

Abstract
To expand our insight into cardiac development, a comparative DNA microarray analysis was performed using tissues from the atrioventricular junction (AVJ) and ventricular chambers of mouse hearts at embryonic day (ED)10.5–11.0. This comparison revealed differential expression of approximately 200 genes, including cartilage link protein 1 (Crtl1). Crtl1 stabilizes the interaction between hyaluronan (HA) and versican, two extracellular matrix components essential for cardiac development. Immunohistochemical studies showed that, initially, Crtl1, versican, and HA are co-expressed in the endocardial lining of the heart, and in the endocardially-derived mesenchyme of the AVJ and outflow tract (OFT). At later stages, this co-expression becomes restricted to discrete populations of endocardially-derived mesenchyme. Histological analysis of the Crtl1-deficient mouse revealed a spectrum of cardiac malformations, including AV septal and myocardial defects, while expression studies showed a significant reduction in versican levels. Subsequent analysis of the hdf mouse, which carries an insertional mutation in the versican gene (CSPG2), demonstrated that haploinsufficient versican mice display septal defects resembling those seen in Crtl1−/− embryos, suggesting that reduced versican expression may contribute to a subset of the cardiac abnormalities observed in the Crtl1−/− mouse. Combined, these findings establish an important role for Crtl1 in heart development.
Keywords: Cartilage Link Protein 1, versican, hyaluronan, atrioventricular cushions, atrioventricular septal defects, ventricular septal defects, thin myocardium
Introduction
In the early steps of heart development, the fusion of two populations of precardiac mesoderm leads to the formation of a primitive cardiac tube. In this primitive heart, the myocardial and endocardial cell layers are separated by the cardiac jelly, an acellular, and extracellular matrix-rich (ECM), space (Markwald et al., 1977; Moorman and Christoffels, 2003; Wessels and Sedmera, 2003). As the heart tube begins to loop, the cardiac jelly accumulates in the atrioventricular (AV) junction and the outflow tract (OFT), a process that is followed by local epithelial-to-mesenchymal transformation (EMT) of the endocardial lining. Combined, these events result in the formation of the mesenchymal cushions in the AVJ and OFT (Krug et al., 1985; Markwald et al., 1977). In addition to the endocardially-derived mesenchyme, the respective cushions also receive mesenchymal contributions from epicardially-derived cells (Dettman et al., 1998; Manner, 1999; Perez-Pomares et al., 2002) and cardiac neural crest-derived cells (Danielian et al., 1998; Nakamura et al., 2006).
The AV cushions are involved in the formation of the mitral and tricuspid valves (Wessels et al., 1996) and contribute, together with the dorsal mesenchymal protrusion (DMP), to the formation of the AV mesenchymal complex, which plays a crucial role in cardiac septation (Snarr et al., 2007; Wessels et al., 2000; Wessels et al., 1996). As many congenital heart malformations involve the derivatives of the AV mesenchyme, elucidation of the mechanisms that govern its development is fundamental to understanding the etiology of congenital heart disease. Over the years, numerous in vitro and in vivo studies have increased our general understanding of the cellular and molecular mechanisms in the respective phases of cushion development. This has led to the general consensus that endocardial cushion formation is a process that relies on a balanced interaction between myocardially- and endocardially/mesenchymally expressed genes, including transcription factors, growth factors, and ECM components (Eisenberg and Markwald, 1995; Lincoln et al., 2006; Schroeder et al., 2003).
To expand our insight into the development of the AV junction, and to create a database of differentially-expressed genes at a critical stage of cushion development, we performed a DNA microarray analysis on AV and ventricular tissues of the mouse heart at embryonic day (ED) 10.5–11.0. In this paper, we focus on one of the genes, cartilage link protein 1 (Crtl1), also known as “hyaluronan and proteoglycan link protein 1 (HAPLN1)”. In non-cardiac tissues, Crtl1 is best known for stabilizing the interaction between HA and proteoglycans such as versican and aggrecan (Binette et al., 1994; Matsumoto et al., 2006), and has recently been described to be expressed in AV cushion mesenchyme by others (Lincoln et al., 2007). The data presented here, demonstrate that Crtl1 plays an important role in cardiac development, as perturbation of Crtl1 expression leads to malformations in the valvuloseptal complex as well as in abnormalities of myocardial development.
Materials and Methods
DNA Microarray Analysis
AV canal and ventricular tissues (VT) from eighty embryonic day (ED)10.5–11.0 C57BL6 mouse hearts were isolated in RNase free PBS, snap-frozen in liquid nitrogen, homogenized, and total RNA was isolated using the Qiagen RNeasy Mini Kit. Total RNA quality was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA) with the RNA 6000 Pico LabChip kit (Agilent Technologies). RNA was precipitated using GenElute Linear Polyacrylamide (Sigma), treated with Promega RQ1 RNase-Free DNase, and purified using the Bio-Rad Aurum Total RNA Mini Kit and then precipitated for a final time. Total RNA was amplified and converted into biotin-labeled RNA using the RiboAmp® HS RNA Amplification Kit (Arcturus Bioscience, Inc., Mountain View, CA). Labeled cRNA was fragmented and then evaluated by Bioanalyzer to ensure appropriate size distribution (~35–200 nt). Each cRNA sample was divided and hybridized to two Mouse Expression 430A GeneChips (Affymetrix, Santa Clara, CA). Hybridization, post hybridization washing, fluorescence staining, and scanning were performed at the MUSC Proteogenomics Facility (http://proteogenomics.musc.edu/) using Affymetrix instrumentation in accordance with Affymetrix protocols. The resulting hybridization data files have been deposited in the MUSC DNA Microarray database (Argraves PMID 14668234; http://proteogenomics.musc.edu/quickSite/musc_madb.php?page=home&act=manage) and can be accessed with the target identifiers _1093467013.966284, _1093467031.924285, _1093467386.772829 and _1093467564.815494. SOFT matrix data and raw hybridization data has also been deposited in the NCBI GEO database (series accession number GSE4903) in accordance with MIAME convention (Brazma: PMID 11726920).
Analysis of DNA microarray data
Absolute detection calls were calculated for all genes (probe sets) using Affymetrix MAS5.0 software. Hybridization data were normalized using the Bioconductor (Gentleman, PMID 15461798) implementation of Robust Multichip Average (Irizarry, PMID 12925520). Differentially expressed genes were defined by the following criteria: 1) the gene was expressed in either the AV or the ventricular region (VT) (i.e., it received “present” calls for both iterations of either AV or VT samples), and 2) the normalized hybridization values for the gene were greater than 2-fold different between AV and VT samples.
Mice
Heterozygous Crtl1 breeder pairs (B6.129-Hapln 1tm1Nid/Ucd; stock#: MMRRC:000041-UCD) were obtained from the Mutant Mouse Regional Resource Centers (MMRRC). Crtl1 mice were genotyped essentially as described before (Czipri et al., 2003) using the following primer sets: LP12 (5′ taa tga cct ttc ctg tct ctc c 3′) and LP13 (5′ ccc aaa acc cgt agt tcc 3′), generating a PCR product of 283bp in wildtype alleles, and Neo1013 (5′ gga tcg gcc att gaa caa g 3′) and Neo1014 (5′ cac cat gat att cgg caa gc 3′) generating a product of 600bp, indicating the cloning vector inserted in the Crtl1 gene to create the “knockout gene” (Watanabe and Yamada, 1999). Hdf mice were genotyped as described previously (Yamamura et al., 1997).
Histology and Immunohistochemistry
Specimens from Crtl1+/−/Crtl1+/− matings, were collected and staged according to Theiler (Theiler, 1989). Embryos were fixed in 4% paraformaldehyde (PFA) for 4 hours. Tissue-processing, hematoxylin/eosin staining, and immunohistochemistry were performed as previously described (Waller and Wessels, 2000). The following monoclonal antibodies were used: 9/30/8-A-4 (Developmental Studies Hybridoma Bank), recognizing Crtl1 (Caterson et al., 1985; Neame et al., 1986), A2172 (Sigma) recognizing sarcomeric actin, and sc-56 (Santa Cruz) recognizing PCNA. The polyclonal antibody AB1033, recognizing the beta GAG domain of versican (Snow et al., 2005), was purchased from Chemicon International. Hyaluronan was detected using biotinylated Hyaluronan Binding Protein (HABP) (Seikagaku Corporation, catalog number 400763) as described previously (Kern et al., 2007; Kern et al., 2006). For fluorescent detection of the primary antibodies, Alexa Fluor 568 (A-11011 goat anti-rabbit IgG from Molecular Probes) and Alexa Fluor 488 (A-11001 Goat anti-mouse IgG from Molecular Probes) were used. HABP binding was visualized with Streptavidin-fluorescein RPN 1232 from Amersham Life Science. Immunofluorescently-stained sections were imaged using the Leica TCS SP2 AOBS confocal microscope system. For colometric visualization of primary antibody binding, rabbit anti-mouse (Sigma A9044) and goat anti-rabbit (Sigma A0545) peroxidase conjugated antibodies were used in combination with the Metal Enhanced Diaminobenzidine (DAB) Substrate Kit (Pierce, Rockford, IL; product number 34065).
Tie2-cre β-galactosidase staining and AMIRA reconstruction
β-galactosidase staining was performed as described before (Snarr et al., 2007). Embryos were isolated and fixed in ice-cold 4% PFA. Embryos were then washed in ice-cold PBS and incubated in permeabilization buffer (0.02% sodium-deoxycholate, 0.01% NP-40, 1× PBS) overnight. Expression was visualized by incubating embryos in X-gal stain solution overnight at 37°C (5µM K-ferricyanide, 5µM K-ferrocyanide, 2µM MgCl2, 1mg/ml X-gal, 1× PBS). The tissue was post-fixed in 4% PFA and processed for histochemistry as described above. Amira three-dimensional reconstruction was performed as described previously (Snarr et al., 2007).
In situ hybridization
Whole mount in situ hybridization was performed as previously reported (Norris et al., 2005). Probes were designed using specific primers for Crtl1 mRNA (CRTL1 forward: 5′ tggaccaggacgcagtgatt; CRTL1 reverse: 5′ gcagcggtcatagcccagaa). Total RNA was isolated from wild type ED10.5 C57BL6 mouse embryos using the Qiagen RNeasy MiniKit. Specific cDNA was made using the Stratagene Stratascript First strand RT-PCR kit. This cDNA served as a template for PCR using the specific mRNA primers described above with addition of T7 or Sp6 RNA polymerase promoters. The final PCR product was labeled with Digoxygenin-UTP using the DIG (Sp6/T7) RNA labeling kit by Roche, and purified using the Qiagen RNeasy MiniKit RNA clean up protocol. ED10.0 and 12.0 hearts were isolated from C57BL6 embryos and fixed in 4%PFA/EGTA, then transferred to 100% Methanol. After proteinase K treatment and post-fixation, the Crtl1 antisense or sense probe was added to a final concentration of 200ng/mL. Hybridization was detected with ImmunO BCIP/NBT liquid substrate by MP Biomedicals.
Quantitative immunofluorescence
To determine relative levels of versican and HA in mesenchymal tissues and sarcomeric actin in adjacent myocardium, semi-quantitative immunofluorescence was performed on Crtl1−/− and Crtl1+/+ ED13.0 embryos from three different litters (for a total of 5 specimens per genotype). Images were collected using the Leica Confocal Microscope System, making sure that emission measurement gain and offset were held constant for all sections. Pixel intensity values were obtained using NIH ImageJ. For each image collected, the pixel count and average pixel intensity were calculated. After collecting the experimental data, a statistical analysis was performed, using the one-tailed t-test. Significance was determined by setting the p-value at p<0.05.
Western blot analysis
Western blot was performed essentially as described before (Kern et al., 2006; Sandy et al., 2001). Briefly, ED13.0 Crtl1−/− and wild type littermate hearts were isolated and snap frozen in liquid nitrogen for storage. Immediately after thawing, the tissue was extracted for 24hr at 4°C in 4 M guanidine-HCl, 10 mM MES, 50 mM sodium acetate, 5 mM ethylenediaminetetraacetic acid [EDTA], containing the protease inhibitor cocktail complete Mini EDTA-free (Roche, Mannheim, Germany) to obtain a proteoglycan-rich extract. The extracts were then centrifuged for 10 min at 12,000 × g at 4°C and supernatants were quantified for total protein (Coomassie Plus Bradford Assay; Pierce). Equal amounts of protein (400 ug) were used for precipitation with ice-cold ethanol for 16 h at −20°C. Precipitates were collected by centrifugation at 13,000 × g for 20 min at 4°C. After centrifugation, the pellets were dried, resuspended in 50 mM Tris, 50 mM sodium acetate, 10 mM EDTA, pH 7.6, and separated into two equal aliquots. One aliquot was digested with chondroitinase ABC (25 milliunits/100µg of GAG, protease-free, Sigma) for 1.5 hr at 37°C to deglycosylate the core proteins of proteoglycans and was used to detect versican and Crtl1. The other aliquot was left untreated and was used for immunodetection of sarcomeric actin to serve as a protein loading control. Two aliquots of each sample were separated on parallel gels (4–15% Tris-HCL gradient SDS-PAGE gels under reducing conditions) and then transferred onto PVDF membrane and probed with a polyclonal versican antibody (AB1033, Chemicon) and the monoclonal sarcomeric actin IgG (A2172, Sigma) antibody, respectively, in 5% milk/TBST. Primary antibody binding was detected by HRP-conjugated secondary antibody (12–348, 12–349, Upstate) followed by visualization with ECL-Plus reagents (Amersham/GE Heathcare). After stripping the membrane used for versican detection using 0.2M Glycine-HCl, pH 2.2 for 2 minutes, and washing three times in TBS, the membrane was then reprobed for Crtl1 expression using the Crtl-1 antibody 9/30/8-A-4. The band densities for versican protein (at approx 366kD) were quantified using Scion software (Scion Corp., Frederick MD) and statistical analysis was performed, using t-test. Significance was determined by setting the p-value at p<0.05.
Results
Microarray analysis identifies Crtl1 as an ECM molecule with abundant expression in the developing atrioventricular junction
To obtain new insights into the formation and subsequent development of the AV mesenchyme, we applied DNA microarray analysis to compare the gene expression profile of AV junctional and ventricular tissues at ED10.5–11.0. Analysis of the DNA microarray data identified approximately 150 genes that were expressed at a 2-fold or higher level in the AV junction as compared to their level of expression in the ventricular tissues (Microarray SOFT matrix data and raw data submitted to NCBI GEO under series accession number GSE4903). As expected, this list contained numerous genes that have previously been reported to be highly expressed in the AV junction and that are known to be involved in cardiac development. This list included Tbx20 (Kraus et al., 2001), Bmp2 (Sugi et al., 2004), periostin (Kruzynska-Frejtag et al., 2001), versican (Mjaatvedt et al., 1998), Tbx2 (Christoffels et al., 2004), Tbx3 (Hoogaars et al., 2004), Pitx2 (Campione et al., 2001), Sox4 (Ya et al., 1998), Bmp4 (Keyes et al., 2003), Nfatc4 (Bushdid et al., 2003), and Has2 (Camenisch et al., 2000).
From the remaining list of genes that were characterized by a relatively high level of expression in the AV junction, we selected a number of candidates for further analysis. In this study we report on one of them, the extracellular matrix molecule, cartilage link protein 1 (Crtl1). Crtl1 is best known for mediating the interaction between hyaluronan (HA) and proteoglycans (PGs), particularly aggrecan, during chondrogenesis, thereby giving strength to the ECM in cartilage (Binette et al., 1994; Schroeder et al., 2003; Shi et al., 2004; Shibata et al., 2003), and has recently been shown to be expressed in the maturing cushion-derived tissues at ED12.5–13.5 and ED18.5 (Lincoln et al., 2007). Putative binding partners of Crtl1 in the AV cushions of the developing heart include the proteoglycan, versican, and the extracellular matrix molecule, hyaluronan, which have both been shown to be of crucial importance for cardiac development (Camenisch et al., 2000; Mjaatvedt et al., 1998; Yamamura et al., 1997). Thus, with the identification of elevated levels of Crtl1 in the AV junction at a critical stage of cushion development, we decided to explore, in more detail, the role of this ECM component in cardiovascular development.
Spatiotemporal expression of Crtl1 in the developing mouse heart
To confirm the differential expression of Crtl1 mRNA in the developing mouse heart at ED10.5, as indicated by the microarray, whole mount in situ hybridization was performed on the developing mouse heart at ED10.0. This experiment confirmed the elevated expression of Crtl1 mRNA in the AV junction. In addition, relatively high Crtl1 expression was observed in the OFT and in the atrial roof, while lower levels of expression were seen in the lining of the atria and ventricles (Fig 1A). At ED12.0 (Fig 1B) Crtl1 mRNA expression was found to be more restricted, with high expression being observed in the AVJ and OFT, while at this stage, Crtl1 mRNA was no longer apparent in the atria and ventricles.
Figure 1. Expression of Crtl1 mRNA in the developing mouse heart.
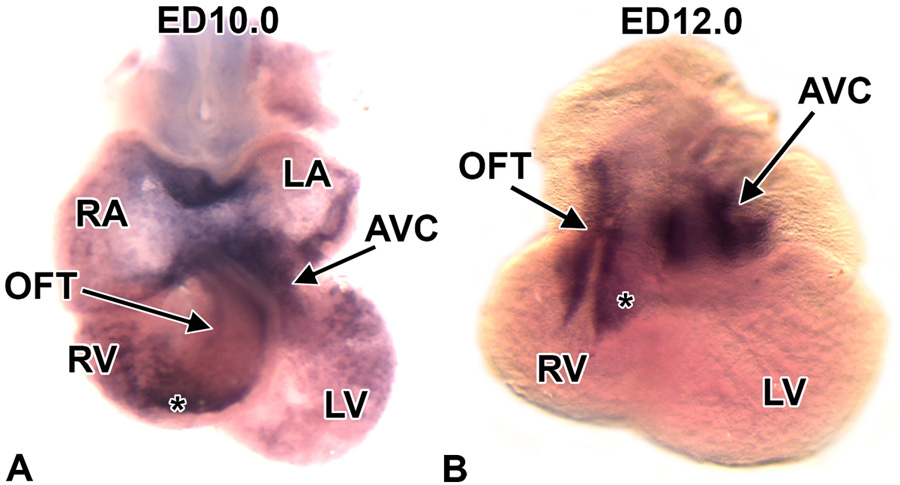
Panels A and B show Crtl1 mRNA expression by whole mount in situ hybridization in the developing mouse heart at ED10.0 (panel A) and ED12.0 (panel B). At ED10.0, Crtl1 mRNA is abundantly expressed in the AVJ and conal region of the OFT (*). Relatively high expression is also seen in the atrial roof. Lower levels of expression are seen in the lining off the atria and ventricles. A ED12.0 (panel B) Crtl1 mRNA expression becomes more restricted, with high expression in the AVJ and OFT (*). At this stage, Crtl1 mRNA expression is no longer detected in the atria and ventricles. LA=left atrium, RA=right atrium, LV=left ventricle, RV=right ventricle, OFT=outflow tract, AVJ=atrioventricular junction, (*)=OFT conal cushions.
To determine the spatiotemporal expression of Crtl1 protein in relation to the expression patterns of its binding partners, versican and hyaluronan, we performed immuno-fluorescent co-labeling on a developmental series of murine embryos.
At ED9.5, Crtl1 expression was seen in the ECM of the AV cushion mesenchyme (Fig 2A,C) and, importantly, also in association with the endocardial lining of the heart, (Fig 2C–F), where it showed significant overlap with the expression of versican. Strong Crtl1 expression was also seen in the reflections of the dorsal mesocardium (Fig 2C). At ED10.5, abundant and overlapping expression of Crtl1 and versican (Fig 2G,H), was observed in AV and OFT cushion mesenchyme. This pattern of versican expression confirms observations in previous studies (Kern et al., 2006).
Figure 2. Expression of Crtl1 and versican at ED9.5 and ED10.5.
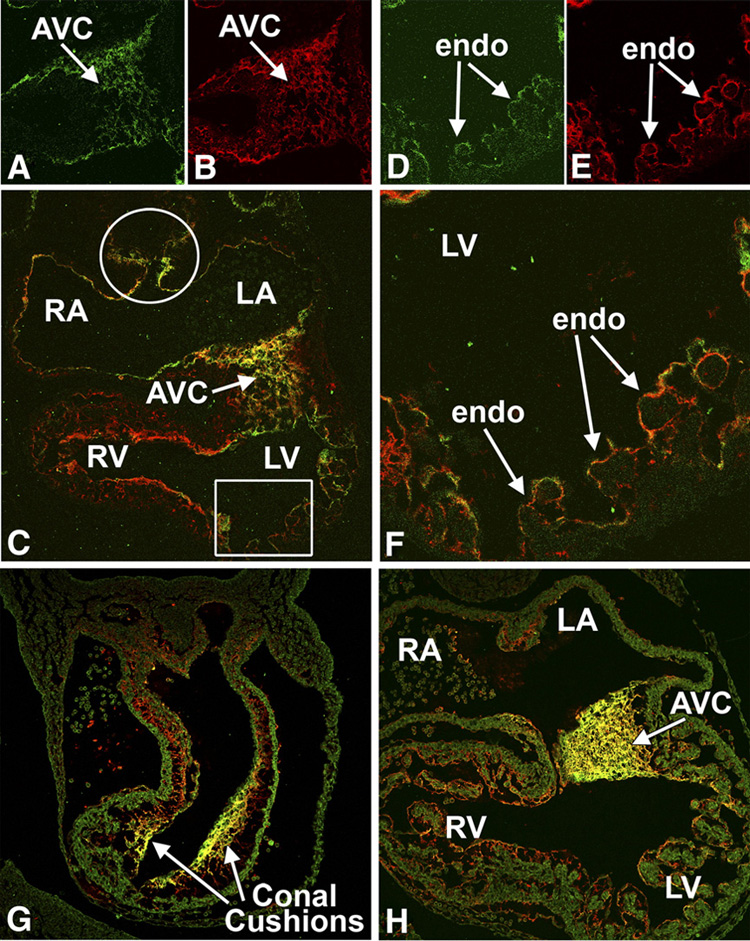
This figure shows immunofluorescently-stained sections co-labeled for Crtl1 (green) and versican (red), (co-exprssion shown in yellow), at ED9.5 (panels A–F) and ED10.5 (G and H). At ED9.5, Crtl1 is expressed in the extracellular matrix surrounding the endocardial and endocardial-derived cells of the AV cushion mesenchyme (panels A and C) and in the lining of the atria (panel C) and ventricles (panels C, D, F). Crtl1 is also expressed in the reflections of the dorsal mesocardium (white circle, panel C). Versican (panels B and E) shows a high degree of co-expression (yellow) with Crtl1 (panels C and F). Panels D–F are higher magnifications of the boxed region in C, showing Crtl1 and versican expression in the extracellular matrix surrounding the endocardial lining of the ventricular trabeculae. At ED10.5, Crtl1 and versican are highly expressed in the sub-endocardial extracellular matrix of the conal cushions in the proximal OFT (panel G). In addition, versican is expressed in the sub-endocardial mesenchyme of the truncal OFT ridges, regions where little Crtl1 expression is detected (panel G). In the AV cushions, Crtl1 and versican also show a high degree of co-expression (panel H). RA=right atrium, LA=left atrium, RV=right ventricle, LV=left ventricle, AVC=atrioventricular cushions, endo=endocardium.
At ED13.0, Crtl1 expression was found to be restricted to two distinct regions in the OFT (Fig 3A,I). Strong expression was seen in the distal-most part of the truncal endocardial ridges (i.e. the upper boundary of the developing semilunar valves) (Fig 3A) and in the most proximal portion of the OFT mesenchyme (OFT conal cushions) (Fig 3A,I). This pattern of Crtl1 expression correlates with the distribution of endocardially-derived cells in the OFT as demonstrated by lacZ-stained sections of Tie2cre/ROSA26Rflox mouse specimens, a mouse model system that allows delineation of endocardial and endocardial-derived cells in the developing heart (Fig 3B,I,J; see (Snarr et al., 2007). Crtl1 was, however, not detected in the proximal truncal ridges (Fig 3A,I), an area known to be heavily populated by neural crest-derived cells (Danielian et al., 1998; de Lange et al., 2004; Nakamura et al., 2006). In contrast to Crtl1, the putative binding partners versican and HA were found to be expressed in all components of the conal and truncal ridges (Fig 3C,D), albeit that expression was most prominent in the endocardially-derived mesenchyme. Recently, we have demonstrated that the AV mesenchymal complex consists of endocardially-and non-endocardially derived structures (Snarr et al., 2007); see Fig 3M). Similar to our finding in the OFT, in the AV junction, Crtl1 expression was only found in the endocardially-derived mesenchymal tissues, i.e. the major and lateral AV cushions but not in the non-endocardially derived mesenchyme of the DMP (Fig 3E,F,K,L). The levels of versican and HA were also lower in the DMP when compared to the other AV mesenchymal tissues (Fig 3G,H).
Figure 3. Overlapping expression of Crtl1, versican, and hyaluronan in the embryonic mouse heart at ED13.0.
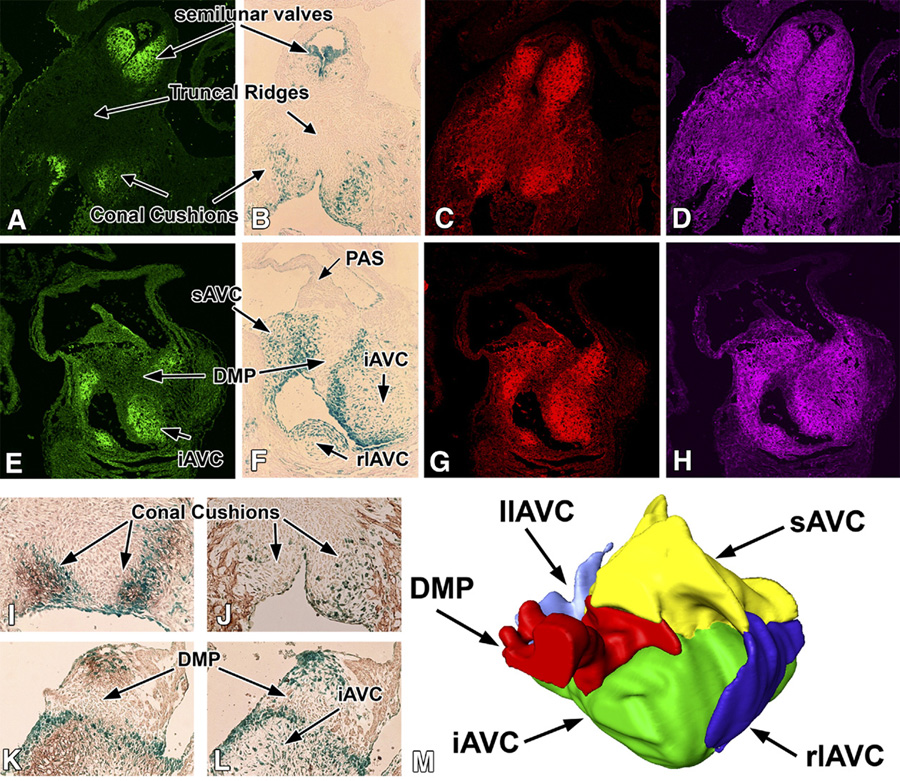
This figure shows the expression of Crtl1, versican and HA in the respective mesenchymal cell populations in the developing heart at ED13.0. Panels A–D and panels I and J show sections of the OFT. Panels E–H and panels K and L show sections of the AV mesenchymal structures. Panels A and E show Crtl1 expression (green), panels C and G show versican staining (red), and panels D and H show hyaluronan staining (purple). Furthermore, panels B, F, I, J, K and L are sections from Tie2-cre/ROSA26 embryos in which endocardially-derived cells are expressing lacZ (blue). The sections in panels I and K are co-stained immunohistochemically for Crtl1 (brown), while the section in J is stained for sarcomeric actin. Combined, panels A, B, I, and J demonstrate that Crtl1 expression is largely confined to the endocardially-derived mesenchyme in the conal cushions and developing semilunar valves. The (proximal) truncal ridges (arrows in A and B) do not express Crtl1. Panels C and D show that in the conal cushions and semilunar valves, the expression of Crtl1, versican and HA overlap. Additionally, versican and HA are also expressed in those parts of the truncal ridges where Crtl1 is absent. Panels E, F, K, and L show that in the AV mesenchymal structures, Crtl1 expression is also restricted to the mesenchymal structures that are of endocardial origin. Thus, Crtl1 is expressed in the inferior, superior, and lateral AV cushions, but not in the dorsal mesenchymal protrusion, which forms a non-endocardially derived mesenchymal wedge in between the inferior and superior cushions (M). Panels G and H show that the DMP expresses lower levels of versican and HA than surrounding mesenchyme. AVC=atrioventricular cushions, sAVC=superior AVC, iAVC=inferior AVC, llAVC=left lateral AVC, rlAVC=right lateral AVC, PAS=primary atrial septum, DMP=dorsal mesenchymal protrusion.
At ED14.0–15.5, the AV mesenchyme is undergoing an extensive remodeling that leads to the formation of the respective leaflets of the mitral and tricuspid AV valves. While Crtl1 and versican continue to be expressed at considerable levels in the developing leaflets of the tricuspid valve (Fig 4A–C), the level of expression in the leaflets of the mitral valve is, in comparison, noticeably lower.
Figure 4. Overlapping expression of Crtl1 and versican at ED14.5.
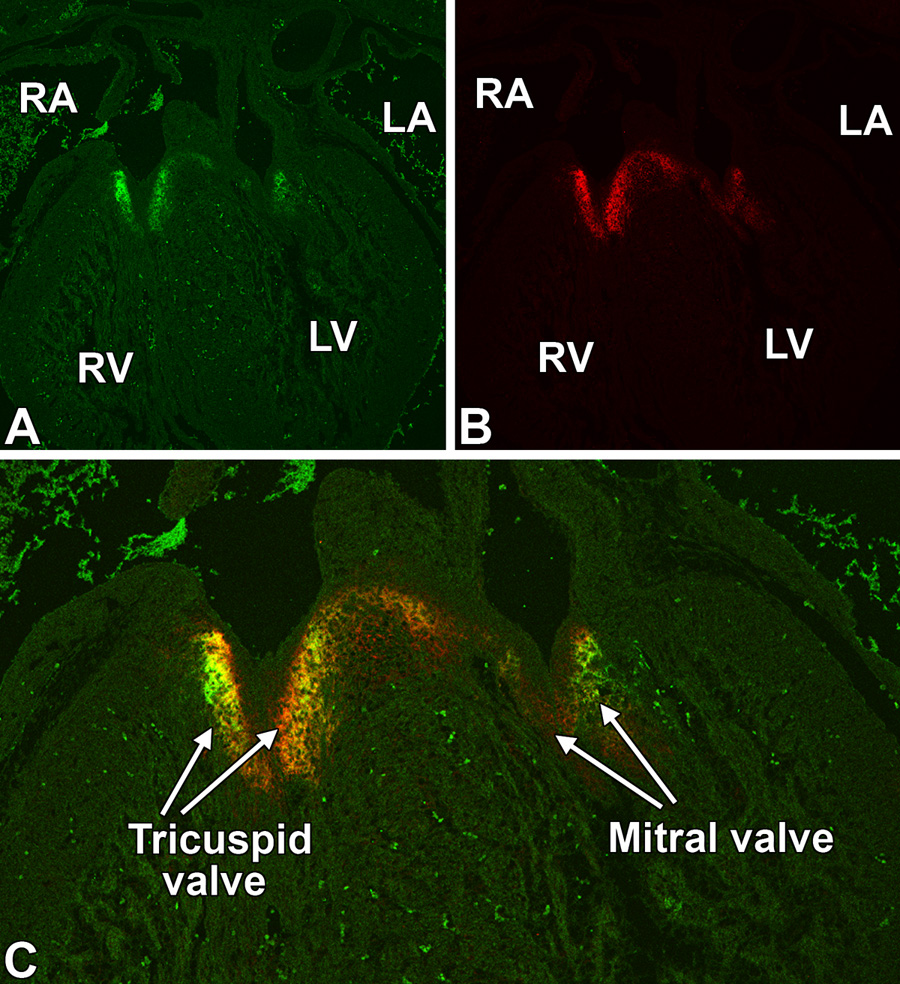
This figure shows a co-labeling for Crtl1 (panel A, green) and versican (panel B, red) expression in a transverse section of a ED14.5 heart. The merged image (panel C) shows the overlapping expression of Crtl1 and versican (yellow) in the leaflets of the tricuspid and mitral AV valves. Note the higher expression of both proteins in the tricuspid valve leaflets when compared to the leaflets of the mitral valve. RA=right atrium, LA=left atrium, RV=right ventricle, LV=left ventricle,
Heart defects in the Crtl1-deficient mouse
Collectively, the spatiotemporal expression pattern of Crtl1, the overlap of Crtl1 expression with that of its putative binding partners (versican and HA), and the well-established functional relationship between these three ECM binding partners, suggested an important role for Crtl1 in cardiac development. The generation of a Crtl1-deficient mouse has previously been published (Czipri et al., 2003; Watanabe and Yamada, 1999). Reportedly, mice homozygous for targeted deletion of Crtl1 die shortly after birth resulting from difficulties in breathing related to the perturbation of tracheal cartilage formation. Importantly, while several phenotypic characteristics of the Crtl1-deficient mouse have been described (Czipri et al., 2003; Watanabe and Yamada, 1999), the cardiac phenotype was never evaluated. To determine the importance of Crtl1 during cardiac development, we examined a total of 55 prenatal and postnatal Crtl1−/− specimens. In this group, we positively identified cardiac abnormalities in 39 Crtl1−/− specimens (71%; Fig 5 and 6). Anomalies of the interventricular/atrioventricular septum were the most prevalent (32/39). Septal defects were found in the fibrous tissue that is derived from the AV cushions and OFT conal cushions (20/32; Fig 5B,D–F,H and Fig 6B–D,F), and also in the muscular component of the ventricular septum (21/32; Fig 5C and 6B,C,E,G). A number of hearts (9/32) exhibited both muscular and fibrous septal defects (e.g Fig.6D–G). Near birth, these fibrous septal defects are typically found at either side of the hingepoint of the septal tricuspid valve leaflet (Fig. 5E,F and Fig 6D,F). At younger stages, prior to leaflet formation (see Fig 5), it is not possible to classify the defects using anatomical classification generally used in describing human congenital heart malformations. Therefore, we refer to these abnormalities as defects of the AV mesenchymal complex. Malformations found in the most severely affected specimens include double outlet right ventricle (DORV), overriding aorta, common AV canal (cAVC; Fig 5B), and double inlet left ventricle (DILV; Fig 5C). The common AV canal defects are associated with hypoplastic cushion tissues and severely impaired development of the DMP, both contributing to the malformation of the AV mesenchymal complex (Fig 5B,G,H). In addition to the structural defects that affect the segmental communications in the developing heart, we also observed a pronounced “thinning” of the compact layer of the myocardium in the ventricular free walls and in the ventricular septum, a phenomenon also known as “thin myocardium syndrome (TMS)” (Jaber et al., 1996) (Fig 5B,C and Fig 7A–F). TMS may result from decreased levels of cell proliferation, or increased apoptosis within the myocardium. TUNEL labeling did not indicate increased levels of apoptosis (data not shown). PCNA labeling experiments, however, showed significantly decreased numbers of proliferating myocardial cells in Crtl1−/− hearts (Fig 7A–F).
Figure 5. Cardiac phenotype of Crtl1-deficient embryos.
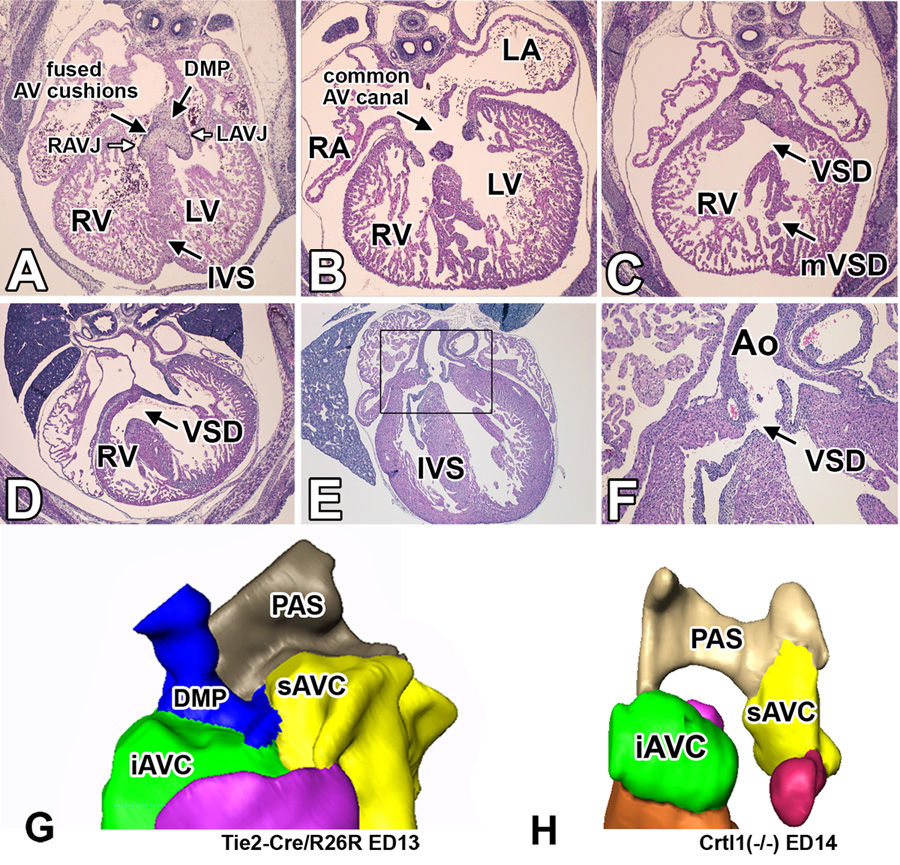
This figure shows hematoxylin-eosin stained transverse sections of wildtype (panel A) and Crtl1-deficient embryos (panels B–F). The section in panel A shows a wildtype mouse at ED13.5 with a well-formed interventricular septum, separating the right and left AV junction. Panels B and C show sections of a severely malformed Crtl1−/− specimen at ED14.0 with common AV canal (B), hypoplastic AV cushion tissues (B), absence of the DMP (B), thin compact myocardium in the RV wall (B,C), and a very disorganized interventricular septum (B,C). Panel D shows a Crtl1−/− specimen at ED16.0 with a large mesenchymal VSD. Panels E and F are sections from an ED17.0 Crtl1−/− specimen showing a small mesenchymal sub-aortic VSD. Panel F is a higher magnification of the boxed region in panel E. Panel G shows a three-dimensional AMIRA reconstruction of the AV mesenchymal complex of a wild type mouse at ED13, demonstrating the position of the DMP within this complex. Panel H is a 3D reconstruction of the ED14.0 Crtl1−/− mouse, shown in panels B and C, demonstrating the severely underdeveloped AV cushion tissues and absent DMP, combined contributing to the common AV canal phenotype. RA=right atrium, LA=left atrium, RV=right ventricle, LV=left ventricle, RAVJ=right atrioventricular junction, LAVJ=left atrioventricular junction, IVS=interventricular septum, VSD=ventricular septal defect, mVSD= muscular VSD, Ao=aorta, PAS=primary atrial septum, DMP=dorsal mesenchymal protrusion, AVC=atrioventricular cushions, iAVC=inferior AVC, sAVC=superior AVC.
Figure 6. Cardiac phenotype of the neonatal Crtl1-deficient mouse.
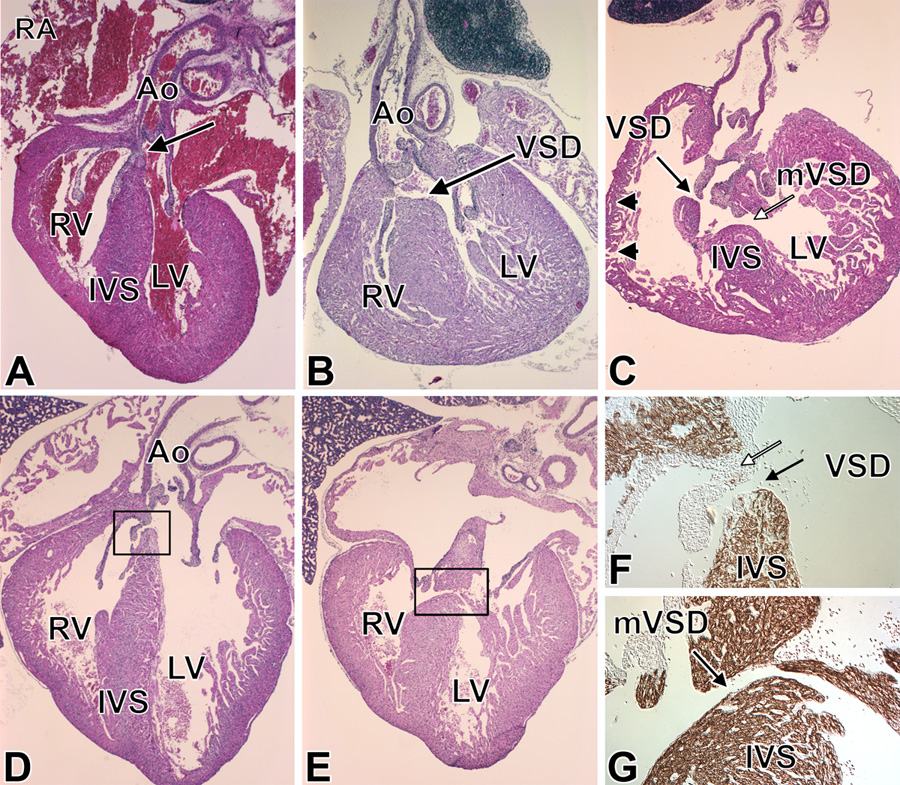
Panel A shows a wild type neonatal heart with a well-formed IVS and AV cushion-derived fibrous tissue (arrow in A). Panel B is an example of a neonatal Crtl1−/− mouse with a sub-aortic VSD affecting both muscular and mesenchymal components of the AV septum. Panel C shows a neonatal Crtl1−/− mouse with severe cardiac malformations including a highly abnormal IVS, with a large muscular VSD (white arrow), a smaller mesenchymal VSD (black arrow), and an abnormally thin right ventricular wall (black arrowheads). Panels D–G are from another severely malformed Crtl1−/− specimen that exhibits both a mesenchymal VSD (D, F) and a muscular VSD (E,G). The sections F and G are stained with sarcomeric actin to delineate myocardial from fibrous structures. Panel F is a sister section of D showing a higher magnification of the VSD (boxed in D). The black arrow demarcates the mesenchymal VSD, the white arrow points to the fibrous, mesenchymal-derived tissues of the tricuspid valve leaflet bordering the superior margin of the defect. Panels E and G show a muscular VSD (black arrow in panel G). Panel G is a sister section of E showing a higher magnification of the boxed region in E. The actin staining demarcates the muscular upper and lower boundaries of the muscular defect. RA=right atrium, RV=right ventricle, LV=left ventricle, Ao=aorta, IVS=interventricular septum, VSD=ventricular septal defect, mVSD=muscular VSD.
Figure 7. Thin myocardium syndrome is associated with reduced myocardial proliferation in the Crtl1-deficient mouse.

Panels A and B are hematoxylin-eosin stained sections of wild-type (A) and Crtl1−/− (B) specimens at ED13.0. The compact myocardium of the Crtl1−/− mouse has approximately half of the number of cell layers as compared to the compact myocardium in the wild type mouse (Panel B; thickness denoted by double-headed white arrows). PCNA labeling of wild type (C, E) and Crtl1−/− (D, F) ED13.0 specimens shows that the level of proliferation, expressed as PCNA-positive nuclei over total nuclei, is significantly reduced (t stat=4.34, p=0.02) in the ventricular wall of the Crtl1−/− specimens (white arrows D, F) when compared to wild type controls (C, E). However proliferation does not appear to be greatly affected in the interventricular septum (C, D, white star) or in the trabeculae (black arrows). LV=left ventricle, (*)= interventricular septum.
Levels of versican are reduced in the Crtl1-deficient mouse
Previous studies reported reduced levels of the Crtl1-binding proteoglycan, aggrecan, in the developing cartilage of Crtl1-deficient mice (Watanabe and Yamada, 1999). To determine whether perturbation of Crtl1 expression would also result in a reduction of the putative cardiac binding partner, versican, we performed semi-quantitative immunofluorescence on the atrioventricular cushions of Crtl1−/− and wildtype hearts at ED13.0. This analysis revealed a significant reduction of versican expression in the AV cushions of the Crtl1−/− specimens as compared to wildtype littermates (p=0.012) (Fig 8A). Subsequent Western Blot analysis, using whole hearts isolated from Crtl1−/− and WT embryos at ED13.0, confirmed these observations (p= 0.02) (Fig 8B). The levels of HA, the other Crtl1 binding partner, in the AV cushions (p=0.164), and sarcomeric actin levels in the adjacent AV myocardium (p=0.296), were not significantly reduced in the Crtl1−/− hearts.
Figure 8. Reduced levels of versican are implicated in the etiology of the cardiac malformations in the Crtl1-deficient and heterozygous hdf mice.
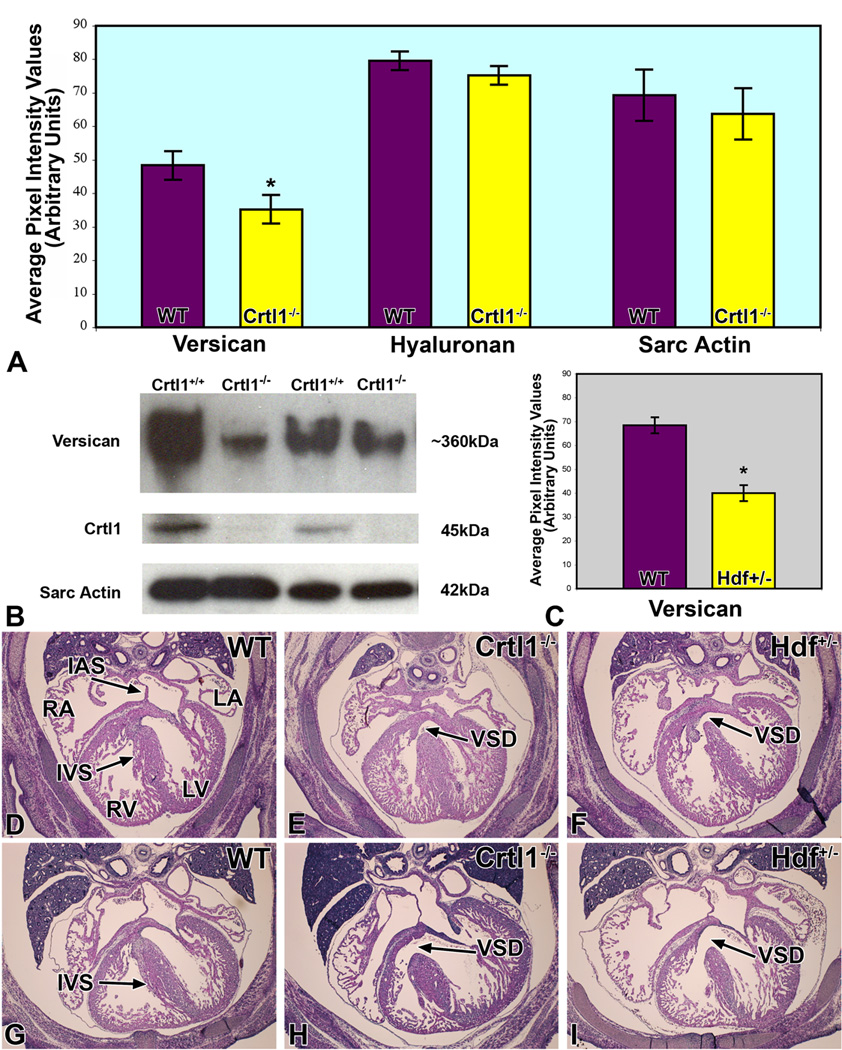
Panel A graphically depicts the levels of versican, hyaluronan, and sarcomeric actin expression, as measured by quantitative immunofluorescence, in the AV cushions of Crtl1−/− mouse embryos at ED13.0. While the levels of versican are significantly reduced (t=2.74; df=8; p=0.012), hyaluronan expression in the AV cushions, and sarcomeric actin expression in the adjacent myocardium, are not significantly changed. Panel B shows Western Blot results, using total hearts of wildtype and Crtl1−/− littermate embryos at ED13.0, confirming the decreased levels of versican in Crtl1−/− mice. Quantification of the western blots indicated a reduction of versican in the Crtl1−/− specimens by approximately 45% (t= 2.66, df= 4, p= 0.02). Crtl1 controls show the absence of Crtl1 in the Crtl1−/− mice, while the sarcomeric actin staining is an independent control of protein loading. Panel C is a graph depicting versican levels in ED13.5 hdf+/− embryos, as measured by quantitative immunofluorescence. The graph demonstrates that versican is significantly reduced, when compared to wild type littermates (p = 0.0005). Panels D–I are hematoxylin-eosin stained sections of wildtype (D and G), Crtl1−/− (E and H), and Hdf+/− (F and I) mice, respectively (D–F: stage ED14.0–14.5; G–I: stage ED15.5–16.0). Panels E, H, F, and I show that Crtl1−/− and Hdf+/− embryos are characterized by very similar ventricular septal defects (black arrows). RA=right atrium, LA=left atrium, RV=right ventricle, LV=left ventricle, IAS=interatrial septum, IVS=interventricular septum, VSD=ventricular septal defect.
Heterozygote hdf embryos have similar cardiac defects as those observed in Crtl1-deficient mice
The decreased levels of versican in the AV cushions of the Crtl1-deficient mouse, the well-documented interactions between Crtl1 and versican, and the reported cardiac abnormalities in versican-deficient (homozygote hdf) embryos, suggested that the decreased levels of versican could play a role in the etiology of the cardiac phenotype in the Crtl1-deficient mouse. This prompted us to examine the phenotype of the heterozygote hdf mouse. As predicted, heterozygote hdf embryos express reduced levels of versican when compared to wild type littermates (p=0.0005) (Fig 8C). Histological analysis of hdf+/− embryos at ED14.5–15.5 showed that 30% displayed ventricular septal defects reminiscent of those seen in the Crtl1−/− mouse (Fig 8E–I).
Discussion
Crtl1 expression in the developing murine heart
To increase our insights into mechanisms of valvuloseptal development, we performed a comparative DNA microarray analysis of gene expression in the AV and ventricular tissues of embryonic mouse hearts at ED10.5–11.0. This approach resulted in a list of roughly 200 differentially-expressed genes, approximately 150 of which were expressed at a significantly higher level in the AV junction than in the ventricles. In this paper, we report on one of them, cartilage link protein 1 (Crtl1).
Crtl1 is a member of the hyaluronan and proteoglycan binding link protein (HAPLN) gene family, and is also known as HAPLN1 (Spicer et al., 2003). Other HAPLN family members include the brain/central nervous system-specific HAPLN family member HAPLN2 (also known as Bral1), the widely expressed HAPLN3 (also known as Lp3), found in a variety of smooth muscle cell types (Ogawa et al., 2004), and HAPLN4 (also known as Bral2). HAPLN proteins stabilize the binding interaction between HA and chondroitin sulfate proteoglycans (CSPGs). These complexes can interact with other matrix molecules (e.g. fibulin-1 and tenascin-C) as well as with cell surface receptors (e.g. CD44 and EGFR’s) (Schroeder et al., 2003; Toole, 2004). Therefore, the HAPLN/HA/CSPG complexes appear to play a significant, yet poorly understood, role in ECM organization and function.
Crtl1 is best known for its role in cartilage formation, where it stabilizes the HA/aggrecan complex (Hardingham, 1979) and the HA/versican complex (Binette et al., 1994; Matsumoto et al., 2006; Matsumoto et al., 2003; Shi et al., 2004; Shibata et al., 2003). The importance of Crtl1 in cartilage formation has previously been demonstrated in studies using the Crtl1-deficient mouse (Czipri et al., 2003; Watanabe and Yamada, 1999). In these studies, it was reported that most Crtl1−/− specimens, characterized by skeletal and craniofacial abnormalities resulting from chondrodysplasia, survive the pre-natal period, but die shortly after birth. Importantly, while a recent paper has also reported the expression of Crtl1 in the endocardial cushions at two important stages of AV valve development (Lincoln et al., 2007), a thorough investigation of Crtl1 expression throughout heart development has not been conducted. Furthermore, none of the published studies on the Crtl1-deficient mice have reported on the effect of perturbation of Crtl1 expression on cardiac development.
Using a panel of specific antibodies, recognizing murine Crtl1 and versican, in combination with a biotinylated hyaluronan-binding protein to detect HA, we studied the spatiotemporal expression of the three putative cardiac binding partners. We determined that, at the youngest stage examined (ED9.5), expression of the binding partners was confined to the endocardial lining of the heart and the endocardially-derived cushion mesenchyme (Fig 2). As development proceeds, the expression of Crtl1 in the endocardial lining of the atria and ventricles sharply declines, Crtl1 expression becoming largely restricted to the cardiac mesenchymal tissues (Fig 3; see also (Lincoln et al., 2007). Here, expression was primarily found in the ECM surrounding the endocardially-derived mesenchyme as indicated by Tie2-cre/ROSA26 lineage studies (Fig 3, see also (de Lange et al., 2004; Kisanuki et al., 2001; Snarr et al., 2007; Snow et al., 2005). We did not see Crtl1 expression in two subpopulations of mesenchyme that are commonly referred to as being “extra-cardiac” derived. The first, and most prominent, population of Crtl1-negative mesenchyme was located in the truncal ridges of the OFT (Fig 3). Numerous studies, specifically those using the Wnt1cre/ROSA26Rflox model, have demonstrated that this Crtl1-negative OFT mesenchyme is, for the most part, of cardiac neural crest origin (Danielian et al., 1998; de Lange et al., 2004; Nakamura et al., 2006). The other subpopulation of mesenchymal cells, characterized by the absence of Crtl1 expression, was found in the venous pole. This non-endocardially derived tissue represents the dorsal mesenchymal protrusion (DMP), an intra-cardiac mesenchymal extension of the dorsal mesocardium (Snarr et al., 2007; Wessels et al., 2000), also known as the spina vestibuli (Mommersteeg et al., 2006).
As the lateral and (fused) major AV cushions remodel into the respective leaflets of the AV valves (Wessels and Sedmera, 2003), we observed that the level of expression of both Crtl1 and versican started to drop sharply in the leaflets of the mitral valve, while the expression of the ECM binding partners in the right AV valve leaflets remained relatively high (Fig 4A–C). This asymmetric pattern, observed as early as ED14.5, suggests that the left and right AV valve do not mature along the same timeline and that the ECM remodeling that accompanies valve development (Kruithof et al., 2007) occurs first in the mitral valve and only later in the tricuspid valve.
Crtl1 deficiency leads to a spectrum of structural cardiac defects
Based on our microarray results, the overlapping expression patterns of Crtl1/versican/HA, and taking into consideration the documented importance of both HA and versican in heart development (Camenisch et al., 2000; Mjaatvedt et al., 1998; Yamamura et al., 1997), we predicted a significant role for Crtl1 in valvuloseptal morphogenesis.
The histological analysis of pre- and postnatal Crtl1−/− hearts revealed a spectrum of structural cardiac defects including atrial and atrioventricular septal defects, muscular ventricular defects, overriding aorta, double outlet right ventricle, and myocardial hypoplasia. The most severely malformed hearts were characterized by common AV canal (cAVC) with severely hypoplastic cushion-derived structures, an underdeveloped DMP, and very thin and flimsy myocardial walls and disorganized muscular IVS. Interestingly, the abnormalities observed were not only found in derivatives of (endocardially derived) cardiac structures that were expressing Crtl1 at one point (e.g. the AV cushions), but also in structures that were adjacent to Crtl1-expressing tissues (e.g. the DMP and the myocardial wall of the ventricles).
Abnormalities in the development of the AV cushions were expected based on the fact that mice that do not produce either of the presumptive cardiac binding partners of Crtl1, i.e. versican or HA, die early in development with poorly-developed AV cushions. Specifically, mice that do not produce HA (Has2 deficient mouse), die at ED10.5. Histological analysis of the Has2−/− hearts revealed that, in the absence of HA, EMT is inhibited, resulting in the failure of AV endocardial cushions to form (Camenisch et al., 2000). A similar phenotype was observed in the hdf mouse, in which the expression of the versican gene, Cspg2, is disrupted by an insertional transgenic mutation (Mjaatvedt et al., 1998; Yamamura et al., 1997). The hdf embryos also die around ED10.5 and are characterized by complete absence of AV cushions, as a result of perturbed EMT (Mjaatvedt et al., 1998; Yamamura et al., 1997).
While the Crtl1-deficient mouse exhibits defects in valvuloseptal structures derived from the AV cushions, the malformations observed are clearly milder than those seen in the Has2 deficient and the hdf mice. Our histological studies indicate that, in the absence of Crtl1, the initial steps of cushion formation are relatively normal and that the onset of EMT is not severely affected. We can, however, not exclude the possibility that the rate of EMT is impaired, a phenomenon that could have consequences for subsequent steps in cushion development. Furthermore, the maturation of endocardial cushion tissues involves migration, proliferation, and differentiation of cushion mesenchyme. Thus, it is plausible that, as a result of the absence of Crtl1, one or more of these events in cushion morphogenesis are compromised. Based on the results of PCNA labeling studies in the myocardium (Fig 7), it is most likely that cushion-related malformations result from perturbation of post-EMT mesenchymal proliferation.
As described above, Crtl1−/− mice exhibited an unexpected spectrum of myocardial defects, including thin ventricular myocardial walls. “Thin myocardium syndrome” is seen in a number of genetically- and/or experimentally-altered animal models and is often reported in association with defects in epicardial development (Gassmann et al., 1995; Moore et al., 1998; Perez-Pomares et al., 2002; Zhao et al., 1998). Given the fact that Crtl1 is not expressed in the epicardium, it is unlikely that the myocardial abnormalities seen in the Crtl1-deficient mouse are epicardium-related. It is more likely that the myocardial proliferation defects in the Crtl1-deficient mouse are associated with the absence of expression of Crtl1 in the adjacent endocardial lining of the looping heart, suggesting that the regulation of myocardial structures is not only reliant upon early signals received from the epicardium, but also on early endocardially-associated ECM signaling. This concept of the convergence of early endocardial and epicardial signals on common proliferative pathways, affecting myocardial wall development, has been previously suggested by others (Kang and Sucov, 2005).
Our histological analysis of the hearts with cAVC shows that the DMP is absent. Recently, we described how the DMP forms an integral component of the AV mesenchymal complex (Snarr et al., 2007). The mechanisms by which Crtl1 may regulate the development of the DMP have not yet been elucidated. It is noteworthy, however, that although Crtl1 is not expressed within the DMP itself, strong expression is found in the mesodermal tissues adjacent to the protruding mesenchyme (Fig 2).
Reduction of versican expression may contribute to the cardiac phenotype of the Crtl1-deficient mouse
Although it has been suggested that Crtl1 can act as a HA/versican independent growth factor (Liu et al., 2000), it is likely that, given the reported binding characteristics of Crtl1, the observed cardiac defects relate to the reported function of Crtl1 in stabilizing the interaction between HA and versican (Matsumoto et al., 2003; Shi et al., 2004). That relationship, together with the finding that another Crtl1 binding partner, aggrecan, is reduced in the cartilage of Crtl1-deficient mice, led us to investigate how Crtl1 deficiency affected the expression of its putative cardiac binding partners. Using a quantitative immunofluorescence approach, confirmed by Western Blot, we determined that the levels of versican protein were significantly reduced in the developing AV cushions of the Crtl1−/− mouse (Fig 8), while there were no significant changes in HA levels. Given the reduction in versican expression, and knowing the severe phenotype of the homozygous hdf mouse, we then re-examined the phenotype of the hdf mouse, focusing on the heterozygous condition. Analysis of the hdf+/− embryos, confirmed that versican was reduced, as compared to wildtypes, to levels similar to those seen in the AV cushions of the Crtl1−/− mice, but that levels of Crtl1 were not reduced (data not shown). Histological inspection of hdf+/− embryos showed AV septal defects in cushion-derived tissues that resembled those seen in a subset of Crtl1−/− specimens. These combined observations suggest that the reduction of versican expression in the developing heart of the Crtl1−/− mouse may contribute to, at least a subset of, the cardiac abnormalities observed in the Crtl1−/− mouse.
While very little is known about the role of Crtl1 in regulating cell behavior, there is significantly more data available regarding the function of versican. Numerous studies have reported on the significance of versican in promoting cell proliferation in a wide variety of cell types (Evanko et al., 2001; Sheng et al., 2005; Wight, 2002). Currently, efforts are underway to further unravel the mechanisms by which Crtl1 and versican are involved in the regulation of heart development. These efforts specifically focus on the question of how these two proteins are involved in controlling proliferation, and hence growth, of the respective mesenchymal (e.g. AV cushions and DMP) and myocardial structures (e.g. ventricular wall and IVS) that were found to be affected in the Crtl1-deficient and versican haploinsufficient mouse.
Acknowledgements
The authors gratefully acknowledge Drs. Yoshihiko Yamada and Tibor Glant for helpful discussions and assistance in relation to the Crtl1 knock out mouse. The authors would also like to thank Dr. Waleed Twal, Sarah Fairey, Joshua Spruill, and Sue Tjepkema-Burrows for expert technical assistance.
Sources of funding We thankfully acknowledge the MUSC Proteogenomics Facility (http://proteogenomics.musc.edu/) which is supported by grants from the National Cancer Institute (CA 095841) and the National Heart Lung and Blood Institute (P20RR016434). This work was supported by NIH Grant funding C06 RR018823 and C06 RR015455 from the Extramural Research Facilities Program of the National Center for Research Resources, American Heart Association Grant-in-aid 0655530U (BSS, AW), NIH T-32 HL07260 (EEW, BSS), NIH HLBI R01-HL084285 (AW, EEW, CHM), and the South Carolina Center of Biomedical Research Excellence (COBRE) NIH NCRR P20-RR016434 (AW, JLB, WSA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest None
References
- Binette F, Cravens J, Kahoussi B, Haudenschild DR, Goetinck PF. Link protein is ubiquitously expressed in non-cartilaginous tissues where it enhances and stabilizes the interaction of proteoglycans with hyaluronic acid. J Biol Chem. 1994;269:19116–19122. [PubMed] [Google Scholar]
- Bushdid PB, Osinska H, Waclaw RR, Molkentin JD, Yutzey KE. NFATc3 and NFATc4 are required for cardiac development and mitochondrial function. Circ Res. 2003;92:1305–1313. doi: 10.1161/01.RES.0000077045.84609.9F. [DOI] [PubMed] [Google Scholar]
- Camenisch TD, Spicer AP, Brehm-Gibson T, Biesterfeldt J, Augustine ML, Calabro A, Jr, Kubalak S, Klewer SE, McDonald JA. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J Clin Invest. 2000;106:349–360. doi: 10.1172/JCI10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campione M, Ros MA, Icardo JM, Piedra E, Christoffels VM, Schweickert A, Blum M, Franco D, Moorman AF. Pitx2 expression defines a left cardiac lineage of cells: evidence for atrial and ventricular molecular isomerism in the iv/iv mice. Dev Biol. 2001;231:252–264. doi: 10.1006/dbio.2000.0133. [DOI] [PubMed] [Google Scholar]
- Caterson B, Baker JR, Christner JE, Lee Y, Lentz M. Monoclonal antibodies as probes for determining the microheterogeneity of the link proteins of cartilage proteoglycan. J Biol Chem. 1985;260:11348–11356. [PubMed] [Google Scholar]
- Christoffels VM, Hoogaars WM, Tessari A, Clout DE, Moorman AF, Campione M. T-box transcription factor Tbx2 represses differentiation and formation of the cardiac chambers. Dev Dyn. 2004;229:763–770. doi: 10.1002/dvdy.10487. [DOI] [PubMed] [Google Scholar]
- Czipri M, Otto JM, Cs-Szabo G, Kamath RV, Vermes C, Firneisz G, Kolman KJ, Watanabe H, Li Y, Roughley PJ, Yamada Y, Olsen BR, Glant TT. Genetic rescue of chondrodysplasia and the perinatal lethal effect of cartilage link protein deficiency. J Biol Chem. 2003;278:39214–39223. doi: 10.1074/jbc.M303329200. [DOI] [PubMed] [Google Scholar]
- Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifeninducible form of Cre recombinase. Curr Biol. 1998;8:1323–1326. doi: 10.1016/s0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- de Lange FJ, Moorman AF, Anderson RH, Manner J, Soufan AT, de Gier-de Vries C, Schneider MD, Webb S, van den Hoff MJ, Christoffels VM. Lineage and morphogenetic analysis of the cardiac valves. Circ Res. 2004;95:645–654. doi: 10.1161/01.RES.0000141429.13560.cb. [DOI] [PubMed] [Google Scholar]
- Dettman RW, Denetclaw W, Jr, Ordahl CP, Bristow J. Common epicardial origin of coronary vascular smooth muscle, perivascular fibroblasts, and intermyocardial fibroblasts in the avian heart. Dev Biol. 1998;193:169–181. doi: 10.1006/dbio.1997.8801. [DOI] [PubMed] [Google Scholar]
- Eisenberg LM, Markwald RR. Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ Res. 1995;77:1–6. doi: 10.1161/01.res.77.1.1. [DOI] [PubMed] [Google Scholar]
- Evanko SP, Johnson PY, Braun KR, Underhill CB, Dudhia J, Wight TN. Platelet-derived growth factor stimulates the formation of versican-hyaluronan aggregates and pericellular matrix expansion in arterial smooth muscle cells. Arch Biochem Biophys. 2001;394:29–38. doi: 10.1006/abbi.2001.2507. [DOI] [PubMed] [Google Scholar]
- Gassmann M, Casagranda F, Orioli D, Simon H, Lai C, Klein R, Lemke G. Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature. 1995;378:390–394. doi: 10.1038/378390a0. [DOI] [PubMed] [Google Scholar]
- Hardingham TE. The role of link-protein in the structure of cartilage proteoglycan aggregates. Biochem J. 1979;177:237–247. doi: 10.1042/bj1770237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogaars WM, Tessari A, Moorman AF, de Boer PA, Hagoort J, Soufan AT, Campione M, Christoffels VM. The transcriptional repressor Tbx3 delineates the developing central conduction system of the heart. Cardiovasc Res. 2004;62:489–499. doi: 10.1016/j.cardiores.2004.01.030. [DOI] [PubMed] [Google Scholar]
- Jaber M, Koch WJ, Rockman H, Smith B, Bond RA, Sulik KK, Ross J, Jr, Lefkowitz RJ, Caron MG, Giros B. Essential role of beta-adrenergic receptor kinase 1 in cardiac development and function. Proc Natl Acad Sci U S A. 1996;93:12974–12979. doi: 10.1073/pnas.93.23.12974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JO, Sucov HM. Convergent proliferative response and divergent morphogenic pathways induced by epicardial and endocardial signaling in fetal heart development. Mech Dev. 2005;122:57–65. doi: 10.1016/j.mod.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Kern CB, Norris RA, Thompson RP, Argraves WS, Fairey SE, Reyes L, Hoffman S, Markwald RR, Mjaatvedt CH. Versican proteolysis mediates myocardial regression during outflow tract development. Dev Dyn. 2007;236:671–683. doi: 10.1002/dvdy.21059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern CB, Twal WO, Mjaatvedt CH, Fairey SE, Toole BP, Iruela-Arispe ML, Argraves WS. Proteolytic cleavage of versican during cardiac cushion morphogenesis. Dev Dyn. 2006;235:2238–2247. doi: 10.1002/dvdy.20838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyes WM, Logan C, Parker E, Sanders EJ. Expression and function of bone morphogenetic proteins in the development of the embryonic endocardial cushions. Anat Embryol (Berl) 2003;207:135–147. doi: 10.1007/s00429-003-0337-2. [DOI] [PubMed] [Google Scholar]
- Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- Kraus F, Haenig B, Kispert A. Cloning and expression analysis of the mouse T-box gene tbx20. Mech Dev. 2001;100:87–91. doi: 10.1016/s0925-4773(00)00499-8. [DOI] [PubMed] [Google Scholar]
- Krug EL, Runyan RB, Markwald RR. Protein extracts from early embryonic hearts initiate cardiac endothelial cytodifferentiation. Dev Bio. 1985;112:414–426. doi: 10.1016/0012-1606(85)90414-2. [DOI] [PubMed] [Google Scholar]
- Kruithof BP, Krawitz SA, Gaussin V. Atrioventricular valve development during late embryonic and postnatal stages involves condensation and extracellular matrix remodeling. Dev Biol. 2007;302:208–217. doi: 10.1016/j.ydbio.2006.09.024. [DOI] [PubMed] [Google Scholar]
- Kruzynska-Frejtag A, Machnicki M, Rogers R, Markwald RR, Conway SJ. Periostin (an osteoblast-specific factor) is expressed within the embryonic mouse heart during valve formation. Mech Dev. 2001;103:183–188. doi: 10.1016/s0925-4773(01)00356-2. [DOI] [PubMed] [Google Scholar]
- Lincoln J, Kist R, Scherer G, Yutzey KE. Sox9 is required for precursor cell expansion and extracellular matrix organization during mouse heart valve development. Dev Biol. 2007 doi: 10.1016/j.ydbio.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln J, Lange AW, Yutzey KE. Hearts and bones: shared regulatory mechanisms in heart valve, cartilage, tendon, and bone development. Dev Biol. 2006;294:292–302. doi: 10.1016/j.ydbio.2006.03.027. [DOI] [PubMed] [Google Scholar]
- Liu H, McKenna LA, Dean MF. An N-terminal peptide from link protein can stimulate biosynthesis of collagen by human articular cartilage. Arch Biochem Biophys. 2000;378:116–122. doi: 10.1006/abbi.2000.1758. [DOI] [PubMed] [Google Scholar]
- Manner J. Does the subepicardial mesenchyme contribute myocardioblasts to the myocardium of the chick embryo heart? A quail-chick chimera study tracing the fate of the epicardial primordium. Anat Rec. 1999;255:212–226. doi: 10.1002/(sici)1097-0185(19990601)255:2<212::aid-ar11>3.3.co;2-o. [DOI] [PubMed] [Google Scholar]
- Markwald RR, Fitzharris TP, Manasek FJ. Structural development of endocardial cushions. Am J Anat. 1977;148:85–119. doi: 10.1002/aja.1001480108. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Kamiya N, Suwan K, Atsumi F, Shimizu K, Shinomura T, Yamada Y, Kimata K, Watanabe H. Identification and characterization of versican/PG-M aggregates in cartilage. J Biol Chem. 2006;281:18257–18263. doi: 10.1074/jbc.M510330200. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Shionyu M, Go M, Shimizu K, Shinomura T, Kimata K, Watanabe H. Distinct interaction of versican/PG-M with hyaluronan and link protein. J Biol Chem. 2003;278:41205–41212. doi: 10.1074/jbc.M305060200. [DOI] [PubMed] [Google Scholar]
- Mjaatvedt CH, Yamamura H, Capehart AA, Turner D, Markwald RR. The Cspg2 gene, disrupted in the hdf mutant, is required for right cardiac chamber and endocardial cushion formation. Dev Biol. 1998;202:56–66. doi: 10.1006/dbio.1998.9001. [DOI] [PubMed] [Google Scholar]
- Mommersteeg MT, Soufan AT, de Lange FJ, van den Hoff MJ, Anderson RH, Christoffels VM, Moorman AF. Two distinct pools of mesenchyme contribute to the development of the atrial septum. Circ Res. 2006;99:351–353. doi: 10.1161/01.RES.0000238360.33284.a0. [DOI] [PubMed] [Google Scholar]
- Moore AW, Schedl A, McInnes L, Doyle M, Hecksher-Sorensen J, Hastie N. YAC transgenic analysis reveals Wilms' tumour 1 gene activity in the proliferating coelomic epithelium, developing diaphragm and limb. Mech Dev. 1998;79:169–184. doi: 10.1016/s0925-4773(98)00188-9. [DOI] [PubMed] [Google Scholar]
- Moorman AF, Christoffels VM. Cardiac chamber formation: development, genes, and evolution. Physiol Rev. 2003;83:1223–1267. doi: 10.1152/physrev.00006.2003. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Colbert MC, Robbins J. Neural crest cells retain multipotential characteristics in the developing valves and label the cardiac conduction system. Circ Res. 2006;98:1547–1554. doi: 10.1161/01.RES.0000227505.19472.69. [DOI] [PubMed] [Google Scholar]
- Neame PJ, Christner JE, Baker JR. The primary structure of link protein from rat chondrosarcoma proteoglycan aggregate. J Biol Chem. 1986;261:3519–3535. [PubMed] [Google Scholar]
- Norris RA, Kern CB, Wessels A, Wirrig EE, Markwald RR, Mjaatvedt CH. Detection of betaig-H3, a TGFbeta induced gene, during cardiac development and its complementary pattern with periostin. Anat Embryol (Berl) 2005;210:13–23. doi: 10.1007/s00429-005-0010-z. [DOI] [PubMed] [Google Scholar]
- Ogawa H, Oohashi T, Sata M, Bekku Y, Hirohata S, Nakamura K, Yonezawa T, Kusachi S, Shiratori Y, Ninomiya Y. Lp3/Hapln3, a novel link protein that co-localizes with versican and is coordinately up-regulated by platelet-derived growth factor in arterial smooth muscle cells. Matrix Biol. 2004;23:287–298. doi: 10.1016/j.matbio.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Perez-Pomares JM, Phelps A, Sedmerova A, Carmona R, Gonzalez-Iriarte M, Munoz-Chapuli R, Wessels A. Experimental studies on the spatiotemporal expression of WT1 and RALDH2 in the embryonic avian heart: a model for the regulation of myocardial and valvuloseptal development by epicardially derived cells (EPDCs) Dev Biol. 2002;247:307–326. doi: 10.1006/dbio.2002.0706. [DOI] [PubMed] [Google Scholar]
- Sandy JD, Westling J, Kenagy RD, Iruela-Arispe ML, Verscharen C, Rodriguez-Mazaneque JC, Zimmermann DR, Lemire JM, Fischer JW, Wight TN, Clowes AW. Versican V1 proteolysis in human aorta in vivo occurs at the Glu441-Ala442 bond, a site that is cleaved by recombinant ADAMTS-1 and ADAMTS-4. J Biol Chem. 2001;276:13372–13378. doi: 10.1074/jbc.M009737200. [DOI] [PubMed] [Google Scholar]
- Schroeder JA, Jackson LF, Lee DC, Camenisch TD. Form and function of developing heart valves: coordination by extracellular matrix and growth factor signaling. J Mol Med. 2003;81:392–403. doi: 10.1007/s00109-003-0456-5. [DOI] [PubMed] [Google Scholar]
- Sheng W, Wang G, Wang Y, Liang J, Wen J, Zheng PS, Wu Y, Lee V, Slingerland J, Dumont D, Yang BB. The roles of versican V1 and V2 isoforms in cell proliferation and apoptosis. Mol Biol Cell. 2005;16:1330–1340. doi: 10.1091/mbc.E04-04-0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S, Grothe S, Zhang Y, O'Connor-McCourt MD, Poole AR, Roughley PJ, Mort JS. Link protein has greater affinity for versican than aggrecan. J Biol Chem. 2004;279:12060–12066. doi: 10.1074/jbc.M310091200. [DOI] [PubMed] [Google Scholar]
- Shibata S, Fukada K, Imai H, Abe T, Yamashita Y. In situ hybridization and immunohistochemistry of versican, aggrecan and link protein, and histochemistry of hyaluronan in the developing mouse limb bud cartilage. J Anat. 2003;203:425–432. doi: 10.1046/j.1469-7580.2003.00226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snarr BS, Wirrig EE, Phelps AL, Trusk TC, Wessels A. A spatiotemporal evaluation of the contribution of the dorsal mesenchymal protrusion to cardiac development. Dev Dyn. 2007 doi: 10.1002/dvdy.21074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow HE, Riccio LM, Mjaatvedt CH, Hoffman S, Capehart AA. Versican expression during skeletal/joint morphogenesis and patterning of muscle and nerve in the embryonic mouse limb. Anat Rec A Discov Mol Cell Evol Biol. 2005;282:95–105. doi: 10.1002/ar.a.20151. [DOI] [PubMed] [Google Scholar]
- Spicer AP, Joo A, Bowling RA., Jr A hyaluronan binding link protein gene family whose members are physically linked adjacent to chondroitin sulfate proteoglycan core protein genes: the missing links. J Biol Chem. 2003;278:21083–21091. doi: 10.1074/jbc.M213100200. [DOI] [PubMed] [Google Scholar]
- Sugi Y, Yamamura H, Okagawa H, Markwald RR. Bone morphogenetic protein-2 can mediate myocardial regulation of atrioventricular cushion mesenchymal cell formation in mice. Dev Biol. 2004;269:505–518. doi: 10.1016/j.ydbio.2004.01.045. [DOI] [PubMed] [Google Scholar]
- Theiler K. The House Mouse: Atlas of Embryonic Development. New York: Springer-Verlag New York Inc.; 1989. [Google Scholar]
- Toole BP. Hyaluronan: from extracellular glue to pericellular cue. Nat Rev Cancer. 2004;4:528–539. doi: 10.1038/nrc1391. [DOI] [PubMed] [Google Scholar]
- Waller BR, 3rd, Wessels A. Cardiac morphogenesis and dysmorphogenesis. An immunohistochemical approach. Methods Mol Biol. 2000;135:151–161. doi: 10.1385/1-59259-685-1:151. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Yamada Y. Mice lacking link protein develop dwarfism and craniofacial abnormalities. Nat Genet. 1999;21:225–229. doi: 10.1038/6016. [DOI] [PubMed] [Google Scholar]
- Wessels A, Anderson RH, Markwald RR, Webb S, Brown NA, Viragh S, Moorman AF, Lamers WH. Atrial development in the human heart: an immunohistochemical study with emphasis on the role of mesenchymal tissues. Anat Rec. 2000;259:288–300. doi: 10.1002/1097-0185(20000701)259:3<288::AID-AR60>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Wessels A, Markman MW, Vermeulen JL, Anderson RH, Moorman AF, Lamers WH. The development of the atrioventricular junction in the human heart. Circ Res. 1996;78:110–117. doi: 10.1161/01.res.78.1.110. [DOI] [PubMed] [Google Scholar]
- Wessels A, Sedmera D. Developmental anatomy of the heart: a tale of mice and man. Physiol Genomics. 2003;15:165–176. doi: 10.1152/physiolgenomics.00033.2003. [DOI] [PubMed] [Google Scholar]
- Wight TN. Versican: a versatile extracellular matrix proteoglycan in cell biology. Curr Opin Cell Biol. 2002;14:617–623. doi: 10.1016/s0955-0674(02)00375-7. [DOI] [PubMed] [Google Scholar]
- Ya J, Schilham MW, de Boer PA, Moorman AF, Clevers H, Lamers WH. Sox4-deficiency syndrome in mice is an animal model for common trunk. Circ Res. 1998;83:986–994. doi: 10.1161/01.res.83.10.986. [DOI] [PubMed] [Google Scholar]
- Yamamura H, Zhang M, Markwald RR, Mjaatvedt CH. A heart segmental defect in the anterior-posterior axis of a transgenic mutant mouse. Dev Biol. 1997;186:58–72. doi: 10.1006/dbio.1997.8559. [DOI] [PubMed] [Google Scholar]
- Zhao YY, Sawyer DR, Baliga RR, Opel DJ, Han X, Marchionni MA, Kelly RA. Neuregulins promote survival and growth of cardiac myocytes. Persistence of ErbB2 and ErbB4 expression in neonatal and adult ventricular myocytes. J Biol Chem. 1998;273:10261–10269. doi: 10.1074/jbc.273.17.10261. [DOI] [PubMed] [Google Scholar]