

Abstract
Achieving long-term expression of a therapeutic gene in a given hematopoietic lineage remains an important goal of gene therapy. Congenital erythropoietic porphyria (CEP) is a severe autosomal-recessive disorder characterized by a deficiency in uroporphyrinogen III synthase (UROS), the fourth enzyme of the heme biosynthetic pathway. We used a recently obtained murine model to check the feasibility of gene therapy in this disease. Lentivirus-mediated transfer of the human UROS cDNA into hematopoietic stem cells (HSCs) from Urosmut248 mice resulted in a complete and long-term enzymatic, metabolic, and phenotypic correction of the disease, favored by a survival advantage of corrected red blood cells. These results demonstrate that the cure of this mouse model of CEP at a moderate transduction level supports the proof of concept of a gene therapy in this disease by transplantation of genetically modified hematopoietic stem cells.
Introduction
Congenital erythropoietic porphyria (CEP [MIM 263700]), or Günther's disease, is a rare disease that is inherited as an autosomal-recessive trait and is characterized by severe cutaneous photosensitivity with mutilating involvement and chronic hemolysis.1,2 The profound uroporphyrinogen III synthase (UROS; EC 4.2.1.75) deficiency3 leads to massive excretion of type I isomers of porphyrins, an excretion that results from excessive synthesis of these compounds in the bone marrow. Mutation analysis has shown a large variety of molecular lesions and has provided genotype/phenotype correlations to predict the severity of the clinical evolution of the disease.4–7 The clinical severity of the anemia and cutaneous lesions is highly variable, and, for some patients, prognosis is poor, with death in early adult life or in the neonatal period.8,9 Classical treatments are only symptomatic and unsatisfactory. Because the predominant site of metabolic expression of the disease is the erythropoietic system, bone marrow (BM) transplantation represents a curative treatment for patients with severe phenotypes, as long as a human leucocyte antigen (HLA)-compatible donor is available.10–19 However, in the absence of HLA-compatible donors, gene therapy with hematopoietic stem cells (HSCs) is a promising curative treatment.
Therapeutic trials targeting hematopoietic cells have been initiated for a number of monogenic diseases, including severe combined immunodeficiency diseases due to common γ chain (SCID-X1)20,21 ([MIM 300400]) or adenosine deaminase (SCID-ADA)22–26 ([MIM 102700]) deficiencies, Gaucher disease27 ([MIM 230800]), and chronic granulomatous disease (CGD)28,29 ([MIM 306400]). In general, the main problem resides in the low percentage of corrected cells in peripheral blood several months following the treatment. One exception is the setting in which a selective advantage is conferred to the corrected target cell. This is the case for SCID-X1, which was the first success of gene therapy with full correction of disease phenotype and clinical benefit, and also for SCID-ADA and CGD. In CGD, a recent clinical trial using retroviral vectors showed significant clinical benefit, owing to an unexpected expansion in vivo of gene-transduced cells by insertional activation of MDS1-EVI1, PRDM16, or SETBP1.29 A recent murine model of CEP30 allowed us to test this approach in this disease. Homozygous Urosmut248/mut248 mice (hereafter called CEP mice) closely mimic the symptoms observed in the human disease. Here, we report a complete enzymatic, metabolic, and phenotypic correction of CEP mice after HSC gene transfer. Moreover, we demonstrate a survival advantage of corrected red blood cells. Thus, our results strongly support the use of HSC gene therapy in clinical trials for CEP patients.
Material and Methods
Construction and Production of Lentiviral Vectors
Vectors were generated from a self-inactivating (SIN) lentiviral vector. The EFpGFP and MNDpGFP vectors derived from the pRRL-PGK-GFP-WPRE lentiviral-vector backbone kindly provided by Dr. D. Trono were previously described.31 ESpUROS vector was derived from EFpGFP by replacement of GFP with the human UROS cDNA and EFp by the chimeric HS40/Ankp.32 Cloning details are available upon request. VSV-G-pseudotyped lentiviral vectors were produced by triple-transient transfection of 293T cells as previously described.33 HIV-1 p24 antigen levels were measured in the concentrated viral supernatants by ELISA assay (Innotest HIV p24 INGEN SA, Rungis, France) to determine viral production. For ESpUROS-EFpGFP and EFpGFP lentiviral supernatant, infectious titers were determined by transduction of erythroblastic K562 cells with serial dilutions of viral supernatant. EGFP expression was quantified 5 days later by flow cytometric analysis. For ESpUROS lentiviral supernatant, viral titers were estimated by comparison of p24 antigen levels of each lentiviral supernatant with a ESpGFP lentiviral supernatant produced simultaneously. Lentiviral-vector supernatants were tested for the presence of replication-competent lentivirus (RCL) as described34 and were found to be free of RCL.
Purification, Transduction, and Transplantation of Murine BM Cells
Donor BALB/cJ CEP mice were injected intraperitoneously with 5-FU (150 mg/kg per mouse). Five days later, nucleated BM cells were harvested by flushing of femurs and tibiae, purified by Ficoll-Paque Plus gradient separation (Amersham Pharmacia Biotech, Orsay, France), and sorted for Sca-1+ cells with the MACS Sca-1 kit (Milteny Biotech, Auburn, CA, USA) (5-FU/Sca-1+ cells). These cells were prestimulated for 6 hr in RM-B00 medium (MABIO-International Laboratories, Tourcoing, France) containing 10% FCS and 50 μM dNTPs and supplemented with the following cytokines: rmSCF (100 ng/mL), rhTPO (100 ng/mL), rhFlt3-L (100 ng/mL), rhIl-6 (10 ng/mL) and rmIl-3 (20 ng/mL). 5-FU/Sca-1+ cells (4 × 105/ml) were transduced with lentiviral vectors at different multiplicity of infections (MOIs) (2–200).
Twenty-four hours after transduction, 5-FU/Sca-1+ cells (7,000 cells in the preliminary experiment and 13,600 to 16,400 cells for the further experiments) were injected into the tail vein of 6–10-week-old female CEP mice that had been conditioned with two doses (25 mg/kg) of Busilvex 24 hr apart. Twenty weeks after bone-marrow transplantation (BMT), animals were sacrificed and BM cells were harvested. For transplantation of secondary mice, 2–3 × 107 BM cells from primary mice were collected, washed twice, and injected into the tail vein of 6–10-week-old female CEP mice that had been conditioned with two doses (25 mg/kg) of Busilvex 24 hr apart. Neither mortality nor malignant events were observed in these primary and secondary experiments.
Ter-119+ and Ter-119− Cell Purification
BM cells from normal, CEP, and secondary mice were collected by flushing of tibiae and femurs. Fifteen to twenty million whole BM cells were collected, washed twice in PBS, and resuspended in PBS containing 2 mM EDTA with 0.5% HSA. Cells were sorted for Ter-119+ and Ter-119− fractions with the MACS TER-119 MicroBeads kit (Miltenyi Biotec, Auburn, CA). More than 90% purity of both Ter-119+ and Ter-119− fractions was obtained as assessed by flow cytometric analyses.
PCR Analysis of Proviral Integration in Colony-Forming Cells
We plated 1500 Sca-1+ cells before graft or 50,000 BM cells from grafted mice in a methylcellulose-based medium (Methocult GF M3434, Stem Cell Technologies). Colony-forming cells (CFCs) were scored on day 10, and 20–60 colonies from each mouse were picked, washed twice with PBS, and frozen. Cells were then digested with proteinase K in lysis buffer (10 mmol/l Tris-Cl, pH 8.0, 50 mmol/l KCl, 2.5 mmol/l MgCl2, 0.5% Tween-20, 100 ng/ml proteinase K) at 50°C for 1 hr, followed by a 10 min exposure at 95°C to inactivate the proteinase K. The presence of the provirus was characterized by PCR with specific primers designed in the woodchuck posttranscriptional regulatory element (WPRE) sequence to generate a 220 bp fragment: W1 5′-TGCTGTCTCTTTATGAGGAG-3′ as a forward primer and W2 5′-GAATTGTCAGTGCCCAACAG-3′ as a reverse primer. Nontransduced colonies were also used as negative controls.
Flow Cytometric Analysis
Flow cytometric analyses (FACS analyses) of GFP expression in peripheral blood (PB) or BM were performed on a FACS Calibur (Becton Dickinson, San Jose, CA). Granulocytes and monocytes in PB were gated by forward and side scatter and then analyzed for GFP expression. Purity of 5-FU/Sca-1+ cells was assessed by labeling of cells with FITC-conjugated anti-Sca-1+ antibody (BD PharMingen, San Diego, CA). BM cells were labeled with PE-conjugated anti-Ter-119 and PE-conjugated anti-GR1 antibodies, (BD PharMingen). The percentage of GFP-positive lineage cells was calculated as the percentage of GFP-expressing cells in the total PE-positive gated regions.
Analysis of Chimerism in Transplanted Mice
The use of males as donors and females as recipients (or reciprocally) made it possible to determine chimerism by fluorescence in situ hybridization (FISH). BM cells were hybridized with a Y FITC-labeled paint probe for the mouse chromosome Y and a cyanin-3-labeled paint probe for the mouse chromosome X (Valbiotech, Paris, France), as described.35 A minimum of 300 cells were examined.
Hematologic Analysis
Blood was collected by puncture of the orbital sinus. Total hemoglobin, red cell counts, and hematocrit were measured on an Animal Blood Counter (vetABC, SCIL, Montpellier, France). Analyses of porphyrin-accumulating cells in PB and BM were performed with a FACS Calibur (BD Biosciences, Le Pont de Claix, France). Porphyrin fluorescence was detected in the FL-3 channel. Reticulocytes were labeled with orange thiazol and counted in the FL-1 channel. Half-life was determined with 3 mg of Biotin-X-NHS (Calbiochem, La Jolla, CA) injected intravenously three times 24 hr apart. RBCs were then detected with a Streptavidin-APC antibody, (BD PharMingen).
Biochemical Measurements
The quantification of the different porphyrins and the uroporphyrin isomers in urines were done by reverse-phase high-performance liquid chromatograph (HPLC).36 Uroporphyrinogen III synthase (UROS) activity was determined by an enzyme-coupled assay as described previously.37 One unit was defined as the amount of enzyme that formed one nmol of uroporphyrinogen III per hour at 37°C.
Skin Photosensitivity and Morphological Studies
Reversal of skin photosensitivity was assayed with an irradiation protocol 5 months after transplantation. Dorsal mouse skin was depilated to render mouse skin accessible to irradiation, and the skin was irradiated at a total dose of 1350 kJ/m2 solar-simulated radiation (SSR) delivered in three sessions with a Sunlight CPS+ Atlas (Moussy le Neuf, France). Skin pictures were taken 5 days after irradiation. Sheets of dorsal mouse skin were fixed in 4% formalin, dehydrated, embedded in paraffin, cut in 5 μm sections, and stained with hematoxylin and eosin to visualize the irradiation-induced damage.
DNA Analysis
DNA was extracted from BM cells, digested with EcoR1 and Kpn1 restriction enzymes, and subjected to Southern-blot analysis in order to confirm the presence of unrearranged provirus and determine the integrated vector copy number. A 32P-labeled WPRE probe was used to hybridize the membrane, and the resulting bands were visualized and quantitated with a Phosphorimager (GE Healthcare, Life Sciences).
Quantitative PCR of Proviral DNA
DNA was extracted from 3 × 106 cells with NucleoSpin Tissue kit (Macherey-Nagel), and real-time PCR was performed on 100 ng of DNA with Taqman technology (Applied Biosystems). DNA from nontransduced mice was isolated and amplified at the same time to demonstrate the absence of contamination. Samples were heated to 50°C for 2 min and to 95°C for 10 min, and then 40 cycles of PCR were performed with 15 s at 95°C and 1 min at 60°C. The following sequences were used: 5′-ATGGCTTTCATTTTCTCCTCCTT-3′ and 5′-CGGGCCACAACTCCTCAT-3′ as primers and 5′-CCAACTGGGACGACATG-3′ as Taqman probe. Real-time PCR was performed on β-actin gene for normalization purposes with 5′-GACCCTGAAGTACCCCATTGAAC-3′ and 5′-CACGCAGCTCATTGTAGAAGGT-3′ as primers and 5′-ATCCTGGTTGCTGTCTC-3′ as Taqman probe. A standard cell line was created by infection of murine 3T3 cells with MNDpGFP vector at an MOI of 0.5 and isolation of a 3T3 clone with one proviral insert. DNA from this cell line was used as a standard for quantification by the ΔCt method.
Statistical Analysis
Student's paired t tests were used to compare untreated and treated mice. Simple linear regression analysis was computed to determine the association between the percentage of GFP+ RBCs and GFP+ WBCs. The regression correlation coefficient R2 allowed us to measure the proportion of variability explained by, or due to, the regression in the sample of paired data observed. Regression coefficient models were compared to demonstrate the selective advantage of corrected cells. All computations were performed with Excel software and Stata version 7.0 software (Stata, College Station, TX). All p values were two-sided, and p values less than 0.05 were considered significant.
Results
Enzymatic, Metabolic, and Phenotypic Correction in GT-Treated CEP Mice
The mouse model of CEP displays erythrodontia, moderate photosensitivity, hepatosplenomegaly, and hemolytic anemia.30 Total porphyrins are highly increased in urine and feces, whereas UROS enzymatic activity is below 1% of the normal level in the different tissues analyzed. Red blood cells (RBCs) accumulate large amounts of uroporphyrin I and are named fluorocytes because of a characteristic red fluorescence analyzed by spectrofluorimetry or flow cytometry. These pathological findings closely mimic the CEP disease in humans.
The present study tested the feasibility of an ex vivo HSC gene therapy of the disease with a lentiviral vector. Considering the predominant erythropoietic expression of the disease, we decided to use the previously characterized erythroid-specific HS-40 enhancer-ankyrin-1 promoter32,38,39 to drive the expression of the UROS cDNA. We transduced 7,000 to 15,0000 5-FU/Sca1+ BM cells from CEP donor mice with the erythroid-specific lentiviral vector ESpUROS (Figure 1A) and injected them into CEP recipient mice conditioned with two doses (25 mg/kg) of busulfan (Busilvex). In a preliminary experiment with four mice (MOI of 200), we obtained 83% CFC integration before grafting, leading to a fully corrected phenotype with a 200-fold decrease in total urinary porphyrins, as well as the disappearance of fluorocytes in peripheral blood. Southern-blot analysis of genomic DNA from BM at sacrifice demonstrated the presence of a single 2.2 kb band, indicating unrearranged provirus with a copy number per cell of 17.8 ± 1.7 (data not shown), similar to that measured by qPCR (15.5 ± 1.3). However, for a clinical trial, it would be important to introduce fewer vector copies per cell to reduce the risk of insertional oncogenesis. We then performed five new experiments (20 mice) with lower MOIs (60 or 20 for group I, 20 or 6 for group II, and 2 for group III), and the results were analyzed as three groups according to a similar transduction efficiency in CFCs before graft: 62.5%–68.2% for group I, 42.9%–45.5% for group II, and 25% for group III (Table 1). Five months after the treatment in the first two groups, there was a complete (group I) or partial restoration (group II) of enzymatic activity in BM (Figure 1B). A dramatic decrease in porphyrin accumulation in urine (Figure 1C; 100–10-fold decrease versus CEP mice) was obtained, leading to the absence of reddish-colored urine. FACS analyses showed a rapid disappearance of fluorocytes as early as one month after transplantation in these first two groups (0.1 ± 0.1% and 2.7 ± 1.9% for groups I and II, respectively, versus 32.5 ± 2.5% in CEP mice; Figures 1D and 1E). However, transduction at a very low level (25% in the CFCs at the time of transduction, group III) led to a partial metabolic correction (43% decrease in urinary porphyrins and 33% decrease in fluorocytes versus CEP mice). Anemia was corrected in groups I and II with a significant increase in hemoglobin concentration and RBC levels (Table 1) rising to the normal range (no significant difference compared with normal Balb/c mice). Erythrocyte hemolysis was dramatically reduced as reflected by the reticulocyte counts, the restoration of RBC half-life, and the considerable reduction in splenomegaly (Table 1). However, these three parameters are not completely normalized for these two groups, because data differ significantly from the wild-type as indicated by the statistical analysis (Table 1). Only a mild improvement occurred in all hematological parameters in group III (Table 1), according to the very low level of vector copy number per cell (0.2 ± 0.1, Table 1).
Figure 1.
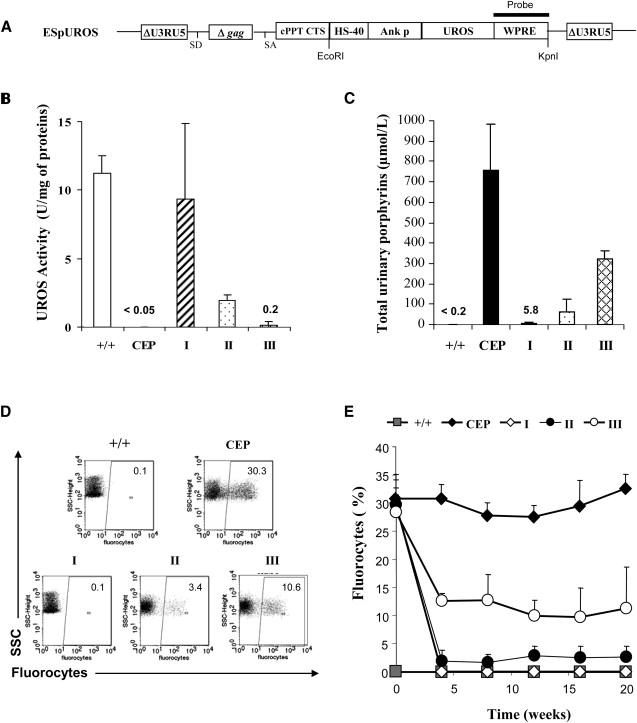
Enzymatic, Metabolic, and Phenotypic Correction of Transplanted Mice
(A) Schematic representation of the proviral form of the erythroid-specific SIN lentiviral vector ESpUROS. The following abbreviations are used: ΔU3, U3 region deleted of tata box and enhancer sequences; cPPT, central polypurine tract; CTS, central termination sequence; Δgag, truncated gag region; SA, splice acceptor site; SD, splice donor site; HS40, enhancer of the human α-globin gene; Ankp, ankyrin promoter of the human ankyrin gene; UROS, cDNA of uroporphyrinogene III synthase; and WPRE, woodchuck posttranscriptional regulatory element. The WPRE probe for Southern-blot analysis is indicated.
(B) UROS activity in BM cells 20 weeks after transplantation from unmanipulated wild-type BALB/cJ mice (+/+), from deficient CEP mice (CEP), and from the three treated groups of mice (groups I to III).
(C) Level of urinary-porphyrin accumulation in the different groups of mice determined by HPLC.
(D) Representative FACS analysis of the percentage of fluorocytes in peripheral blood from one mouse in each group of controls or treated mice.
(E) Percentage of fluorocytes monitored over time by FACS in all groups.
Values represent mean ± standard deviation (SD).
Table 1.
Proviral Integration and Hematological Parameters in Mice 20 Weeks after BM Transplantation
| Mice | % CFC Integration |
Donor Cells (%) | Proviral Copy Number (at sacrifice) | Hematocrit (%) | Hb (g/dL) | RBCs (106/ml) | Reticulocytes (%) | RBC half life (days) | Spleen weight (mg) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Before Graft | At Sacrifice | |||||||||
| Normal (n = 8) | na | na | na | na | 48.2 ± 4.4 | 14.3 ± 1.1 | 9.4 ± 0.7 | 3.8 ± 0.4 | 8.5 ± 0.5 | 130 ± 10 |
| Deficient (n = 8) | na | na | na | na | 29.2 ± 3.1 | 8.1 ± 0.4 | 6.5 ± 0.6 | 46.7 ± 7.0 | 4.9 ± 0.2 | 1500 ± 200 |
| Deficient treated | ||||||||||
| Group I (n = 8) | 62.5–68.2 | 90.4 ± 10.5 | 92.2 ± 9.2 | 3.8 ± 1.4 | 44.6 ± 2.1∗ | 13.3 ± 0.6∗ | 9.4 ± 0.4∗ | 5.4 ± 0.7∗# | 8.0 ± 1.5∗# | 260 ± 50∗ |
| Group II (n = 8) | 42.9–45.5 | 58.7 ± 18.7 | 97.1 ± 3.4 | 0.6 ± 0.2 | 41.6 ± 3.6∗# | 12.0 ± 1.1∗# | 9.2 ± 0.7∗ | 11.0 ± 5.1∗# | 7.9 ± 0.8∗# | 470 ± 90∗ |
| Group III (n = 4) | 25 | 22.1 ± 16.1 | 98.9 ± 0.5 | 0.2 ± 0.1 | 30.2 ± 1.2# | 8.9 ± 0.7# | 6.7 ± 0.7# | 27.0 ± 4.3∗# | 7.2 ± 0.8∗# | 1000 ± 480 |
| Secondary mice (n = 6) from group I | 92.2 ± 4.0 | 93.1 ± 2.2 | 90.9 ± 10 | 4.3 ± 2.1 | 48.9 ± 2.7∗ | 14.2 ± 0.8∗ | 10.1 ± 0.7∗ | 5.0 ± 0.8∗ | nd | 240 ± 30∗ |
The following abbreviations are used: Hb, hemoglobin; RBCs, red blood cells; na, not applicable; and nd, not done. ∗indicates p < 0.001 versus deficient mice. # indicates p < 0.01 versus normal mice. Note: Deficient treated mice were grafted between 6 and 10 weeks of age. All the mice were 26–30 weeks old at sacrifice. Primary recipient mice (groups I, II, and III) received between 13,600 and 16,400 5-FU/Sca-1+ transduced cells. Secondary recipient mice received 2–3 × 107 whole BM cells from group I mice.
For evaluation of the clinical correction, a photosensitivity assay was performed 20 weeks after BM transplantation. Normal mice did not demonstrate any macroscopic or microscopic skin lesions 5 days after UV exposure, whereas macroscopic lesions and immune cell infiltrates were observed in nontransplanted CEP mice. Skin photosensitivity completely disappeared in mice treated by gene therapy, except those in group III, which showed a moderate skin photosensitivity (not shown). From these cohorts of treated mice, we concluded that our erythroid-specific lentiviral vector was relevant for efficient correction of the disease.
Long-Term Stability and Erythroid-Specific Expression
To study the long-term expression of the therapeutic gene in HSCs, we carried out eight secondary transplants with BM cells from the eight group I primary recipients. Twenty weeks after BM transplantation, two secondary mice presented a very low chimerism (3% and 10.6%, respectively) and were not included in the analysis. For the six other secondary mice (90.9 ± 10% chimerism, Table 1), a complete correction of the disease was obtained. Indeed, these highly engrafted mice did not present any skin lesions, a finding in accordance with the restoration of BM UROS activity (13.5 ± 1.9 U/mg of proteins for secondary mice versus 12.1 ± 0.3 for normal control mice in total BM; Figure 2A). These mice also presented a decrease in the percentage of fluorocytes in RBCs (Figure 2B) and in urinary porphyrins (Figure 2C). Finally, hematological parameters were normalized (Table 1, last line). To demonstrate that the correction occurred with a specific expression of the transgene in the erythroid lineage, we isolated Ter-119-positive and -negative BM cells from normal, CEP, and secondary mice and measured UROS activity (Figure 2A). Specific activity in normal mice was 15-fold higher in erythroid Ter-119+ compared to Ter-119− cells, thereby confirming the preferential expression of the mouse endogenous UROS in the erythropoietic compartment. As expected, UROS activity was very low in both cellular populations from CEP mice. In contrast, in secondary mice, UROS activity in Ter-119+ cells was completely restored (31.2 ± 2.1 U/mg of proteins for CEPII mice versus 33.6 ± 0.3 U/mg of proteins in normal mice, Figure 2A). On the other hand, we detected a very low activity in Ter-119− compared with Ter-119+ cells (p < 0.001), demonstrating the tissue-specific expression of the vector in vivo. Therefore, UROS expression limited to the erythroid lineage was sufficient to correct photosensitivity in CEP mice.
Figure 2.
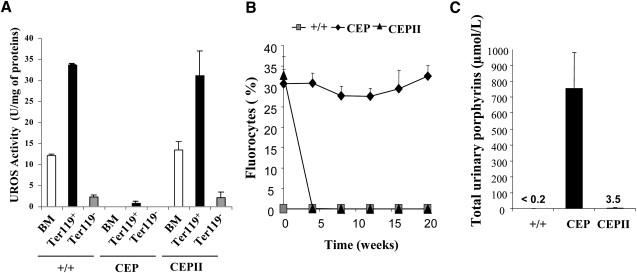
Erythroid-Specific Expression in Secondary Mice
(A) UROS activity in whole BM and in Ter-119+- and Ter-119−-sorted BM cells 20 weeks after transplantation from wild-type (+/+), CEP-deficient mice (CEP), and secondary recipients (CEPII).
(B) Percentage of fluorocytes monitored over time by FACS from all groups.
(C) Level of urinary-porphyrin accumulation in different groups of mice determined by HPLC.
Values represent mean ± SD.
Selective Advantage of Corrected Cells
Interestingly, in these experiments, the same full correction was observed for a percentage of CFC integration varying between 43% and 68% (first two groups). These results suggested a selective advantage of corrected cells due to a survival advantage of red blood cells and/or a proliferative advantage of erythroid progenitors. To confirm the presence of a selective advantage, we performed cell-therapy experiments with mixed proportions of deficient and normal cells (Figure 3). With a high proportion of normal cells (45% and 60%), we obtained a complete metabolic correction as shown by the low percentage of fluorocytes (Figure 3A) and the correction of anemia (Figure 3B). Interestingly, a mixture of 30% of normal and 70% of deficient cells also led to a high correction with a 77% decrease in fluorocytes. A mixture of 15% of normal and 85% of deficient cells led to a partial correction with a 33% decrease in fluorocytes.
Figure 3.
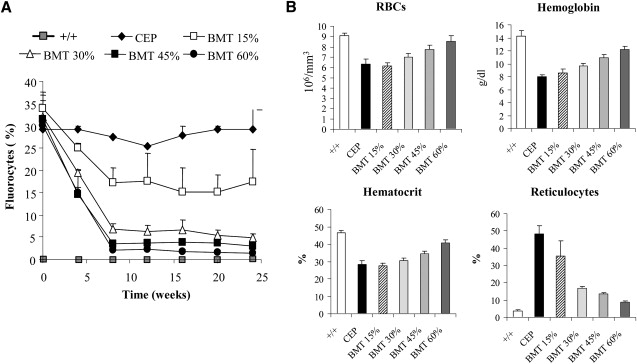
Cell-Therapy Experiments
(A) Percentage of fluorocytes monitored over time by FACS in the CEP-deficient mice (CEP) and mice treated by BM transplantation: BMT 15% (15% of normal cells mixed with 85% of deficient cells), BMT 30%, BMT 45% and BMT 60%. (B) Hematological parameters (RBCs, hemoglobin, hematocrit and reticulocyte counts) were analyzed for the different groups of mice 24 weeks after transplantation.
To investigate the selective advantage mechanism, we designed experiments with two new vectors: One is a marker GFP vector under the control of the ubiquitous elongation factor 1 alpha (EF1α) promoter, named EFpGFP; the other is a bipromoter vector expressing both the UROS cDNA driven by the erythroid-specific promoter and the GFP under the control of the EF1α promoter, named ESpUROS-EFpGFP (Figure 4A). Using the simple marker vector EFpGFP, we first studied the level of GFP expression in the erythroid and nonerythroid compartments at different MOIs (Figure 4B). Analysis of CEP mice at 4 and 12 weeks showed a lower expression of GFP in RBCs compared to white blood cells (WBCs), with a ratio of 0.36 (slope of the linear regression curve). This different level of expression was not due to the disease itself because a similar ratio (0.42–0.48) was found with normal mice. Rather, it was due to a characteristic of the promoter, which is weaker in RBCs compared to WBCs. In contrast, experiments performed with the therapeutic vector ESpUROS-EFpGFP showed a higher GFP expression in RBCs with an inverted ratio (2.29–2.34, p < 0.001) (Figure 4B). This result is due to the presence of the therapeutic gene, with a survival advantage of genetically corrected cells compared to the deficient red blood cells. As expected, Figure 4C shows the inverse correlation between the percentage of fluorocytes (corresponding to noncorrected cells) and the percentage of GFP-positive RBCs.
Figure 4.
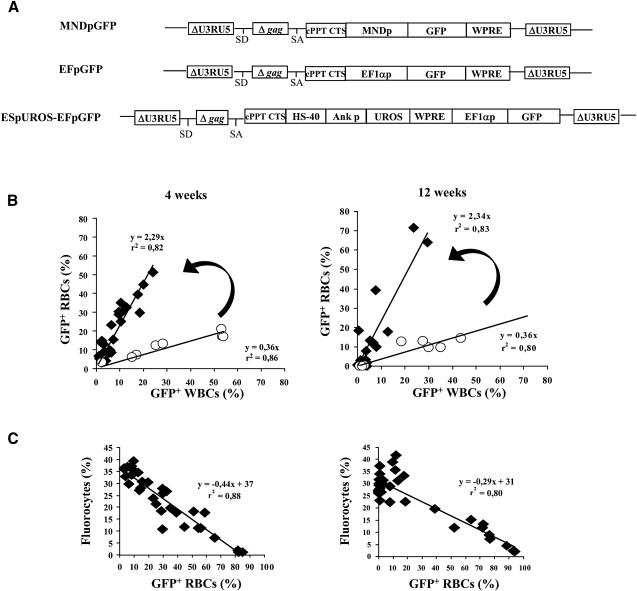
Selective Advantage of Corrected Erythropoietic Cells
(A) Schematic representation of the proviral form of the three SIN lentiviral vectors. The first two are constitutive vectors expressing GFP from the LTR-derived MND or the human cellular EF1α promoter. The third is a bipromoter vector coexpressing UROS cDNA from the erythroid HS40/Ank promoter and GFP from the EF1α promoter.
(B) GFP expression in red blood cells (RBCs) and white blood cells (WBCs), 4 and 12 weeks after BM transplantation. The dots correspond to individual CEP mice transplanted with HSCs transduced at five different MOIs (2, 6, 20, 60, and 80) either with the EFpGFP (○) or with the ESpUROS-EFpGFP (◆) vector. Arrows emphasize the difference in the slopes of the curves obtained with the two vectors, reflecting the survival advantage of corrected RBCs.
(C) Relation between the percentage of fluorocytes and the percentage of GFP+ RBCs at 4 and 12 weeks. CEP mice were transplanted with HSCs transduced with the therapeutic ESpUROS-EFpGFP vector.
For determining whether a selective advantage occurred in the bone marrow at an early stage of the erythropoietic differentiation, cells were gated on Ter-119 expression (Figure 5A) and three different groups were distinguished according to their side scatter (SSC) and forward scatter (FSC) profiles: (III) early normoblasts, (II) intermediate normoblasts, and (I) RBCs (Figure 5A). The percentage of GFP-positive cells in these three groups was compared with that in the myeloid compartment (Gr-1-positive cells). When the simple EFpGFP marker vector in normal and deficient mice was used, GFP expression was lower in the different populations of the erythroid compartment compared to Gr-1-positive cells (Figure 5B). This was observed previously in peripheral blood when we compared the percentages of GFP-positive cells between RBCs and WBCs (Figure 4B). This is a characteristic of the EF1α promoter given that the use of another constitutive promoter (MNDp) led to an identical level of expression in all lineages (Figure 5B, left part). Using the therapeutic ESpUROS-EFpGFP vector, we observed a 2-fold increase in GFP expression in early normoblasts compared to the use of the simple vector (EFpGFP), suggesting that a moderate proliferative advantage exists at an early stage of erythroid differentiation. In addition, a 5-fold increase in the percentage of GFP-expressing cells was observed in peripheral RBCs between EFpGFP-treated and ESpUROS-EFpGFP-treated mice. This result is in accordance with the results of Figure 4B, demonstrating the survival advantage of peripheral corrected cells.
Figure 5.
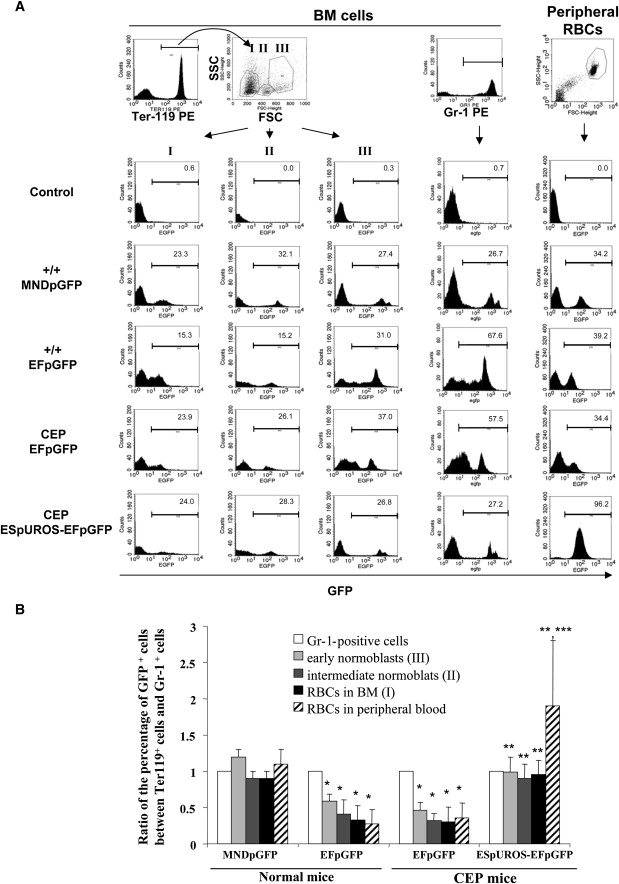
Selective Advantage of Corrected Cells of the Erythroid Lineage
(A) Representative FACS analysis of BM cells and peripheral RBCs from each group of mice. BM cells were analyzed for Ter-119 expression, and three different groups were differentiated by their SSC and FSC profiles (upper left part): (I) RBCs, (II) intermediate normoblasts, and (III) early normoblasts. BM cells were also analyzed for Gr-1 expression (upper middle part). GFP expression was then analyzed in each population of erythroid (Ter-119) and myeloid (Gr-1) BM cells and in peripheral RBCs (right part).
(B) Ratio of the percentage of GFP expression between the different populations of erythropoietic cells and Gr-1-positive cells. ∗p < 0.01 versus MNDpGFP vector in normal mice; ∗∗p < 0.001 versus EFpGFP vector in normal and CEP mice; ∗∗∗p < 0.001 versus ESpUROS-EFpGFP vector in the different populations of erythropoietic cells of CEP mice.
Values for (B) represent mean ± SD.
Level of Transduction Necessary to Obtain a Complete Correction of CEP Mice
To assess the level of transduction necessary for a metabolic correction, we analyzed the percentage of GFP+ WBCs (which reflects the initial percentage of HSC transduction) versus the percentage of fluorocytes at 4 and 12 weeks. The population of mice corresponding to a percentage of transduction ≥ 40% presented a total metabolic and phenotypic correction (Figure 6). These results are in agreement with the initial experiments obtained with the first two groups of mice, which presented a complete or near-complete metabolic correction (see Table 1). In contrast, mice grafted with a percentage of genetically modified cells ≤ 10% had neither metabolic nor phenotypic correction: They showed a percentage of fluorocytes between 20% and 40% and remained photosensitive (Figure 6). Experiments performed with a percentage of transduction between 10% and 40% led to a partial correction (fluorocytes between 5% and 15%, mild photosensitivity, Figure 6 and group III in Table 1).
Figure 6.
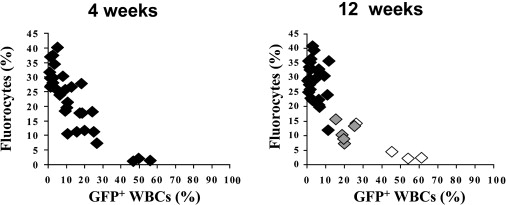
Level of Transduction Necessary to Obtain a Complete Correction of the Phenotype
CEP mice were transplanted with HSCs transduced with the therapeutic ESpUROS-EFpGFP vector (same experiments as in Figure 4C), and the percentages of fluorocytes and GFP+ WBCs were determined for each mouse at 4 and 12 weeks. In the right part, skin photosensitivity was assayed 20 weeks after transplantation: (◆), mice with severe skin photosensitivity; ( ), mice with mild photosensitivity; and (◇) mice without any macroscopic or microscopic lesions.
), mice with mild photosensitivity; and (◇) mice without any macroscopic or microscopic lesions.
Discussion
The evaluation of the efficacy and safety of lentiviral-mediated gene therapy for genetic diseases requires an animal model. We have now developed a knock-in mouse model of CEP (Urosmut248) that reproduces the missense mutation (pP248Q) observed in the human disease.30 Homozygous urosmut248/mut248 mice presented with skin photosensitivity and hemolytic anemia as observed in the disease in humans. Recently, Bishop et al.40 reported a new viable homozygous knock-in mouse model of CEP in which the C73R/V99A mutations are responsible for a milder phenotype. Our new model allowed us to test an ex vivo gene therapy consisting in the autograft of genetically modified stem cells. By using a lentiviral vector to deliver an erythroid-specific expression cassette for human UROS cDNA, we achieved persistent and long-term correction of porphyrinuria and hemolytic anemia and a complete cure of the CEP phenotype, i.e., photosensitivity, in this new model. Correction of the disease in secondary mice (in which engrafted cells are provided by the first transplanted mice) clearly demonstrates that the gene correction occurs at the stem cell level. This correction was efficient at a moderate transduction level of HSCs in the absence of a selection system. Because of shortened red cell survival in our disease, gene-corrected erythroid cells are likely to be preferentially amplified. Here we clearly demonstrate a survival advantage of corrected red blood cells with 5-fold amplification and restoration of their half-life. A significant amplification of the erythroid lineage derived from the genetically normal HSCs was also observed in a murine model of β-thalassemia.41,42 These findings constitute a significant advance over other hematologic diseases requiring a selection system or a high transduction level of HSCs for a complete cure. Our results contrast with the data obtained with another porphyria, erythropoietic protoporphyria (EPP, [MIM 177000]), which is characterized by an accumulation of protoporphyrin in RBCs and the absence of hemolytic anemia. During the course of gene therapy of this murine model, no selective advantage was found.33,43,44 Other studies will be needed to test whether the survival advantage is sufficient to allow a milder conditioning regimen, as is the case for gene therapy in other genetic diseases. For example, Aiuti et al. successfully transplanted autologous ADA-transduced stem cells into two children with ADA-SCID after nonmyeloablative conditioning with busulfan (4 mg/kg).22 However, if we consider the necessity to use low multiplicity of infection in order to avoid multiple integration events, the transduction efficiency will not reach more than 40%–60%. In this paper, we demonstrate that a minimum of 40% corrected cells is required to obtain a complete metabolic and phenotypic correction, suggesting that a full chimerism is likely to be necessary. In the case of partial conditioning, chimerism can be reinforced by conferring a selective advantage to transduced hematopietic stem cells. It would be interesting to evaluate a lentiviral vector that coexpresses UROS and a selective gene such as the O6-methylguanine-DNA-methyltransferase (MGMT),31,45 chemical inducers of dimerization,46 or the truncated Epo receptor.47
The recent reports of insertional leukemogenesis in 4 of 11 patients who were treated by gene therapy for SCID-X148 due to activation of the LMO2 gene by the promoter of the integrated provirus underscore the need for safer vectors that would be lineage specific and have enhancer-blocking effects. In the present study, the use of a cellular promoter instead of a retroviral promoter reduced the level of gene deregulation in the neighborhood of proviral integration.49 In addition, the use of erythroid-specific lentiviral vectors would therefore confine the risk of oncogene activation to a single lineage. The probability of promoter transactivation by vector-encoded enhancers could be further reduced by the incorporation into the vectors of insulators such as cHS4.50–52
The clinical course of CEP disease is rapidly devastating because of a rapid extension of cutaneous impairment including loss of digits, eyelids, nostrils, and ears. Anemia due to erythrocyte hemolysis can be life threatening if the marrow cannot compensate, and severely affected patients are transfusion dependent. In the past, death often occurs before the age of 40 as a result of multiple organ failure. In the most severe forms, the disease develops in utero, leading to premature abortion or death in the neonatal period.8,9 BM transplantation, which is proposed in severe forms of the disease diagnosed in the first years of life, has proven curative in patients with CEP: 11 cases have been reported.10–19 However, this treatment is limited by the need for an HLA-identical donor and is associated with a significant risk of morbidity and mortality. In the case of a gene-therapy clinical trial, the survival advantage of corrected cells is likely to be dependent on the severity of hemolysis. This point can be a problem for patients with severe photosensitivity, which may in itself justify gene therapy, but with absence of (or well-compensated) hemolysis. A clinical trial will concern in the first place severe cases with hemolytic anemia. Splenomegaly as a consequence of hemolytic anemia is a nearly obligate sign in CEP and sometimes results in leukopenia and thrombocytopenia (hypersplenism). Splenectomy may minimize the hemolytic anemia and reduce transfusion requirement in some patients, but it is not always beneficial.8 It will be interesting to test the effect of splenectomy on the hemolytic anemia in our CEP mouse model.
The success of BM transplantation in alleviating the severe manifestations of the disease provides the rationale for hematopoietic stem cell (HSC) gene therapy. Previous experiments performed into Epstein-Barr-virus-transformed B cell lines37 showed the absence of transcellular complementation between transduced and nontransduced cells. This is not surprising because uroporphyrin I, which is the main formed compound in the absence of the enzyme UROS, corresponds to a metabolic dead end. We very efficiently transduced human CD34+ cells isolated from mobilized peripheral blood (mPB).53 The experiments performed in vitro with lentiviral vectors demonstrated that the level of enzymatic correction obtained by gene transfer was sufficient to allow total metabolic correction of the cells in the short and long term.54 The evaluation of UROS gene-transfer efficiency, expression, and safety in an immunodeficient mouse model should reinforce these preclinical studies.
In conclusion, the cure of the mouse model of CEP supports the proof of concept of clinical testing of gene therapy in this severe disease. The survival advantage of corrected red blood cells is a major argument for a therapeutic benefit in human because not all the stem cells need to be corrected.
Web Resources
The URL for data presented herein is as follows:
Online Mendelian Inheritance in Man (OMIM): http://www.ncbi.nlm.nih.gov/Omim/ (for UROS gene and congenital erythropoietic porphyria)
Acknowledgments
We would like to thank D. Trono (Geneva, Switzerland) for providing the SIN-lentiviral backbone pRRL-PGK-WPRE, D. Kohn (Childrens Hospital, Los Angeles, CA) for MNDp, C. Moya and C. Pain for technical assistance, and T. Barnetche for statistical analyses. We thank E. Richard for critical reading of the manuscript. This work was supported by the Association Française contre les Myopathies (AFM), the Agence Nationale de la Recherche (ANR), and the Conseil Régional d'Aquitaine.
References
- 1.Anderson K.E., Sassa S., Bishop D.F., Desnick R.J. Disorders of heme biosynthesis: X-linked sideroblastic anemia and the porphyrias. In: Scriver R., Beaudet A.L., Valle E., Sly W.S., editors. The Metabolic and Molecular Bases of Inherited Disease. C McGraw-Hill; New York: 2001. pp. 2961–3062. [Google Scholar]
- 2.de Verneuil H., Ged C., Moreau-Gaudry F. Congenital erythropoietic porphyria. In: Kadish K.M., Smith K.M., Guilard R., editors. The Porphyrin Handbook. Academic Press; New York: 2003. pp. 43–63. [Google Scholar]
- 3.Romeo G., Levin E.Y. Uroporphyrinogen III cosynthase in human congenital erythropoietic porphyria. Biochemistry. 1969;63:856–863. doi: 10.1073/pnas.63.3.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deybach J.C., de Verneuil H., Boulechfar S., Grandchamp B., Nordmann Y. Point mutations in the uroporphyrinogen III synthase gene in congenital erythropoietic porphyria. Blood. 1990;75:1763–1765. [PubMed] [Google Scholar]
- 5.Xu W., Warner C.A., Desnick R.J. Congenital erythropoietic porphyria: Identification and expression of 10 mutations in the uroporphyrinogen III synthase gene. J. Clin. Invest. 1995;95:905–912. doi: 10.1172/JCI117742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fontanellas A., Bensidhoum M., Enriquez de Salamanca R., Moruno Tirado A., de Verneuil H., Ged C. A systematic analysis of the mutations of the uroporphyrinogen III synthase gene in congenital erythropoietic porphyria. Eur. J. Hum. Genet. 1996;4:274–282. doi: 10.1159/000472214. [DOI] [PubMed] [Google Scholar]
- 7.Shady A.A., Colby B.R., Cunha L.F., Astrin K.H., Bishop D.F., Desnick R.J. Congenital erythropoietic porphyria: Identification and expression of eight novel mutations in the uroporphyrinogen III synthase gene. Br. J. Haematol. 2002;117:980–987. doi: 10.1046/j.1365-2141.2002.03558.x. [DOI] [PubMed] [Google Scholar]
- 8.Fritsch C., Bolsen K., Ruzicka T., Goerz G. Congenital erythropoietic porphyria. J. Am. Acad. Dermatol. 1997;36:594–610. doi: 10.1016/s0190-9622(97)70249-4. [DOI] [PubMed] [Google Scholar]
- 9.de Verneuil H., Moreau-Gaudry F., Ged C., Bensidhoum M., Hombrados I., Tricoire J., Rolland M. Congenital erythropoietic porphyria. A propos of a fatal case in the neonatal period due to acute hemolysis with hepatic failure. Arch. Pediatr. 1995;2:755–761. doi: 10.1016/0929-693x(96)81246-2. [DOI] [PubMed] [Google Scholar]
- 10.Kauffman L., Evans D.I.K., Stevens R., Weinkove C. Bone marrow transplantation for congenital erythropoietic porphyria. Lancet. 1991;337:1510–1511. doi: 10.1016/0140-6736(91)93198-i. [DOI] [PubMed] [Google Scholar]
- 11.Thomas C., Ged C., Nordmann Y., de Verneuil H., Pellier I., Fischer A., Blanche S. Correction of congenital erythropoietic porphyria by bone marrow transplantation. J. Pediatr. 1996;129:453–456. doi: 10.1016/s0022-3476(96)70082-3. [DOI] [PubMed] [Google Scholar]
- 12.Lagarde C., Hamel-Teillac D., De Prost Y., Blanche S., Thomas C., Fischer A., Nordmann Y., de Verneuil H. Allogeneic bone marrow transplantation in congenital erythropoietic porphyria. Gunther's disease. Ann. Dermatol. Venereol. 1998;125:114–117. [PubMed] [Google Scholar]
- 13.Zix-Kieffer I., Langer B., Eyer D., Acar G., Racadot E., Schlaeder G., Oberlin F., Lutz P. Successful cord blood stem cell transplantation for congenital erythropoietic porphyria (Gunther's disease) Bone Marrow Transplant. 1996;18:217–220. [PubMed] [Google Scholar]
- 14.Tezcan I., Xu W., Gurgey A., Tuncer M., Cetin M., Oner C., Yetgin S., Ersoy F., Aizencang G., Astrin K.H., Desnick R.J. Congenital erythropoietic porphyria successfully treated by allogeneic bone marrow transplantation. Blood. 1998;92:4053–4058. [PubMed] [Google Scholar]
- 15.Fritsch C., Lang K., Neuse W., Ruzicka T., Lehmann P. Photodynamic diagnosis and therapy in dermatology. Skin Pharmacol. Appl. Skin Physiol. 1998;11:358–373. doi: 10.1159/000029858. [DOI] [PubMed] [Google Scholar]
- 16.Shaw P.H., Mancini A.J., McConnell J.P., Brown D., Kletzel M. Treatment of congenital erythropoietic porphyria in children by allogeneic stem cell transplantation: A case report and review of the literature. Bone Marrow Transplant. 2001;27:101–105. doi: 10.1038/sj.bmt.1702738. [DOI] [PubMed] [Google Scholar]
- 17.Harada F.A., Shwayder T.A., Desnick R.J., Lim H.W. Treatment of severe congenital erythropoietic porphyria by bone marrow transplantation. J. Am. Acad. Dermatol. 2001;45:279–282. doi: 10.1067/mjd.2001.114730. [DOI] [PubMed] [Google Scholar]
- 18.Dupuis-Girod S., Akkari V., Ged C., Galambrun C., Kebaili K., Deybach J.C., Claudy A., Geburher L., Philippe N., de Verneuil H., Bertrand Y. Successful match-unrelated donor bone marrow transplantation for congenital erythropoietic porphyria (Gunther disease) Eur. J. Pediatr. 2005;164:104–107. doi: 10.1007/s00431-004-1575-x. [DOI] [PubMed] [Google Scholar]
- 19.Phillips J.D., Steensma D.P., Pulsipher M.A., Spangrude G.J., Kushner J.P. Congenital erythropoietic porphyria due to a mutation in GATA-1: The first trans-acting mutation causative for a human porphyria. Blood. 2007;109:2618–2621. doi: 10.1182/blood-2006-06-022848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cavazzana-Calvo M., Hacein-Bey S., de Saint Basile G., Gross F., Yvon E., Nusbaum P., Selz F., Hue C., Certain S., Casanova J.L. A Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288:669–672. doi: 10.1126/science.288.5466.669. [DOI] [PubMed] [Google Scholar]
- 21.Gaspar H.B., Parsley K.L., Howe S., King D., Gilmour K.C., Sinclair J., Brouns G., Schmidt M., Von Kalle C., Barington T. Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet. 2004;364:2181–2187. doi: 10.1016/S0140-6736(04)17590-9. [DOI] [PubMed] [Google Scholar]
- 22.Aiuti A., Slavin S., Aker M., Ficara F., Deola S., Mortellaro A., Morecki S., Andolfi G., Tabucchi A., Carlucci F. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296:2410–2413. doi: 10.1126/science.1070104. [DOI] [PubMed] [Google Scholar]
- 23.Aiuti A., Vai S., Mortellaro A., Casorati G., Ficara F., Andolfi G., Ferrari G., Tabucchi A., Carlucci F., Ochs H.D. Immune reconstitution in ADA-SCID after PBL gene therapy and discontinuation of enzyme replacement. Nat. Med. 2002;8:423–425. doi: 10.1038/nm0502-423. [DOI] [PubMed] [Google Scholar]
- 24.Muul L.M., Tuschong L.M., Soenen S.L., Jagadeesh G.J., Ramsey W.J., Long Z., Carter C.S., Garabedian E.K., Alleyne M., Brown M. Persistence and expression of the adenosine deaminase gene for 12 years and immune reaction to gene transfer components: Long-term results of the first clinical gene therapy trial. Blood. 2003;101:2563–2569. doi: 10.1182/blood-2002-09-2800. [DOI] [PubMed] [Google Scholar]
- 25.Schmidt M., Carbonaro D.A., Speckmann C., Wissler M., Bohnsack J., Elder M., Aronow B.J., Nolta J.A., Kohn D.B., von Kalle C. Clonality analysis after retroviral-mediated gene transfer to CD34+ cells from the cord blood of ADA-deficient SCID neonates. Nat. Med. 2003;9:463–468. doi: 10.1038/nm844. [DOI] [PubMed] [Google Scholar]
- 26.Gaspar H.B., Bjorkegren E., Parsley K., Gilmour K.C., King D., Sinclair J., Zhang F., Giannakopoulos A., Adams S., Fairbanks L.D. Successful Reconstitution of Immunity in ADA-SCID by Stem Cell Gene Therapy Following Cessation of PEG-ADA and Use of Mild Preconditioning. Mol. Ther. 2006;14:505–513. doi: 10.1016/j.ymthe.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 27.Dunbar C.E., Kohn D.B., Schiffmann R., Barton N.W., Nolta J.A., Esplin J.A., Pensiero M., Long Z., Lockey C., Emmons R.V. Retroviral transfer of the glucocerebrosidase gene into CD34+ cells from patients with Gaucher disease: In vivo detection of transduced cells without myeloablation. Hum. Gene Ther. 1998;9:2629–2640. doi: 10.1089/hum.1998.9.17-2629. [DOI] [PubMed] [Google Scholar]
- 28.Malech H.L., Maples P.B., Whiting-Theobald N., Linton G.F., Sekhsaria S., Vowells S.J., Li F., Miller J.A., DeCarlo E., Holland S.M. Prolonged production of NADPH oxidase-corrected granulocytes after gene therapy of chronic granulomatous disease. Proc. Natl. Acad. Sci. USA. 1997;94:12133–12138. doi: 10.1073/pnas.94.22.12133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ott M.G., Schmidt M., Schwarzwaelder K., Stein S., Siler U., Koehl U., Glimm H., Kühlcke K., Schilz A., Kunkel H. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1–EVI1, PRDM16 or SETBP1. Nat. Med. 2006;12:401–409. doi: 10.1038/nm1393. [DOI] [PubMed] [Google Scholar]
- 30.Ged C., Mendez M., Robert E., Lalanne M., Lamrissi-Garcia I., Costet P., Daniel J.Y., Dubus P., Mazurier F., Moreau-Gaudry F., de Verneuil H. A knock-in mouse model of congenital erythropoietic porphyria. Genomics. 2006;87:84–92. doi: 10.1016/j.ygeno.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 31.Richard E., Robert E., Cario-André M., Ged C., Géronimi F., Gerson S.L., de Verneuil H., Moreau-Gaudry F. Hematopoietic stem cell gene therapy of murine protoporphyria by methylguanine-DNA-methyltransferase-mediated in vivo drug selection. Gene Ther. 2004;11:1638–1647. doi: 10.1038/sj.gt.3302335. [DOI] [PubMed] [Google Scholar]
- 32.Moreau-Gaudry F., Xia P., Jiang G., Perelman N.P., Bauer G., Ellis J., Surinya K.H., Mavilio F., Shen C.K., Malik P. High-level erythroid-specific gene expression in primary human and murine hematopoietic cells with self-inactivating lentiviral vectors. Blood. 2001;98:2664–2672. doi: 10.1182/blood.v98.9.2664. [DOI] [PubMed] [Google Scholar]
- 33.Richard E., Mendez M., Mazurier F., Morel C., Costet P., Xia P., Fontanellas A., Geronimi F., Cario-Andre M., Taine L. Gene therapy of a mouse model of protoporphyria with a self-inactivating erythroid-specific lentiviral vector without preselection. Mol. Ther. 2001;4:331–338. doi: 10.1006/mthe.2001.0467. [DOI] [PubMed] [Google Scholar]
- 34.Case S.S., Price M.A., Jordan C.T., Yu X.J., Wang L., Bauer G., Haas D.L., Xu D., Stripecke R., Naldini L. Stable transduction of quiescent CD34+CD38- human hematopoietic cells by HIV-1-based lentiviral vectors. Proc. Natl. Acad. Sci. USA. 1999;96:2988–2993. doi: 10.1073/pnas.96.6.2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fontanellas A., Mazurier F., Landry M., Taine L., Morel C., Larou M., Daniel J.Y., Montagutelli X., Enriquez de Salamanca R., de Verneuil H. Reversion of hepatobiliary alterations by bone marrow transplantation in a murine model of erythropoietic protoporphyria. Hepatology. 2000;32:73–81. doi: 10.1053/jhep.2000.8531. [DOI] [PubMed] [Google Scholar]
- 36.Lim C.K., Peters T.J. Urine and fecal porphyrin profiles by reversed-phase high performance liquid chromatography in the porphyries. Clin. Chim. Acta. 1984;139:55–63. doi: 10.1016/0009-8981(84)90192-x. [DOI] [PubMed] [Google Scholar]
- 37.Moreau-Gaudry F., Mazurier F., Bensidhoum M., Ged C., de Verneuil H. Metabolic correction of Congenital Erythropoietic Porphyria by retroviral-mediated gene transfer into lymphoblastoid cell lines. Blood. 1995;85:1449–1453. [PubMed] [Google Scholar]
- 38.Ren S., Wong B.Y., Li J., Luo X.N., Wong P.M., Atweh G.F. Production of genetically stable high-titer retroviral vectors that carry a human gamma-globin gene under the control of the alpha-globin locus control region. Blood. 1996;87:2518–2524. [PubMed] [Google Scholar]
- 39.Sabatino D.E., Seidel N.E., Aviles-Mendoza G.J., Cline A.P., Anderson S.M., Gallagher P.G., Bodine D.M. Long-term expression of gamma-globin mRNA in mouse erythrocytes from retrovirus vectors containing the human gamma-globin gene fused to the ankyrin-1 promoter. Proc. Natl. Acad. Sci. USA. 2000;97:13294–13299. doi: 10.1073/pnas.230453097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bishop D.F., Johansson A., Phelps R., Shady A.A., Ramirez M.C.M., Yasuda M., Caro A., Desnick R.J. Uroporphyrinogen III synthase knock-in mice have the human congenital erythropoietic porphyria phenotype, including the characteristic light-induced cutaneous lesions. Am. J. Hum. Genet. 2006;78:645–658. doi: 10.1086/502667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Persons D.A., Allay E.R., Sabatino D.E., Kelly P., Bodine D.M., Nienhuis A.W. Functional requirements for phenotypic correction of murine β-thalassemia: Implications for human gene therapy. Blood. 2001;97:3275–3282. doi: 10.1182/blood.v97.10.3275. [DOI] [PubMed] [Google Scholar]
- 42.Rivella S., May C., Chadburn A., Riviere I., Sadelain M. A novel murine model of Cooley anemia and its rescue by lentiviral-mediated human beta-globin gene transfer. Blood. 2003;101:2932–2939. doi: 10.1182/blood-2002-10-3305. [DOI] [PubMed] [Google Scholar]
- 43.Pawliuk R., Bachelot T., Wise R.J., Mathews-Roth M.M., Leboulch P. Long-term cure of the photosensitivity of murine erythropoietic protoporphyria by preselective gene therapy. Nat. Med. 1999;5:768–773. doi: 10.1038/10488. [DOI] [PubMed] [Google Scholar]
- 44.Fontanellas A., Mendez M., Mazurier F., Cario-André M., Navarro S., Ged C., Taine L., Géronimi F., Richard E., Moreau-Gaudry F. Successful therapeutic effect in a mouse model of erythropoietic protoporphyria by partial genetic correction and fluorescence-based selection of hematopoietic cells. Gene Ther. 2001;8:618–626. doi: 10.1038/sj.gt.3301427. [DOI] [PubMed] [Google Scholar]
- 45.Trobridge G., Beard B.C., Kiem H.P. Hematopoietic stem cell transduction and amplification in large animal models. Hum. Gene Ther. 2005;16:1355–1366. doi: 10.1089/hum.2005.16.1355. [DOI] [PubMed] [Google Scholar]
- 46.Richard R.E., Weinreich M., Chang K.H., Ieremia J., Stevenson M.M., Blau C.A. Modulating erythrocyte chimerism in a mouse model of pyruvate kinase deficiency. Blood. 2004;103:4432–4439. doi: 10.1182/blood-2003-10-3705. [DOI] [PubMed] [Google Scholar]
- 47.Urbinati F., Lotti F., Facchini G., Montanari M., Ferrari G., Mavilio F., Grande A. Competitive engraftment of hematopoietic stem cells genetically modified with a truncated erythropoietin receptor. Hum. Gene Ther. 2005;16:594–608. doi: 10.1089/hum.2005.16.594. [DOI] [PubMed] [Google Scholar]
- 48.Baum C. What are the consequences of the fourth case? Mol. Ther. 2007;15:1401–1402. doi: 10.1038/sj.mt.6300263. [DOI] [PubMed] [Google Scholar]
- 49.Fischer A., Abina S.H., Thrasher A., von Kalle C., Cavazzana-Calvo M. LMO2 and gene therapy for severe combined immunodeficiency. N. Engl. J. Med. 2004;350:2526–2527. doi: 10.1056/NEJM200406103502422. [DOI] [PubMed] [Google Scholar]
- 50.Robert-Richard E., Richard E., Malik P., Ged C., de Verneuil H., Moreau-Gaudry F. Murine Retroviral but not Human Cellular Promoters Induce In vivo deregulation that can be Prevented by Insulators. Mol. Ther. 2007;15:173–182. doi: 10.1038/sj.mt.6300030. [DOI] [PubMed] [Google Scholar]
- 51.Malik P., Arumugam P.I., Yee J.K., Puthenveetil G. Successful correction of the human Cooley's anemia beta-thalassemia major phenotype using a lentiviral vector flanked by the chicken hypersensitive site 4 chromatin insulator. Ann. N Y Acad. Sci. 2005;1054:238–249. doi: 10.1196/annals.1345.030. [DOI] [PubMed] [Google Scholar]
- 52.Puthenveetil G., Scholes J., Carbonell D., Qureshi N., Xia P., Zeng L., Li S., Yu Y., Hiti A.L., Yee J.K., Malik P. Successful correction of the human beta-thalassemia major phenotype using a lentiviral vector. Blood. 2004;104:3445–3453. doi: 10.1182/blood-2004-04-1427. [DOI] [PubMed] [Google Scholar]
- 53.Géronimi F., Richard E., Redonnet-Vernhet I., Lamrissi-Garcia I., Lalanne M., Ged C., Moreau-Gaudry F., de Verneuil H. Highly efficient lentiviral gene transfer in CD34+ and CD34+/38-/lin- cells from mobilized peripheral blood after cytokine prestimulation. Stem Cells. 2003;21:472–480. doi: 10.1634/stemcells.21-4-472. [DOI] [PubMed] [Google Scholar]
- 54.Géronimi F., Richard E., Lamrissi-Garcia I., Lalanne M., Ged C., Redonnet-Vernhet I., Moreau-Gaudry F., de Verneuil H. Lentivirus-mediated gene transfer of uroporphyrinogen III synthase fully corrects the porphyric phenotype in human cells. J. Mol. Med. 2003;81:310–320. doi: 10.1007/s00109-003-0438-7. [DOI] [PubMed] [Google Scholar]