

Abstract
A family of genetically-encoded metabolite sensors has been constructed using bacterial periplasmic binding proteins (PBPs) linearly fused to protein fluorophores. The ligand-induced conformational change in a PBP allosterically regulates the relative distance and orientation of a fluorescence resonance energy transfer (FRET)-compatible protein pair. Ligand binding is transduced into a macroscopic FRET observable, providing a reagent for in vitro and in vivo ligand-measurement and visualization. Sensors with a higher FRET signal change are required to expand the dynamic range and allow visualization of subtle analyte changes under high noise conditions. Various observations suggest that factors other than inter-fluorophore separation contribute to FRET transfer efficiency and the resulting ligand-dependent spectral changes. Empirical and rational protein engineering leads to enhanced allosteric linkage between ligand binding and chromophore rearrangement; modifications predicted to decrease chromophore rotational averaging enhance the signal change, emphasizing the importance of the rotational freedom parameter κ2 to FRET efficiency. Tighter allosteric linkage of the PBP and the fluorophores by linker truncation or by insertion of chromophores into the binding protein at rationally designed sites gave rise to sensors with improved signal change. High-response sensors were obtained with fluorescent proteins attached to the same binding PBP lobe, suggesting that indirect allosteric regulation during the hinge-bending motion is sufficient to give rise to a FRET response. The optimization of sensors for glucose and glutamate, ligands of great clinical interest, provides a general framework for the manipulation of ligand-dependent allosteric signal transduction mechanisms.
Keywords: conformational changes, structure/function studies, new methods, molecular mechanics/dynamics, glucose, glutamate, nanosensor, neurotransmitter, biosensor, protein engineering, FRET
Biosensors are molecular machines linking the highly evolved ligand-binding properties of a biological macromolecule to the production of a physical observable (Nakamura and Karube 2003; Looger et al. 2005). Many classes of biomolecules have been employed as “recognition elements” of biosensors, although proteins have emerged as the gold standard due to their superior properties, including specificity, affinity, and stability. Moreover, such sensors may be genetically encodable, thus providing significant advantages for in vivo applications: (1) stable expression, (2) efficient introduction into a wide spectrum of cell types and organisms, (3) targeting for measuring changes within subcellular compartments, and (4) efficient control over sensor production.
Most protein-based biosensors exploit the allosteric linkage between ligand binding and conformation, transducing binding-coupled structural changes into the altered function of a “reporter element.” Biosensors have been successfully fabricated with diverse reporter elements: ionic current flowing through an oligomeric protein pore complex (Bayley and Jayasinghe 2004); displacement of a nanofabricated cantilever attached to the protein (Ziegler 2004) or surface plasmon resonance of a monolayer to which the protein is adsorbed (Haes and Van Duyne 2004); the activity of a small molecule covalently attached to the protein, either fluorescence emission of an environmentally sensitive metal or small molecule dye (de Lorimier et al. 2002; Hoffmann et al. 2005) or current flux to a redox cofactor (Benson et al. 2001); the regulation of the activity of an enzyme by binding at a region distal from the active site (Villaverde 2003), or in a hybrid enzyme in which the binding domain and enzyme sequences are interwoven by nonhomologous recombination (Guntas et al. 2004); and the fluorescence resonance energy transfer (FRET) between a pair of attached small molecules (Medintz et al. 2003). The phenomenon of FRET is uniquely suited to the detection and visualization of events in the 1–10-nm (10–100 Å) range (Jares-Erijman and Jovin 2003) and meets the criteria for robust imaging in a variety of formats and environments (Clegg 1995). FRET is a high-sensitivity indicator of conformational change, which has been exploited to create a number of FRET-based ligand sensors (Gaits and Hahn 2003; Zaccolo 2004).
For small molecule and metal detection, the enhanced cyan fluorescent protein/enhanced yellow fluorescent protein (ECFP/EYFP) FRET pair has been grafted onto the termini of several proteins that undergo conformational changes upon ligand binding, thus generating a small family of genetically encoded nanosensors. A genetically-encoded calcium sensor version made use of a conformational actuator (an M13 calmodulin-binding peptide which binds to calcium-loaded calmodulin) to magnify the degree of relative fluorophore rearrangement and thus the FRET change upon ligand binding (Miyawaki et al. 1997; Romoser et al. 1997).
Members of the superfamily of bacterial periplasmic binding proteins (PBPs) (Tam and Saier 1993; Fukami-Kobayashi et al. 1999) bind to a wide spectrum of ligands with high affinity and specificity, and undergo a dramatic conformational change in concert with binding (Quiocho and Ledvina 1996; Mowbray and Sandgren 1998). For these reasons, PBPs function well as recognition elements in biosensors, in conjunction with a variety of reporter elements.
We have shown that terminally fused, genetically encoded FRET sensors derived from PBPs have moderate signal-to-noise, with FRET ratio change values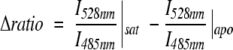 ; I528nm is the peak acceptor emission; I485nm is the peak donor emission) in the range 0.2–0.3 (Fehr et al. 2002, 2003, 2005; Lager et al. 2003; Okumoto et al. 2005). Although structurally diverse, the PBP sensors for maltose, glucose, ribose, and glutamate show a similar ligand-dependent FRET change. To date, sensors have been constructed as terminal fusions of full-length protein chromophores (ECFP, EYFP, and their improved variants), attached via short linkers to binding protein termini chosen either on the basis of crystal structure analysis (the sugar sensors; N- and C-terminal residues selected to complete well-formed secondary structure elements) or by sequence homology to functional sensors (the glutamate sensor). Identical short flexible linkers were used for the construction of all sensors so far. The overall diameter (longitudinal elliptic axis) of PBPs is in the range of 6–7 nm. The distance between N and C termini as taken from the crystal structure of the ligand-free form is ~4–5 nm for MBP and RBP (Sharff et al. 1992; Bjorkman and Mowbray 1998). The N and C termini move closer together upon ligand binding by 0.7 nm in case of MBP, while in the case of RBP the termini move further apart by 0.2 nm (cf. Okumoto et al. 2004). The similar FRET ratio change values may thus be due to the use of flexible linkers leading to rotational averaging of the fluorophore dipole orientation ensemble. We thus hypothesized that linker truncation and rigidification may increase signal change via tighter allosteric linkage and/or reduced rotational averaging. We employed a combination of rational and empirical protein engineering methods to better elucidate the allosteric coupling mechanism and to create a set of optimized sensors.
; I528nm is the peak acceptor emission; I485nm is the peak donor emission) in the range 0.2–0.3 (Fehr et al. 2002, 2003, 2005; Lager et al. 2003; Okumoto et al. 2005). Although structurally diverse, the PBP sensors for maltose, glucose, ribose, and glutamate show a similar ligand-dependent FRET change. To date, sensors have been constructed as terminal fusions of full-length protein chromophores (ECFP, EYFP, and their improved variants), attached via short linkers to binding protein termini chosen either on the basis of crystal structure analysis (the sugar sensors; N- and C-terminal residues selected to complete well-formed secondary structure elements) or by sequence homology to functional sensors (the glutamate sensor). Identical short flexible linkers were used for the construction of all sensors so far. The overall diameter (longitudinal elliptic axis) of PBPs is in the range of 6–7 nm. The distance between N and C termini as taken from the crystal structure of the ligand-free form is ~4–5 nm for MBP and RBP (Sharff et al. 1992; Bjorkman and Mowbray 1998). The N and C termini move closer together upon ligand binding by 0.7 nm in case of MBP, while in the case of RBP the termini move further apart by 0.2 nm (cf. Okumoto et al. 2004). The similar FRET ratio change values may thus be due to the use of flexible linkers leading to rotational averaging of the fluorophore dipole orientation ensemble. We thus hypothesized that linker truncation and rigidification may increase signal change via tighter allosteric linkage and/or reduced rotational averaging. We employed a combination of rational and empirical protein engineering methods to better elucidate the allosteric coupling mechanism and to create a set of optimized sensors.
The systematic truncation of the composite linker sequences connecting binding protein with chromophore (defined as the sum of the two sets of terminal FP residues not absolutely required for folding or fluorescence, plasmid-derived linker residues, and the terminal binding protein residues not absolutely required for folding or binding) gave rise to glucose sensors with an up to threefold improved signal change. Enhanced FRET change was effected beyond a minimal linker truncation, and maximal truncations were found for both N- and C-terminal linkers beyond which sensor stability and function were compromised. As an alternative approach, the binding proteins were scanned for permissive solvent-exposed sites (Martineau et al. 1992; Guntas et al. 2004), at which insertion of a chromophore sequence was tolerated, as determined by retention of both binding and fluorescence signal change. Small libraries of internally inserted chromophore-binding protein chimeras were screened for fluorescence in bacteria. Multiple sites were found to be permissive. Subsequently, a second chromophore was added to either the N or the C terminus of permissive chimeras, and the resulting constructs were screened for ligand binding. Many constructs yielded functional sensors, with up to a fivefold improvement in signal change for both the glucose and the glutamate sensor. For both proteins, sensors with both chromophores on the same binding protein lobe were found to be functional, confirming the importance of indirect allosteric regulation and the orientation component of energy transfer efficiency.
Taken together, these complementary engineering techniques provide a systematic framework for the optimization of multicomponent protein chimeras. We demonstrate the technique by iteratively improving the fluorescence properties of two genetically encoded nanosensors, in the process creating valuable reagents for the detection of the clinically relevant analytes glucose and glutamate.
Results
Two approaches were followed to systematically modulate the allosteric linkage between binding and energy transfer: (1) variation of the composite linkers connecting the regions absolutely required for binding and fluorescence (the “core” structures) in the FLIPglu sensor and (2) insertion of the donor chromophore ECFP into permissive sites of FLIPglu and FLIPE.
FLIPglu linker variation
The glucose nanosensor FLIPglu consists of the mature glucose/galactose-binding protein MglB from Escherichia coli linearly fused to ECFP and EYFP via six-amino-acid linkers at the N and C termini, respectively (Fehr et al. 2003). The linker and less well structured domains at the termini of MglB and the GFP variants (together comprising the “composite linker”) would be assumed to allow flexible (if not free) rotation of the fluorophores relative to the binding protein and one another. Composite linker regions were systematically truncated in an attempt to decrease rotational averaging and to enhance the allosteric coupling. Fluorescent proteins (FPs) possess terminal regions not absolutely required for folding and fluorescence (an N-terminal helix and a C-terminal coil) (Li et al. 1997). Furthermore, five amino acids may be deleted from the C-terminal region of the MglB binding protein without affecting binding. This together yields 32 amino acids, the removal of which might a priori be expected to preserve binding and fluorescence, assuming that the FPs are sterically accommodated at their respective BP termini (Fig. 1A ▶). Composite linker regions were deleted from FLIPglu in a stepwise manner (Supplemental Fig. 1 ▶). Deletions of up to 20 amino acids did not influence sensor properties; larger deletions significantly affected the ratio change. Of the 13 constructs tested, six showed an increased response, with the best (FLIPglu- 600μΔ13, 31-amino-acid truncation; Δratio=−0.7 [28% decrease]) showing a threefold improvement relative to the original terminally fused sensor (FLIPglu-600μ, Δratio=−0.25 [8% decrease]) (Table 1; Fig. 1B,C ▶). Consistent with the importance of tight coupling, insertion of additional flexible or triple-helical rigid domains did not lead to improved sensors: Insertion of a six-amino-acid region corresponding to an introduced restriction site into the original FLIPglu-600μ had no effect on the signal change, while insertion of a 127-amino-acid triple helical region into the N-terminal composite linker abolished the signal change (Table 1). TheKd of the sensors was unaffected by the linker engineering.
Figure 1.
A glucose sensor with enhanced signal change due to linker truncations. (A) FLIPglu construct consisting of N-terminal His-tag, ECFP (cyan), an N-terminal composite linker (CL-N), the mature MglB (red), a C-terminal composite linker (CL-C), and EYFP (yellow). (B) Emission spectra of FLIPglu-600μΔ13 in the presence and the absence of 10 mM glucose (excitation 433 nm). (C) Glucose binding isotherms of FLIPglu-600μ and FLIPglu-600μΔ13. (D) Correlation of Δratio and the number of amino acids deleted from the composite linkers. After deletion of >20 amino acids from the two composite linkers, increased FRET changes are observed.
Table 1.
Effect on ratio change of deletion from and insertion into composite linker sequences of linearly fused FLIPglu sensor
| Sensor | CL-N deletiona | CL-C deletiona | Δ ratio | Δ ratio (%) |
| FLIPglu-600μ | 0 | 0 | −0.25 | −8 |
| FLIPglu-600μΔ1 | 0 | 5 | −0.25 | −8 |
| FLIPglu-600μΔ2 | 3 | 2 | −0.26 | −8 |
| FLIPglu-600μΔ3 | 10 | 2 | −0.26 | −8 |
| FLIPglu-600μΔ4 | 10 | 10 | −0.27 | −8 |
| FLIPglu-600μΔ5 | 13 | 10 | −0.15 | −4 |
| FLIPglu-600μΔ5 | 14 | 10 | −0.27 | −10 |
| FLIPglu-600μΔ7 | 15 | 10 | −0.12 | −4 |
| FLIPglu-600μΔ8 | 13 | 13 | −0.5 | −20 |
| FLIPglu-600μΔ9 | 14 | 13 | −0.25 | −9 |
| FLIPglu-600μΔ10 | 15 | 13 | −0.45 | −15 |
| FLIPglu-600μΔ11 | 13 | 16 | −0.6 | −23 |
| FLIPglu-600μΔ12 | 14 | 16 | −0.5 | −19 |
| FLIPglu-600μΔ13 | 15 | 16 | −0.7 | −28 |
| CL-N insertiona | CL-C insertiona | Δ ratio | Δ ratio (%) | |
| FLIPglu-600μLI1 | 6 | 0 | −0.25 | −8 |
| FLIPglu-600μLI2 | 127 | 0 | 0 | 0 |
a Number of amino acids deleted from, or inserted into, N- and C-terminal composite linker sequences, respectively.
Internally fused glucose sensors
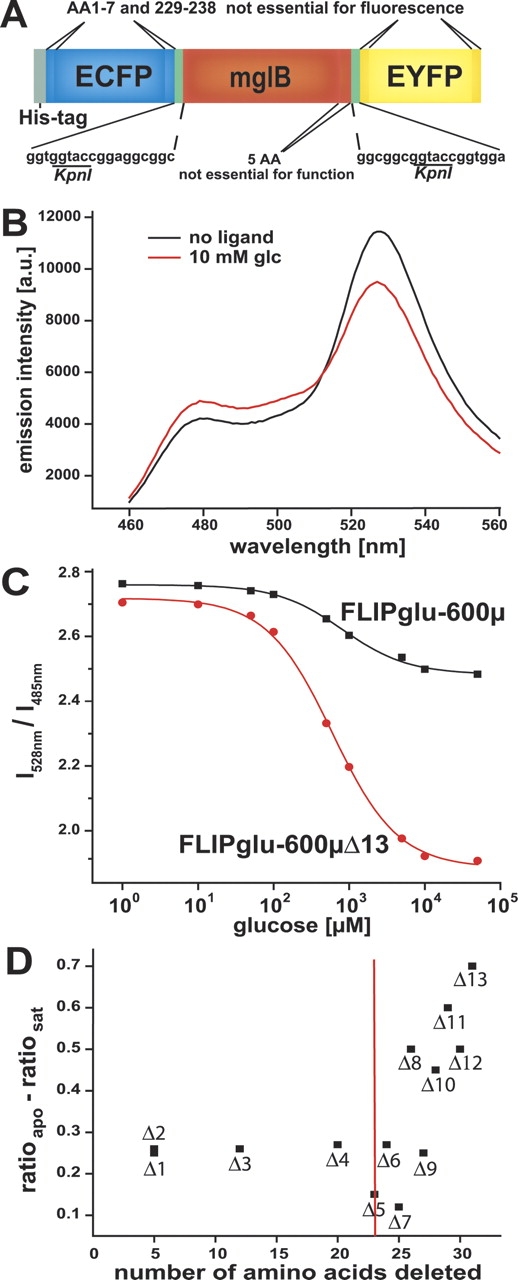
Fourteen sites in MglB were selected for ECFP insertion; sites were chosen to be solvent-exposed, between regions of well-formed secondary structure, and capable of sterically accommodating the FP domain. Nine sites were identified on the N-terminal, and five on the C-terminal, lobe of MglB (Fig. 2 ▶). An NruI restriction site was introduced into the predicted permissive sites of the F16A mutant of mglB, thus converting the wild-type sequence of each site to Ser-Arg (Supplemental Table 2). An ECFP sequence (amino acids 1–238) was produced by PCR with N-terminal Ala-Gly and C-terminal Gly-Ser linkers. This ECFP fragment was inserted by blunt-end ligation into the NruI-digested plasmids. In-frame insertions were screened for fluorescence in bacterial colonies. For each ECFP insertion site, constructs were generated with Citrine (a yellow fluorescent protein [YFP] version with reduced environmental sensitivity) (Griesbeck et al. 2001) attached either N- or C-terminally, totaling 28 sensors. Fusions were screened for FRET in bacterial colonies. All 28 proteins showed FRET (data not shown), indicating that the two chromophore domains and binding protein are not severely impaired by the internal insertions; 19 showed a Citrine:ECFP ratio of at least 1.5, a threshold arbitrarily set to exclude less well-folded proteins, which are expected to have a low FRET transfer efficiency. ECFP insertions were tolerated at 11 of the 14 sites tested, as determined by either the N- or C-terminal Citrine fusion having FRET efficiency above this threshold. The sensors with T102S-ECFP-D103R, G198S-ECFP-P199R, and N226S-ECFP-K227R insertion sites showed a ratio below this threshold, suggesting that these sites did not accommodate the FP internal insertion. Of the 28 putative sensor proteins constructed, 14 carried both chromophores on the same binding protein lobe, and 14 featured them on opposite lobes (Fig. 2 ▶). Sensors were given the nomenclature FLIIXPglu-Y (fluorescent indicator-ECFP insertionposition X protein for glucose; Y is affinity for glucose, if measurable [e.g., Y= 600μ indicates a binding constant of 600μM]).
Figure 2.

Fluorophore insertion sites in MglB. (A) MglB structure, in which cyan spheres indicate the sites (position relative to first codon in mature MglB is given) at which ECFP was inserted. N and C termini are labeled. (B) MglB structure with Citrine attached to the C terminus, indicated by a yellow sphere, and two permissive insertion sites of ECFP, marked by cyan spheres. (C) MglB structure with Citrine attached to the N terminus, indicated by a yellow sphere, and three permissive insertion sites of ECFP, marked by cyan spheres.
Emission spectra were recorded for all proteins, without ligand, and with 10 mM glucose. Five proteins showed an absolute glucose-dependent FRET signal change greater than that of the full-length terminally fused sensor FLIPglu-600μ (12% decrease in FRET ratio) (Table 2). Of these five, three sensors have ECFP and Citrine attached to the same BP lobe: FLII32Pglu (13% increase), FLII275NPglu (42% increase), and FLII282Pglu (21% increase), all with both FPs attached to the N-terminal lobe of MglB. Two sensors carry the two FPs on opposite lobes: FLII12Pglu (58% increase) and FLII275CPglu (17% decrease), both with N-terminal ECFP and C-terminal Citrine. Glucose titrations were performed for these five sensors, with the affinities determined as FLII32Pglu-2.2m, FLII275NPglu-4.6m, FLII282Pglu-4m, FLII12Pglu-600μ, and FLII275CPglu-13.8m, respectively (Fig. 3 ▶). It was discovered that FLII12Pglu-600μ contains the wild-type phenylalanine at position 16 instead of alanine; thus all ECFP insertions have decreased the affinity of the sensor for glucose.
Table 2.
Signal change and glucose affinity of the FLIIXPglu sensors
| Sensor | EYFP terminus | Ratio absence | Δ ratio | Δ ratio (%) | Kd (mM) |
| FLII12Pglu-600μ | C | 4.55 | 2.66 | 58 | 0.6 |
| FLII275NPglu-4.6m | N | 1.63 | 0.69 | 42 | 4.6 |
| FLII282Pglu-4m | N | 2.11 | 0.44 | 21 | 4.0 |
| FLII32Pglu-2.2m | N | 3.4 | 0.44 | 13 | 2.2 |
| FLIPglu-600μ | C | 2.95 | −0.35 | −12 | 0.6 |
| FLII275CPglu-13.8m | C | 1.93 | −0.33 | −17 | 13.8 |
Figure 3.

Signal change of internally fused glucose sensors. (A) Ratio change for the five FLIIxPglu sensors (relative to the original FLIPglu-600μ [red]) with a signal change greater than FLIPglu-600μ. Positive values indicate an increase in fluorescence intensity ratio upon ligand addition; negative values indicate a reduction in ratio. (B) Normalized glucose binding isotherms for the five FLIIxPglu sensors relative to the original FLIPglu-600μ (red). Note the differences in affinity (all except FLII12Pglu-600μ carry the F16A mutation).
Internally fused glutamate sensor
The E. coli glutamate/aspartate-binding protein YbeJ (also GltI; gene ID eco:b0655) has been shown to function as a glutamate sensor (de Lorimier et al. 2002). The terminally fused YbeJ FRET nanosensor (FLIPE-600n) has a glutamate-dependent EYFP/ECFP ratio decrease of 0.27, sufficient signal-to-noise for in vivo measurements in rat neuronal cell culture (Okumoto et al. 2005).
Currently, there is no structural information about YbeJ; thus the sequence of the predicted mature polypeptide was submitted for automated structure prediction using the Robetta server (http://robetta.bakerlab.org); the E. coli glutamine-binding protein GlnH was used as a template for the sequence threading calculation (Okumoto et al. 2005). The highest-ranking model structure suggests that YbeJ has an elongated C-terminal region relative to periplasmic amino acid–binding proteins of known structure, and thus the assignment of this part of the model is particularly uncertain (Fig. 4 ▶). The five highest-scoring models for YbeJ, however, all predict that this C-terminal extension associates with the N-terminal lobe, suggesting that YbeJ is a type II PBP (Tam and Saier 1993; Fukami-Kobayashi et al. 1999), as are all periplasmic hydrophilic amino acid–binding proteins of known structure.
Figure 4.
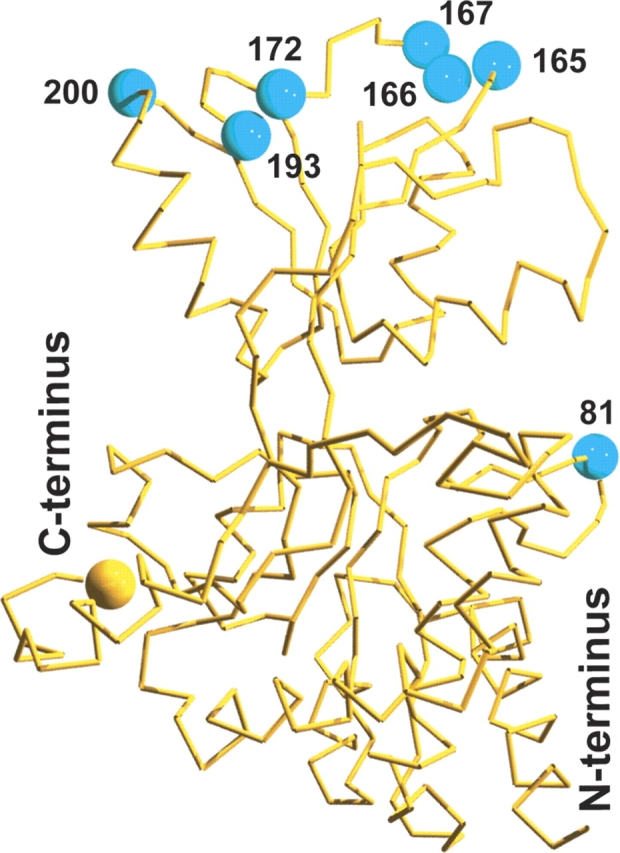
Fluorophore insertion sites in YbeJ. Structural model of YbeJ produced by the Robetta server. (Yellow sphere) C-terminal Venus; (cyan spheres) YbeJ sites tested for permissiveness of ECFP insertion.
Sites predicted to permit ECFP insertion were chosen from the YbeJ model and unique blunt-end restriction sites were introduced by site-directed mutagenesis at these locations (Table 3). A truncated ECFP (amino acids 1–232) was inserted into these sites and clones were screened for fluorescence in bacterial colonies. Only the protein with the N81V-ECFP-Q82N insertion was found to be highly fluorescent. A C-terminal Venus (a YFP with reduced environmental sensitivity) (Nagai et al. 2002) gene was fused to this construct, producing a high-FRET ratio chimeric protein. Upon glutamate saturation, the FRET ratio was found to decrease by 48%, significantly more than the terminally fused FLIPE-600n sensor (11% decrease) (Fig. 5 ▶). This internally fused sensor shows an affinity for glutamate of 1 μM (the sensor is thus denoted as FLII81PE-1μ), similar to the terminally fused sensor (Fig. 5 ▶).
Table 3.
ECFP insertion sites in YbeJ
| YbeJ position | Original sequence | Altered sequence (restriction site) | ECFPa fluorescence |
| N81V-Q82N | aatcag | gttaac (HpaI) | + |
| G165-G166A | ggcggc | ggcgcc (NarI) | N.D. |
| G166-D167A | ggcgat | ggcgcc (NarI) | N.D. |
| D167-I168 | gatatc | gatatc (EcoRV)b | N.D. |
| A172V-D173N | gccgac | gttaac (HpaI) | N.D. |
| D173E-L174 | gacctg | gagctc (Ec1136II) | N.D. |
| M200-N201H | atgaat | atgcat (BfrBI) | N.D. |
a Visual comparison of colonies at 470/40nm excitation.
b Native site, unaltered sequence.
N.D., not detectable.
Figure 5.
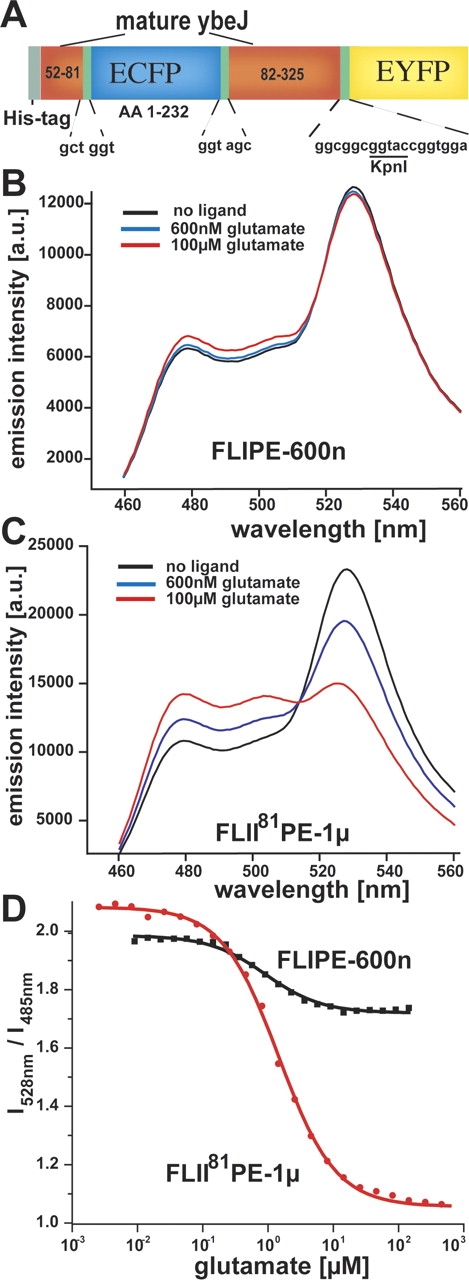
Internal insertion of ECFP into YbeJ. (A) FLII81PE construct consisting of an N-terminal His-tag, ECFP (cyan), an N-terminal composite linker (CL-N), the mature YbeJ (red domains; numbers indicate amino acid positions relative to initiation codon), a C-terminal composite linker (CL-C), and Venus (yellow). (B) Emission spectra of FLIPE-600n in the presence of 0, 600 nM, and 100 μM glutamate (excitation 433 nm). (C) Emission spectra of FLII81PE-1μ in the presence of 0, 600 nM, and 100 μM glutamate (excitation 433 nm). (D) Glutamate titration curves of FLIPE-600n and FLII81PE-1μ.
Measurements of ratio and ratio change in phosphate buffer yield lower values compared to those done in MOPS buffer (e.g., cf. FLIPglu-600μ in Fig. 1B,C ▶ and Fig. 3A,B ▶). The lower ratio is potentially due to quenching in phosphate buffer. Similarly, the absolute ratio observed for FLII81PE-1μ in the absence of glutamate in MOPS buffer is 5.0 and the ratio change is 3.8 (data not shown).
Discussion
Distance and orientation components of FRET transfer efficiency
Förster showed that the FRET transfer efficiency between a donor and an acceptor fluorophore occurs nonradiatively via long-range dipole–dipole coupling and is given by
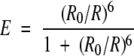 |
where E is transfer efficiency, R is donor–acceptor separation, and R0 is the donor–acceptor separation at which transfer efficiency is half-maximal (Jares-Erijman and Jovin 2003). The “Förster distance” R0 of a fluorophore pair depends on intrinsic properties of the fluorophores, as well as context-dependent parameters, and is given by R06 = c0κ2Jn−4Q0, where c0 is a constant, κ2 is the “orientation factor” of the donor-acceptor pair, J is the overlap integral of donor emission and acceptor absorption spectra, n−4 is the refractive index of the medium, and Q0 is the donor quantum yield (Lakowicz 1999; Jares-Erijman and Jovin 2003). The parameter κ2 captures the orientation-dependence of transfer efficiency and as such may vary with conformation, in concert with the separation R. For randomly oriented donor and acceptor, undergoing rapid and isotropic rotation during the lifetime of the donor excited state, this parameter evaluates to 2/3. The value of R0 at κ2=2/3 for the commonly used ECFP/EYFP FRET pair (the cyan and the yellow spectral variants of the Aequorea victoria green fluorescent protein) is in the range of 4.9 nm (Patterson et al. 2000). However, the assumption of rapid isotropic rotation is unlikely to hold, particularly for protein fluorophores (Stryer 1978), because the fluorescent lifetimes of protein chromophores are typically ~1/5 of their rotational correlation times (Jares- Erijman and Jovin 2003), greatly limiting the amount of rotational relaxation during the lifetime of the donor excited state. In most settings, particularly if donor and acceptor are covalently attached, the parameter κ2 may vary between 0 and 4, as different molecular configurations and conformations are accessed.
Thus, allosterically regulated changes in resonance energy transfer between two fluorophores covalently attached to a PBP recognition element can be due to distance or orientation effects, or more likely both. Such a change in the relative chromophore separation and orientation may be a simple result of the action of the hinge bend of the PBP on one chromophore relative to the other. Alternatively, steric constraints placed on one or both of the reporter groups (interaction of one reporter group with the other, or with the recognition element) by the global conformational change may alter the ensemble of accessible states of the system, leading to substantial changes in separation, orientation, and conformational flexibility.
FRET measurements (except for single molecules) are ensemble averages over large numbers of donor-acceptor pairs, and over the time frame of the experiment, which is typically long relative to the time scale of molecular motions. The Förster theory of energy transfer has been extended to explicitly handle such considerations (Van Der Meer et al. 1994; Scholes 2003). The functional form of the FRET transfer efficiency dictates that high-transfer orientations are rare relative to low- or zero-transfer orientations (Van Der Meer et al. 1994), and thus orientational degeneracy will dampen energy transfer. Thus a structure such as a flexible linker tethering a chromophore to a recognition element will serve both to increase separation (thus decreasing transfer efficiency) and to expose the chromophore to transfer-depressing orientational averaging. The rational optimization of biosensor signal-to-noise thus involves (at least implicitly) the consideration of the ensemble of allowable chromophore relative orientations, both in terms of a significant change in the transfer efficiency of the ensemble-averaged orientations of the free and ligand-bound states, and a low degree of ensemble entropy.
Rational engineering of nanosensor proteins
It has previously been demonstrated that protein engineering may be applied to the systematic optimization of biosensor protein properties. The affinity (Marvin and Hellinga 2001; Fehr et al. 2002, 2003; Lager et al. 2003) and binding specificity may be altered by computational protein design (Looger et al. 2003) or by directed evolution (Korndorfer et al. 2003). The stability and robustness of the sensors may be improved, as well (Nagai and Miyawaki 2004). Finally, the allosteric linkage between recognition and reporter elements may be optimized, by rational (Dattelbaum et al. 2005) or empirical (Guntas and Ostermeier 2004; Guntas et al. 2004) means. The signal change of genetically encoded FRET sensors in particular has been optimized, e.g., by linker engineering (Nagai and Miyawaki 2004) and by circular chromophore permutation (Baird et al. 1999; Nagai et al. 2004), probably affecting ensemble distance and orientation.
It was initially discovered that the linear fusion of the full-length wild-type E. coli maltose-binding protein malE to ECFP and EYFP failed to produce a functional sensor (Fehr et al. 2002). Deletion of five amino acids from the N terminus of the malE protein gave rise to a functional sensor with a ratio change in the interval 0.2–0.3, similar to that of other PBP nanosensors (Fehr et al. 2002, 2003, 2004, 2005; Lager et al. 2003). A small terminal deletion is unlikely to significantly change chromophore separation in the absence of a dramatic steric constraint placed by the shortened composite linker on the ensemble of allowable chromophore positions (Fig. 6 ▶). Similarly, the linear fusion of the full-length wild-type E. coli glutamate/aspartate-binding protein YbeJ to ECFP and EYFP produced a sensor of similar overall signal change, even though YbeJ is predicted (Okumoto et al. 2005) to feature both N and C termini on a single protein lobe (a type II PBP) (Tam and Saier 1993), with little or no relative motion between them upon ligand binding. Thus, the successful engineering of the FLIPE sensor is best explained by allosteric regulation of the ensemble of relative chromophore positions by the interaction of the chromophores with one another, or by the interaction of one or both with the recognition element (Fig. 6D ▶). For both FLIPmal and FLIPE, the ensemble averaging of the orientation component is expected to contribute significantly to the overall transfer efficiency, because of the exaggerated effect of averaging on this component relative to chromophore separation (Van Der Meer et al. 1994).
Figure 6.
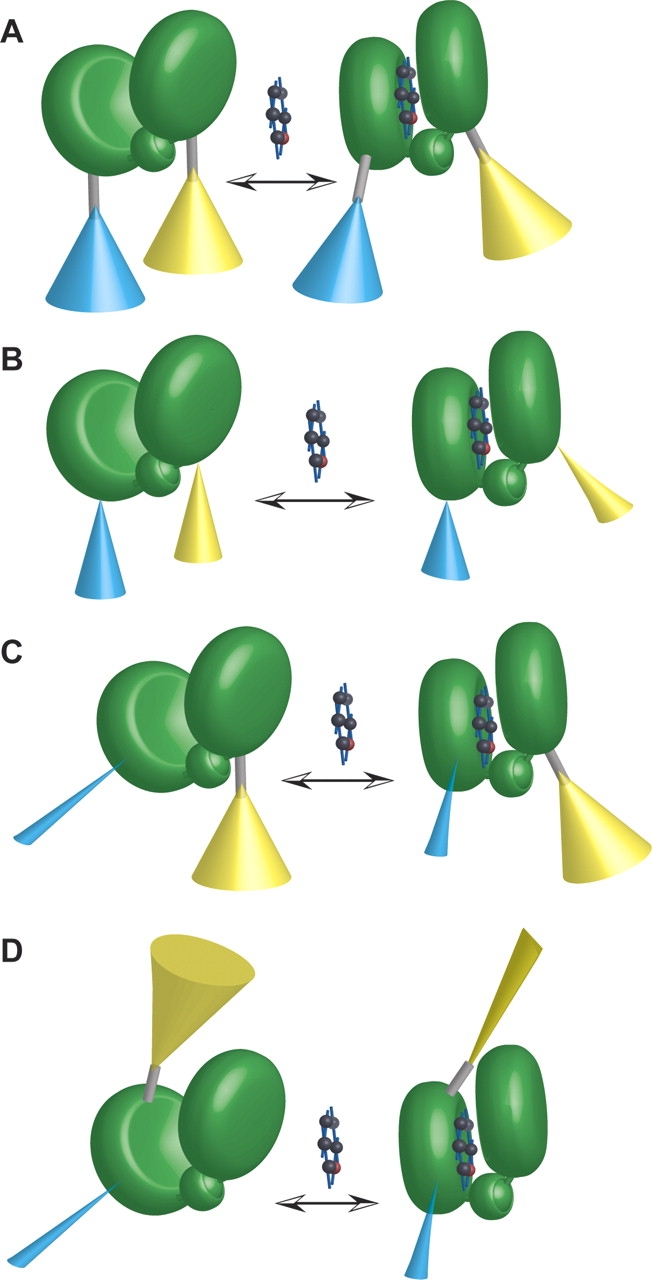
Hypothetical models of sensor function. (A) First-generation FLIPglu glucose sensor model. Fluorophores (ECFP [cyan], EYFP [yellow]) attached via flexible linkers (gray rod) to the recognition element (MglB protein, consisting of two lobes connected by a hinge [green]). The ligand (glucose) is indicated by a space-filling model. Cones represent a simplified model of the hypothetical ensemble space of fluorophore dipole. Cones are depicted comparatively wide to indicate large freedom of rotation around peptide bonds in the linkers. Thus FRET changes are expected to be relatively small. (B) Sensors with truncated linkers. Truncation reduces rotational freedom as indicated by smaller cones (reduced rotation will probably also lead to distortion of cones, not depicted). Thus FRET changes are expected to be larger. (C) Fluorophore insertions. ECFP (cyan) insertion into the recognition element leads to a dramatic reduction in rotational freedom. EYFP (yellow) is attached to the respective other lobe. Thus FRET changes are expected to be larger. (D) Fluorophore insertions. ECFP (cyan) insertion into the recognition element leads to a dramatic reduction in rotational freedom. EYFP (yellow) is attached to the same lobe carrying the insertion. A scenario is depicted in which the conformational change in the recognition element upon ligand binding restricts the rotational freedom of EYFP, providing one potential mechanism for the observation of ratio changes in the case of reporter attachment on the same lobe.
The sensors developed so far were used successfully to answer specific biological questions, such as the analysis of glucose levels in the cytosol and nuclei of mammalian cells (Fehr et al. 2003, 2004), the elucidation of a high-capacity glucose transport system at the ER membrane (M. Fehr and W. Frommer, in prep.), the ability of mammalian cells to transport ribose (Lager et al. 2003), and the visualization of glutamate release from neurons (Okumoto et al. 2005). However, the existing set of sensors may not be sensitive enough to detect subtle changes in steady-state metabolite levels, or to detect changes in high-background environments such as plants (Looger et al. 2005). Sensors with an improved signal change will have a lower relative error on the measured metabolite concentrations (Marvin et al. 1997), resulting in a larger useful dynamic range. Furthermore, improved sensor signal-to-noise will increase the efficacy of the reagents in formats such as high-throughput compound screening and validation.
We sought to optimize sensor signal change by two routes, linker truncation and internal chromophore insertion, both aimed at reducing the entropy of the chromophore relative positional ensemble. Truncation of the linker sequences of the glucose nanosensor FLIPglu-600μ had no effect below a threshold of 20 amino acids. Above this threshold, however, sensor response increased with linker truncation, up to a more than threefold increase, relative to the original sensor. The threshold is more consistent with a steric restriction of the chromophore, and a concomitant decrease in orientational averaging, than with a gradual modulation of chromophore separation in the responsive range of the ECFP-EYFP pair (Fig. 6A,B ▶).
Screening of the binding proteins MglB and YbeJ for permissive sites for ECFP insertion yielded 11/14 MglB sites and 1/7 YbeJ sites, at which chromophore insertion neither disturbed folding nor fluorescence to a large extent. The ECFP inserted into YbeJ was truncated by six amino acids relative to that inserted into MglB, potentially explaining the lower incidence of permissive sites. Of the internally fused FLIIxPglu sensors, five showed an up to fivefold enhancement of the signal relative to the linearly fused construct. Of these five sensors, four showed a glucose-dependent increase in transfer efficiency. All showed a decreased affinity for glucose relative to the linearly fused sensor, indicating that steric interactions between the inserted chromophore and the recognition element are less favorable in the closed, ligand-bound state (Marvin and Hellinga 2001). The single permissive YbeJ site discovered in this study yielded a glutamate sensor with fivefold improved signal-to-noise, and a less than twofold effect on affinity. Taken together, these results suggest a model in which the internally inserted chromophore is greatly restricted in its orientational averaging (Fig. 6C ▶), yielding high-response sensors. Many of the best sensors feature both chromophores on the same recognition element lobe, indicating that allosteric regulation by features of the recognition element during the hinge-bending motion is sufficient to drive substantial chromophore rearrangement (Fig. 6D ▶), and to produce an optimized genetically encoded sensor. It is likely that the combination of the fluorophore insertion approach with the linker deletion or the internal insertion of both reporter elements will lead to further improved sensor proteins.
Conclusions
Taken together, the data support a model in which local allosteric regulation, particularly of reporter element orientation, plays a significant role in the resonance energy transfer of a family of genetically encoded nanosensor proteins. Testing of this hypothesis by rational protein design produced sensors with greatly improved signal-tonoise, enabling a wide array of in vivo applications. Molecular modeling may provide a route to further sensor improvement, and may prove useful in the optimization of other signal transduction mechanisms, such as allosteric enzymatic switches (Guntas et al. 2004). These findings may be relevant for the optimization of other types of FRET sensors as well as the generation of novel sensors.
Materials and methods
Optimization of linearly fused constructs
FLIPglu linker variation
To lengthen the terminal linkers of pFLIPglu-600μ (Fehr et al. 2003), an MfeI site was first introduced into the N-terminal KpnI site by cassette mutagenesis (primers 1, 2), resulting in pFLIPglu-600μLI1. Subsequently, a 398-base-pair-product encoding the triple helix region from rat neuronal T-Snare syntaxin-1A (GenBank accession no. AF217191; PDB accession no. 1EZ3) was amplified with MfeI sticky ends (primers 3, 4) from Rattus norvegicus cDNA (generously supplied by Dr. Axel Brunger, Stanford University). The PCR product was cleaved with MfeI and ligated into the MfeI-linearized pFLIPglu-600μLI1. Correct orientation was verified by restriction with BamHI and BglI; the resulting construct was denoted pFLIPglu-600μLI2. To shorten the binding protein-derived linkers of the sensor, shortened versions of the mglB gene were amplified with KpnI sticky ends (primers 5–8) from pFLIPglu-600μ. The resulting PCR products were cleaved with KpnI and inserted in place of the full-length mglB gene of pFLIPglu-600μ (resulting in pFLIPglu-600μΔ1 and Δ2); correct orientation was verified by restriction with EcoRI and PstI. To shorten the N-terminal fluorophore-derived linkers of the sensor, the ECFP gene was amplified with BamHI and KpnI sticky ends from pFLIPglu-600μ (primers 9, 10) and inserted in place of the ECFP and mglB genes of pFLIPglu-600μ; the resulting plasmid was restricted with KpnI, and a shortened mglB gene (primers 7, 8) was inserted, resulting in pFLIPglu-600μΔ3. To shorten the C-terminal fluorophore-derived linkers of the sensor, the EYFP gene with KpnI and HindIII sticky ends was amplified from pFLIPglu-600μ (primers 11, 12) and inserted in place of the mglB and EYFP genes of pFLIPglu-600μ; the resulting plasmid was restricted with KpnI, and a shortened mglB gene (primers 7, 13) was inserted, resulting in pFLIPglu-600μΔ4. The N-terminal fluorophore-derived linker sequence was further shortened by site-directed mutagenesis of pFLIPglu-600μΔ4 (primer 14), resulting in pFLIPglu-600μΔ5; mutagenesis of pFLIPglu-600μΔ5 (primers 15–18) decreased the size of the fluorophore-derived linkers even more, yielding pFLIPglu-600μΔ6 through pFLIPglu-600μΔ13. All constructs are shown in Supplemental Figure 1 ▶.
Optimization of internally fused constructs
FLIPglu internal fusions
The nucleotide sequence encoding the mature MglB protein without periplasmic leader sequence (base pairs 70–927 relative to the initiator ATG) was amplified by PCR from E. coli K12 genomic DNA, with a BamHI site for EYFP cloning at either the N (primers 19, 20) or the C (primers 21, 22) terminus, and cloned into the KpnI site of the pRSETb vector (Invitrogen). All genes additionally encoded the F16A (numbered relative to the mature protein sequence) affinity mutation, to facilitate the production of sensors with dynamic range appropriate for detection of high glucose concentrations (Fehr et al. 2003). Site-directed mutagenesis (primers 23–36) was used to introduce unique NruI sites at the 14 regions of mglB selected for ECFP insertion. An ECFP PCR product (amino acids 1–238) was created with N-terminal Ala-Gly (gctggt) and C-terminal Gly-Ser (ggtagc) linkers, and Eco47III terminal restriction sites (primers 37, 38). Following Eco47III restriction, the ECFP insertion was introduced into NruI-digested plasmids by blunt-end ligation. A PCR product encoding Citrine (EYFP with the additional mutation Q69M) was amplified (amino acids 1–240; primers 39, 40) and ligated into the BamHI site of the mglB-ECFP chimeras. This resulted in a six-amino acid linker connecting MglB and Citrine: Gly-Gly-Thr-Gly-Gly-Ala (ggtggtaccggaggcgcc) for N-terminal Citrine; Gly-Ala-Gly-Thr-Gly-Gly (ggcgccggtaccggtgga) for C-terminal Citrine. Cassettes were designed to yield in-frame products at all stages to enable fluorescence screening in bacterial colonies.
FLIPE internal fusions
A nucleotide sequence encoding the mature glutamate/aspartate-binding protein (YbeJ, also GltI; protein accession no. NP_415188) without periplasmic leader sequence (as predicted by sequence alignment with closely related bacterial amino acid–binding proteins [de Lorimier et al. 2002]; amino acid positions are given relative to the initiator methionine because of the uncertainty of the signal peptide) was amplified with 5′ BamHI and 3′ HindIII sticky ends by PCR from E. coli K12 genomic DNA (primers 41, 42), and cloned into pRSETb. Site-directed mutagenesis (primers 44–50) was used to introduce unique blunt-end restriction sites at seven separation locations in the gene. A truncated ECFP PCR product (amino acids 1–232) was created with N- and C-terminal Gly (ggt) linkers, and Eco47III terminal restriction sites (primers 51, 52). The in-frame clones were screened in colonies, and only the insertion at N81V-Q82N was found to be highly fluorescent. Subsequently, a nucleotide sequence encoding Venus (EYFP with the additional mutations F46L, F64L, M153T, V163A, and S175G) (Nagai et al. 2002) was amplified (amino acids 1–240, primers 39, 40) and ligated to the 3′-end of the ybeJ-ECFP chimera using KpnI/HindIII sites; this resulted in a six-amino acid linker (Gly-Ala-Gly-Thr-Gly-Gly) connecting YbeJ with the C-terminal Venus.
In vitro analysis of sensors
Constructs were transferred to E. coli BL21(DE3)Gold (Stratagene) using electroporation, extracted and purified as described (Fehr et al. 2002). Emission spectra and ligand titration curves were obtained by using a monochromator microplate reader (Safire). The excitation filter was 433/12 nm; emission filters for ECFP and EYFP (also Citrine and Venus) emission were 485/12 and 528/12 nm, respectively. All analyses for FLIPE constructs and linearly fused FLIPglu constructs were performed in 20 mM sodium phosphate buffer (pH 7.0); analyses of FLIIXPglu were done in 20 mM MOPS buffer (pH 7.0). The Kd of each FLIPE sensor was determined by fitting to a single site binding isotherm: S=(r − rapo)/(rsat − rapo)=[L]/(Kd+[L]), where S is saturation; [L], ligand concentration; r, ratio; rapo, ratio in the absence of ligand; and rsat, ratio at saturation with ligand. Measurements were performed with at least three independent protein extracts. ECFP emission is characterized by two peaks at 485 nm and at 502 nm; the ratio was defined here as the uncorrected fluorescence intensity at 528 nm divided by the intensity at 485 nm.
Acknowledgments
We thank Atsushi Miyawaki (Wako City, Japan) for providing the Venus gene. This work was made possible by grants to W.B.F. from the NIH (Roadmap Initiative “Metabolomics Technology Development” R33DK070272), HFSP (RGP0041/2004C), and DOE (DE-FG02-04ER15542).
Abbreviations
FLIPglu, fluorescent indicator protein for glucose
FLIPE, fluorescent indicator protein for glutamate
FRET, fluorescence resonance energy transfer
FP, fluorescent protein
ECFP, enhanced cyan fluorescent protein
EYFP, enhanced yellow fluorescent protein
BP, binding protein
PBP, periplasmic binding protein
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051508105.
Supplemental material: see www.proteinscience.org
References
- Baird, G.S., Zacharias, D.A., and Tsien, R.Y. 1999. Circular permutation and receptor insertion within green fluorescent proteins. Proc. Natl. Acad. Sci. 96 11241–11246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayley, H. and Jayasinghe, L. 2004. Functional engineered channels and pores. Mol. Membr. Biol. 21 209–220. [DOI] [PubMed] [Google Scholar]
- Benson, D.E., Conrad, D.W., de Lorimier, R.M., Trammell, S.A., and Hellinga, H.W. 2001. Design of bioelectronic interfaces by exploiting hinge-bending motions in proteins. Science 293 1641–1644. [DOI] [PubMed] [Google Scholar]
- Bjorkman, A.J. and Mowbray, S.L. 1998. Multiple open forms of ribosebinding protein trace the path of its conformational change. J. Mol. Biol. 279 651–664. [DOI] [PubMed] [Google Scholar]
- Clegg, R.M. 1995. Fluorescence resonance energy transfer. Curr. Opin. Biotech. 6: 103–110. [DOI] [PubMed] [Google Scholar]
- Dattelbaum, J.D., Looger, L.L., Benson, D.E., Sali, K.M., Thompson, R.B., and Hellinga, H.W. 2005. Analysis of allosteric signal transduction mechanisms in an engineered fluorescent maltose biosensor. Protein Sci. 14 284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lorimier, R.M., Smith, J.J., Dwyer, M.A., Looger, L.L., Sali, K.M., Paavola, C.D., Rizk, S.S., Sadigov, S., Conrad, D.W., Loew, L., et al. 2002. Construction of a fluorescent biosensor family. Protein Sci. 11 2655–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehr, M., Frommer, W.B., and Lalonde, S. 2002. Visualization of maltose uptake in living yeast cells by fluorescent nanosensors. Proc. Natl. Acad. Sci. 99 9846–9851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehr, M., Lalonde, S., Lager, I., Wolff, M.W., and Frommer, W.B. 2003. In vivo imaging of the dynamics of glucose uptake in the cytosol of COS-7 cells by fluorescent nanosensors. J. Biol. Chem. 278 19127–19133. [DOI] [PubMed] [Google Scholar]
- Fehr, M., Lalonde, S., Ehrhardt, D.W., and Frommer, W.B. 2004. Live imaging of glucose homeostasis in nuclei of COS-7 cells. J. Fluoresc. 14 603–609. [DOI] [PubMed] [Google Scholar]
- Fehr, M., Okumoto, S., Deuschle, K., Lager, I., Looger, L.L., Persson, J., Kozhukh, L., Lalonde, S., and Frommer, W.B. 2005. Development and use of fluorescent nanosensors for metabolite imaging in living cells. Biochem. Soc. Trans. 33 287–290. [DOI] [PubMed] [Google Scholar]
- Fukami-Kobayashi, K., Tateno, Y., and Nishikawa, K. 1999. Domain dislocation: A change of core structure in periplasmic binding proteins in their evolutionary history. J. Mol. Biol. 286 279–290. [DOI] [PubMed] [Google Scholar]
- Gaits, F. and Hahn, K. 2003. Shedding light on cell signaling: Interpretation of FRET biosensors. Science STKE 2003 PE3. [DOI] [PubMed] [Google Scholar]
- Griesbeck, O., Baird, G.S., Campbell, R.E., Zacharias, D.A., and Tsien, R.Y. 2001. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J. Biol. Chem. 276 29188–29194. [DOI] [PubMed] [Google Scholar]
- Guntas, G. and Ostermeier, M. 2004. Creation of an allosteric enzyme by domain insertion. J. Mol. Biol. 336 263–273. [DOI] [PubMed] [Google Scholar]
- Guntas, G., Mitchell, S.F., and Ostermeier, M. 2004. A molecular switch created by in vitro recombination of nonhomologous genes. Chem. Biol. 11 1483–1487. [DOI] [PubMed] [Google Scholar]
- Haes, A.J. and Van Duyne, R.P. 2004. A unified view of propagating and localized surface plasmon resonance biosensors. Anal. Bioanal. Chem. 14 920–930. [DOI] [PubMed] [Google Scholar]
- Hoffmann, C., Gaietta, G., Bunemann, M., Adams, S.R., Oberdorff- Maass, S., Behr, B., Vilardaga, J.P., Tsien, R.Y., Ellisman, M.H., and Lohse, M.J. 2005. A FlAsH-based FRET approach to determine G protein-coupled receptor activation in living cells. Nat. Methods 2 171–176. [DOI] [PubMed] [Google Scholar]
- Jares-Erijman, E.A. and Jovin, T.M. 2003. FRET imaging. Nat. Biotechnol. 21 1387–1395. [DOI] [PubMed] [Google Scholar]
- Korndorfer, I.P., Beste, G., and Skerra, A. 2003. Crystallographic analysis of an “anticalin” with tailored specificity for fluorescein reveals high structural plasticity of the lipocalin loop region. Proteins 53 121–129. [DOI] [PubMed] [Google Scholar]
- Lager, I., Fehr, M., Frommer, W.B., and Lalonde, S. 2003. Development of a fluorescent nanosensor for ribose. FEBS Lett. 553 85–89. [DOI] [PubMed] [Google Scholar]
- Lakowicz, J.R. 1999. Principles of fluorescence spectroscopy, 2nd ed. Kluwer Academic/Plenum Publishers, New York.
- Li, X., Zhang, G., Ngo, N., Zhao, X., Kain, S.R., and Huang, C.C. 1997. Deletions of the Aequorea victoria green fluorescent protein define the minimal domain required for fluorescence. J. Biol. Chem. 272 28545–28549. [DOI] [PubMed] [Google Scholar]
- Looger, L.L., Dwyer, M.A., Smith, J.J., and Hellinga, H.W. 2003. Computational design of receptor and sensor proteins with novel functions. Nature 423 185–190. [DOI] [PubMed] [Google Scholar]
- Looger, L.L., Lalonde, S., and Frommer, W.B. 2005. Genetically encoded FRET sensors for visualizing metabolites with subcellular resolution in living cells. Plant Physiol. 138 555–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martineau, P., Guillet, J.G., Leclerc, C., and Hofnung, M. 1992. Expression of heterologous peptides at two permissive sites of the MalE protein: Antigenicity and immunogenicity of foreign B-cell and T-cell epitopes. Gene 113 35–46. [DOI] [PubMed] [Google Scholar]
- Marvin, J.S. and Hellinga, H.W. 2001. Manipulation of ligand binding affinity by exploitation of conformational coupling. Nat. Struct. Biol. 8 795–798. [DOI] [PubMed] [Google Scholar]
- Marvin, J.S., Corcoran, E.E., Hattangadi, N.A., Zhang, J.V., Gere, S.A., and Hellinga, H.W. 1997. The rational design of allosteric interactions in a monomeric protein and its applications to the construction of biosensors. Proc. Natl. Acad. Sci. 94 4366–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medintz, I.L., Goldman, E.R., Lassman, M.E., and Mauro, J.M. 2003. A fluorescence resonance energy transfer sensor based on maltose binding protein. Biocon. Chem. 14 909–918. [DOI] [PubMed] [Google Scholar]
- Miyawaki, A., Llopis, J., Heim, R., McCaffery, J.M., Adams, J.A., Ikura, M., and Tsien, R.Y. 1997. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 388 882–887. [DOI] [PubMed] [Google Scholar]
- Mowbray, S.L. and Sandgren, M.O. 1998. Chemotaxis receptors: A progress report on structure and function. J. Struct. Biol. 124 257–275. [DOI] [PubMed] [Google Scholar]
- Nagai, T. and Miyawaki, A. 2004. A high-throughput method for development of FRET-based indicators for proteolysis. Biochem. Biophys. Res. Commun. 319 72–77. [DOI] [PubMed] [Google Scholar]
- Nagai, T., Ibata, K., Park, E.S., Kubota, M., Mikoshiba, K., and Miyawaki, A. 2002. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 20 87–90. [DOI] [PubMed] [Google Scholar]
- Nagai, T., Yamada, S., Tominaga, T., Ichikawa, M., and Miyawaki, A. 2004. Expanded dynamic range of fluorescent indicators for Ca2+ by circularly permuted yellow fluorescent proteins. Proc. Natl. Acad. Sci. 101 10554–10559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, H. and Karube, I. 2003. Current research activity in biosensors. Anal. Bioanal. Chem. 377 446–468. [DOI] [PubMed] [Google Scholar]
- Okumoto, S., Deuschle, K., Fehr, M., Hilpert, M., Lager, I., Lalonde, S., Looger, L.L., Persson, J., Schmidt, A., and Frommer, W.B. 2004. Genetically encoded sensors for ions and metabolites. Soil. Sci. Plant Nutr. 50 947–953. [Google Scholar]
- Okumoto, S., Looger, L.L., Micheva, K.D., Reimer, R.J., Smith, S.J., and Frommer, W.B. 2005. Detection of glutamate release from neurons by genetically encoded surface-displayed FRET nanosensors. Proc. Natl. Acad. Sci. 102 8740–8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson, G.H., Piston, D.W., and Barisas, B.G. 2000. Förster distances between green fluorescent protein pairs. Anal. Biochem. 284 438–440. [DOI] [PubMed] [Google Scholar]
- Quiocho, F.A. and Ledvina, P.S. 1996. Atomic structure and specificity of bacterial periplasmic receptors for active transport and chemotaxis: Variation of common themes. Mol. Microbiol. 20 17–25. [DOI] [PubMed] [Google Scholar]
- Romoser, V.A., Hinkle, P.M., and Persechini, A. 1997. Detection in living cells of Ca2+-dependent changes in the fluorescence emission of an indicator composed of two green fluorescent protein variants linked by a calmodulin-binding sequence. A new class of fluorescent indicators. J. Biol. Chem. 272 13270–13274. [DOI] [PubMed] [Google Scholar]
- Scholes, G.D. 2003. Long-range resonance energy transfer in molecular systems. Annu. Rev. Phys. Chem. 54 57–87. [DOI] [PubMed] [Google Scholar]
- Stryer, L. 1978. Fluorescence energy transfer as a spectroscopic ruler. Annu. Rev. Biochem. 47 819–846. [DOI] [PubMed] [Google Scholar]
- Tam, R. and Saier Jr., M.H. 1993. Structural, functional, and evolutionary relationships among extracellular solute-binding receptors of bacteria. Microbiol. Rev. 57 320–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Meer, B.W., Cooker, G.I., and Chen, S.Y.S. 1994. Resonance energy transfer. VCH Publishers, New York.
- Villaverde, A. 2003. Allosteric enzymes as biosensors for molecular diagnosis. FEBS Lett. 554 169–172. [DOI] [PubMed] [Google Scholar]
- Zaccolo, M. 2004. Use of chimeric fluorescent proteins and fluorescence resonance energy transfer to monitor cellular responses. Circ. Res. 94 866–873. [DOI] [PubMed] [Google Scholar]
- Ziegler, C. 2004. Cantilever-based biosensors. Anal. Bioanal. Chem. 379 946–959. [DOI] [PubMed] [Google Scholar]