completely reconcile the disparate conclusions reached in previous studies of SDF1α structure. NMR structural studies detected only the monomeric SDF1α species at pH 4.9 in acetate buffer (Crump et al. 1997), solution conditions in which SDF1α will not self-associate since neither of the requirements described above is satisfied. In contrast, both X-ray structures of SDF1α revealed a dimeric arrangement similar to other CXC chemokines (Dealwis et al. 1998; Ohnishi et al. 2000), but in each case the protein was crystallized in buffers at or above pH 7.0 in the presence of 1.5–2 M ammonium sulfate. Thus, both the pH and counterion requirements were met, and we would expect dimer formation to occur. Finally, the SDF1α monomer–dimer equilibrium characterized previously by analytical ultracentrifugation and dynamic light scattering with a dimer Kd of 150 ± 30 μM at pH 7.4 in phosphate buffer (Holmes et al. 2001) agrees with the Kd of 140 ± 19 μM that we determined in similar buffer conditions by FP.
The pH dependence of the SDF1α monomer–dimer equilibrium was rationalized in terms of the charge of the His25 side chain, which is paired with the positively charged Lys27 side chain of the opposing subunit. We confirmed this hypothesis by replacing His25 with a series of other amino acids, each of which eliminated the effect of pH on dimerization. At low pH, protonation of the His25 side chain creates two unfavorable ion pairs at the dimer interface (Fig. 4 ▶). This increase in positive charge when the pH is below the pKa of His25 is hypothesized to result in electrostatic repulsion between the two subunits and disruption of the SDF1α dimer. A comparison with other CXC chemokines revealed that His is found at this sequence position only in SDF1α. While pH-dependent aggregation behavior has been reported for other chemokines, such as RANTES (Skelton et al. 1995), this typically involves high order aggregation and precipitation at near-neutral pH, and low pH (< 4) is required to preserve a homogeneous, nonaggregated species in solution, which remains dimeric irrespective of pH. Thus, the strong dependence of dimerization on pH values near the physiological range observed for SDF1α seems unique among the chemokine family.
Functional role of chemokine dimerization
Even though dimerization is an essential element of in vivo activity for RANTES, MIP-1β, and MCP-1 (Proudfoot et al. 2003a), in the absence of other factors, each of these proteins will be completely monomeric at concentrations of 1–10 nM, and the same is true of SDF1α. The Kd values of ~100–250 μM observed in the presence of 100 mM phosphate, sulfate, or citrate were the lowest we observed for SDF1α, and increased concentrations of counterions failed to provide further dimer stabilization. Thus, under optimal solution conditions SDF1α self-association remains a low-affinity interaction, and weak in comparison to other dimeric chemokines. For example, interleukin-8 (Burrows et al. 1994; Lowman et al. 1997), MIP-1β (Laurence et al. 2000), and MCP-1 (Lau et al. 2004), typically dimerize with Kd values in the 0.1–10 μM range. However, SDF1α dimerization may still be important for its activity in vivo. Chemokines bind and activate their GPCR targets in vitro at concentrations where the monomeric species must predominate, suggesting that dimerization is not required to induce GPCR signaling. Thus, to be involved in chemokine signaling at concentrations below the dimer Kd, oligomerization must be functionally coupled to another intermolecular interaction, like GAG binding.
SDF1α dimerization is coupled to GAG binding
Chemokine oligomerization and GAG binding have been linked in numerous biochemical and functional studies of other CXC and CC chemokines. Heparin binding induces the formation of MCP-1 tetramers (Lau et al. 2004), as was previously suggested for MCP-1, RANTES, and interleukin-8 (Hoogewerf et al. 1997). NMR studies of the dimeric CC chemokine MIP-1β and a monomeric MIP-1β variant show that heparin disaccharides bind preferentially to the dimeric protein (McCornack et al. 2003) and shift the MIP-1β monomer–dimer equilibrium toward dimer (McCornack et al. 2004). Disruption of either chemokine dimer formation or GAG binding abrogates the in vivo recruitment of cells by RANTES, MIP-1β, and MCP-1 (Proudfoot et al. 2003a). These interactions seem to be functionally coupled, given that a nonheparin binding variant of RANTES inhibits the activity of endogenous RANTES in vivo by disrupting GAG-associated chemokine multimers (Johnson et al. 2004). Because binding of heparin disaccharide stabilizes the dimeric structure (Fig. 5 ▶), we hypothesize that SDF1α self-association may be similarly essential for its in vivo activity.
Interestingly, in the presence of heparin SDF1α becomes a more potent inhibitor of CXCR4-mediated T cell entry by X4 strains of HIV-1 (Valenzuela-Fernandez et al. 2001). In the context of the present results, this may suggest that dimerization improves the ability of SDF1α to serve as an antagonist of HIV-1 gp120. A complete understanding of the relationship between GAG binding and the monomer–dimer equilibrium will require characterization of an SDF1α–heparin complex. Titration experiments using heparin tetra-, hexa-, and octasaccharides suggest that longer oligosaccharides further stabilize the SDF1α dimer (C.T. Veldkamp and B.F. Volkman, unpubl.) and will form the basis of future structural analysis of SDF1α–GAG complexes.
Our studies reconcile previous results which described SDF1α as a either monomer, dimer, or in a monomer–dimer equilibrium (Crump et al. 1997; Dealwis et al. 1998; Ohnishi et al. 2000; Holmes et al. 2001) by identifying the factors that control the oligomeric state of SDF1α. We showed that the SDF1α dimer forms only at nonacidic pH and requires stabilizing counterions including sulfated binding partners like GAGs. We speculate that heparin mediated SDF1α oligomerization is essential for signaling in vivo, and that this intermolecular interface may represent a novel target for altering SDF1α/CXCR4 signaling, as suggested for the CC chemokine RANTES (Proudfoot et al. 2003a; Johnson et al. 2004).
Materials and methods
Cloning and mutagenesis
PCR was used to generate a full-length SDF1α fragment with BamHI and HindIII sites at the 5′ and 3′ ends, respectively, to facilitate insertion into a modified pQE30 vector (Qiagen) that incorporates an N-terminal His6 tag and tobacco etch virus (TEV) protease cleavage site (Dougherty et al. 1989; Peterson et al. 2004). As a result of this cloning strategy, the SDF1α protein produced from this expression construct contains an additional Gly-Ser dipeptide at the amino terminus after digestion with TEV protease. Site-directed mutagenesis was performed using pairs of complementary primers and the QuikChange kit (Stratagene). All expression vectors were verified by DNA sequencing.
Protein expression and purification
The SDF1α expression plasmid was transformed into Escherichia coli strain SG13009[pRPEP4] (Qiagen). Cells were grown at 37°C in either Luria-Bertani or M9 minimal medium. Isotopically labeled proteins for NMR were produced using M9 medium containing 15NH4Cl and [U-13C]-glucose as the sole nitrogen and carbon sources, respectively. Protein expression was induced by the addition of isopropyl-β-D-thiogalactopyranoside (IPTG) to a final concentration of 1 mM when the culture reached an OD600 of 0.7. After incubation at 37°C for 6 h, cells were pelleted at 5000g and stored at −80°C until further processing.
Cell pellets were resuspended in 10 mL of a buffer containing 50 mM Na2PO4 (pH 8.0), 300 mM NaCl, 10 mM imidazole, 1 mM phenylmethylsufonyl fluoride, and 0.1% (v/v) 2-mercaptoethanol. Resuspended cells were lysed by two to three passages through a French pressure cell at 16,000 psi. Inclusion bodies containing SDF1α were collected by centrifugation at 15,000g and the supernatant was discarded. The insoluble inclusion body pellet was dissolved in buffer AD (6 M guanidinium chloride, 50 mM Na2PO4 (pH 8.0), 300 mM NaCl, 10 mM imidazole) and batch loaded onto 2 mL of Ni-NTA resin (Qiagen). After 30 min the column was washed with 4 × 10 mL of buffer AD followed by SDF1α elution with a buffer containing 6 M guanidinium chloride, 50 mM sodium acetate (pH 4.5), 300 mM NaCl, and 10 mM imidazole. Pooled SDF1α fractions were dialyzed against 3 × 4 L of 0.3% acetic acid. After dialysis, Na2HPO4 and NaCl were each added to a concentration of 50 mM, the pH was adjusted to 6.75 with NaOH, and TEV protease (~1:1000 [w/w]) was added to remove the His6 tag. SDF1α disulfide bond formation was performed by subsequently diluting the cleavage reaction to 150 mL in 20 mM Tris (pH 8.0) and dialyzing against the same buffer. Refolded SDF1α was acidified with HCl (pH < 3.0), concentrated by ultrafiltration (MWCO 3500), and purified to >98% homogeneity using reverse phase HPLC with a 30-min gradient from 21% to 42% CH3CN in aqueous 0.1% TFA. SDF1α was frozen, lyophilized, and stored at −20°C. Purity, identity, and molecular weight were verified by matrix-assisted laser desorption ionization mass spectrometry and NMR.
Tissue culture
SUP-T1 cells were obtained from ATCC and grown in RPMI 1640 supplemented with 10% FBS (heat-inactivated at 56°C for 1 h), MEM/Sodium Pyruvate (5 mL of 100× stock per 500 mL medium [Invitrogen]), and 2 mM L-glutamine. Cells were grown and maintained at 37°C with 5% CO2 at densities of 3–20 × 105 cells/mL.
Chemotaxis assay
The SUP-T1 cell line is routinely used for investigations of SDF1–CXCR4 signaling due to its high CXCR4 expression levels and responsiveness to SDF1α (Princen et al. 2003). SUP-T1 cells (2 × 107) were spun down at 400g at room temperature for 5 min. Cells were washed with PBS, and then washed in migration buffer (RPMI 1640 without phenolphthalein containing bovine serum albumin, 1 mg/mL). Cells were resuspended in migration buffer at 5 × 106 cells/mL. Chemotaxis was assayed using Transwells (5 μm pore [Costar]). Migration buffer containing 30 nM SDF1α (600 μL) was added to the lower chamber of a 24-well plate, a transwell insert was added to each well, and 5 × 105 cells in 100 μL of migration buffer were placed in the top chamber. The covered plate was incubated for 3 h at 37°C under 5% CO2 atmosphere. Inserts were removed and the number of cells that migrated into the in the lower chamber were counted using a hematocytometer. Assays were also performed with no SDF1α in the lower chamber or with SDF1α present in both the lower and upper chamber as controls for random migration and chemokinesis, respectively. Recombinant SDF1α (30 nM) induced chemotaxis in SUP-T1 cells with a chemotactic index of 3.2 ± 0.9.
NMR spectroscopy
NMR experiments were performed on a Bruker DRX 600 equipped with a 1H/15N/13C Cryoprobe or a conventional 1H/15N/13C probe equipped with three axis gradients. NMR samples contained 90% H2O, 10% D2O, and 0.02% NaN3 with various buffer, pH, and protein concentrations as specified in the text. Complete 1H, 15N, and 13C resonance assignments for SDF1α (1.2 mM, 25 mM MES [pH 6.8]) were obtained using the following experiments: 15N-1H HSQC (Mori et al. 1995), 3D SE HNCO (Grzesiek and Bax 1992; Muhandiram and Kay 1994), 3D SE HNCA (Grzesiek and Bax 1992; Kay et al. 1994), 3D SE HN(CO)CA (Grzesiek and Bax 1992), 3D 15N SE NOESY-HSQC (Talluri and Wagner 1996), 3D 15N SE TOCSY-HSQC (Zhang et al. 1994), 3D SE C(CO)NH (Grzesiek et al. 1993), 3D HCCH TOCSY (Kay et al. 1993), 2D 13C constant time HSQC (Santoro and King 1992), and 3D 13C SE NOESY-HSQC (one each for aromatic and aliphatic regions) (Kay et al. 1993).
Incremental dilutions of SDF1α (20 mM sodium phosphate [pH 6.0]) from 1.2 to 0.01 mM were monitored using 1D 1H and 2D 15N-1H HSQC spectra. Under these buffer conditions SDF1α monomer–dimer interconversion occurs in fast exchange on the chemical shift time scale, allowing most chemical shift assignments to be easily transferred by inspection of the series of 2D spectra. Initial chemical shift assignments were confirmed using 3D 15N SE NOESY-HSQC and 3D 15N SE TOCSY-HSQC experiments. Chemical shift perturbations were computed as [(5ΔδNH)2 + (ΔδN)2]1/2, where ΔδNH and Delta;δN are the changes in backbone amide 1H and 15N chemical shifts, respectively. A pH titration of 250 μM SDF1α in 20 mM sodium phosphate from pH 8.0 to 6.0 was similarly monitored with 1H and 15N-1H HSQC spectra. Titrations of SDF1α at 250 μM and 10 μM in 20 mM MES at pH 6.8 with sodium phosphate or sodium sulfate (0–100 mM) or I-S heparin disaccharide (0–1 mM) were also monitored by 1D 1H and 2D 15N-1H HSQC spectra. The pH of all NMR samples remained constant throughout titrations with sodium phosphate, sodium sulfate, and I-S heparin disaccharide.
Diffusion coefficient measurements
Diffusion coefficients were measured using a pulse field gradient water-suppressed longitudinal encode-decode (Water-SLED) experiment (Altieri et al. 1995) at various SDF1α concentrations in either 20 mM sodium phosphate (pH 7.4) or 20 mM MES (pH 7.4). Since the pKa of MES is 6.1, the pH of the SDF1α samples in MES was monitored closely. The diffusion delay was 80 msec and the gradient pulse length was 5 msec. The gradient strength was varied from 10% to 80% in 1% intervals with 57.5 G/cm as the maximum (100%) gradient strength. A 1% (w/v) solution of β-cyclodextrin in 90% H2O and 10% D2O, with a diffusion coefficient 3.239 × 10−6 cm2/sec at 25°C (Uedaira and Uedaira 1970), was used as a standard for gradient strength calibration. Nonlinear least-squares fitting was used to obtain self-diffusion coefficients (Ds) from the following equation:
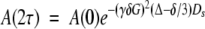 |
(1) |
where γ is the magnetogyric ratio of 1H, δ is the gradient pulse length, G is the gradient intensity, and Δ is the diffusion delay.
Fluorescence polarization assay
SDF1α has a single conserved tryptophan residue, and we monitored its fluorescence polarization (FP) as a function of protein concentration in order to measure the equilibrium dissociation constant (Kd) of the dimer. All samples and buffers used for FP measurements were filtered (0.2 μm) and degassed. Lyophilized SDF1α or mutant chemokines were dissolved in phosphate, HEPES, or MES buffers, and the pH was adjusted with NaOH. FP values were measured at 25°C on a PTI spectrofluorometer equipped with quartz polarizers using the time-based polarization method in the program FeliX32. The excitation wavelength was 295 nm, to avoid tyrosine excitation, and the emission was monitored at 324 nm, near the empirically determined tryptophan emission maximum for SDF1α. Background fluorescence was subtracted from all measurements and g-factors were measured and calculated for each sample. Dimer dissociation constants (Kd) were obtained by nonlinear fitting of fluorescence polarization measurements at protein concentrations ranging from 0.01 to 1 mM to an equation describing a monomer–dimer equilibrium as reviewed by Martin (1996). Starting with the following assumptions (Equations 2–4), the equation (Equation 5) for fitting the dimer dissociation Kd, FPmonomer, and FPdimer values was derived.
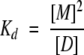 |
(2) |
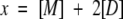 |
(3) |
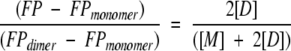 |
(4) |
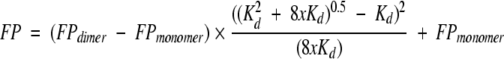 |
(5) |
[M] is the concentration of monomer, [D] is the concentration of dimer, and x is the total concentration of SDF1α monomers. The mol fraction dimer is shown in Equation 4 with FP being the measured value and FPmonomer and FPdimer as the polarization values for pure monomer or dimer, respectively. All nonlinear fits were performed using ProFit 5.6.6 (Quantum Soft).
Acknowledgments
This work was supported by NIH grant R01 AI058072 to B.F.V. We gratefully acknowledge Steven Schwarze for assistance with chemotaxis assays.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041219505.
References
- Altieri, A.S., Hinton, D.P., and Byrd, R.A. 1995. Association of biomolecular systems via pulsed field gradient NMR self-diffusion measurements. J. Am. Chem. Soc. 117 7566–7567. [Google Scholar]
- Amara, A., Lorthioir, O., Valenzuela, A., Magerus, A., Thelen, M., Montes, M., Virelizier, J.L., Delepierre, M., Baleux, F., Lortat-Jacob, H., et al. 1999. Stromal cell-derived factor-1α associates with heparan sulfates through the first β-strand of the chemokine. J. Biol. Chem. 274 23916–23925. [DOI] [PubMed] [Google Scholar]
- Ara, T., Nakamura, Y., Egawa, T., Sugiyama, T., Abe, K., Kishimoto, T., Matsui, Y., and Nagasawa, T. 2003. Impaired colonization of the gonads by primordial germ cells in mice lacking a chemokine, stromal cell-derived factor-1 (SDF-1). Proc. Natl. Acad. Sci. 100 5319–5323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleul, C.C., Farzan, M., Choe, H., Parolin, C., Clark-Lewis, I., Sodroski, J., and Springer, T.A. 1996. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature 382 829–833. [DOI] [PubMed] [Google Scholar]
- Burrows, S.D., Doyle, M.L., Murphy, K.P., Franklin, S.G., White, J.R., Brooks, I., McNulty, D.E., Scott, M.O., Knutson, J.R., Porter, D., et al. 1994. Determination of the monomer–dimer equilibrium of interleukin-8 reveals it is a monomer at physiological concentrations. Biochemistry 33 12741–12745. [DOI] [PubMed] [Google Scholar]
- Crump, M.P., Gong, J.H., Loetscher, P., Rajarathnam, K., Amara, A., Arenzana-Seisdedos, F., Virelizier, J.L., Baggiolini, M., Sykes, B.D., and Clark-Lewis, I. 1997. Solution structure and basis for functional activity of stromal cell- derived factor-1; Dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 16 6996–7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dealwis, C., Fernandez, E.J., Thompson, D.A., Simon, R.J., Siani, M.A., and Lolis, E. 1998. Crystal structure of chemically synthesized [N33A] stromal cell-derived factor 1α, a potent ligand for the HIV-1 “fusin” coreceptor. Proc. Natl. Acad. Sci. 95 6941–6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doitsidou, M., Reichman-Fried, M., Stebler, J., Koprunner, M., Dorries, J., Meyer, D., Esguerra, C.V., Leung, T., and Raz, E. 2002. Guidance of primordial germ cell migration by the chemokine SDF-1. Cell 111 647–659. [DOI] [PubMed] [Google Scholar]
- Dougherty, W.G., Cary, S.M., and Parks, T.D. 1989. Molecular genetic analysis of a plant virus polyprotein cleavage site: A model. Virology 171 356–364. [DOI] [PubMed] [Google Scholar]
- D’Souza, M.P., Cairns, J.S., and Plaeger, S.F. 2000. Current evidence and future directions for targeting HIV entry: Therapeutic and prophylactic strategies. JAMA 284 215–222. [DOI] [PubMed] [Google Scholar]
- Grzesiek, S. and Bax, A. 1992. Inproved 3D triple-resonance NMR techniques applied to a 31 kDa protein. J. Magn. Reson. 96 432–440. [Google Scholar]
- Grzesiek, S., Anglister, J., and Bax, A. 1993. Correlation of backbone amide and aliphatic side-chain resonances in 13C/15N-enriched proteins by isotropic mixing of 13C magnetization. J. Magn. Reson. B 101 114–119. [Google Scholar]
- Helbig, G., Christopherson 2nd, K.W., Bhat-Nakshatri, P., Kumar, S., Kishimoto, H., Miller, K.D., Broxmeyer, H.E., and Nakshatri, H. 2003. NF-κB promotes breast cancer cell migration and metastasis by inducing the expression of the chemokine receptor CXCR4. J. Biol. Chem. 278 21631–21638. [DOI] [PubMed] [Google Scholar]
- Holmes, W.D., Consler, T.G., Dallas, W.S., Rocque, W.J., and Willard, D.H. 2001. Solution studies of recombinant human stromal-cell-derived factor-1. Protein Expr. Purif. 21 367–377. [DOI] [PubMed] [Google Scholar]
- Hoogewerf, A.J., Kuschert, G.S., Proudfoot, A.E., Borlat, F., Clark-Lewis, I., Power, C.A., and Wells, T.N. 1997. Glycosaminoglycans mediate cell surface oligomerization of chemokines. Biochemistry 36 13570–13578. [DOI] [PubMed] [Google Scholar]
- Johnson, Z., Kosco-Vilbois, M.H., Herren, S., Cirillo, R., Muzio, V., Zaratin, P., Carbonatto, M., Mack, M., Smailbegovic, A., Rose, M., et al. 2004. Interference with heparin binding and oligomerization creates a novel anti-inflammatory strategy targeting the chemokine system. J. Immunol. 173 5776–5785. [DOI] [PubMed] [Google Scholar]
- Kay, L.E., Xu, G.-Y., Singer, A.U., Muhandiram, D.R., and Forman-Kay, J.D. 1993. A gradient-enhanced HCCH-TOCSY experiment for recording sidechain 1H and 13C correlations in H2O samples of proteins. J. Magn. Reson. B 101 333–337. [Google Scholar]
- Kay, L.E., Xu, G.Y., and Yamazaki, T. 1994. Enhanced-sensitivity triple-resonance spectroscopy with minimal H2O saturation. J. Magn. Reson. A 109 129–133. [Google Scholar]
- Knaut, H., Werz, C., Geisler, R., and Nusslein-Volhard, C. 2003a. A zebrafish homologue of the chemokine receptor Cxcr4 is a germ-cell guidance receptor. Nature 421 279–282. [DOI] [PubMed] [Google Scholar]
- Knaut, H., Werz, C., Geisler, R., Nusslein-Volhard, C., and Tubingen 2000 Screen Consortium. 2003b. A zebrafish homologue of the chemokine receptor Cxcr4 is a germ-cell guidance receptor. Nature 421 279–282. [DOI] [PubMed] [Google Scholar]
- Kuloglu, E.S., McCaslin, D.R., Markley, J.L., and Volkman, B.F. 2002. Structural rearrangement of human lymphotactin, a C chemokine, under physiological solution conditions. J. Biol. Chem. 277 17863–17870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunwar, P.S. and Lehmann, R. 2003. Developmental biology: Germ-cell attraction. Nature 421 226–227. [DOI] [PubMed] [Google Scholar]
- Lau, E.K., Paavola, C.D., Johnson, Z., Gaudry, J.P., Geretti, E., Borlat, F., Kungl, A.J., Proudfoot, A.E., and Handel, T.M. 2004. Identification of the glycosaminoglycan binding site of the CC chemokine, MCP-1: Implications for structure and function in vivo. J. Biol. Chem. 279 22294–22305. [DOI] [PubMed] [Google Scholar]
- Laurence, J.S., LiWang, A.C., and LiWang, P.J. 1998. Effect of N-terminal truncation and solution conditions on chemokine dimer stability: Nuclear magnetic resonance structural analysis of macrophage inflammatory protein 1β mutants. Biochemistry 37 9346–9354. [DOI] [PubMed] [Google Scholar]
- Laurence, J.S., Blanpain, C., Burgner, J.W., Parmentier, M., and LiWang, P.J. 2000. CC chemokine MIP-1 β can function as a monomer and depends on Phe13 for receptor binding. Biochemistry 39 3401–3409. [DOI] [PubMed] [Google Scholar]
- Lowman, H.B., Fairbrother, W.J., Slagle, P.H., Kabakoff, R., Liu, J., Shire, S., and Hebert, C.A. 1997. Monomeric variants of IL-8: Effects of side chain substitutions and solution conditions upon dimer formation. Protein Sci. 6 598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, R.B. 1996. Comparisons of indefinite self-association models. Chem. Rev. 96 3043–3064. [DOI] [PubMed] [Google Scholar]
- Mbemba, E., Gluckman, J.C., and Gattegno, L. 2000. Glycan and glycosaminoglycan binding properties of stromal cell-derived factor (SDF)-1α. Glycobiology 10 21–29. [DOI] [PubMed] [Google Scholar]
- McCornack, M.A., Cassidy, C.K., and LiWang, P.J. 2003. The binding surface and affinity of monomeric and dimeric chemokine macrophage inflammatory protein 1 β for various glycosaminoglycan disaccharides. J. Biol. Chem. 278 1946–1956. [DOI] [PubMed] [Google Scholar]
- McCornack, M.A., Boren, D.M., and LiWang, P.J. 2004. Glycosaminoglycan disaccharide alters the dimer dissociation constant of the chemokine MIP-1β. Biochemistry 43 10090–10101. [DOI] [PubMed] [Google Scholar]
- Moepps, B., Braun, M., Knopfle, K., Dillinger, K., Knochel, W., and Gierschik, P. 2000. Characterization of a Xenopus laevis CXC chemokine receptor 4: Implications for hematopoietic cell development in the vertebrate embryo. Eur. J. Immunol. 30 2924–2934. [DOI] [PubMed] [Google Scholar]
- Molyneaux, K.A., Zinszner, H., Kunwar, P.S., Schaible, K., Stebler, J., Sunshine, M.J., O’Brien, W., Raz, E., Littman, D., Wylie, C., et al. 2003. The chemokine SDF1/CXCL12 and its receptor CXCR4 regulate mouse germ cell migration and survival. Development 130 4279–4286. [DOI] [PubMed] [Google Scholar]
- Mori, S., Abeygunawardana, C., Johnson, M.O., and van Zijl, P.C.M. 1995. Improved sensitivity of HSQC spectra of exchanging protons at short interscan delays using a new fast HSQC (FHSQC) detection scheme that avoids water saturation. J. Magn. Reson. B 105 94–98. [DOI] [PubMed] [Google Scholar]
- Muhandiram, D.R. and Kay, L.E. 1994. Gradient-enhanced triple-resonance three-dimensional NMR experiments with improved sensitivity. J. Magn. Reson. B 103 203–216. [Google Scholar]
- Muller, A., Homey, B., Soto, H., Ge, N., Catron, D., Buchanan, M.E., Mc-Clanahan, T., Murphy, E., Yuan, W., Wagner, S.N., et al. 2001. Involvement of chemokine receptors in breast cancer metastasis. Nature 410 50–56. [DOI] [PubMed] [Google Scholar]
- Murdoch, C. 2000. CXCR4: Chemokine receptor extraordinaire. Immunol. Rev. 177 175–184. [DOI] [PubMed] [Google Scholar]
- Nagasawa, T., Hirota, S., Tachibana, K., Takakura, N., Nishikawa, S., Kitamura, Y., Yoshida, N., Kikutani, H., and Kishimoto, T. 1996. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 382 635–638. [DOI] [PubMed] [Google Scholar]
- Ohnishi, Y., Senda, T., Nandhagopal, N., Sugimoto, K., Shioda, T., Nagal, Y., and Mitsui, Y. 2000. Crystal structure of recombinant native SDF-1α with additional mutagenesis studies: An attempt at a more comprehensive interpretation of accumulated structure–activity relationship data. J. Interferon Cytokine Res. 20 691–700. [DOI] [PubMed] [Google Scholar]
- Paavola, C.D., Hemmerich, S., Grunberger, D., Polsky, I., Bloom, A., Freedman, R., Mulkins, M., Bhakta, S., McCarley, D., Wiesent, L., et al. 1998. Monomeric monocyte chemoattractant protein-1 (MCP-1) binds and activates the MCP-1 receptor CCR2B. J. Biol. Chem. 273 33157–33165. [DOI] [PubMed] [Google Scholar]
- Peterson, F.C., Elgin, E.S., Nelson, T.J., Zhang, F., Hoeger, T.J., Linhardt, R.J., and Volkman, B.F. 2004. Identification and characterization of a glycosaminoglycan recognition element of the C chemokine lymphotactin. J. Biol. Chem. 279 12598–12604. [DOI] [PubMed] [Google Scholar]
- Princen, K., Hatse, S., Vermeire, K., De Clercq, E., and Schols, D. 2003. Evaluation of SDF-1/CXCR4-induced Ca2+ signaling by fluorometric imaging plate reader (FLIPR) and flow cytometry. Cytometry 51A 35–45. [DOI] [PubMed] [Google Scholar]
- Proudfoot, A.E., Handel, T.M., Johnson, Z., Lau, E.K., LiWang, P., Clark-Lewis, I., Borlat, F., Wells, T.N., and Kosco-Vilbois, M.H. 2003a. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. 100 1885–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot, A.E., Power, C.A., Rommel, C., and Wells, T.N. 2003b. Strategies for chemokine antagonists as therapeutics. Semin. Immunol. 15 57–65. [DOI] [PubMed] [Google Scholar]
- Rajarathnam, K., Sykes, B.D., Kay, C.M., Dewald, B., Geiser, T., Baggiolini, M., and Clark-Lewis, I. 1994. Neutrophil activation by monomeric interleukin-8. Science 264 90–92. [DOI] [PubMed] [Google Scholar]
- Rajarathnam, K., Clark-Lewis, I., and Sykes, B.D. 1995. 1H NMR solution structure of an active monomeric interleukin-8. Biochemistry 34 12983–12990. [DOI] [PubMed] [Google Scholar]
- Rossi, D. and Zlotnik, A. 2000. The biology of chemokines and their receptors. Annu. Rev. Immunol. 18 217–242. [DOI] [PubMed] [Google Scholar]
- Sadir, R., Baleux, F., Grosdidier, A., Imberty, A., and Lortat-Jacob, H. 2001. Characterization of the stromal cell-derived factor-1α–heparin complex. J. Biol. Chem. 276 8288–8296. [DOI] [PubMed] [Google Scholar]
- Santoro, J. and King, G.C. 1992. A constant-time 2D overbodenhausen experiment for inverse correlation of isotopically enriched species. J. Magn. Reson. 97 202–207. [Google Scholar]
- Skelton, N.J., Aspiras, F., Ogez, J., and Schall, T.J. 1995. Proton NMR assignments and solution conformation of RANTES, a chemokine of the C-C type. Biochemistry 34 5329–5342. [DOI] [PubMed] [Google Scholar]
- Tachibana, K., Hirota, S., Iizasa, H., Yoshida, H., Kawabata, K., Kataoka, Y., Kitamura, Y., Matsushima, K., Yoshida, N., Nishikawa, S., et al. 1998. The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature 393 591–594. [DOI] [PubMed] [Google Scholar]
- Talluri, S. and Wagner, G. 1996. An optimized 3D NOESY-HSQC. J. Magn. Reson. B 112 200–205. [DOI] [PubMed] [Google Scholar]
- Uedaira, H. and Uedaira, H. 1970. Translational frictional coefficients of molecules in aqueous solution. J. Phys. Chem. 74 2211–2214. [Google Scholar]
- Valenzuela-Fernandez, A., Palanche, T., Amara, A., Magerus, A., Altmeyer, R., Delaunay, T., Virelizier, J.L., Baleux, F., Galzi, J.L., and Arenzana-Seisdedos, F. 2001. Optimal inhibition of X4 HIV isolates by the CXC chemokine stromal cell-derived factor 1 α requires interaction with cell surface heparan sulfate proteoglycans. J. Biol. Chem. 276 26550–26558. [DOI] [PubMed] [Google Scholar]
- Zhang, O., Kay, L.E., Olivier, J.P., and Forman-Kay, J.D. 1994. Backbone 1H and 15N resonance assignments of the N-terminal SH3 domain of drk in folded and unfolded states using enhanced-sensitivity pulsed field gradient NMR techniques. J. Biomol. NMR 4 845–858. [DOI] [PubMed] [Google Scholar]
- Zou, Y.R., Kottmann, A.H., Kuroda, M., Taniuchi, I., and Littman, D.R. 1998. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 393 595–599. [DOI] [PubMed] [Google Scholar]