

Abstract
Polycyclic aromatic hydrocarbons (PAH) form stable and depurinating DNA adducts in mouse skin to induce preneoplastic mutations. Some mutations transform cells, which then clonally expand to establish tumors. Strong clues about the mutagenic mechanism can be obtained if the PAH-DNA adducts can be correlated with both preneoplastic and tumor mutations. To this end, we studied mutagenesis in PAH-treated early preneoplastic skin (1 day after exposure) and in the induced papillomas in SENCAR mice. Papillomas were studied by PCR amplification of the H-ras gene and sequencing. For benzo[a]pyrene (BP), BP-7,8-dihydrodiol (BPDHD), 7,12-dimethylbenz[a]anthracene (DMBA) and dibenzo[a,l]pyrene (DB[a,l]P), the codon 13 (GGC to GTC) and codon 61 (CAA to CTA) mutations in papillomas corresponded to the relative levels of Gua and Ade-depurinating adducts, despite BP and BPDHD forming significant amounts of stable DNA adducts. Such a relationship was expected for DMBA and DB[a,l]P, as they formed primarily depurinating adducts. These results suggest that depurinating adducts play a major role in forming the tumorigenic mutations. To validate this correlation, preneoplastic skin mutations were studied by cloning H-ras PCR products and sequencing individual clones. DMBA- and DB[a,l]P-treated skin showed primarily A.T to G.C mutations, which correlated with the high ratio of the Ade/Gua-depurinating adducts. Incubation of skin DNA with T.G-DNA glycosylase eliminated most of these A.T to G.C mutations, indicating that they existed as G.T heteroduplexes, as would be expected if they were formed by errors in the repair of abasic sites generated by the depurinating adducts. BP and its metabolites induced mainly G.C to T.A mutations in preneoplastic skin. However, PCR over unrepaired anti-BPDE-N2dG adducts can generate similar mutations as artifacts of the study protocol, making it difficult to establish an adduct-mutation correlation for determining which BP-DNA adducts induce the early preneoplastic mutations. In conclusion, this study suggests that depurinating adducts play a major role in PAH mutagenesis.
Keywords: Polycyclic aromatic hydrocarbons, SENCAR mouse skin, H-ras, Mutations
1. Introduction
Polycyclic aromatic hydrocarbons (PAH) are environmental carcinogens [1–3] associated with skin, lung, pharynx, oral and other cancers [4–6]. Initiation of these diseases is often associated with mutations in H-, K- and N-ras genes [7–13]. These ras mutations are thought to be induced by the PAH-DNA adducts generated in exposed cells [14–18].
In the cell, PAH are oxidized by cytochrome P450s to form electrophilic derivatives (e.g., diolepoxides and radical cations) that react with DNA to form adducts [19]. PAH react primarily with the purine bases in DNA, forming bulky stable and depurinating adducts [20–24]. The adduct-forming reaction is moderately sequence-selective [25–32]. In the cell, PAH-DNA adduct formation blocks replication and induces base and nucleotide excision repair (BER and NER) activities during a 24 h-long period [33–39]. The stable adducts undergo NER, whereas the abasic sites generated from the spontaneous loss of the depurinating adducts undergo BER [38,40–42]. During this period, some PAH-induced abasic sites are converted into heteroduplex mutations by BER errors [39,43,44], and a subpopulation of the stable adducts are removed by error-free NER [45,46]. As cells re-enter S-phase, the heteroduplex mutations are “fixed” in daughter DNA molecules by one round of replication [44]. In addition, some cells carrying the unrepaired stable adducts acquire mutations as DNA polymerases go over the adducted bases, incorporate incorrect bases and fail to remove these mistakes by proofreading [47–57]. Two rounds of replication are needed to fix this latter type of mutations into daughter DNA molecules [58,59].
Studies in the SENCAR mouse skin cancer model [14–16] have led to suggestions that stable and depurinating PAH-DNA adducts can both contribute to the formation of the tumorigenic preneoplastic mutations [16,42,60,61]. These ideas were mainly inspired by correlating PAH-DNA adducts (which form within hours of exposure) with oncogenic H-ras mutations found in mouse skin tumors (which form 7–9 weeks later) [14–16]. However, such correlations can provide limited information about the nature of mutagenesis. PAH induce both oncogenic and non-oncogenic mutations in the preneoplastic stage [43,58,59]. Cells that acquire the oncogenic mutations become clonally-expanded (based on their transforming potentials and tissue specificities) to establish the tumors [62–65]. Therefore, oncogenic mutations found in the tumors cannot describe the mutational specificity of PAH-DNA adducts. Knowing the mutational specificity in the preneoplastic tissue is important, because it can reveal the critical lesions for cancer. In vitro, the mutational specificity is obtained from studies that examine polymerase errors during the filling synthesis of nicks or gaps similar to those generated by the excision steps of NER and BER [66,67], or those during bypass over PAH-DNA stable adducts (translesion synthesis), similar to those left unrepaired by NER [68,69]. The knowledge obtained from such studies is limited to the specific DNA polymerases, and sometimes cannot explain mutagenesis in vivo. For example, mouse skin papillomas induced by benzo[a]pyrene (BP) show both codon 13 (GGC to GTC) and 61 (CAA to CTA) mutations in the H-ras gene [16]. Translesional synthesis errors over BP diolepoxide (BPDE)-N2dG adducts at the central G of GGC form the GTC mutation [49], but the BPDE-N6dA adducts do not form the CTA mutation at the central A of CAA [69,70].
A number of studies in PAH-treated cells in culture or PAH-exposed tissues of animals have been conducted to understand the in vivo mutational specificity (discussed in Section 4). Results of these studies need to be interpreted with caution, because cell death from DNA damage-induced apoptosis and necrosis, and from fatal mutations [71–73], as well as selective proliferation and regression of oncogenically-mutated cells [74–76], can induce dramatic changes in preneoplastic mutation spectra. For example, some of the initial mutations (6 h- 1 d) observed in dibenzo[a,l]pyrene (DB[a,l]P)-treated mouse skin are enriched or lost in the later preneoplastic period (days 2–7) [43,44]. Therefore, early preneoplastic spectra can provide important information about PAH mutational specificity in the target cells. In both DB[a,l]P- and DB[a,l]P-diolepoxide-treated mouse skin, the early mutation period appears to be between 6 h - 1 d [43,44]. In the present study, we have used the 1 d spectra as a time point for learning about PAH mutational specificity in vivo.
Errors made by repair and by translesion synthesis may produce quite different spectra of mutations. For example, if error-prone BER induces mutations from PAH-induced abasic sites, the spectra will be determined primarily by the specificities of gap-filling errors of BER polymerases. If DNA polymerase β or ι is involved, the majority of base substitutions should be A to G transitions [66,77,78]. On the other hand, translesional synthesis errors of DNA polymerases over PAH-DNA stable adducts can produce a greater variety of base substitution mutations, dictated by factors such as the structure and conformation of the adducts, their DNA sequence contexts, and the DNA polymerase [79,80]. The classical replicative polymerases are inefficient in bypassing bulky stable adducts or extending DNA synthesis beyond the adducted sites. Therefore, bypass polymerases are now thought to be more important for generating mutations by this mechanism. For example, DNA polymerases η/ζ/ι may incorporate an A or a G opposite BPDE-N2dG adducts in the template strand (leading to G to T or G to C mutations) [57,81,82]. In comparison, BPDE-N6dA adducts induce fewer mistakes and show little misincorporation preference [82].
The extent to which mutagenesis by errors in repair or translesion synthesis contributes to cancer can be understood from studies that examined the roles of quiescent or proliferating cells in tumor formation. Since ~95% of the epidermal cells in telogen skin are quiescent, this question can be easily addressed in vivo [74]. One study reported that 7,12-dimethylbenz[a]anthracene (DMBA) induces 3-fold (males) and 13-fold (females) more skin tumors when the skin of the mice is treated (initiation only) in the resting (telogen) phase rather than the proliferative (anagen) phase [83]. In another study, mouse skin was treated with 5-fluorouracil either 1 d before or 1 d after treatment with DMBA [84]. In that study, 5-fluorouracil (stops DNA replication and selectively kills cycling cells) only marginally reduced the yield of benign tumors (papillomas) and did not reduce the yield of carcinomas. This result suggests that following DMBA treatment, either the non-proliferating cells acquire the oncogenic mutations, or DNA replication/cell proliferation in the first couple of days is not critical for tumor formation. If the first idea is correct, DMBA-treated quiescent skin cells would be expected to have mutations induced by erroneous repair. In a third study, DMBA did not show a relationship between stable DNA adducts and tumor latency [85]. Our studies with DB[a,l]P, which forms predominantly depurinating DNA adducts [23,24], suggest that quiescent skin cells form preneoplastic mutations by error-prone BER [39,43,44]. DMBA also forms predominantly depurinating DNA adducts [21]; therefore, it would be expected to form preneoplastic mutations by error-prone repair.
Biological evidence for the role of proliferating cells in tumorigenesis was obtained from a study with BPDE in the regenerating rat liver model [48]. BPDE forms predominantly stable DNA adducts [22,86] and it formed the most number of tumors when early S-phase liver cells were exposed [48]. This result is consistent with studies suggesting that NER of the BPDE-induced stable adducts is error-free [45,54], and that mutations are induced in the proliferating cells by errors in translesion synthesis. Taken together, it appears that the PAH or their metabolites that form predominantly depurinating adducts may form preneoplastic mutations by error-prone repair in quiescent cells, whereas the PAH that form mainly stable adducts would induce mutations by errors in translesion synthesis in proliferating cells. PAH such as BP, which forms significant amounts of both types of adducts [22,86], appear to induce mutations from both stable and depurinating adducts. For example, the relative proportions of BP-induced depurinating adducts show a strong correlation with the relative abundance of Ade- or Gua-specific oncogenic H-ras mutations in skin tumors [16]. In addition, BP-induced stable DNA adducts do not show a linear relationship with tumor yield [75,87]. On the other hand, deficiencies in some NER genes increase BP-induced mutagenesis and tumor formation [88–90].
In view of the complexities in mutagenesis by PAH, we have conducted a comprehensive study to examine PAH-induced mutations in preneoplastic mouse skin and in papillomas, both to examine the mutations and their relationship with DNA adducts, and to obtain insights into the mutagenic mechanisms.
2. Materials and Methods
2.1. Chemicals
The PAH and their derivatives (BP, BPDHD, anti-BPDE, DMBA, DB[a,l]P and anti-DB[a,l]PDE) were either purchased from the Chemical Carcinogen Repository of the National Cancer Institute or synthesized in our laboratory as described previously [91,92]. These PAH are hazardous and handled according to NIH guidelines [93].
2.2. Treatment of SENCAR mice and harvesting preneoplastic skin
Eight wk-old female SENCAR mice (National Cancer Institute-Frederick Cancer Center, Frederick, MD) were treated once on an area of shaved dorsal skin with 100 μL acetone (solvent) or a PAH in 100 μL acetone (initiation) at doses indicated in Table 1. Animals (3 mice/treatment group) were sacrificed 24 h later by asphyxiation under CO2, and the treated area of dorsal skin harvested for the analysis of preneoplastic mutations. In addition, samples of unexposed ventral skin were also harvested from these mice to verify the absence of pre-existing H-ras mutations. Harvested skin samples were flash-frozen in liquid N2, wrapped in aluminum foil and stored at −80 °C until use.
Table 1.
Summary of PAH-DNA adducts and H-ras mutations in papillomas induced by PAH initiation and TPA promotion and in PAH-treated preneoplastic skin
| PAH (nmol) | Papillomas/Mouse a | H-ras mutations in papillomas | DNA adducts in skin (4h after PAH treatment) c | Preneoplastic H-ras mutations (24 h after PAH treatment) | |||||
|---|---|---|---|---|---|---|---|---|---|
| Codon | Sequence | No./total papillomas * | Stable (% total) | Depur. (% total) | Mutation type | No.of mutations/total mutations/total clones * | Distribution | ||
| BP (400) | 82/24 | 13
61 |
GGC to GTC
CAA to CTA |
11/22b
5/22b |
G (23)
Other (6) |
G (46)
A (25) |
G.C to T.A
A.T to G.C |
4/5/71
1/5/71 |
G.C (80%)
A.T (20%) |
| BPDHD (400) | 62/28 | 13
61 |
GGC to GTC
CAA to CTA |
2/12
4/12 |
G (61)
Other (2) |
G (12)
A (25) |
G.C to T.A | 5/5/32
- |
G.C (100%) |
| Anti-BPDE (400) | 1/27 | 13 | GGC to GTC | 1/1 | G (98)
Other (0.6) |
G (0.1)
A (1.2) |
G.C to A.T
G.C to T.A A.T to G.C A.T to T.A |
1/9/42
6/9/42 1/9/42 1/9/42 |
G.C (78%)
A.T (22%) |
| DMBA (100) | 397/24 |
61 |
CAA to CTA |
10/10b |
Other (0.9) | G (20)
A (79) |
G.C to A.T
G.C to T.A A.T to G.C A.T to T.A |
2/21/39
4/21/39 14/21/39 1/21/39 |
G.C (6/21)
A.T (15/21) |
| DB[a,l]P (200) | 129/30 |
61 61 |
CAA to CTA CAA to CTT |
10/12b 1/12b |
G (0.5)
A (0.5) |
G (18)
A (81) |
G.C to A.T
G.C to T.A G.C to C.G A.T to G.C A.T to T.A |
2/17/50
1/17/50 1/17/50 12/17/50 1/17/50 |
G.C (4/17)
A.T (13/17) |
2.3. Papillomas
Tumors induced by the initiation-promotion protocol in previously described experiments and stored frozen at −80 °C [16,91,94] were analyzed for mutations. To obtain these tumors the PAH-treated mice were promoted by twice weekly doses of 3 nmol 12-O-tetradecanoyl phorbol 13-acetate (TPA) in 100 μL acetone, until papilloma induction was complete (7–9 wks).
2.4 Analysis of H-ras mutations
Chromosomal DNA from the papillomas and preneoplastic skin samples was isolated as previously described [16,43,44]. For analysis of the clonal H-ras mutations in the papillomas, a 550 bp-long segment spanning the exon 1-2 region was PCR amplified with primers MRF and MRR and the product directly sequenced as previously described [16]. Direct sequencing of PCR products from tumors detects mutations that exist in at least 10% of the DNA molecules [95]. This approach provides adequate sensitivity, since clonally expanded oncogenic H-ras mutations are typically present in 14–47 % of cells in PAH-induced papillomas [43]. Linear amplification reactions for sequencing were conducted with dye-deoxy nucleotides and previously described primers [16] and the reaction products were resolved in a Beckman/Coulter CEQ2000XL 8-capillary DNA sequencer (Genomics Core Research Facility, University of Nebraska, Lincoln). When a clonally-enriched H-ras mutation was present in papilloma DNA, the sequencing electropherogram showed an overlapped peak corresponding to the mutant and wild-type bases.
To examine the induction of preneoplastic mutations in PAH-treated mouse skin, the same H-ras gene exon 1-2 segment was PCR amplified with primers MFPS and MRER from skin DNA as previously described [44,96]. MRER (contains an EcoRI site) is an altered MRR, and MFPS (contains a PstI site) is an altered MRF, and they were useful for restriction digesting the PCR products and direct cloning into the EcoRI/PstI sites of pUC 18 to form recombinant plasmids, which were then transformed into E.coli Top10 cells (Invitrogen Life Technologies, Carlsbad, CA). Single colonies carrying the recombinant plasmids were identified on the basis of the IPTG/X-gal blue-white colony color assay, grown in small cultures, re-verified for recombinant plasmid content by dot-blot experiments conducted with a non-radioactively-labeled H-ras PCR product as probe (ECL direct labeling and detecting system, Amersham Pharmacia, Piscataway, NJ), and a ZetaProbe membrane (Biorad Laboratories, Hercules, CA). Cultures grown from ligation experiments that resulted in 80% or greater cloning efficiency were used to isolate recombinant plasmids and sequence the H-ras inserts. Sequencing reactions were conducted with M13 forward and reverse primers.
In addition to preneoplastic skin, two DB[a,l]P-induced papillomas (A43 and B32) were examined by the PCR-cloning-sequencing procedure. This procedure can detect mutations present in lower frequency than direct sequencing. For example, the detection limit is 2% when one mutation is scored in 50 recombinant plasmids. Mutation frequency was calculated by dividing the number of mutations by the product of the number of plasmids and the length of the sequenced region, as described previously [44,96,97].
2.5 T.G-DNA glycosylase assay for the detection of heteroduplex mutations induced by repair errors
Errors in excision repair would form heteroduplex mutations. For example, A.T to G.C mutations would be formed either as G.T heteroduplexes (substitution of A with G across T) or as A.C heteroduplexes (substitution of T with C across A). Since PAH react primarily with purines, repair will excise the purine lesions and resynthesize the gap. Thus, A.T to G.C mutations would really be A to G mutations, i.e., G.T heteroduplexes. To detect G.T heteroduplexes in skin, we employed the T.G-DNA glycosylase (TDG) assay [44,96,97]. TDG removes T residues from G.T heteroduplexes to form G.AP sites [44,96]. We used the Methanobacterium thermoautotrophicum TDG (Trevigen, Gaithersburg, MD), which removes T residues from G.T heteroduplexes, and G residues from G.G duplexes (G.C to C.G mutations, formed by incorporation of G in a C-gap), but has no activity on A.C heteroduplexes.
After TDG treatment, skin DNA is re-purified by phenol:chloroform extraction and ethanol precipitation to remove any remaining enzyme and subjected to mutation analysis by the PCR-cloning-sequencing procedure. Abasic site-containing DNA is refractory to PCR [44,98]. By generating abasic sites, TDG renders G.T and G.G heteroduplex-containing DNA refractory to PCR, which would result in a drastic reduction in the proportion of A.T to G.C (and G.C to C.G) mutations, if present. The reduction of A.T to G.C mutations is taken as evidence for the presence of G.T heteroduplexes. In DB[a,l]P-treated mouse skin, the effect of TDG on A.T to G.C mutations is only observed until 1 d (the repair period) [39]. In the post-repair period, DNA replication converts the G.T heteroduplexes into A.T and G.C pairs in daughter DNA molecules, and TDG does not alter the frequency of A.T to G.C mutations [44,96,97]. Similarly, TDG does not reduce mutations generated in vitro by PCR over bulky adducted bases [44,96,97].
2.6 Statistical analysis
Mutation frequencies were compared using Fisher’s Exact test or a Chi-square test for linear association. These tests assume that each mutation observed is an independent event. This test has been used previously for similar analyses [97,99].
3. Results and Discussion
3.1. Mutagenesis by BP and metabolites
3.1.1 Papillomas
An earlier study reported that relatively few (23%, 3 out of 13) BP-induced SENCAR mouse skin papillomas contained oncogenic H-ras mutations (one GGCGly to CGCArg and two GGCGly to GTCVal at codon 13) [15]. In contrast, similar experiments in the CD-1 mouse showed a high percentage (90%, 9 out of 10) of skin papillomas carry oncogenic H-ras mutations, and a greater variety of mutations (two GGAGly to GTAVal at codon 12, five GGC to GTC at codon 13 and two CAAGln to CTALeu at codon 61) [14]. Our previous study in SENCAR mice also showed that a high percentage (69 %, 9 out of 13) of BP-induced skin papillomas carry oncogenic H-ras mutations (seven GGC to GTC at codon 13 and two CAA to CTA at codon 61) [16]. In mouse skin, BP forms 2.5-fold more depurinating (34% BP-6-C8G, 10% BP-6-N7G, 2% BPDE-10-N7G, 22% BP-6-N7A, 3% BPDE-10-N7A) than stable DNA adducts (23% BPDE-10-N2dG, and 6% other adducts, including small amounts of BPDE-10-N6dA) [22,86]. The distribution of clonal H-ras mutations (which, due to their abundance, are detectable by direct sequencing, see Materials and Methods, section 2.4) in the papillomas correlated better with BP-induced depurinating adducts (54% codon 13 G to T mutations with 46% G-depurinating adducts, and 15% codon 61 A to T mutations with 25% A-depurinating adducts) than with the stable adducts. To make a similar correlation with stable adducts, the BPDE-N6dA stable adducts should be highly mutagenic and account for the codon 61 A to T mutations. However, the BPDE-N6dA adduct does not appear to be exceptionally mutagenic [82] and forms A to G mutations in codon 61 [69].
To re-examine the correlation between BP-DNA adducts and mutations, and to determine whether other types of oncogenic H-ras mutations can be found in BP-induced papillomas, we have analyzed nine additional tumors, bringing the total to 22 (Table 1). Taken together, eleven papillomas (50%) showed the codon 13 GGC to GTC mutation and five papillomas (23%) showed the codon 61 CAA to CTA mutation. This result strengthened the relationship between the clonally-enriched oncogenic mutations in papillomas and the depurinating adducts.
To our knowledge, there are no reports of mutation analysis in BPDE-induced skin papillomas. However, one study reported that anti-BPDE forms the H-ras codon 12 GGC to GTC mutation in NIH3T3 cells to induce anchorage independence [100]. The solitary BPDE-induced papilloma obtained in our study contained the H-ras codon 13 GGC to GTC mutation (Table 1). In mouse skin, anti-BPDE forms 100-fold more stable (98% BPDE-10-N2dG and 0.6% other) than depurinating adducts (0.1% BPDE-10-N7G, 1.2% BPDE-10-N7A) [22,86]. Studies have shown that the BPDE-N2dG stable adduct mainly forms G to T mutations [49,82,101]. Thus, anti-BPDE would be expected to form the codon 13 G to T mutation in papillomas. We did not have enough tumors for this study; therefore, this result should be considered anecdotal.
To obtain greater insight into the role of the diolepoxide-DNA adducts in inducing oncogenic mutations, we examined twelve papillomas induced by the immediate precursor of BPDE, BP-7,8-dihydro-9,10-diol (BPDHD). BPDHD forms more stable adducts (61% BPDE-10-N2dG, 2% unidentified) than BP and a reverse profile of depurinating adducts (25% BPDE-10-N7A, 12% BPDE-10-N7G) compared to BP (25% Ade- and 46% Gua-depurinating adducts) (Table 1) [22]. Two of the BPDHD-induced papillomas contained the H-ras codon 13 GGC to GTC mutation, while another four contained the codon 61 CAA to CTA mutation. The relative proportions of the codon 13 G to T (17%) and codon 61 A to T (33%) mutations in BPDHD-induced papillomas made a strong correlation with depurinating adducts. The relative proportions of the H-ras mutations in BP- and BPDHD-induced papillomas followed those of the depurinating adducts, even though these carcinogens induce opposite profiles of these adducts. In contrast, the abundant N2dG stable adduct did not correlate with the relatively few codon 13 mutations. These results suggest that the depurinating adducts may make a greater contribution than the stable adducts in inducing the oncogenic H-ras mutations. However, these correlations do not exclude the possible role of the BPDE-N2dG stable adducts in inducing the codon 13 GGC to GTC mutation.
3.1.2 Preneoplastic mutations
Forward mutation assays in the Hprt and Aprt genes in Chinese hamster cells showed that BPDE induces 65–75% substitution and 26–36% deletion mutations [102–105]. Among the substitution mutations, G.C to T.A/C.G/A.T mutations were abundant (79–86%); however, significant numbers of A.T to G.C/T.A/C.G mutations (14–21%) were also observed. As the BPDE dose was lowered, the relative proportions of the A.T mutations increased [102,106,107]. Both BP and BPDE induced essentially similar spectra of Aprt mutations in Chinese hamster ovary cells [108]. This similarity was cited as evidence that BPDE is the chief mutagenic metabolite of BP. However, this similarity has not been explored in reporter gene assays, which score more PAH-induced mutations than other forward mutation assays that score mutations based on survival [109] and may show a somewhat different mutational spectrum. For example, analysis of transfected SupF plasmid in monkey cells [108,110] and a transgenic lacI in the skin of BB mice [111] shows that BPDE induces essentially a similar pattern of G.C-specific mutations as in the survival-based forward mutation assays, but far fewer (0–7%) A.T-specific mutations. Information about the mutagenic contributions of BP-DNA adducts in preneoplastic mouse skin has been lacking.
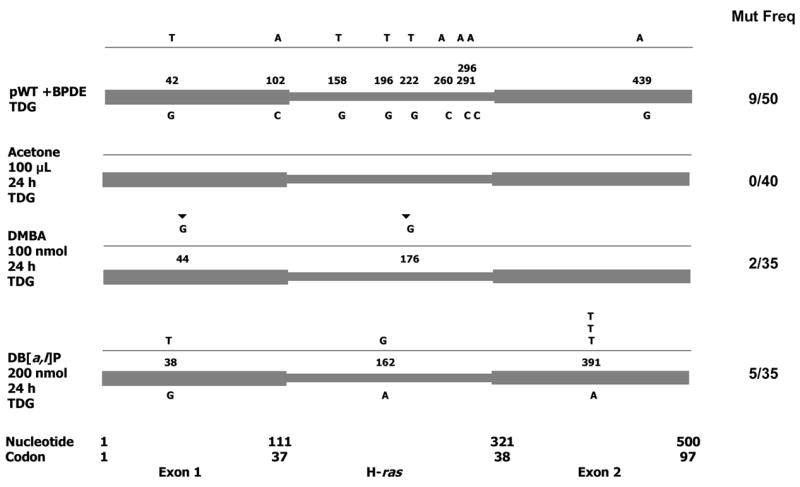
To examine the preneoplastic mutations in mouse skin, we have amplified the H-ras exon 1-2 region with a high fidelity mixture of DNA polymerases (8:1 combination of Tth and Vent DNA polymerases) [43,44,96,97]. Previous studies showed that this enzyme cocktail induces occasional misincorporations over untreated DNA (5.5–6.8 × 10−5) [43,44,96,97]. To determine PCR-induced spontaneous errors in vitro, a plasmid clone (pWT) containing the mouse H-ras exon 1-2 segment was PCR amplified, cloned and sequenced (Fig. 1). No mutations in 32 plasmids were observed (mutation frequency <3% of clones or <5.7 × 10−5 sequenced bases).
Figure 1. H-ras mutations induced in mouse skin 24 h after treatment with various PAH.

The nucleotide numbers correspond to GenBank U89950. The cartoons depict the location of the mutations in the H-ras gene, the wild-type bases are shown underneath and the mutations above. In vitro control: pWT, pWT + acid, pWT (4 μg) + anti-BPDE (1 μg in acetonitrile). Experimental: acetone, BP, BPDHD, anti-BPDE, DMBA, DB[a,l]P.
In addition to spontaneous misincorporations, unrepaired PAH-DNA stable adducts and abasic sites [42] in DNA can induce base incorporation errors during PCR. PCR-induced misincorporations over unrepaired lesions may make it difficult to identify the mutations formed by the cell. To test the nature of PCR-induced misincorporations over depurinated or stably-adducted DNA, pWT was reacted in vitro with a depurination buffer (10 mM NaCl, 10 mM sodium citrate, 10 mM Na3 PO4, pH 2.0) [44] or with anti-BPDE (1 μg in 10 μL acetonitrile to 4 μg pWT for 15 min). As observed previously [44], PCR amplification of the depurinated pWT resulted in 0 mutations in 33 plasmids (mutation frequency <3%, <5.5 × 10−5). The lack of mutations from the depurinated DNA could be related to the refractory nature of abasic site-containing DNA to PCR amplification [44,98]. On the other hand, anti-BPDE-treated pWT yielded nine mutations in 42 plasmids (mutation frequency = 21.4%, 38.9 × 10−5), all of which were G.C to T.A transversions. This result was expected because anti-BPDE forms principally (98%) the N2dG stable adduct, which is known to induce these mutations [49]. Therefore, under our experimental conditions, if skin DNA treated with BP or its metabolites contained unrepaired anti-BPDE-stable DNA adducts, chiefly G.C to T.A misincorporations would be introduced by the PCR polymerases (8:1 mixture of Tth and Vent DNA polymerases).
Next, we examined whether acetone (used as solvent for the PAH treatments) induces any mutations in mouse skin DNA (Fig. 1). Analysis of 35 plasmids from acetone (100 μL)-treated DNA showed one A to G mutation (mutation frequency = 2.8% of the clones or, 5.2 × 10−5 of sequenced bases) (Table 2). This result indicates that acetone may form a very low frequency of A.T to G.C mutations in mouse skin.
Table 2.
Preneoplastic mutation frequencies in PAH-treated mouse skin
| Treatment | Mutation frequency | ||
|---|---|---|---|
| Total mutations/Total no. of plasmids (%) | Mutations/Sequenced bases | ||
| Acetone (solvent)
Acetone + TDG |
100 μL | 2.8
0 |
5.2 × 10−5
0 |
| BP | 400 nmol | 7 | 12.8 × 10−5 |
| BPDHD | 400 nmol | 15.6 | 28.4 × 10−5 |
| Anti-BPDE | 400 nmol | 21.4 | 38.8 × 10−5 |
| DMBA
DMBA + TDG |
100 nmol | 53.8
5.7 |
97.9 × 10−5
10.3 × 10−5 |
| DB[a,l]P
DB[a,l]P + TDG |
200 nmol | 34
14.3 |
61.8 × 10−5
25.9 × 10−5 |
Treatment of mouse skin with BP resulted in five mutations in 71 plasmids (mutation frequency = 7%, 12.8 × 10−5) (Table 2), four of which were G.C to T.A transversions and one was an A.T to G.C transition (Fig. 1). Similar treatment with BPDHD induced five mutations in 32 plasmids (mutation frequency = 15.6%, 28.4 × 10−5) (Table 2), all of which were G.C to T.A transversions. Anti-BPDE induced nine mutations in 42 plasmids (21.4%, 38.9 × 10−5) (Table 2), six of which were G.C to T.A transversions, and one each of G.C to A.T, A.T to G.C and A.T to T.A mutations (Fig. 1).
The increasing mutagenesis by BP < BPDHD < anti-BPDE (7% vs. 15.6% vs. 21.4%, p = 0.026) in mouse skin supports the idea that BP metabolism is an important factor for generating these preneoplastic mutations. None of these three compounds showed either the codon 13 (GGC to GTC) or the codon 61 (CAA to CTA) transforming mutation at 1 d. However, these mutations are found in the papillomas, which suggests that BP and its metabolites induce the transforming mutations in preneoplastic skin. Under our experimental conditions, G.C to T.A mutations were most abundant in the mouse skin spectra induced by BP, BPDHD or anti-BPDE. These mutations are similar to that formed by PCR bypass errors over BPDE-N2dG adducts (Fig. 1). Therefore, the G.C to T.A mutations cannot be readily identified as ‘real’ (physiologically formed) mutations. On the other hand, skin mutations other than G.C to T.A transversions (G.C to A.T, A.T to G.C and A.T to T.A mutations) could be physiological. Since we have examined preneoplastic mutations in the pre-replication repair period [33–39], these results raise the possibility that excision repair errors in the skin cells contributed to generating some of these mutations.
In summary, we found a strong relationship between depurinating adducts and clonally-expanded papilloma mutations. We are unable to make a correlation between preneoplastic mutations and BP-DNA adducts, because of the difficulties in identifying the abundant G.C to T.A mutations by origin (physiological vs. PCR bypass misincorporations). A meaningful relationship should consider only the physiological mutations. The clonally-expanded, oncogenic mutations in the papillomas are physiological, because these mutations are needed to establish the tumors and therefore cannot be PCR artifacts.
3.2. Mutagenesis by DB[a,l]P
3.2.1 Papillomas
We have previously reported that four out of five DB[a,l]P-induced papillomas contained the clonal H-ras codon 61 (CAA to CTA) mutation, and the fifth (A43) contained an uncharacterized codon 61 clonal mutation [16]. To identify the clonal mutation in A43, we amplified its H-ras PCR product, cloned it in pUC18, and sequenced recombinant plasmids from individual colonies (Fig. 2). This papilloma contained the clonal CAA to CTT mutation. Both CTA and CTT code for Leu. In addition, A43 contained a variety of non-clonal mutations. These were mostly transitions (eight A.T→G.C and threeG.C→A.T out of twelve point mutations). In addition, there were two G-deletions (ΔG28, ΔG29). To examine whether similar non-clonal mutations can be found in other papillomas, we examined another DB[a,l]P-induced papilloma (B32) (Fig. 2). This papilloma contained the CAA to CTA as its clonal H-ras mutation. B32 also showed a similar spectrum of non-clonal mutations (six A.T to G.C transitions in a total of eight point mutations, and a C276-deletion). Except for one plasmid clone from B32 (which contained both the clonal CAA to CTA mutation and a non-clonal A431 to G mutation), all plasmid clones from these two papillomas contained only one mutation. The frequencies of clonal codon 61 mutations in the two papillomas [B32, 6 among 26 plasmids (23%) and A43, 11 in 26 plasmids (42%)] are consistent with frequencies of clonal mutations in other DB[a,l]P-induced papillomas (14–47%) previously determined by direct quantification [43]. Therefore, mutation frequencies calculated from the spectra match those obtained by direct quantification.
Figure 2. H-ras mutations in mouse skin papillomas induced by DB[a,l]P.

The two papillomas (A43 and B32) were obtained by treating the dorsal skin of SENCAR mice with DB[a,l]P (200 nmol) and promoting with chronic twice-weekly treatments with 3 nmol phorbol ester. No. of mutations/Total No. of plasmids: 15/17 (B32) and 25/26 (A43). ▲, deletion. Control: A single treatment with TPA resulted in seven mutations in fortyeight clones (50% A.T to G.C transitions), suggesting that some of the mutations in the tumors may be due to the chronic TPA treatment.
Our previous studies of mutations in DB[a,l]P-treated mouse skin suggested that most cells carrying the early mutations (12 h-4 d) are eliminated in the later preneoplastic period (5–7 d) [44,74]. Therefore, we investigated whether promoting treatments with phorbol ester (TPA) could be the origin of the non-clonal mutations in DB[a,l]P-induced papillomas. TPA is known to induce infiltration of neutrophils in mouse skin, which release reactive oxygen to form oxidative DNA damage [112]. We verified that 12 h after TPA treatment there was a significant increase in myeloperoxidase activity (a neutrophil marker) [113] and visible neutrophil infiltration (hematoxylin and eosin staining of skin sections) (results not shown). At this time point, TPA induced seven mutations in 48 clones, of which 50% were A.T to G.C mutations (Fig. 2). This result is consistent with the idea that some of the non-clonal mutations in the papillomas may have been generated by the chronic TPA treatment.
Tandem base mutations (GG to TT) in the K-ras gene have been reported in lung cancer [114]. Since PAH contribute to lung cancer, the observation of the tandem base mutation in H-ras codon 61 (CAA to CTT) in a DB[a,l]P-induced papilloma is interesting. To determine how frequently DB[a,l]P induces the tandem base codon 61 mutations, we analyzed seven additional DB[a,l]P-induced papillomas by direct sequencing of the H-ras PCR product (Table 1). Of these, six contained the H-ras codon 61 (CAA to CTA) clonal mutation, whereas the seventh tumor did not carry any clonal mutations in the exon 1-2 region. Taken together with previous results [16], it appears that DB[a,l]P induces the single-base codon 61 mutations at a much higher frequency (83%, ten out of twelve papillomas) than the tandem base mutation (8%, one out of twelve papillomas).
In mouse skin, DB[a,l]P forms 99% depurinating adducts (49% N3A, 14% N7A, 10% C8G and 2% N7G, generated by the DB[a,l]P radical cation, and 18% N7A, 6% N7G generated by DB[a,l]PDE) and 1% stable adducts (0.5% N2dG, 0.45% N6dA, generated by DB[a,l]PDE)[23,24]. Taken together, DB[a,l]P forms 81% Ade-depurinating adducts, 18% Gua-depurinating adducts and 1% stable (equal amounts of A and G) adducts. Thus, the H-ras codon 61 CAA to CTA/CTT mutations in DB[a,l]P-induced skin papillomas strongly correlated with the abundant A-depurinating adducts. It is worth noting that a different profile of mutations is found in the DB[a,l]P-induced adenomas in the A/J mouse lung. These adenomas carry a variety of K-ras mutations (44% carry codon 12 GGT to TGT/GTT/CGT mutations and 56% carry codon 61 CAA to CTA/CGA/CAT/CAC mutations) [115,116]. A correlation of these adenoma mutations with DB[a,l]P-induced stable adducts has been suggested [115,116]. To our knowledge, depurinating adduct formation by DB[a,l]P in the A/J mouse lung has not been studied.
3.2.2 Preneoplastic mutations
DB[a,l]P treatment of mouse skin resulted in 17 mutations in 50 plasmids (mutation frequency = 34%, 61.8 × 10−5) (Fig. 1 & Table 2). Twelve of these mutations (70%) were A.T to G.C transitions. This result is similar to that obtained in a previous study [44]. The PCR-cloning-sequencing procedure cannot determine whether these mutations are A to G or T to C transitions. However, the T.G-DNA glycosylase (TDG) assay indicates that these mutations are A to G transitions, formed as G.T heteroduplexes (see Section 3.4). If so, the abundant A.T to G.C mutations can be readily correlated with the abundant Ade-depurinating adducts (81% of all adducts).
These mutations do not show a ready correlation with DB[a,l]P-induced stable adducts. Our studies show that DB[a,l]P forms its stable adducts through its diolepoxides (equal amounts of Ade and Gua adducts) [24]. We had previously observed that anti-DB[a,l]PDE treatment of mouse skin results in the formation of 53% A.T-specific (16 out of 30) and 47% G.C-specific (14 out of 30) mutations [44]. This spectrum of mutations is correlated with the relative proportions of stable adducts. Fourteen of these A.T-specific mutations were A.T to G.C transitions. Most of the DB[a,l]PDE-induced N7Ade-depurinating adducts are contributed by syn-DB[a,l]PDE; the anti-isomer produces only 6% of the total. The TDG assay did not identify the anti-DB[a,l]PDE mutations as G.T heteroduplexes [44]. This is in contrast with mutagenesis by DB[a,l]P itself. These results differentiated the A.T to G.C mutations induced by DB[a,l]P from those induced by DB[a,l]PDE.
It is worth noting that depending on the cell type, DB[a,l]P or its metabolites may induce different adduct profiles. For example, in MCF-7 cells, DB[a,l]PDE forms predominantly Ade-specific stable adducts [117]. In Chinese hamster V79 cells, anti-DB[a,l]PDE forms both G.C-specific and A.T-specific mutations in the Hprt gene [99]. At lower anti-DB[a,l]PDE doses, A.T mutations predominated, whereas at higher doses, G.C mutations were more abundant. On the other hand, DB[a,l]PDE forms A.T to T.A > G.C to T.A mutations in the cII gene of Big Blue mouse cells [118].
3.3 Mutagenesis by DMBA
3.3.1 Papillomas
Like DB[a,l]P, DMBA forms mainly (99%) depurinating adducts (79% N7A and 20% N7G from the radical cation) and 1% stable adducts generated by DMBADE [21,61]. Even though the DMBADEs form mainly A-specific stable adducts [61,119], in mouse skin, DMBA itself forms only ~23% more A than G stable adducts [61].
A high percentage (90–100%) of mouse skin papillomas induced by DMBA contain the H-ras codon 61 CAA to CTA mutation [16,120,121]. DMBA can also induce other types of mutations in papillomas. For example, H-ras codon 61 CAA to CGA and CAA to CAT mutations have been reported in 4% and 2% of DMBA-induced papillomas [122]. Similar results were obtained with transgenic C57BL/6 mice expressing ornithine decarboxylase. The majority (50%) of DMBA-induced papillomas were found to contain the H-ras codon 61 CAA to CTA mutation, whereas 4% contained K-ras codon 12 GGT to TGT, and another 29% contained K-ras codon 61 CAA to CTA mutations [123]. Therefore, in all these studies, A-specific mutations predominated. All six DMBA-induced papillomas newly reported here contained the H-ras codon 61 CAA to CTA mutation (Table 1). In a previous study, four DMBA-induced papillomas were found to contain the H-ras codon 61 (CAA to CTA) mutation [16]. Taken together with our previous results, ten out of ten DMBA-induced papillomas contained this mutation.
Therefore, DMBA showed a strong stochastic relationship between the major Ade-depurinating adducts and the codon 61 A to T mutations in papillomas (Table 1). Tang et al. have suggested that these mutations originate from the stable adducts induced by DMBADE [61]. DMBADE induces 5–9-fold fewer papillomas than DMBA, but 94–96% of these papillomas also contain the codon 61 CAA to CTA mutation [61]. Even though DMBADE forms mainly A-specific stable adducts, its reaction with H-ras codon 12 GGA, codon 13 GGC and codon 61 CAA sequences has been demonstrated [124], which makes it difficult to explain why its papillomas only show A-specific mutations. DMBADE also forms depurinating adducts (N7A and N7G) [125], but how much of these adducts are formed in mouse skin is yet to be determined.
Although different mutations in the same oncogenic site are not equally expandable by phorbol ester (cells carrying the CAA to CTA mutation at codon 61 are favored over cells carrying the CAA to CGA or CAA to CAT mutations) [59], it is not very likely that the absence of codon 13 mutations among DMBA papillomas is a result of their inefficient clonal expansion. After all, H-ras codon 13 GGC to GTC mutations were found in a majority of BP-induced papillomas in the same species of mouse (Table 1). It is more likely that relatively few codon 13 GGC to GTC mutations are formed in DMBA-treated skin. To address these questions, we analyzed H-ras mutations in DMBA-treated preneoplastic skin.
3.3.2 Preneoplastic mutations
Analysis of the H-ras gene 1 d after DMBA treatment of SENCAR mouse skin also showed predominantly A.T to G.C mutations (Fig. 1). Of the 21 mutations, six were G.C-specific (two G.C to A.T and four G.C to T.A) and the remaining 15 were A.T-specific (fourteen A.T to G.C and one A.T to T.A). The overall mutation frequency was 53.8% or 97.9 × 10−5. This result is consistent with earlier observations reporting preneoplastic mutation frequency (~10−4) in DMBA-treated mouse skin [58]. However, the DMBA mutation frequency was higher than that observed with DB[a,l]P, which is a stronger carcinogen than the former [94]. It is worth noting that at the chosen dose (200 nmol), DB[a,l]P shows significant skin toxicity, which makes it difficult to compare mutagenesis by these two PAH.
Earlier studies also reported that DMBA forms mainly A.T-specific mutations. For example, in DMBA-treated Big Blue rats, spleen lymphocytes and mammary glands show predominantly A.T to T.A (42–44%) and some G.C to T.A (11–24%) mutations in the Hprt and lacI genes [126,127]. Since in mouse skin, DMBA forms 23% more A than G stable adducts of DMBADE [61], in the absence of evidence that DMBADE-G stable adducts should show much weaker mutagenic properties compared to the DMBADE-A stable adducts, it would be difficult to correlate the abundant A.T to G.C mutations with DMBADE-A stable adducts. It is worth noting that DB[a,l]PDE also induces similar mutational spectra (A.T to T.A > G.C to T.A) in the cII gene of Big Blue mouse cells, and it is difficult to explain the minority of G mutations by the stable adducts, even after exploring factors such as differences in their removal by NER [118]. In contrast, there is better correlation between the BPDE-N2dG stable adduct and the abundant G.C to A.T/T.A/C.G mutations observed in a shuttle vector assay in monkey cells or in the lacI gene of Big Blue mice [110,111]. Our observation that DMBA forms abundant amounts of A.T to G.C mutations in preneoplastic mouse skin (Fig. 1) does permit a straightforward correlation with the abundant A-depurinating adducts (Table 1).
3.4 DB[a,l]P and DMBA form mismatched heteroduplex mutations
DB[a,l]P and DMBA preneoplastic mutations were abundantly A.T to G.C transitions, and thus, were suitable for the T.G-DNA glycosylase (TDG) assay to determine whether these mutations are induced as G.T heteroduplexes [44,96,128]. In this assay, skin DNA is incubated with TDG, the DNA re-purified, and subjected to mutation analysis by the PCR-cloning-sequencing protocol. TDG converts G.T heteroduplexes to G.abasic sites. Since abasic site-containing DNA is refractory to PCR amplification, TDG treatment of G.T heteroduplex-containing DNA drastically reduces the frequency of A.T to G.C mutations in the spectra (see Section 2.5).
TDG treatment of anti-BPDE-treated pWT did not significantly reduce the frequency of mutations in the PCR misincorporation spectra (p = 0.79) [nine out 42 plasmids (21.4%, 38.9 × 10−5) before and nine among 50 plasmids (18%, 32.7 × 10−5) after TDG treatment] (Figs 1 and 3). Acetone-treated skin DNA showed a background frequency of mutations (one A to G mutation in 35 plasmids) (Fig. 1), whereas TDG treatment of the same DNA did not show any mutations (no mutations in 40 plasmids) (Fig. 3). These results suggest that TDG does not act on BPDE-induced stable adducts, or form new mutagenic lesions. In contrast, TDG treatment of DB[a,l]P- or DMBA-treated skin DNAs caused drastic reductions in the frequency of A.T to G.C mutations (Figs. 1 and 3). For example, DB[a,l]P formed 17 mutations in 50 plasmids (34%, including 12 A.T to G.C mutations), which were reduced by TDG treatment to five mutations in 35 plasmids (14.3%) (p = 0.048) (Table 2). Three of the five mutations in the DB[a,l]P-TDG spectrum were the oncogenic codon 61 CAA to CTA (nt 391) mutation (Fig. 3). The reduction of A.T to G.C mutations indicates that the A.T to G.C mutations are formed as G.T heteroduplexes, as would be expected if these mutations are formed by errors in repair. We think that the increased frequency of codon 61 mutations observed after TDG treatment is due to PCR enrichment of mutations present in DNA that did not contain the A.T to G.C mutations. This result also confirms that DB[a,l]P forms the tumor-forming codon 61 oncogene mutations (Table 1) in the early preneoplastic repair period. Similarly, DMBA formed 21 mutations in 39 plasmids (53.8%, including 14 A.T to G.C mutations), which were largely eliminated by TDG treatment (2 insertions in 35 plasmids, 5.7%) (p <0.001) (Table 2). Therefore, like DB[a,l]P, DMBA also forms G.T heteroduplex mutations in the early preneoplastic repair period. Papillomas formed by DMBA contain the codon 61 mutation (Table 1); however, these mutations were not detected in preneoplastic DMBA mutation spectra with or without TDG treatment. This result, however, does not exclude the possibility that DMBA also forms the codon 61 mutations in this period, but targets this codon at lower affinity than DB[a,l]P. This idea is worth consideration, because multiple studies have not produced evidence that overall mutagenic efficiency is a good indicator of tumor-forming ability. Indeed, we find that compared to BP, overall mutagenesis with equimolar amounts of anti-BPDE- or BPDHD-treated skin was greater, which is contrary to their tumor-forming activities.
Figure 3. H-ras mutations after treatment of mouse skin DNA with T.G-DNA glycosylase.
Incubation of PAH-treated skin DNA with this glycosylase (TDG) results in drastic reduction of A.T to G.C mutations present as G.T heteroduplexes. Mutations/total No. of plasmids: Controls: pWT + anti-BPDE + TDG, acetone-treated skin + TDG. Experimental: DMBA + TDG (2 ins/35), DB[a,l]P + TDG (5/35). ▼, insertion.
3.5 Sequence contexts of the A.T to G.C mutations
If the abundant A.T to G.C mutations are formed by errors in BER of abasic sites generated by the de-adenylation of the appropriate depurinating adducts, they should correspond to gap-filling error preferences of BER polymerases. As noted in Section 1, this mutational specificity is consistent with the error preferences of DNA polymerases β and ι [66,77]. If so, analysis of the sequence contexts of these mutations may provide further characteristics of the sequence selectivity for inducing this type of mutation.
Alignment of the sequences of the DMBA- or DB[a,l]P-induced preneoplastic A.T to G.C mutations revealed that they frequently occurred at putatively conserved sequences (Fig. 4). In both cases, these mutations frequently occurred at adenines flanked by doublets at 5′ or 3′, or both. The sequence preference can be dictated at three levels. First, they reflect the preferences of PAH intercalation [129]. Second, they should be among the hotspots of PAH-DNA reaction [25–32]. Third, they should be among the sequences in which BER polymerases make frequent errors [66,77].
Figure 4. Sequence contexts of PAH-induced A.T to G.C mutations.

Alignment of the sequences that showed A.T to G.C mutations in mouse skin treated with DMBA or DB[a,l]P. Some of the mutations were scored as A to G and others as T to C, the latter are also presented as A to G (italicized). The results show a frequent presence of doublet sequences flanking the mutated Adenines (underlined) either on the 5′, 3′ or both sides.
Previous studies with DMBA showed the formation of abundant amounts of A.T-specific mutations, but they were chiefly A.T to T.A transversions [126,127]. These mutations may arise by a different mechanism, as they frequently occurred at 5′CA sites [126]. If these mutations are also generated from PAH-induced abasic sites, bypass errors over unrepaired abasic sites could be a possible mechanism, especially since it would satisfy the A-rule [130,131]. In our studies, however, A.T to T.A mutations were rare in DMBA- or DB[a,l]P-treated preneoplastic skin. It would be interesting to examine whether these differences in mutagenesis are a reflection of gene-specific differences in DNA damage repair, such as those known among active and inactive genes in G1 and G2 phase cells [132].
4. Conclusions
Our previous study with DB[a,l]P was the only one in which preneoplastic skin and papilloma mutations were correlated with DNA adducts [44]. That study showed that depurinating adducts exhibit a strong stochastic relationship with both skin and papilloma mutations in the initiating oncogene. The current study expands on those findings. The results suggest that depurinating adducts induced by DMBA and DB[a,l]P play a major role in generating the H-ras mutations that lead to tumors. These results also suggest that the mutations are generated by errors in the repair of the abasic sites generated by the spontaneous dissociation of the depurinating adducts from DNA. Available biological evidence about the role of quiescent cells in DMBA tumorigenesis supports this idea. Although a strong relationship between BP-depurinating adducts and H-ras mutations in papillomas was found, we were unable to validate this relationship in preneoplastic skin, because the mutation analysis protocol used in this study cannot identify the abundant G.C to T.A mutations as either PCR misincorporation artifacts over BP-stable adducts or as ‘real’ mutations generated physiologically. Nevertheless, the current study highlights the value of studying PAH-induced mutations in both preneoplastic and tumor tissue. We propose that the role of DNA adducts in forming the mutations that lead to tumors can be described with greater certainty when adducts can be correlated with mutations in both types of studies.
Acknowledgments
We thank Dr. Leo Kirnarskiy and Oleg Shats of the Eppley Institute Molecular Modeling Core Facility for assistance with modeling with the H-ras gene. This research was supported by US PHS grants P01 CA49210 from the National Cancer Institute and P20 RR17675 from the National Center for Research Resources. Core support at the Eppley Institute was funded by NCI Laboratory Cancer Research Support (Core) grant CA 36727.
Abbreviations
- BP
benzo[a]pyrene
- BPDHD
benzo[a]pyrene-7,8-dihydrodiol
- anti-BPDE
(±)-anti- benzo[a]pyrene-7,8-dihydrodiol-9,10-epoxide
- DB[a,l]P
dibenzo[a,l]pyrene
- DMBA
7,12-dimethylbenz[a]anthracene
- PAH
polycyclic aromatic hydrocarbons
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Snook ME, Sevenson RF, Arrendale RF, Higman HC, Chortyk OT. The identification of high molecular weight polynuclear aromatic hydrocarbons in a biologically active fraction of cigarette smoke condensate. Beitrag Tabacforsch. 1977;9:79–101. [Google Scholar]
- 2.Mumford JL, Li X, Hu F, Lu XB, Chuang JC. Human exposure and dosimetry of polycyclic aromatic hydrocarbons in urine from Xuan Wei, China with high lung cancer mortality associated with exposure to unvented coal smoke. Carcinogenesis. 1995;16:3031–3036. doi: 10.1093/carcin/16.12.3031. [DOI] [PubMed] [Google Scholar]
- 3.Yu S, Campiglia AD. Direct determination of dibenzo[a,l]pyrene and its four dibenzopyrene isomers in water samples by solid-liquid extraction and laser-excited time-resolved Shpol’skii spectrometry. Anal Chem. 2005;77:1440–1447. doi: 10.1021/ac048310d. [DOI] [PubMed] [Google Scholar]
- 4.Everall JD, Dowd PM. Influence of environmental factors excluding ultra violet radiation on the incidence of skin cancer. Bull Cancer. 1978;65:241–247. [PubMed] [Google Scholar]
- 5.Hoffmann D, Hecht SS. Advances in tobacco carcinogenesis. In: Cooper CS, Grover PL, editors. Chemical carcinogenesis and mutagenesis I. Springer-Verlag; Berlin: 1990. pp. 63–102. [Google Scholar]
- 6.Hecht SS. Tobacco smoke carcinogens and breast cancer. Environ Mol Mutagen. 2002;39:119–126. doi: 10.1002/em.10071. [DOI] [PubMed] [Google Scholar]
- 7.Bos JL. Ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 8.van der Schroeff JG, Evers LM, Boot AJ, Bos JL. Ras oncogene mutations in basal cell carcinomas and squamous cell carcinomas of human skin. J Invest Dermatol. 1990;94:423–425. doi: 10.1111/1523-1747.ep12874504. [DOI] [PubMed] [Google Scholar]
- 9.Barbacid M. Ras oncogenes: their role in neoplasia. Eur J Clin Invest. 1990;20:225–235. doi: 10.1111/j.1365-2362.1990.tb01848.x. [DOI] [PubMed] [Google Scholar]
- 10.Grendys EC, Jr, Barnes WA, Weitzel J, Sparkowski J, Schlegel R. Identification of H, K, and N-ras point mutations in stage IB cervical carcinoma. Gynecol Oncol. 1997;65:343–347. doi: 10.1006/gyno.1997.4649. [DOI] [PubMed] [Google Scholar]
- 11.Shiraishi T, Muneyuki T, Fukutome K, Ito H, Kotake T, Watanabe M, Yatani R. Mutations of ras genes are relatively frequent in Japanese prostate cancers: pointing to genetic differences between populations. Anticancer Res. 1998;18:2789–2792. [PubMed] [Google Scholar]
- 12.Esapa CT, Johnson SJ, Kendall-Taylor P, Lennard TW, Harris PE. Prevalence of Ras mutations in thyroid neoplasia. Clin Endocrinol (Oxf) 1999;50:529–535. doi: 10.1046/j.1365-2265.1999.00704.x. [DOI] [PubMed] [Google Scholar]
- 13.Semczuk A, Schneider-Stock R, Berbec H, Marzec B, Jakowicki JA, Roessner A. K-ras exon 2 point mutations in human endometrial cancer. Cancer Lett. 2001;164:207–212. doi: 10.1016/s0304-3835(01)00380-9. [DOI] [PubMed] [Google Scholar]
- 14.Colapietro AM, Goodell AL, Smart RC. Characterization of benzo[a]pyrene-initiated mouse skin papillomas for Ha-ras mutations and protein kinase C levels. Carcinogenesis. 1993;14:2289–2295. doi: 10.1093/carcin/14.11.2289. [DOI] [PubMed] [Google Scholar]
- 15.DiGiovanni J, Beltran L, Rupp A, Harvey RG, Gill RD. Further analysis of c-Ha-ras mutations in papillomas initiated by several polycyclic aromatic hydrocarbons and papillomas from uninitiated, promoter-treated skin in SENCAR mice. Mol Carcinog. 1993;8:272–279. doi: 10.1002/mc.2940080410. [DOI] [PubMed] [Google Scholar]
- 16.Chakravarti D, Pelling JC, Cavalieri EL, Rogan EG. Relating aromatic hydrocarbon-induced DNA adducts and c-H-ras mutations in mouse skin papillomas: the role of apurinic sites. Proc Natl Acad Sci U S A. 1995;92:10422–10426. doi: 10.1073/pnas.92.22.10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vahakangas KH, Bennett WP, Castren K, Welsh JA, Khan MA, Blomeke B, Alavanja MC, Harris CC. p53 and K-ras mutations in lung cancers from former and never-smoking women. Cancer Res. 2001;61:4350–4356. [PubMed] [Google Scholar]
- 18.Li D, Firozi PF, Zhang W, Shen J, DiGiovanni J, Lau S, Evans D, Friess H, Hassan M, Abbruzzese JL. DNA adducts, genetic polymorphisms, and K-ras mutation in human pancreatic cancer. Mutat Res. 2002;513:37–48. doi: 10.1016/s1383-5718(01)00291-1. [DOI] [PubMed] [Google Scholar]
- 19.Cavalieri EL, Rogan EG. Central role of radical cations in metabolic activation of polycyclic aromatic hydrocarbons. Xenobiotica. 1995;25:677–688. doi: 10.3109/00498259509061885. [DOI] [PubMed] [Google Scholar]
- 20.Paules RS, Cordeiro-Stone M, Mass MJ, Poirier MC, Yuspa SH, Kaufman DG. Benzo[a]pyrene diol epoxide I binds to DNA at replication forks. Proc Natl Acad Sci U S A. 1988;85:2176–2180. doi: 10.1073/pnas.85.7.2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Devanesan PD, RamaKrishna NV, Padmavathi NS, Higginbotham S, Rogan EG, Cavalieri EL, Marsch GA, Jankowiak R, Small GJ. Identification and quantitation of 7,12-dimethylbenz[a]anthracene-DNA adducts formed in mouse skin. Chem Res Toxicol. 1993;6:364–371. doi: 10.1021/tx00033a018. [DOI] [PubMed] [Google Scholar]
- 22.Chen L, Devanesan PD, Higginbotham S, Ariese F, Jankowiak R, Small GJ, Rogan EG, Cavalieri EL. Expanded analysis of benzo[a]pyrene-DNA adducts formed in vitro and in mouse skin: their significance in tumor initiation. Chem Res Toxicol. 1996;9:897–903. doi: 10.1021/tx960004a. [DOI] [PubMed] [Google Scholar]
- 23.Cavalieri EL, Rogan EG, Li KM, Todorovic R, Ariese F, Jankowiak R, Grubor N, Small GJ. Identification and quantification of the depurinating adducts formed in mouse skin treated with dibenzo[a,l]pyrene (DB[a,l]P) or its metabolites and in rat mammary gland treated with DB[a,l]P. Chem Res Toxicol. 2005;18:976–983. doi: 10.1021/tx049682k. [DOI] [PubMed] [Google Scholar]
- 24.Todorovic R, Devanesan P, Rogan EG, Cavalieri EL. Identification and quantification of stable DNA adducts formed from dibenzo[a,l]pyrene or its metabolites in vitro and in mouse skin and rat mammary gland. Chem Res Toxicol. 2005;18:984–990. doi: 10.1021/tx049681s. [DOI] [PubMed] [Google Scholar]
- 25.Lobanenkov VV, Plumb M, Goodwin GH, Grover PL. The effect of neighbouring bases on G-specific DNA cleavage mediated by treatment with the anti-diol epoxide of benzo[a]pyrene in vitro. Carcinogenesis. 1986;7:1689–1695. doi: 10.1093/carcin/7.10.1689. [DOI] [PubMed] [Google Scholar]
- 26.Reardon DB, Bigger CA, Strandberg J, Yagi H, Jerina DM, Dipple A. Sequence selectivity in the reaction of optically active hydrocarbon dihydrodiol epoxides with rat H-ras DNA [letter] Chem Res Toxicol. 1989;2:12–14. doi: 10.1021/tx00007a002. [DOI] [PubMed] [Google Scholar]
- 27.Rill RL, Marsch GA. Sequence preferences of covalent DNA binding by anti-(+)- and anti-(−)- benzo[a]pyrene diol epoxides. Biochemistry. 1990;29:6050–6058. doi: 10.1021/bi00477a024. [DOI] [PubMed] [Google Scholar]
- 28.Dittrich KA, Krugh TR. Mapping of (+/−)-anti-benzo[a]pyrene diol epoxide adducts to human c-Ha-ras1 protooncogene. Chem Res Toxicol. 1991;4:277–281. doi: 10.1021/tx00021a003. [DOI] [PubMed] [Google Scholar]
- 29.Hruszkewycz AM, Canella KA, Peltonen K, Kotrappa L, Dipple A. DNA polymerase action on benzo[a]pyrene-DNA adducts. Carcinogenesis. 1992;13:2347–2352. doi: 10.1093/carcin/13.12.2347. [DOI] [PubMed] [Google Scholar]
- 30.Bigger CA, Cheh A, Latif F, Fishel R, Canella KA, Stafford GA, Yagi H, Jerina DM, Dipple A. DNA strand breaks induced by configurationally isomeric hydrocarbon diol epoxides. Drug Metab Rev. 1994;26:287–299. doi: 10.3109/03602539409029798. [DOI] [PubMed] [Google Scholar]
- 31.Chakravarti D, Cavalieri EL, Rogan EG. Linear amplification mapping of polycyclic aromatic hydrocarbon-reactive sequences in H-ras gene. DNA Cell Biol. 1998;17:529–539. doi: 10.1089/dna.1998.17.529. [DOI] [PubMed] [Google Scholar]
- 32.Chakravarti D. Polycyclic Aromatic Compounds Mapping. In: Meyers RA, editor. Encyclopedia of Analytical Chemistry: Instrumentation and Application. John Wiley & Sons, Ltd; England: 2000. pp. 5144–5159. [Google Scholar]
- 33.Slaga TJ, Bowden GT, Shapas BG, Boutwell RK. Macromolecular synthesis following a single application of polycyclic hydrocarbons used as initiators of mouse skin tumorigenesis. Cancer Res. 1974;34:771–777. [PubMed] [Google Scholar]
- 34.Stewart BW. Generation and persistence of carcinogen-induced repair intermediates in rat liver DNA in vivo. Cancer Res. 1981;41:3238–3243. [PubMed] [Google Scholar]
- 35.Sawyer TW, Gill RD, Smith-Oliver T, Butterworth BE, DiGiovanni J. Measurement of unscheduled DNA synthesis in primary cultures of adult mouse epidermal keratinocytes. Carcinogenesis. 1988;9:1197–1202. doi: 10.1093/carcin/9.7.1197. [DOI] [PubMed] [Google Scholar]
- 36.Gill RD, Butterworth BE, Nettikumara AN, DiGiovanni J. Relationship between DNA adduct formation and unscheduled DNA synthesis (UDS) in cultured mouse epidermal keratinocytes. Environ Mol Mutagen. 1991;18:200–206. doi: 10.1002/em.2850180307. [DOI] [PubMed] [Google Scholar]
- 37.Li R, Waga S, Hannon GJ, Beach D, Stillman B. Differential effects by the p21 CDK inhibitor on PCNA-dependent DNA replication and repair. Nature. 1994;371:534–537. doi: 10.1038/371534a0. [DOI] [PubMed] [Google Scholar]
- 38.Braithwaite E, Wu X, Wang Z. Repair of DNA lesions induced by polycyclic aromatic hydrocarbons in human cell-free extracts: involvement of two excision repair mechanisms in vitro. Carcinogenesis. 1998;19:1239–1246. doi: 10.1093/carcin/19.7.1239. [DOI] [PubMed] [Google Scholar]
- 39.Khan GA, Chakravarti D. Imbalanced induction of short-patch base excision repair genes in dibenzo[a,l]pyrene-treated mouse skin and estradiol-3,4-quinone-treated rat mammary gland is associated with XRCC1 insufficiency. Proc Am Assoc Cancer Res. 2005;46:719. [Google Scholar]
- 40.Wei D, Maher VM, McCormick JJ. Site-specific rates of excision repair of benzo[a]pyrene diol epoxide adducts in the hypoxanthine phosphoribosyltransferase gene of human fibroblasts: correlation with mutation spectra. Proc Natl Acad Sci U S A. 1995;92:2204–2208. doi: 10.1073/pnas.92.6.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mukherjee JJ, Gupta SK, Kumar S, Sikka HC. Effects of cadmium(II) on (+/−)-anti-benzo[a]pyrene-7,8-diol-9,10-epoxide-induced DNA damage response in human fibroblasts and DNA repair: a possible mechanism of cadmium’s cogenotoxicity. Chem Res Toxicol. 2004;17:287–293. doi: 10.1021/tx034229e. [DOI] [PubMed] [Google Scholar]
- 42.Chakravarti D, Badawi AF, Venugopal D, Meza JL, Crandall LZ, Rogan EG, Cavalieri EL. Improved measurement of dibenzo[a,l]pyrene-induced abasic sites by the aldehyde-reactive probe assay. Mutat Res. 2005;588:158–165. doi: 10.1016/j.mrgentox.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 43.Chakravarti D, Mailander P, Franzen J, Higginbotham S, Cavalieri EL, Rogan EG. Detection of dibenzo[a,l]pyrene-induced H-ras codon 61 mutant genes in preneoplastic SENCAR mouse skin using a new PCR-RFLP method. Oncogene. 1998;16:3203–3210. doi: 10.1038/sj.onc.1201853. [DOI] [PubMed] [Google Scholar]
- 44.Chakravarti D, Mailander PC, Cavalieri EL, Rogan EG. Evidence that error-prone DNA repair converts dibenzo[a,l]pyrene-induced depurinating lesions into mutations: formation, clonal proliferation and regression of initiated cells carrying H-ras oncogene mutations in early preneoplasia. Mutat Res. 2000;456:17–32. doi: 10.1016/s0027-5107(00)00102-0. [DOI] [PubMed] [Google Scholar]
- 45.Watanabe M, Maher VM, McCormick JJ. Excision repair of UV- or benzo[a]pyrene diol epoxide-induced lesions in xeroderma pigmentosum variant cells is ‘error free’. Mutat Res. 1985;146:285–294. doi: 10.1016/0167-8817(85)90070-7. [DOI] [PubMed] [Google Scholar]
- 46.Choi DJ, Roth RB, Liu T, Geacintov NE, Scicchitano DA. Incorrect base insertion and prematurely terminated transcripts during T7 RNA polymerase transcription elongation past benzo[a]pyrenediol epoxide-modified DNA. J Mol Biol. 1996;264:213–219. doi: 10.1006/jmbi.1996.0635. [DOI] [PubMed] [Google Scholar]
- 47.Maher VM, McCormick JJ. Role of DNA lesions and repair in the transformation of human cells. In: Grunberger D, Groff SP, editors. Mechanisms of Cellular Transformation by Carcinogenic agents. Pergamon Press; New York: 1987. pp. 135–149. [Google Scholar]
- 48.Kaufmann WK. Cell cycle checkpoints and DNA repair preserve the stability of the human genome. Cancer Metastasis Rev. 1995;14:31–41. doi: 10.1007/BF00690209. [DOI] [PubMed] [Google Scholar]
- 49.Moriya M, Spiegel S, Fernandes A, Amin S, Liu T, Geacintov N, Grollman AP. Fidelity of translesional synthesis past benzo[a]pyrene diol epoxide-2′-deoxyguanosine DNA adducts: marked effects of host cell, sequence context, and chirality. Biochemistry. 1996;35:16646–16651. doi: 10.1021/bi9608875. [DOI] [PubMed] [Google Scholar]
- 50.McGregor WG, Wei D, Chen RH, Maher VM, McCormick JJ. Relationship between adduct formation, rates of excision repair and the cytotoxic and mutagenic effects of structurally-related polycyclic aromatic carcinogens. Mutat Res. 1997;376:143–152. doi: 10.1016/s0027-5107(97)00037-7. [DOI] [PubMed] [Google Scholar]
- 51.Zhang Y, Yuan F, Wu X, Rechkoblit O, Taylor JS, Geacintov NE, Wang Z. Error-prone lesion bypass by human DNA polymerase eta. Nucleic Acids Res. 2000;28:4717–4724. doi: 10.1093/nar/28.23.4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y, Yuan F, Wu X, Wang M, Rechkoblit O, Taylor JS, Geacintov NE, Wang Z. Error-free and error-prone lesion bypass by human DNA polymerase kappa in vitro. Nucleic Acids Res. 2000;28:4138–4146. doi: 10.1093/nar/28.21.4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perlow RA, Broyde S. Evading the proofreading machinery of a replicative DNA polymerase: induction of a mutation by an environmental carcinogen. J Mol Biol. 2001;309:519–536. doi: 10.1006/jmbi.2001.4674. [DOI] [PubMed] [Google Scholar]
- 54.Perlow RA, Kolbanovskii A, Hingerty BE, Geacintov NE, Broyde S, Scicchitano DA. DNA adducts from a tumorigenic metabolite of benzo[a]pyrene block human RNA polymerase II elongation in a sequence- and stereochemistry-dependent manner. J Mol Biol. 2002;321:29–47. doi: 10.1016/s0022-2836(02)00593-4. [DOI] [PubMed] [Google Scholar]
- 55.Simhadri S, Kramata P, Zajc B, Sayer JM, Jerina DM, Hinkle DC, Wei CS. Benzo[a]pyrene diol epoxide-deoxyguanosine adducts are accurately bypassed by yeast DNA polymerase zeta in vitro. Mutat Res. 2002;508:137–145. doi: 10.1016/s0027-5107(02)00211-7. [DOI] [PubMed] [Google Scholar]
- 56.Schinecker TM, Perlow RA, Broyde S, Geacintov NE, Scicchitano DA. Human RNA polymerase II is partially blocked by DNA adducts derived from tumorigenic benzo[c]phenanthrene diol epoxides: relating biological consequences to conformational preferences. Nucleic Acids Res. 2003;31:6004–6015. doi: 10.1093/nar/gkg771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao B, Wang J, Geacintov NE, Wang Z. Poleta, Polzeta and Rev1 together are required for G to T transversion mutations induced by the (+)- and (−)-trans-anti-BPDE-N2-dG DNA adducts in yeast cells. Nucleic Acids Res. 2006;34:417–425. doi: 10.1093/nar/gkj446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nelson MA, Futscher BW, Kinsella T, Wymer J, Bowden GT. Detection of mutant Ha-ras genes in chemically initiated mouse skin epidermis before the development of benign tumors [published erratum appears in Proc Natl Acad Sci U S A 1993 Jan 15;90(2):781] Proc Natl Acad Sci U S A. 1992;89:6398–6402. doi: 10.1073/pnas.89.14.6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Finch JS, Albino HE, Bowden GT. Quantitation of early clonal expansion of two mutant 61st codon c-Ha- ras alleles in DMBA/TPA treated mouse skin by nested PCR/RFLP. Carcinogenesis. 1996;17:2551–2557. doi: 10.1093/carcin/17.12.2551. [DOI] [PubMed] [Google Scholar]
- 60.Melendez-Colon VJ, Smith CA, Seidel A, Luch A, Platt KL, Baird WM. Formation of stable adducts and absence of depurinating DNA adducts in cells and DNA treated with the potent carcinogen dibenzo[a,l]pyrene or its diol epoxides. Proc Natl Acad Sci U S A. 1997;94:13542–13547. doi: 10.1073/pnas.94.25.13542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tang MS, Vulimiri SV, Viaje A, Chen JX, Bilolikar DS, Morris RJ, Harvey RG, Slaga TJ, DiGiovanni J. Both (+/−)syn- and (+/−)anti-7,12-dimethylbenz[a]anthracene-3,4-diol-1,2-epoxides initiate tumors in mouse skin that possess -CAA- to -CTA- mutations at Codon 61 of c-H-ras. Cancer Res. 2000;60:5688–5695. [PubMed] [Google Scholar]
- 62.Willumsen BM, Papageorge AG, Kung HF, Bekesi E, Robins T, Johnsen M, Vass WC, Lowy DR. Mutational analysis of a ras catalytic domain. Mol Cell Biol. 1986;6:2646–2654. doi: 10.1128/mcb.6.7.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sukumar S, Barbacid M. Specific patterns of oncogene activation in transplacentally induced tumors. Proc Natl Acad Sci U S A. 1990;87:718–722. doi: 10.1073/pnas.87.2.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Karen J, Wang Y, Javaherian A, Vaccariello M, Fusenig NE, Garlick JA. 12-O-tetradecanoylphorbol-13-acetate induces clonal expansion of potentially malignant keratinocytes in a tissue model of early neoplastic progression. Cancer Res. 1999;59:474–481. [PubMed] [Google Scholar]
- 65.Yamakage K, Omori Y, Zaidan-Dagli ML, Cros MP, Yamasaki H. Induction of Skin Papillomas, Carcinomas, and Sarcomas in Mice in Which the Connexin 43 Gene is Heterologously Deleted. J Invest Dermatol. 2000;114:289–294. doi: 10.1046/j.1523-1747.2000.00873.x. [DOI] [PubMed] [Google Scholar]
- 66.Kunkel TA. The mutational specificity of DNA polymerase β during in vitro DNA synthesis. Production of frameshift, base substitution, and deletion mutations. J Biol Chem. 1985;260:5787–5796. [PubMed] [Google Scholar]
- 67.Thomas DC, Roberts JD, Sabatino RD, Myers TW, Tan CK, Downey KM, So AG, Bambara RA, Kunkel TA. Fidelity of mammalian DNA replication and replicative DNA polymerases. Biochemistry. 1991;30:11751–11759. doi: 10.1021/bi00115a003. [DOI] [PubMed] [Google Scholar]
- 68.Chary P, Lloyd RS. In vitro replication by prokaryotic and eukaryotic polymerases on DNA templates containing site-specific and stereospecific benzo[a]pyrene- 7,8-dihydrodiol-9,10-epoxide adducts. Nucleic Acids Res. 1995;23:1398–1405. doi: 10.1093/nar/23.8.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chary P, Latham GJ, Robberson DL, Kim SJ, Han S, Harris CM, Harris TM, Lloyd RS. In vivo and in vitro replication consequences of stereoisomeric benzo[a]pyrene-7,8-dihydrodiol 9,10-epoxide adducts on adenine N6 at the second position of N-ras codon 61. J Biol Chem. 1995;270:4990–5000. doi: 10.1074/jbc.270.10.4990. [DOI] [PubMed] [Google Scholar]
- 70.Lavrukhin OV, Lloyd RS. Mutagenic replication in a human cell extract of DNAs containing site-specific and stereospecific benzo(a)pyrene-7,8-diol-9,10-epoxide DNA adducts placed on the leading and lagging strands. Cancer Res. 1998;58:887–891. [PubMed] [Google Scholar]
- 71.Solhaug A, Refsnes M, Lag M, Schwarze PE, Husoy T, Holme JA. Polycyclic aromatic hydrocarbons induce both apoptotic and anti-apoptotic signals in Hepa1c1c7 cells. Carcinogenesis. 2004;25:809–819. doi: 10.1093/carcin/bgh069. [DOI] [PubMed] [Google Scholar]
- 72.Huc L, Rissel M, Solhaug A, Tekpli X, Gorria M, Torriglia A, Holme JA, Dimanche-Boitrel MT, Lagadic-Gossmann D. Multiple apoptotic pathways induced by p53-dependent acidification in benzo[a]pyrene-exposed hepatic F258 cells. J Cell Physiol. 2006;208:527–537. doi: 10.1002/jcp.20686. [DOI] [PubMed] [Google Scholar]
- 73.Stout GJ, Oosten M, Acherrat FZ, Wit J, Vermeij WP, Mullenders LH, Gruijl FR, Backendorf C. Selective DNA damage responses in murine Xpa(−/−), Xpc(−/−) and Csb(−/−) keratinocyte cultures. DNA Repair (Amst) 2005;4:1337–1344. doi: 10.1016/j.dnarep.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 74.Khan GA, Bhattacharya G, Mailander PC, Meza JL, Hansen LA, Chakravarti D. Harvey-ras gene expression and epidermal cell proliferation in dibenzo[a,l]pyrene-treated early preneoplastic SENCAR mouse skin. J Invest Dermatol. 2005;125:567–574. doi: 10.1111/j.0022-202X.2005.23845.x. [DOI] [PubMed] [Google Scholar]
- 75.Albert RE, Miller ML, Cody T, Andringa A, Shukla R, Baxter CS. Benzo[a]pyrene-induced skin damage and tumor promotion in the mouse. Carcinogenesis. 1991;12:1273–1280. doi: 10.1093/carcin/12.7.1273. [DOI] [PubMed] [Google Scholar]
- 76.Corominas M, Leon J, Kamino H, Cruz-Alvarez M, Novick SC, Pellicer A. Oncogene involvement in tumor regression: H-ras activation in the rabbit keratoacanthoma model. Oncogene. 1991;6:645–651. [PubMed] [Google Scholar]
- 77.Zhang Y, Yuan F, Wu X, Wang Z. Preferential incorporation of G opposite template T by the low-fidelity human DNA polymerase iota. Mol Cell Biol. 2000;20:7099–7108. doi: 10.1128/mcb.20.19.7099-7108.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sobol RW, Watson DE, Nakamura J, Yakes FM, Hou E, Horton JK, Ladapo J, Van Houten B, Swenberg JA, Tindall KR, Samson LD, Wilson SH. Mutations associated with base excision repair deficiency and methylation-induced genotoxic stress. Proc Natl Acad Sci U S A. 2002;99:6860–6865. doi: 10.1073/pnas.092662499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Seo KY, Jelinsky SA, Loechler EL. Factors that influence the mutagenic patterns of DNA adducts from chemical carcinogens. Mutat Res. 2000;463:215–246. doi: 10.1016/s1383-5742(00)00047-8. [DOI] [PubMed] [Google Scholar]
- 80.Yan SF, Wu M, Geacintov NE, Broyde S. Altering DNA polymerase incorporation fidelity by distorting the dNTP binding pocket with a bulky carcinogen-damaged template. Biochemistry. 2004;43:7750–7765. doi: 10.1021/bi0499516. [DOI] [PubMed] [Google Scholar]
- 81.Zhang Y, Wu X, Guo D, Rechkoblit O, Geacintov NE, Wang Z. Two-step error-prone bypass of the (+)- and (−)-trans-anti-BPDE-N2-dG adducts by human DNA polymerases eta and kappa. Mutat Res. 2002;510:23–35. doi: 10.1016/s0027-5107(02)00249-x. [DOI] [PubMed] [Google Scholar]
- 82.Rechkoblit O, Zhang Y, Guo D, Wang Z, Amin S, Krzeminsky J, Louneva N, Geacintov NE. Trans-lesion synthesis past bulky benzo[a]pyrene diol epoxide N2-dG and N6-dA lesions catalyzed by DNA bypass polymerases. J Biol Chem. 2002;277:30488–30494. doi: 10.1074/jbc.M201167200. [DOI] [PubMed] [Google Scholar]
- 83.Andreasen E. Cyclic changes in the skin of the mouse. Acta Pathol Scand. 1953;32:157–164. doi: 10.1111/j.1699-0463.1953.tb00237.x. [DOI] [PubMed] [Google Scholar]
- 84.Morris RJ, Coulter K, Tryson K, Steinberg SR. Evidence that cutaneous carcinogen-initiated epithelial cells from mice are quiescent rather than actively cycling. Cancer Res. 1997;57:3436–3443. [PubMed] [Google Scholar]
- 85.Fischer WH, Lutz WK. Correlation of individual papilloma latency time with DNA adducts, 8- hydroxy-2′-deoxyguanosine, and the rate of DNA synthesis in the epidermis of mice treated with 7,12-dimethylbenz[a]anthracene. Proc Natl Acad Sci U S A. 1995;92:5900–5904. doi: 10.1073/pnas.92.13.5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rogan EG, Devanesan PD, RamaKrishna NV, Higginbotham S, Padmavathi NS, Chapman K, Cavalieri EL, Jeong H, Jankowiak R, Small GJ. Identification and quantitation of benzo[a]pyrene-DNA adducts formed in mouse skin. Chem Res Toxicol. 1993;6:356–363. doi: 10.1021/tx00033a017. [DOI] [PubMed] [Google Scholar]
- 87.Talaska G, Jaeger M, Reilman R, Collins T, Warshawsky D. Chronic, topical exposure to benzo[a]pyrene induces relatively high steady-state levels of DNA adducts in target tissues and alters kinetics of adduct loss. Proc Natl Acad Sci U S A. 1996;93:7789–7793. doi: 10.1073/pnas.93.15.7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.de Vries A, van Oostrom CT, Dortant PM, Beems RB, van Kreijl CF, Capel PJ, van Steeg H. Spontaneous liver tumors and benzo[a]pyrene-induced lymphomas in XPA- deficient mice. Mol Carcinog. 1997;19:46–53. [PubMed] [Google Scholar]
- 89.de Vries A, Dolle ME, Broekhof JL, Muller JJ, Kroese ED, van Kreijl CF, Capel PJ, Vijg J, van Steeg H. Induction of DNA adducts and mutations in spleen, liver and lung of XPA- deficient/lacZ transgenic mice after oral treatment with benzo[a]pyrene: correlation with tumour development. Carcinogenesis. 1997;18:2327–2332. doi: 10.1093/carcin/18.12.2327. [DOI] [PubMed] [Google Scholar]
- 90.Wijnhoven SW, Kool HJ, van Oostrom CT, Beems RB, Mullenders LH, van Zeeland AA, van der Horst GT, Vrieling H, van Steeg H. The relationship between benzo[a]pyrene-induced mutagenesis and carcinogenesis in repair-deficient Cockayne syndrome group B mice. Cancer Res. 2000;60:5681–5687. [PubMed] [Google Scholar]
- 91.Gill HS, Kole PL, Wiley JC, Li KM, Higginbotham S, Rogan EG, Cavalieri EL. Synthesis and tumor-initiating activity in mouse skin of dibenzo[a,l]pyrene syn- and anti-fjord-region diolepoxides. Carcinogenesis. 1994;15:2455–2460. doi: 10.1093/carcin/15.11.2455. [DOI] [PubMed] [Google Scholar]
- 92.Hanson AA, Li KM, Lin CH, Jankowiak R, Small GJ, Rogan EG, Cavalieri EL. Synthesis and structure determination of 6-methylbenzo[a]pyrene-deoxyribonucleoside adducts and their identification and quantitation in vitro and in mouse skin. Chem Biol Interact. 2000;128:65–90. doi: 10.1016/s0009-2797(00)00189-7. [DOI] [PubMed] [Google Scholar]
- 93.NIH Guidelines for the Laboratory Use of Chemical Carcinogens, US Government Printing Office, Washington, DC, 1981.
- 94.Higginbotham S, RamaKrishna NV, Johansson SL, Rogan EG, Cavalieri EL. Tumor-initiating activity and carcinogenicity of dibenzo[a,l]pyrene versus 7,12-dimethylbenz[a]anthracene and benzo[a]pyrene at low doses in mouse skin. Carcinogenesis. 1993;14:875–878. doi: 10.1093/carcin/14.5.875. [DOI] [PubMed] [Google Scholar]
- 95.Sun Y, Hegamyer G, Cheng YJ, Hildesheim A, Chen JY, Chen IH, Cao Y, Yao KT, Colburn NH. An infrequent point mutation of the p53 gene in human nasopharyngeal carcinoma. Proc Natl Acad Sci U S A. 1992;89:6516–6520. doi: 10.1073/pnas.89.14.6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chakravarti D, Mailander PC, Li KM, Higginbotham S, Zhang HL, Gross ML, Meza JL, Cavalieri EL, Rogan EG. Evidence that a burst of DNA depurination in SENCAR mouse skin induces error-prone repair and forms mutations in the H-ras gene. Oncogene. 2001;20:7945–7953. doi: 10.1038/sj.onc.1204969. [DOI] [PubMed] [Google Scholar]
- 97.Mailander PC, Khan GA, Meza JL, Higginbotham S, Chakravarti D. Induction of abundant A.T to G.C mutations by erroneous repair of estrogen-induced depurinated DNA in the mammary gland of ACI rats. J Steroid Biochem Mol Biol. 2006;101:205–215. doi: 10.1016/j.jsbmb.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 98.Fromenty B, Demeilliers C, mansouri A, Pessayre D. Escherichia coli exonuclease III enhances long PCR amplification of damaged DNA templates. Nucleic Acids Res. 2000;28:e50. doi: 10.1093/nar/28.11.e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mahadevan B, Dashwood WM, Luch A, Pecaj A, Doehmer J, Seidel A, Pereira C, Baird WM. Mutations induced by (−)-anti-11R,12S-dihydrodiol 13S,14R-epoxide of dibenzo[a,l]pyrene in the coding region of the hypoxanthine phosphoribosyltransferase (Hprt) gene in Chinese hamster V79 cells. Environ Mol Mutagen. 2003;41:131–139. doi: 10.1002/em.10136. [DOI] [PubMed] [Google Scholar]
- 100.Stevens CW, Manoharan TH, Fahl WE. Characterization of mutagen-activated cellular oncogenes that confer anchorage independence to human fibroblasts and tumorigenicity to NIH 3T3 cells: sequence analysis of an enzymatically amplified mutant HRAS allele. Proc Natl Acad Sci U S A. 1988;85:3875–3879. doi: 10.1073/pnas.85.11.3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Seo KY, Nagalingam A, Tiffany M, Loechler EL. Mutagenesis studies with four stereoisomeric N2-dG benzo[a]pyrene adducts in the identical 5′-CGC sequence used in NMR studies: G→T mutations dominate in each case. Mutagenesis. 2005;20:441–448. doi: 10.1093/mutage/gei061. [DOI] [PubMed] [Google Scholar]
- 102.Wei SJ, Chang RL, Hennig E, Cui XX, Merkler KA, Wong CQ, Yagi H, Jerina DM, Conney AH. Mutagenic selectivity at the HPRT locus in V-79 cells: comparison of mutations caused by bay-region benzo[a]pyrene 7,8-diol-9,-10-epoxide enantiomers with high and low carcinogenic activity. Carcinogenesis. 1994;15:1729–1735. doi: 10.1093/carcin/15.8.1729. [DOI] [PubMed] [Google Scholar]
- 103.Schiltz M, Cui XX, Lu YP, Yagi H, Jerina DM, Zdzienicka MZ, Chang RL, Conney AH, Wei SJ. Characterization of the mutational profile of (+)-7R,8S-dihydroxy-9S, 10R-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene at the hypoxanthine (guanine) phosphoribosyltransferase gene in repair-deficient Chinese hamster V-H1 cells. Carcinogenesis. 1999;20:2279–2286. doi: 10.1093/carcin/20.12.2279. [DOI] [PubMed] [Google Scholar]
- 104.Yang H, Mazur-Melnyk M, de Boer JG, Glickman BW. A comparison of mutational specificity of mutations induced by S9-activated B[a]P and benzo[a]pyrene-7,8-diol-9,10-epoxide at the endogenous aprt gene in CHO cells. Mutat Res. 1999;423:23–32. doi: 10.1016/s0027-5107(98)00221-8. [DOI] [PubMed] [Google Scholar]
- 105.Conney AH, Chang RL, Cui XX, Schiltz M, Yagi H, Jerina DM, Wei SJ. Dose-dependent differences in the profile of mutations induced by carcinogenic (R,S,S,R) bay- and fjord-region diol epoxides of polycyclic aromatic hydrocarbons. Adv Exp Med Biol. 2001;500:697–707. doi: 10.1007/978-1-4615-0667-6_102. [DOI] [PubMed] [Google Scholar]
- 106.Wei SJ, Chang RL, Bhachech N, Cui XX, Merkler KA, Wong CQ, Hennig E, Yagi H, Jerina DM, Conney AH. Dose-dependent differences in the profile of mutations induced by (+)- 7R,8S-dihydroxy-9S,10R-epoxy-7,8,9,10-tetrahydrobenzo(a)pyrene in the coding region of the hypoxanthine (guanine) phosphoribosyltransferase gene in Chinese hamster V-79 cells. Cancer Res. 1993;53:3294–3301. [PubMed] [Google Scholar]
- 107.Shukla R, Liu T, Geacintov NE, Loechler EL. The major, N2-dG adduct of (+)-anti-B[a]PDE shows a dramatically different mutagenic specificity (predominantly, G → A) in a 5′-CGT-3′ sequence context. Biochemistry. 1997;36:10256–10261. doi: 10.1021/bi970541+. [DOI] [PubMed] [Google Scholar]
- 108.Yang JL, Maher VM, McCormick JJ. Kinds of mutations formed when a shuttle vector containing adducts of benzo[a]pyrene-7,8-diol-9,10-epoxide replicates in COS7 cells. Mol Cell Biol. 1987;7:1267–1270. doi: 10.1128/mcb.7.3.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Skopek TR, Kort KL, Marino DR, Mittal LV, Umbenhauer DR, Laws GM, Adams SP. Mutagenic response of the endogenous hprt gene and lacI transgene in benzo[a]pyrene-treated Big Blue B6C3F1 mice. Environ Mol Mutagen. 1996;28:376–384. doi: 10.1002/(SICI)1098-2280(1996)28:4<376::AID-EM11>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 110.Roilides E, Gielen JE, Tuteja N, Levine AS, Dixon K. Mutational specificity of benzo[a]pyrene diolepoxide in monkey cells. Mutat Res. 1988;198:199–206. doi: 10.1016/0027-5107(88)90055-3. [DOI] [PubMed] [Google Scholar]
- 111.Miller ML, Vasunia K, Talaska G, Andringa A, de Boer J, Dixon K. The tumor promoter TPA enhances benzo[a]pyrene and benzo[a]pyrene diolepoxide mutagenesis in Big Blue mouse skin. Environ Mol Mutagen. 2000;35:319–327. doi: 10.1002/1098-2280(2000)35:4<319::aid-em6>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 112.Frenkel K, Wei L, Wei H. 7,12-dimethylbenz[a]anthracene induces oxidative DNA modification in vivo. Free Radic Biol Med. 1995;19:373–380. doi: 10.1016/0891-5849(95)00046-z. [DOI] [PubMed] [Google Scholar]
- 113.Kensler TW, Egner PA, Moore KG, Taffe BG, Twerdok LE, Trush MA. Role of inflammatory cells in the metabolic activation of polycyclic aromatic hydrocarbons in mouse skin. Toxicol Appl Pharmacol. 1987;90:337–346. doi: 10.1016/0041-008x(87)90341-3. [DOI] [PubMed] [Google Scholar]
- 114.Keohavong P, DeMichele MA, Melacrinos AC, Landreneau RJ, Weyant RJ, Siegfried JM. Detection of K-ras mutations in lung carcinomas: relationship to prognosis. Clin Cancer Res. 1996;2:411–418. [PubMed] [Google Scholar]
- 115.Prahalad AK, Ross JA, Nelson GB, Roop BC, King LC, Nesnow S, Mass MJ. Dibenzo[a,l]pyrene-induced DNA adduction, tumorigenicity, and Ki-ras oncogene mutations in strain A/J mouse lung. Carcinogenesis. 1997;18:1955–1963. doi: 10.1093/carcin/18.10.1955. [DOI] [PubMed] [Google Scholar]
- 116.Nesnow S, Ross JA, Mass MJ, Stoner GD. Mechanistic relationships between DNA adducts, oncogene mutations, and lung tumorigenesis in strain A mice. Exp Lung Res. 1998;24:395–405. doi: 10.3109/01902149809087376. [DOI] [PubMed] [Google Scholar]
- 117.Ralston SL, Seidel A, Luch A, Platt KL, Baird WM. Stereoselective activation of dibenzo[a,l]pyrene to (−)-anti(11R,12S,13S,14R)- and (+)-syn(11S,12R,13S,14R)-11,12-diol-13,14-epoxides which bind extensively to deoxyadenosine residues of DNA in the human mammary carcinoma cell line MCF-7. Carcinogenesis. 1995;16:2899–2907. doi: 10.1093/carcin/16.12.2899. [DOI] [PubMed] [Google Scholar]
- 118.Yoon JH, Besaratinia A, Feng Z, Tang MS, Amin S, Luch A, Pfeifer GP. DNA damage, repair, and mutation induction by (+)-syn and (−)-anti-dibenzo[a,l]pyrene-11,12-diol-13,14-epoxides in mouse cells. Cancer Res. 2004;64:7321–7328. doi: 10.1158/0008-5472.CAN-04-1094. [DOI] [PubMed] [Google Scholar]
- 119.Cheng SC, Prakash AS, Pigott MA, Hilton BD, Lee H, Harvey RG, Dipple A. A metabolite of the carcinogen 7,12-dimethylbenz[a]anthracene that reacts predominantly with adenine residues in DNA. Carcinogenesis. 1988;9:1721–1723. doi: 10.1093/carcin/9.9.1721. [DOI] [PubMed] [Google Scholar]
- 120.Balmain A, Ramsden M, Bowden GT, Smith J. Activation of the mouse cellular Harvey-ras gene in chemically induced benign skin papillomas. Nature. 1984;307:658–660. doi: 10.1038/307658a0. [DOI] [PubMed] [Google Scholar]
- 121.Fujiki H, Suganuma M, Yoshizawa S, Kanazawa H, Sugimura T, Manam S, Kahn SM, Jiang W, Hoshina S, Weinstein IB. Codon 61 mutations in the c-Harvey-ras gene in mouse skin tumors induced by 7,12-dimethylbenz[a]anthracene plus okadaic acid class tumor promoters. Mol Carcinog. 1989;2:184–187. doi: 10.1002/mc.2940020403. [DOI] [PubMed] [Google Scholar]
- 122.Brown K, Buchmann A, Balmain A. Carcinogen-induced mutations in the mouse c-Ha-ras gene provide evidence of multiple pathways for tumor progression. Proc Natl Acad Sci U S A. 1990;87:538–542. doi: 10.1073/pnas.87.2.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Megosh L, Halpern M, Farkash E, O’Brien TG. Analysis of ras gene mutational spectra in epidermal papillomas from K6/ODC transgenic mice. Mol Carcinog. 1998;22:145–149. doi: 10.1002/(sici)1098-2744(199807)22:3<145::aid-mc1>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 124.Chen JX, Pao A, Zheng Y, Ye X, Kisleyou AS, Morris R, Slaga TJ, Harvey RG, Tang MS. Sequence preference of 7,12-dimethylbenz[a]anthracene-syn-diol epoxide-DNA binding in the mouse H-ras gene detected by UvrABC nucleases. Biochemistry. 1996;35:9594–9602. doi: 10.1021/bi9604136. [DOI] [PubMed] [Google Scholar]
- 125.Cavalieri EL, Rogan EG. Mechanisms of tumor initiation by polycyclic aromatic hydrocarbons in mammals. In: Neilson AH, editor. The handbook of environmental chemistry. Springer-Verlag; Heidelberg: 1998. pp. 81–117. [Google Scholar]
- 126.Gorelick NJ, Andrews JL, Gu M, Glickman BW. Mutational spectra in the lacl gene in skin from 7,12- dimethylbenz[a]anthracene-treated and untreated transgenic mice. Mol Carcinog. 1995;14:53–62. doi: 10.1002/mc.2940140110. [DOI] [PubMed] [Google Scholar]
- 127.Manjanatha MG, Shelton SD, Culp SJ, Blankenship LR, Casciano DA. DNA adduct formation and molecular analysis of in vivo lacI mutations in the mammary tissue of Big Blue rats treated with 7, 12- dimethylbenz[a]anthracene. Carcinogenesis. 2000;21:265–273. doi: 10.1093/carcin/21.2.265. [DOI] [PubMed] [Google Scholar]
- 128.Mailander PC, Meza JL, Higginbotham S, Chakravarti D. Induction of A.T to G.C mutations by erroneous repair of depurinated DNA following estrogen treatment of the mammary gland of ACI rats. J Steroid Biochem Mol Biol. 2006;101:204–215. doi: 10.1016/j.jsbmb.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 129.Geacintov NE, Shahbaz M, Ibanez V, Moussaoui K, Harvey RG. Base-sequence dependence of noncovalent complex formation and reactivity of benzo[a]pyrene diol epoxide with polynucleotides. Biochemistry. 1988;27:8380–8387. doi: 10.1021/bi00422a013. [DOI] [PubMed] [Google Scholar]
- 130.Strauss BS. The ‘A rule’ of mutagen specificity: a consequence of DNA polymerase bypass of non-instructional lesions? Bioessays. 1991;13:79–84. doi: 10.1002/bies.950130206. [DOI] [PubMed] [Google Scholar]
- 131.Shibutani S, Takeshita M, Grollman AP. Translesional synthesis on DNA templates containing a single abasic site. A mechanistic study of the “A rule”. J Biol Chem. 1997;272:13916–13922. doi: 10.1074/jbc.272.21.13916. [DOI] [PubMed] [Google Scholar]
- 132.Petersen LN, Orren DK, Bohr VA. Gene-specific and strand-specific DNA repair in the G1 and G2 phases of the cell cycle. Mol Cell Biol. 1995;15:3731–3737. doi: 10.1128/mcb.15.7.3731. [DOI] [PMC free article] [PubMed] [Google Scholar]