

Abstract
The recent structural elucidation of about one dozen channels (in which we include transporters) has provided further evidence that these membrane proteins typically undergo large movements during their function. However, it is still not well understood how these proteins achieve the necessary trade-off between stability and mobility. To identify specific structural properties of channels, we compared the helix-packing and hydrogen-bonding patterns of channels with those of membrane coils; the latter is a class of membrane proteins whose structures are expected to be more rigid. We describe in detail how in channels, helix pairs are usually arranged in packing motifs with large crossing angles (|τ| ≈ 40°), where the (small) side chains point away from the packing core and the backbones of the two helices are in close contact. We found that this contributes to a significant enrichment of Cα–H···O bonds and to a packing geometry where right-handed parallel (τ = −40° ± 10°) and antiparallel (τ = +140° ± 25°) arrangements are equally preferred. By sharp contrast, the interdigitation and hydrogen bonding of side chains in helix pairs of membrane coils results in narrowly distributed left-handed antiparallel arrangements with crossing angles τ = −160° ± 10° (|τ| ≈ 20°). In addition, we show that these different helix-packing modes of the two types of membrane proteins correspond to specific hydrogen-bonding patterns. In particular, in channels, three times as many of the hydrogen-bonded helix pairs are found in parallel right-handed motifs than are non-hydrogen-bonded helix pairs. Finally, we discuss how the presence of weak hydrogen bonds, water-containing cavities, and right-handed crossing angles may facilitate the required conformational flexibility between helix pairs of channels while maintaining sufficient structural stability.
INTRODUCTION
Membrane proteins are embedded into the hydrophobic environment of lipid bilayers where they modulate the exchange of information and mass between the different partitions of cells and tissues. This class of proteins includes highly biomedically and pharmaceutically relevant proteins such as G-protein coupled receptors, channels, and transporters. Most human membrane proteins are assemblies of hydrophobic transmembrane helices that bind coenzymes or ligands or, alternatively, form protein channels. This type of membrane channel can be distinguished from the β-stranded membrane proteins that form the rather unselective pores in membranes that originate from bacterial or mitochondrial outer membranes (1). These β-barrels are constructed from β-sheets in which the polar amino acids lining the pores and the hydrophobic residues that face the membrane are arranged in an alternating manner. By contrast, the amino acid composition of helical membrane proteins is primarily hydrophobic, with ∼90%–95% of the membrane-spanning residues being nonpolar (2).
Another effect of the lipid bilayer is to cause a weakening of the hydrophobic effect for the protein, which is the main driving force for the folding of water-soluble globular proteins (3). Consequently, it has been proposed that optimized van der Waals interactions between transmembrane helices could compensate for the lack of the hydrophobic effect and provide the driving force for membrane protein folding (4). However, recent analysis has revealed that helical membrane proteins are not more tightly packed than water-soluble proteins (5,6). Therefore, it remains an important outstanding question how the assembly of transmembrane helices is actually accomplished (7).
Stable transmembrane helices are formed as a result of regular patterns of hydrogen bonds between polar main-chain atoms (2,3). However, since the helical backbone is only partially shielded by the side chains (8), electrostatic interactions between polar backbone atoms are likely also to play an important role in the interactions between transmembrane helices. For example, packing interfaces containing the small amino acids Gly or Ala allow for the formation of Cα–H···O hydrogen bonds in helical membrane proteins (9). Since there is no entropic cost in burying the backbone atoms of small amino acids, such positions may even serve as initial points for the folding of helical membrane proteins. Such Cα–H···O bonds probably have a stabilizing effect of up to −1 kcal/mol (10) and accordingly could be nearly as strong as classical amide hydrogen bonds of globular proteins (11), where the strength of hydrogen bonds is diminished by the high dielectric effect in water and the competition from water for hydrogen bonds (7). Nevertheless, the role of Cα–H···O hydrogen bonds in the stability of the tertiary structure of helical membrane proteins has been somewhat controversial (10,12,13).
For classical hydrogen bonds, the free energy of formation is much higher within the hydrophobic core of the lipid bilayer, because of the low dielectric constant: estimates of this value range from −2 to −5 kcal mol−1 per bond (3,14). Consistent with this observation, it has been shown that a single polar amino acid such as Asn or Glu can drive homomeric association of model transmembrane peptides (15,16). However, such charged and polar amino acids are rare in helical membrane proteins, probably because of the free energy cost of desolvating polar side chains. Nevertheless, about half of all helix pairs in membrane proteins are stabilized by classical hydrogen bonds (5,17), and most of these are found in motifs involving medium polar residues such as Ser or Thr. Indeed, it has been shown that such Ser/Thr motifs can also drive the association of model transmembrane helices (18). Thus, classical hydrogen bonds are clearly important in helix-helix interactions in membrane proteins.
The strength of both classical and Cα–H···O hydrogen bonds depends on the distance, the chemistry of the donor and acceptor atoms, the relative arrangement of donor and acceptor atoms, and the nature of the surrounding milieu (19,20). As a consequence, even small conformational changes may result in the breaking of hydrogen bonds (13). Conformational changes such as the shifting, rotating, or tilting of helices are believed to occur frequently in some membrane proteins and are suggested to be facilitated by specific structural characteristics such as helix kinks, the smoothness of helical surfaces, or local packing defects (6,21,22). The analysis of the packing and the geometrical features of helix pairs that are also involved in hydrogen bonds could therefore provide clues to understanding the structure-function relationship of membrane proteins.
We have recently shown that the prevalent motif of helix-helix interactions in channels and transporters (collectively referred to as channels) is the right-handed motif, whereas this type of contact is clearly underrepresented in other membrane proteins (collectively referred to as membrane coils) (6). It is also known that multiple Cα–H···O hydrogen bonds tend to cluster at such right-handed contacts (9), which is consistent with the fact that channels are rich in sequence patterns containing small and medium polar amino acids (23). Moreover, channels are packed significantly less densely than membrane coils (6). These packing defects are likely to be functionally important, because they cluster at the proposed hinge regions of transporters or in the pores of channels. Together, these results suggest that the distribution of hydrogen bonds in membrane proteins may correlate (via specific architectural features such as handedness of helix-helix interactions or packing density) with the function of the protein.
To test this hypothesis we report here a comprehensive analysis of the abundance, types, and location of hydrogen bonds, including Cα–H···O hydrogen bonds, in known membrane protein structures. We first correlate differences in hydrogen-bond distributions with the two classes of membrane proteins: channels and membrane coils. In an earlier study, we observed that many crystal structures of membrane proteins include cavities that presumably contain water molecules not resolved in the electron density (6). Thus, we also inspect the regions around hydrogen bonds for large cavities to identify whether the presence of hydrogen bonds correlates with nearby putative buried waters. Finally, we show how the differences in helix-helix interactions (i.e., crossing angles) in the two protein classes correlate with the hydrogen-bonding patterns. More broadly, because of the importance of hydrogen bonding in the membrane interior, the detailed description of hydrogen bonds obtained from this analysis can be considered an important step toward a solution to the membrane protein folding problem (7).
MATERIALS AND METHODS
We used a nonredundant data set of 27 high resolution membrane protein structures for the analysis of hydrogen bonds; these can be divided into 13 channels and 14 membrane coils structures (Table 1). To differentiate between those amino acids located within or outside the lipophilic environment, two parallel planes were drawn to delineate the hydrophobic part of the lipid bilayer, applying the criteria described in our previous analysis (2). Polar interactions were considered for this analysis only if both hydrogen-bonding partners are located between these two planes. The Ca2+-ATPase is the only member of the channel group for which the transport mechanism has been revealed by the structural elucidation of four different states (24). The ATP-bound (Protein Data Bank (PDB) code: 1vfp) form of the channel was taken for the statistics of hydrogen bonds. To estimate the number of hydrogen bonds preserved during transport, the Ca2+-bound and -unbound forms (1su4, 1iwo) and the intermediate state with hydrolyzed ATP (1wpg) were also studied (24).
TABLE 1.
Interhelical hydrogen bonds in membrane proteins with different functions
| Cα–H···O
|
Classical
|
All
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Channels | +/h-h | NoC/No | /h | /aa | NoC/No | /h | /aa | NoC/No | /h | /aa | |
| 1fx8 | Glycerol facilitator | 6/6 | 1/22 | 2.8 | 0.17 | 1/17 | 2.1 | 0.13 | 2/39 | 4.9 | 0.30 |
| 1iwg | AcrB* | 15/19 | 15/29 | 2.4 | 0.13 | 21/40 | 3.3 | 0.18 | 36/69 | 5.8 | 0.30 |
| 1j4n | AQP1* | 10/10 | 1/21 | 2.6 | 0.20 | 1/21 | 2.6 | 0.20 | 2/42 | 5.3 | 0.40 |
| 1jvm | KcsA* | 1/1 | 0/1 | 0.5 | 0.03 | 0/0 | 0.0 | 0.00 | 0/1 | 0.5 | 0.03 |
| 1kpl | ClC* | 14/18 | 22/42 | 3.0 | 0.21 | 9/20 | 1.4 | 0.10 | 31/62 | 4.4 | 0.32 |
| 1l7v | BtuCD* | 6/8 | 8/18 | 1.8 | 0.13 | 1/9 | 0.9 | 0.06 | 9/27 | 2.7 | 0.19 |
| 1msl | MscL* | 2/2 | 0/1 | 0.5 | 0.03 | 0/1 | 0.5 | 0.03 | 0/2 | 1.0 | 0.06 |
| 1okc | ADP/ATP carrier | 4/4 | 2/6 | 1.0 | 0.08 | 2/3 | 0.5 | 0.04 | 4/9 | 1.5 | 0.12 |
| 1pv7 | Lactose permease | 4/10 | 12/18 | 1.5 | 0.10 | 19/28 | 2.3 | 0.16 | 31/46 | 3.8 | 0.26 |
| 1pw4 | GlpT* | 4/10 | 25/28 | 2.3 | 0.16 | 22/25 | 2.1 | 0.14 | 47/53 | 4.4 | 0.29 |
| 1rh5 | Translocon | 8/14 | 15/26 | 2.0 | 0.13 | 16/35 | 2.7 | 0.17 | 31/61 | 4.7 | 0.29 |
| 1vfp | Calcium ATPase | 5/7 | 11/15 | 1.5 | 0.11 | 27/49 | 4.9 | 0.35 | 38/64 | 6.4 | 0.46 |
| 2bl2 | V-type ATPase | 1/4 | 10/16 | 4.0 | 0.25 | 4/9 | 2.3 | 0.14 | 14/25 | 6.3 | 0.39 |
| All | 78/113 | 122/243 | 2.2 | 0.14 | 133/257 | 2.3 | 0.15 | 255/500 | 4.4 | 0.29 | |
| Cα–H···O
|
Classical
|
All
|
|||||||||
| Membrane coils
|
+/h-h
|
NoC/No
|
/h
|
/aa
|
NoC/No
|
/h
|
/aa
|
NoC/No
|
/h
|
/aa
|
|
| 1ar1 | Cytochrome c oxidase | 4/11 | 5/10 | 0.8 | 0.05 | 19/38 | 3.2 | 0.20 | 24/48 | 4.0 | 0.25 |
| 1c3w | Bacteriorhodopsin | 1/9 | 0/7 | 1.0 | 0.07 | 1/15 | 2.1 | 0.15 | 1/22 | 3.1 | 0.22 |
| 1e12 | Halorhodopsin | 1/11 | 8/17 | 2.4 | 0.12 | 8/24 | 3.4 | 0.17 | 16/41 | 5.9 | 0.29 |
| 1eys | Reaction center | 0/4 | 0/2 | 0.2 | 0.01 | 2/8 | 0.8 | 0.04 | 2/10 | 1.0 | 0.06 |
| 1f88 | Rhodopsin | 2/5 | 6/8 | 1.1 | 0.07 | 5/15 | 2.1 | 0.13 | 11/23 | 3.3 | 0.21 |
| 1h2s | Sensory rhodopsin | 3/9 | 1/12 | 1.3 | 0.07 | 2/18 | 2.0 | 0.10 | 3/30 | 3.3 | 0.17 |
| 1jb0 | Photosystem I | 5/20 | 12/62 | 1.9 | 0.10 | 10/55 | 1.7 | 0.09 | 22/117 | 3.5 | 0.19 |
| 1kqf | Formate dehydrogenase | 1/3 | 1/3 | 0.6 | 0.04 | 3/9 | 1.8 | 0.11 | 4/12 | 2.4 | 0.15 |
| 1l0v | Fumarate reductase | 3/8 | 3/7 | 1.4 | 0.13 | 10/15 | 3.0 | 0.27 | 13/22 | 4.4 | 0.40 |
| 1nek | Succinate dehydrogenase | 1/4 | 2/5 | 0.8 | 0.05 | 4/11 | 1.8 | 0.11 | 6/16 | 2.7 | 0.17 |
| 1ppj | Cytochrome bc1 | 2/5 | 3/10 | 0.8 | 0.05 | 5/10 | 0.8 | 0.05 | 8/20 | 1.7 | 0.10 |
| 1q16 | NarGHI* | 0/3 | 4/8 | 1.6 | 0.10 | 0/7 | 1.4 | 0.09 | 4/15 | 3.0 | 0.19 |
| 1qla | Fumerate reductase | 1/5 | 0/3 | 0.6 | 0.04 | 1/11 | 2.2 | 0.14 | 1/14 | 2.8 | 0.18 |
| 2occ | Cytochrome c oxidase | 11/32 | 8/41 | 2.0 | 0.11 | 17/55 | 2.6 | 0.15 | 25/96 | 4.6 | 0.25 |
| All | 35/129 | 53/195 | 1.4 | 0.08 | 87/291 | 2.0 | 0.12 | 140/486 | 3.4 | 0.19 | |
No. of h-bonds: /h, per helix; /aa, per residue; NoC, at cavities; +/h-h, No. of right-handed/hydrogen-bonded helix pairs .
AcrB, bacterial multidrug efflux transporter; AQP1, aquaporin 1; KcsA, potassium channel; ClC, chloride transporter; BtuCD, ABC transporter; MscL, mechanosensitive channel; GlpT, glycerol-3-phosphate transporter; and NarGHI, nitrate reductase.
The Cα–H···O and classical (N, O, Cβ−ζ)–H···O hydrogen bonds were calculated using the programs TMCHbond and HBexplore, respectively (9,20). The hydrogen coordinates were generated according to standard geometrical rules in a first step, taking into account the hybridization of the donor atoms and the atomic environment (25). For the calculation of classical hydrogen bonds, only those interactions were considered with distances between the acceptor O and the donor H equal to or shorter than 2.5 Å and with bonding angles ζ larger than 90°. We used the standard parameters recommended by the authors to calculate the Cα–H···O bonds with TMCHbond (9). That is, the maximum distance between the acceptor O and the donor H was 3.5 Å with bond angles ζ > 120° or 3.0 Å with ζ > 90°.
For the calculation of packing densities, the Voronoi cell method was applied (26). This method uses curved instead of planar interfaces between atoms to create more reasonable assignments. Another advantage of this method is that it also works for atoms located in protein regions with large packing defects, which are frequently found in channels (6). Hydrogen bonds were referred to as “in close proximity to” such an internal cavity if at least one atom lying within a radius of 6 Å around the center of the bond is in contact with the cavity. To evaluate the atomic packing densities, all buried atoms of two neighboring transmembrane helices with their van der Waals radii closer than 1.5 Å were considered. Atoms are classified as buried when less than 40% of the atoms' surface contacts the surrounding milieu (6).
For the evaluation of crossing angles, we first identified pairs of helices with at least two residues with atoms less than 1.5 Å from one another. Transmembrane helices are regularly kinked or curved (2). The crossing angle was therefore determined using the local axes of the two helical sections that are actually involved in the contact after extending each helical section by an additional turn at both termini. Thus helices are only considered in their entirety for helix pairs with virtually parallel arrangements.
The Shannon entropy H is a well-established measure to estimate the conservation of a certain amino acid position during evolution (27). It was taken from the corresponding position of the multiple sequence alignment of the PDB-associated HSSP file (28). The absolute value of the Shannon entropy depends on the average similarity of all the sequences used for the alignment. Thus, only homologous proteins with a sequence similarity above 30% were considered. To estimate the variability of a certain amino acid, a reasonable amount of distantly related proteins is required for the alignment. Protein alignments with an average pair-wise sequence identity above 50% (1ppj and 2occ) were not taken into account. The entropy of residues exposed to lipid, Hexposed, is generally high in all helical membrane proteins (29–31). The average value of Hexposed over all residues in each protein was used as a reference to facilitate comparison across different proteins. Thus, for an amino acid, a, the conservation of the buried amino acids is defined as
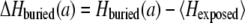 |
and the conservation of the hydrogen-bonded amino acids is defined as
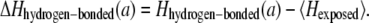 |
RESULTS
Channels are enriched in hydrogen bonds
Analysis of interhelical hydrogen bonding in membrane proteins reveals significant differences between channels and membrane coils. Specifically, in channel proteins, there are 0.29 hydrogen bonds per amino acid, whereas residues in membrane coils are stabilized on average by 0.19 hydrogen bonds (Table 1). Hence, channels are stabilized by 50% more hydrogen bonds than membrane coils. This statistically highly significant difference (t-test: 99.9% probability) is mainly caused by the higher number of Cα–H···O hydrogen bonds in channels. Almost two times as many Cα–H···O hydrogen bonds per residue are formed in channels (0.14) compared to membrane coils (0.08). In addition, the number of classical hydrogen bonds per residue is slightly increased in channels (0.15 vs. 0.12).
Another way to interpret these results is to say that approximately every third amino acid in channels and every fifth amino acid in membrane coils are involved in interhelical hydrogen bonding. Since approximately half of the residues of the investigated membrane proteins are exposed to lipid (23), this would mean that, on average, approximately every second buried amino acid is hydrogen bonded. It is important to note that we considered only interactions with both donor and acceptor atoms located inside the hydrophobic part of the lipid bilayer. In that region, the value of the dielectric constant is low and electrostatic interactions are of considerable strength (3). Thus, hydrogen bonding appears to play a key role in the stabilization of the tertiary structure of helical membrane proteins.
To estimate the strengths of the hydrogen bonds in the two data sets, the distances between acceptor and donor atoms were compared. At longer distances (2.6 Å) the number of classical hydrogen bonds begins to decrease in both data sets (Fig. 1). This distribution is also characteristic of hydrogen bonding in water-soluble globular proteins (19,32), suggesting that the number of classical hydrogen bonds does not depend on the surrounding milieu (5).
FIGURE 1.

Distribution (%) of different lengths (Å) centered at each value of classical (solid) and Cα–H···O (hatched) hydrogen bonds in channels (black columns) and membrane coils (white columns).
At distances shorter than 2.0 Å, we observe that the fraction of all hydrogen-bond types is higher in membrane coils than in channels. Conversely, the fraction of hydrogen bonds longer than 2.0 Å is considerably higher in channels. This trend is enhanced by the higher occurrence of Cα–H···O hydrogen bonds in channels, also starting at bond lengths of ∼2.0 Å. As a consequence, the fraction of helix-helix contacts stabilized by the shortest—and hence the strongest—hydrogen bonds is considerably lower in channels than in membrane coils (Fig. 1).
The analysis of interhelical hydrogen bonding of membrane proteins therefore suggests that channels are enriched in hydrogen bonds, and particularly in bonds with weak to medium strength.
Residues forming interhelical hydrogen bonds are conserved
The composition of residues forming interhelical hydrogen bonds varies significantly between channels and membrane coils. In channels, the smallest amino acids (Gly and Ala) form the majority of the interhelical hydrogen bonds (Fig. 2), whereas in membrane coils the medium polar amino acids Thr and Ser form most bonds. This difference is caused by the higher content of these residues in the respective protein types (2) as well as the higher likelihood that Gly will form Cα–H···O hydrogen bonds in channels (t-test: 95% probability). Specifically, 37% of Gly residues are involved in Cα–H···O hydrogen bonds in these proteins. By contrast, in membrane coils only 22% of the Gly residues contribute to Cα–H···O hydrogen bonds. Although the statistical significance is not very high, these results suggest that Gly may be of particular importance for stabilizing channels via hydrogen bonding.
FIGURE 2.

Proportion (%) of amino acid types involved in classical (solid) and Cα–H···O (hatched) hydrogen bonds in channels (black columns; total = 342) and in membrane coils (white columns; total = 358).
The variability of a certain amino acid position during evolution indicates whether it plays a vital role for the protein in terms of folding, stability, or function. The Shannon entropy, calculated from a sample of homologous protein sequences, is a well-established measure for this variability. Indeed, since exposed residues are generally less conserved than buried residues, this measure has been used to effectively predict buried versus exposed residues (30,31,33). Since the Shannon entropy largely depends on the quality of each alignment, the differential to an internal standard has to be used to compare the residual variability of different proteins (see Materials and Methods). Using this measure we found that buried residues in helical membrane proteins are conserved up to 25% more than exposed residues (Fig. 3). This is in agreement with previous analyses (29–31).
FIGURE 3.

Relative decrease in Shannon entropy of buried or hydrogen-bonded residues versus exposed residues in channels (black columns) and membrane coils (white columns). Error bars signify the 95% confidence levels.
When considering the Shannon entropy of residues that form hydrogen bonds, we find that in both protein types these residues are noticeably more conserved than buried residues as a whole (Fig. 3). In fact, hydrogen-bonding residues are ∼40% more conserved than exposed residues. We previously observed similar results in an analysis of classical hydrogen bonds in membrane proteins (5). The impact of Cα–H···O hydrogen bonds on the stability of helical membrane proteins, however, is somewhat more controversial (12,13). Therefore, it is important to note that, in both protein types, amino acids involved in this type of hydrogen bond are significantly more conserved (t-test: 99% probability) than buried residues in general. Indeed, in channels, residues taking part in Cα–H···O hydrogen bonding are slightly more conserved even than residues involved in classical hydrogen bonds (Fig. 3). The analysis of the per-residue variability, therefore, supports the notion that Cα–H···O hydrogen bonds are important for the stabilization and function of helical membrane proteins (9).
Hydrogen bonds in channels are frequently adjacent to putative internal water
It has previously been stated that the presence of hydrogen bonds correlates with higher density helix-helix contacts in all types of membrane proteins (17). This finding is only partially supported by our analysis. In membrane coils (which account for 10 of the 13 membrane protein structures considered in the analysis of Adamian and Liang (17)), this increase is indeed highly significant for those helix pairs that are stabilized by multiple hydrogen bonds (Fig. 4, AB). By contrast, in channels the packing density of hydrogen-bonded helix-helix contacts is only marginally increased compared to non-hydrogen-bonded pairs. We have pointed out recently that helix-helix contacts in membrane coils are packed more densely than in channels (6). This observation also holds for hydrogen-bonded helix pairs of membrane coils, which are packed significantly (t-test: 99.5% probability) more densely than those of channels.
FIGURE 4.

Comparison of packing densities of non-hydrogen-bonded and hydrogen-bonded helix pairs (A, classical; B, Cα–H···O; AB, both types) in channels (black columns) and membrane coils (white columns). Error bars signify the 99% confidence levels.
The lower packing density in channels may be due to the presence of cavities that contain internal waters (6). Since the stability of interhelical hydrogen bonds does not depend only on geometry but also on the chemical environment (3), we investigated the portion of hydrogen bonds that is located in close proximity to protein voids or pockets (see Materials and Methods). In membrane coils, only about one quarter (28%) of the hydrogen bonds are located closer than 6 Å to such cavities (Table 1). By contrast, one half (51%) of the hydrogen bonds of channels are found in close proximity to a cavity. The breaking of interhelical hydrogen bonds in channels during their function could therefore be facilitated by interactions with water molecules in nearby internal cavities.
In the Ca2+-ATPase, we found that 59% of the hydrogen bonds are in close proximity to water-sized cavities (Table 1, Noc/No). We detected a total of 130 different hydrogen bonds that are formed during the reaction cycle (data not shown). The only two classical and six Cα–H···O hydrogen bonds that are preserved during the entire reaction cycle are located in the anchor domain, in which the helices do not change conformation (24). Furthermore, water-sized cavities in the anchor domain are practically absent (only 1 of 38 cavities of the Ca2+-bound state is located there). Accordingly, in the Ca2+-ATPase, there seems to be a correlation between the presence of water-sized cavities and the ability of interhelical hydrogen bonds to break.
Hydrogen-bonding patterns correlate with the different architectures of channels and membrane coils
An analysis of helix crossing angles reveals that 69% of the hydrogen-bonded helix pairs of channels cross at right-handed angles (Table 1). This is significantly more than in membrane coils, where only 27% of the hydrogen-bonded helix pairs cross at right-handed angles (χ2: 99.9% probability). Another striking difference is that the helix crossing angles of hydrogen-bonded helices cluster around |τ| ≈ 40° for channels and |τ| ≈ 20° for membrane coils (Fig. 5). The preference for certain packing modes in helical membrane proteins has also been observed by others (5,34,35). However, the subdivision of the membrane proteins into channels and membrane coils shows that these differences are correlated with protein function.
FIGURE 5.

Proportion (%) of different helix crossing angles (τ) stabilized by classical (solid) and Cα–H···O (hatched) hydrogen bonds in channels (black columns) and membrane coils (white columns).
The right-handed and left-handed packing motifs prevalent in channels and membrane coils match the class a and class c “knobs-into-holes” packing motifs, respectively, derived from a helical lattice superposition model (36). Walther and co-workers stated in that work that there should be greater flexibility in class a helix packing since small amino acids are frequently involved in the packing core. Consistent with that hypothesis, the distribution of helix-helix crossing angles for the antiparallel right-handed (class a) helix pairs in channels is very broad (Fig. 5). Moreover, hydrogen-bonded helix pairs with |τ| ≈ 40° are preferred in both parallel and antiparallel helix-helix interactions of channels. By contrast, antiparallel (class c) helix-helix interactions are strongly preferred in membrane coils for which the distribution of helix crossing angles is extremely narrow.
The two different types of helix-helix interactions characteristic for channels and membrane coils are illustrated schematically in Fig. 6. The class c packing (36) of helices in membrane coils (|τ| ≈ 20°) results in a parallel arrangement of the helix turns and the clear preference for antiparallel helix packing motifs, where the bulky side chains of one helix interlock between two bulky residues of the other helix (Fig. 7). In the class a packing (36) of helices in channels (|τ| ≈ 40°), the turns of the contacting helices are positioned nearly orthogonal to each other, where the Cα-Cβ vectors of the buried residues point away from the packing core. The preferred burial of small residues at helix-helix interfaces of channels (23,37) together with the geometry of the packing of side chains in right-handed contacts (Fig. 7) allows the helices to arrange in both parallel and antiparallel motifs.
FIGURE 6.

Schematic representation of the left-handed knobs-into-hole packing of membrane coils (left) with parallel arrangement of the helix turns and the right-handed knobs-into-holes packing of channels (right) with orthogonal arrangement of the helix turns. The parallel arrangement of the helix turns and the resultant arrangements of side chains (Fig. 7) lead to a preference for antiparallel interactions in membrane coils. The dotted lines indicate the angle of the turn on the rear of the helices.
FIGURE 7.

Schematic representation of the knobs-into-holes packing of an antiparallel left-handed helix pair of membrane coils (PDB code: 1c3w) (left) and of a right-handed parallel helix pair of channels (1kpl) (right). The Cα-Cβ vectors of the core residues (in magenta) in right-handed interactions point away from the contact, where small residues are again preferred. The knobs are therefore articulately flattened, and the sterical restrictions imposed on the conformation of these helix pairs are limited. For the interdigitations of side chains, the conformation of left-handed helix pairs is sterically constrained and antiparallel arrangements are strongly preferred.
Finally, there is a clear preference for parallel class a packing (τ = −40° ± 10°) and antiparallel class c packing (τ = −160° ± 10°) in hydrogen-bonded helix pairs of channels and membrane coils, respectively, compared to non-hydrogen-bonded helix pairs. Exactly 50% of all hydrogen-bonded helix pairs of membrane coils are found in the antiparallel class c packing motif (Fig. 5). This is two times as many as in non-hydrogen-bonded helix pairs, where the same motif accounts for only 22% of the pairs (data not shown). In channels, three times as many (or 23%) of the hydrogen-bonded helix pairs are found in the parallel class a packing motif than in non-hydrogen-bonded helix pairs (8%). Our results therefore highlight that the different types of helix packing in membrane proteins correlate with the presence of hydrogen bonds.
DISCUSSION
Using a comprehensive analysis of geometrical features and hydrogen bonding, we found that there are significant differences between the two classes of membrane proteins studied here. In terms of helix packing, as shown also in our previous work (23), it is clear that helices in channels form right-handed contacts, whereas helices in membrane coils tend to form left-handed crossing angles. Interestingly, DeGrado and co-workers recently illustrated that if helix pairs of membrane proteins are categorized according to their structural similarity (35), clusters very similar to those obtained from the classifications in channels and membrane coils are achieved (Fig. 5). That is, the antiparallel left-handed helix pairs (typical for membrane coils) and the parallel and antiparallel right-handed helix pairs (typical for channels) form the biggest clusters of structurally similar helix pairs. The subdivisions of helical membrane proteins according to structural (35) or functional (Fig. 5) characteristics, therefore, refer to the same building blocks.
Here, we have expanded on this analysis to show how these different helix-packing arrangements may contribute to the function of helical membrane proteins. In particular, in right-handed packing arrangements the helices adopt a much broader range of crossing angles than in the left-handed packing motifs (Fig. 5). Moreover, due to the nature of the side-chain packing, these right-handed helix pairs—which predominate in channels—are not interlocked the same way as the left-handed helix pairs of membrane coils (Fig. 7). It therefore seems that there is a higher conformational freedom in the right-handed helix pairs that are clearly overrepresented in channels. Since the function of channels frequently involves large structural rearrangements, we propose that this higher conformational freedom is important for their function.
The low packing density of the helix pairs of channels (Fig. 4) may also facilitate greater conformational freedom. It has recently been proposed that the energetic barrier in separating the surfaces of two hydrophobic helices may be very high (38). This separation involves energetically highly unfavorable transition states, as void volumes form and steric dewetting occurs (39,40). This result suggests that a “tight” packing of transmembrane helices (4), i.e., via optimized van der Waals contacts, would therefore lead to rather rigid contacts. This energetic barrier, however, does not exist for helix pairs separated by individual water molecules (38). We have previously shown that solvated helix pairs are typically found in channels (6). It is therefore possible that the water that accumulates at the functionally important regions of channels is necessary to facilitate the required conformational switching.
One of the most significant findings of this analysis is that the different types of helix packing in membrane proteins correlate with the presence of hydrogen bonds of specific types (Fig. 5). This is notable since hydrogen bonds are particularly strong in the hydrophobic environment of the lipid bilayer. Specifically, in right-handed crossings the “knobs” of one helix that pack into the “holes” of the other helix are typically flattened, resulting in a close packing of backbones (Fig. 7). Channels are therefore clearly enriched in Cα–H···O hydrogen bonds (Table 1). Moreover, helix pairs stabilized by hydrogen bonds are also found to arrange in “knobs-onto-knobs” packing patterns (36) in which the side chains of adjacent helices are typically not interlocked. Such a motif where Gly and Ala residues of contacting helices are directly opposed is vital for the assembly and the opening mechanism of the mechanosensitive channel of small conductance (41). Indeed, the conservation of residues involved in hydrogen bonds (Fig. 3) argues for a central role of hydrogen bonds for the folding, stabilization, and function of helical membrane proteins in general (9,17,42).
Hydrogen bonds are frequently found close to water-sized cavities (Table 1). By providing alternative hydrogen-bonding partners, the nearby water may be important to stabilize transition states in which the hydrogen-bonding network is significantly altered, as shown here for the Ca2+-ATPase. The requirement for easy breaking and reformation of hydrogen bonds is also consistent with the fact that channels are enriched in hydrogen bonds of medium strengths, such as Cα–H···O hydrogen bonds, rather than the short, and thus presumably strong, hydrogen bonds found in membrane coils (Fig. 1). Accordingly, by the use of hydrogen bonds, channels may achieve a high flexibility while preserving energetic stability.
Finally, in addition to providing a functional explanation for the presence of specific helix packing and hydrogen-bonding types, the results provide new approaches for the prediction of the structure and function of helical membrane proteins. For example, the observation that certain helix-packing architectures are observed more frequently reduces the conformational search space required for low-resolution modeling. Furthermore, it will be interesting to see whether such helix-packing arrangements can be predicted via the detection of specific sequence patterns. Alternatively, the identification of water-containing cavities and particular structure motifs in proteins of known structure may enable identification of regions that are involved in conformational changes.
CONCLUSION
We have described several basic geometrical features characteristic of helical membrane proteins with different functions. Hydrogen bonds are clearly overrepresented in the structural motifs that are common to channels and tend to originate from highly conserved residues. The fact that channels are significantly enriched in Cα–H···O hydrogen bonds is consistent with the proposal that the enrichment of short interhelical distances between transmembrane helices (due to an enrichment of small amino acid residue types) may be important for energetic stabilization of membrane proteins (5,6,9). The presumed weakness of the hydrogen bonds in channels, along with the prevalence of nearby cavities that may contain water molecules, together support a hypothesis that breaking and reformation of different networks of hydrogen bonds may facilitate conformational changes in channels. As more structures of channels in different functional states become available, it should become clear whether this hypothesis holds as a general rule.
Acknowledgments
We thank Dr. Senes and Dr. Suhnel for kindly providing their programs and Dr. Honig and Dr. Holzhütter for helpful suggestions.
Funding was provided by National Science Foundation grant MCB-0416708, the Bundesministerium für Bildung und Forschung (Berlin Center for Genome Based Bioinformatics), the European Union (Programm zur Förderung von Forschung, Innovationen und Technologien), and the Deutsche Forschungsgemeinschaft (Sonderforschungsbereich 449, Z1).
Editor: Gregory A. Voth.
References
- 1.Wimley, W. C. 2003. The versatile beta-barrel membrane protein. Curr. Opin. Struct. Biol. 13:404–411. [DOI] [PubMed] [Google Scholar]
- 2.Hildebrand, P. W., R. Preissner, and C. Frömmel. 2004. Structural features of transmembrane helices. FEBS Lett. 559:145–151. [DOI] [PubMed] [Google Scholar]
- 3.White, S. H., and W. C. Wimley. 1999. Membrane protein folding and stability: physical principles. Annu. Rev. Biophys. Biomol. Struct. 28:319–365. [DOI] [PubMed] [Google Scholar]
- 4.Eilers, M., S. C. Shekar, T. Shieh, S. O. Smith, and P. J. Fleming. 2000. Internal packing of helical membrane proteins. Proc. Natl. Acad. Sci. USA. 97:5796–5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gimpelev, M., L. R. Forrest, D. Murray, and B. Honig. 2004. Helical packing patterns in membrane and soluble proteins. Biophys. J. 87:4075–4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hildebrand, P. W., K. Rother, A. Goede, R. Preissner, and C. Frommel. 2005. Molecular packing and packing defects in helical membrane proteins. Biophys. J. 88:1970–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bowie, J. U. 2005. Solving the membrane protein folding problem. Nature. 438:581–589. [DOI] [PubMed] [Google Scholar]
- 8.Baldwin, R. L. 2003. In search of the energetic role of peptide hydrogen bonds. J. Biol. Chem. 278:17581–17588. [DOI] [PubMed] [Google Scholar]
- 9.Senes, A., I. Ubarretxena-Belandia, and D. M. Engelman. 2001. The Cα–H···O hydrogen bond: a determinant of stability and specificity in transmembrane helix interactions. Proc. Natl. Acad. Sci. USA. 98:9056–9061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arbely, E., and I. T. Arkin. 2004. Experimental measurement of the strength of a Cα–H···O bond in a lipid bilayer. J. Am. Chem. Soc. 126:5362–5363. [DOI] [PubMed] [Google Scholar]
- 11.Ben-Tal, N., D. Sitkoff, I. A. Topol, A. Yang, S. K. Burt, and B. Honig. 1997. Free energy of amide hydrogen bond formation in vacuum, in water, and in liquid alkane solution. J. Phys. Chem. B. 101:450–457. [Google Scholar]
- 12.Yohannan, S., S. Faham, D. Yang, D. Grosfeld, A. K. Chamberlain, and J. U. Bowie. 2004. A Cα-H···O hydrogen bond in a membrane protein is not stabilizing. J. Am. Chem. Soc. 126:2284–2285. [DOI] [PubMed] [Google Scholar]
- 13.Mottamal, M., and T. Lazaridis. 2005. The contribution of Cα-H···O hydrogen bonds to membrane protein stability depends on the position of the amide. Biochemistry. 44:1607–1613. [DOI] [PubMed] [Google Scholar]
- 14.Faham, S., D. Yang, E. Bare, S. Yohannan, J. P. Whitelegge, and J. U. Bowie. 2004. Side-chain contributions to membrane protein structure and stability. J. Mol. Biol. 335:297–305. [DOI] [PubMed] [Google Scholar]
- 15.Zhou, F. X., M. J. Cocco, W. P. Russ, A. T. Brunger, and D. M. Engelman. 2000. Interhelical hydrogen bonding drives strong interactions in membrane proteins. Nat. Struct. Biol. 7:154–160. [DOI] [PubMed] [Google Scholar]
- 16.Gratkowski, H., J. D. Lear, and W. F. DeGrado. 2001. Polar side chains drive the association of model transmembrane peptides. Proc. Natl. Acad. Sci. USA. 98:880–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adamian, L., and J. Liang. 2002. Interhelical hydrogen bonds and spatial motifs in membrane proteins: polar clamps and serine zippers. Proteins. 47:209–218. [DOI] [PubMed] [Google Scholar]
- 18.Dawson, J. P., J. S. Weinger, and D. M. Engelman. 2002. Motifs of serine and threonine can drive association of transmembrane helices. J. Mol. Biol. 316:799–805. [DOI] [PubMed] [Google Scholar]
- 19.Baker, E. N., and R. E. Hubbard. 1984. Hydrogen bonding in globular proteins. Prog. Biophys. Mol. Biol. 44:97–179. [DOI] [PubMed] [Google Scholar]
- 20.Lindauer, K., C. Bendic, and J. Suhnel. 1996. HBexplore—a new tool for identifying and analysing hydrogen bonding patterns in biological macromolecules. Comput. Appl. Biosci. 12:281–289. [DOI] [PubMed] [Google Scholar]
- 21.Perozo, E., A. Kloda, D. M. Cortes, and B. Martinac. 2002. Physical principles underlying the transduction of bilayer deformation forces during mechanosensitive channel gating. Nat. Struct. Biol. 9:696–703. [DOI] [PubMed] [Google Scholar]
- 22.Cordes, F. S., J. N. Bright, and M. S. Sansom. 2002. Proline-induced distortions of transmembrane helices. J. Mol. Biol. 323:951–960. [DOI] [PubMed] [Google Scholar]
- 23.Hildebrand, P. W., S. Lorenzen, A. Goede, and R. Preissner. 2006. Analysis and prediction of helix-helix interactions in membrane channels and transporters. Proteins. 64:253–262. [DOI] [PubMed] [Google Scholar]
- 24.Toyoshima, C., and G. Inesi. 2004. Structural basis of ion pumping by Ca2+-ATPase of the sarcoplasmic reticulum. Annu. Rev. Biochem. 73:269–292. [DOI] [PubMed] [Google Scholar]
- 25.McDonald, I. K., and J. M. Thornton. 1994. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 238:777–793. [DOI] [PubMed] [Google Scholar]
- 26.Goede, A., R. Preissner, and C. Frömmel. 1997. Voronoi cell: new method for allocation of space among atoms: elimination of avoidable errors in calculation of atomic volume and density. J. Comput. Chem. 18:1113–1123. [Google Scholar]
- 27.Strait, B. J., and T. G. Dewey. 1996. The Shannon information entropy of protein sequences. Biophys. J. 71:148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dodge, C., R. Schneider, and C. Sander. 1998. The HSSP database of protein structure-sequence alignments and family profiles. Nucleic Acids Res. 26:313–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor, W. R., D. T. Jones, and N. M. Green. 1994. A method for α-helical integral membrane protein fold prediction. Proteins. 18:281–294. [DOI] [PubMed] [Google Scholar]
- 30.Fleishman, S. J., and N. Ben-Tal. 2006. Progress in structure prediction of alpha-helical membrane proteins. Curr. Opin. Struct. Biol. 16:496–504. [DOI] [PubMed] [Google Scholar]
- 31.Park, Y., and V. Helms. 2007. On the derivation of propensity scales for predicting exposed transmembrane residues of helical membrane proteins. Bioinformatics. 23:701–708. [DOI] [PubMed] [Google Scholar]
- 32.Preissner, R., U. Egner, and W. Saenger. 1991. Occurrence of bifurcated three-center hydrogen bonds in proteins. FEBS Lett. 288:192–196. [DOI] [PubMed] [Google Scholar]
- 33.Liao, H., W. Yeh, D. Chiang, R. L. Jernigan, and B. Lustig. 2005. Protein sequence entropy is closely related to packing density and hydrophobicity. Protein Eng. Des. Sel. 18:59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bowie, J. U. 1997. Helix packing in membrane proteins. J. Mol. Biol. 272:780–789. [DOI] [PubMed] [Google Scholar]
- 35.Walters, R. F., and W. F. DeGrado. 2006. Helix-packing motifs in membrane proteins. Proc. Natl. Acad. Sci. USA. 103:13658–13663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walther, D., F. Eisenhaber, and P. Argos. 1996. Principles of helix-helix packing in proteins: the helical lattice superposition model. J. Mol. Biol. 255:536–553. [DOI] [PubMed] [Google Scholar]
- 37.Kim, S., T. J. Jeon, A. Oberai, D. Yang, J. J. Schmidt, and J. U. Bowie. 2005. Transmembrane glycine zippers: physiological and pathological roles in membrane proteins. Proc. Natl. Acad. Sci. USA. 102:14278–14283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacCallum, J. L., M. S. Moghaddam, H. S. Chan, and D. P. Tieleman. 2007. Hydrophobic association of alpha-helices, steric dewetting, and enthalpic barriers to protein folding. Proc. Natl. Acad. Sci. USA. 104:6206–6210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheung, M. S., A. E. Garcia, and J. N. Onuchic. 2002. Protein folding mediated by solvation: water expulsion and formation of the hydrophobic core occur after the structural collapse. Proc. Natl. Acad. Sci. USA. 99:685–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Daggett, V., and A. Fersht. 2003. The present view of the mechanism of protein folding. Nat. Rev. Mol. Cell Biol. 4:497–502. [DOI] [PubMed] [Google Scholar]
- 41.Edwards, M. D., Y. Li, S. Kim, S. Miller, W. Bartlett, S. Black, S. Dennison, I. Iscla, P. Blount, J. U. Bowie, and I. R. Booth. 2005. Pivotal role of the glycine-rich TM3 helix in gating the MscS mechanosensitive channel. Nat. Struct. Mol. Biol. 12:113–119. [DOI] [PubMed] [Google Scholar]
- 42.Curran, A. R., and D. M. Engelman. 2003. Sequence motifs, polar interactions and conformational changes in helical membrane proteins. Curr. Opin. Struct. Biol. 13:412–417. [DOI] [PubMed] [Google Scholar]