

Abstract
A new and sensitive molecular probe, 2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridine (HPIP), for monitoring structural changes in lipid bilayers is presented. Migration of HPIP from water into vesicles involves rupture of hydrogen (H) bonds with water and formation of an internal H bond once the probe is inside the vesicle. These structural changes of the dye allow the occurrence of a photoinduced intramolecular proton-transfer reaction and a subsequent twisting/rotational process upon electronic excitation of the probe. The resulting large Stokes-shifted fluorescence band depends on the twisting motion of the zwitterionic phototautomer and is characterized in vesicles of dimyristoyl-phosphatidylcholine and in dipalmitoyl-phosphatidylcholine at the temperature range of interest and in the presence of cholesterol. Because the fluorescence of aqueous HPIP does not interfere in the emission of the probe within the vesicles, HPIP proton-transfer/twisting motion fluorescence directly allows us to monitor and quantify structural changes within bilayers. The static and dynamic fluorescence parameters are sensitive enough to such changes to suggest this photostable dye as a potential molecular probe of the physical properties of lipid bilayers.
Several fluorescent organic molecules have been described as molecular probes of structural and dynamical changes in lipid bilayers (1–3). These changes generally are observed by monitoring the variation of photophysical properties, such as fluorescence anisotropy and lifetime, spectral position, and fluorescence quantum yield of the dye within the bilayers (1–4). Recently, molecules showing excited-state proton-transfer (ESPT) reactions have been suggested as probes for the study of protein conformation and binding-sites (5–8). ESPT reactions are very fast elementary processes (9–11) involved in various area of physics, chemistry, and biology (6–18). Because of the observed large Stokes-shifted fluorescence of the proton-transfer band in these systems, such molecules might show some advantages with respect to others having a normal Stokes-shifted emission band.
Very recently, we have shown that the ESPT cycle can be coupled to a cis–trans isomerization reaction resulting in the formation of a rotamer of the phototautomer and opening a possible way to store information at a molecular level (15). We also reported on the photophysics and cyclodextrin inclusion of a dye, 2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridine (HPIP, Fig. 1). HPIP shows ESPT in fluid (16) and restricted media (cyclodextrins, CDx) (17). More specifically, we showed that the proton motion within the internal H bond of this dye in the first electronically excited state generates a zwitterionic phototautomer emitting a large Stokes-shifted fluorescence band. In rigid media [poly(methyl methacrylate) films or in CDx] the yellow-green fluorescence comes from the tautomer 2 or one of its close rotamers (17). In fluid media 2 gives rise to the red-emitting species 3 through a rotational process around the C2—C1′ single bond (Fig. 1). The spectral shift of the fluorescence going from 2 to 3 is about 70 nm (2,300 cm−1). In H-bonding solvents, the intramolecular H bond of the dye is broken and the resulting solvated species (such as 4 in water, Fig. 1) emits at around 390 nm (17).
Figure 1.

Molecular structures of the different species of HPIP in hydrophobic (1) and in aqueous (4) solutions. Tautomer 2 is the intramolecular proton-transfer phototautomer of 1, and 3 is a rotamer of 2.
We describe herein the quantitative study of the structure/dynamics of lipid bilayers of dimyristoyl-phosphatidylcholine (DMPC) and dipalmitoyl-phosphatidylcholine (DPPC) at different temperatures and in the presence of cholesterol based on the spectral shift of the ESPT fluorescence band and steady-state anisotropy and fluorescence lifetime measurements.
MATERIALS AND METHODS
Materials.
HPIP was synthesized and purified as described elsewhere (16). Spin labels 5-doxyl-stearic acid (5NS), 16-doxyl-stearic acid (16NS), and lipids DMPC, DPPC, and cholesterol were obtained from Sigma–Aldrich. The paraffin oil, PRIMOL ESSO 352 (viscosity at 20°C, η = 1.9 poise), free of fluorescent impurities in the wavelength range of interest (>330 nm), was used as received.
Preparation and Labeling of Spherical Lipid Bilayers.
Multilamellar vesicles were prepared by resuspending the appropriate amount of dried phospholipid in triple-distilled water (≈0.25 mg/ml) or in Tris⋅HCl buffer (pH = 7.0). This suspension was kept at a temperature above the lipid thermal phase transition and vortexed. Large, unilamellar vesicles (average diameter ≈90 nm) were prepared from a suspension of multilamellar vesicles by pressure extrusion through 0.1-μm polycarbonate filters. To prepare lipid bilayers containing cholesterol, dried cholesterol–phospholipid mixtures were dissolved in a small amount of chloroform and the solvent was evaporated with a stream of nitrogen.
The incorporation of the probe to the lipid bilayers was made by adding microliter amounts of a stock solution of 10−3 M HPIP in ethanol to the vesicle suspension (probe/lipid molar ratio ≈1/200).
Spectroscopic Measurements.
Absorption spectra were recorded on a Cary 3E spectrophotometer (Varian). Fluorescence spectra, quenching, and steady-state anisotropy [<r> = (I∥ − GI⊥ )/(I∥ + 2GI⊥), where G is correction factor (19), I∥, and I⊥ are the vertically and horizontally polarized emission intensities, respectively] measurements were carried out with a SLM-8000D fluorimeter (SLM–Aminco, Urbana, IL). Emission quantum yields were measured by reference to quinine sulfate in 0.05 M H2SO4 (φF = 0.51). Fluorescence lifetimes with ≈0.5-ns resolution were recorded with a time-correlated, single-photon counting system (16). The samples were excited at 357 nm with a nanosecond flash lamp (Edinburgh Instruments, EI199) filled with nitrogen. The emission was detected through a band-pass filter (Schott KV) at 500 nm. To eliminate rotational depolarization effects, polarizers were inserted in both channels. The fluorescence decays were analyzed by using a nonlinear least-squares iterative convolution method (19).
Quenching Measurements.
The transverse location of a fluorescent probe in lipid bilayers can be estimated by means of lipophilic spin probes that place a quenching nitroxide group in a defined location within the membrane (20). To this end, microliter aliquots of a 10−3 M stock ethanol solution of the nitroxide quenchers 5NS or 16NS were added to 3 ml of the vesicle suspension.
Quenching data were analyzed by means of Stern–Volmer plots of I0/I versus [Q]L, where I0 and I stand for the fluorescence intensities in the absence and in the presence of the quencher, respectively. [Q]L is the quencher (spin label) molar concentration within the lipid phase, given by
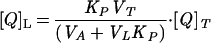 |
where Kp = [Q]L/[Q]A is the partition coefficient of the quencher between the lipid and the aqueous phases. [Q]T is the total concentration of the quencher in the total volume VT = VL + VA, where VL and VA are, respectively, the volume of lipid and aqueous phases (20). The values of Kp for 5NS and 16NS in the fluid phase are 8.9 × 103 and 9.73 × 103, respectively (21).
RESULTS AND DISCUSSION
HPIP Spectroscopy in Homogeneous Fluid Solutions.
The UV-visible absorption and emission spectra of HPIP in cyclohexane are characterized by a structured absorption band with maxima at 340 and 357 nm, and a large structureless emission band of around 590 nm (16). The absorption band is assigned to the first electronic transition of 1, whereas the emission is attributed to the rotamer 3 of the phototautomer 2, which results from an ESPT reaction of 1 (16, 17). Changing the temperature from 10°C (η = 1.3 × 10−2 poise) to 40°C (η = 0.65 × 10−2 poise) does not modify appreciably the position of either the absorption or fluorescence spectra in cyclohexane. The molar absorption coefficient determined at 340 nm is ≈22,000 M−1 ⋅ cm−1, and the emission quantum yield at room temperature is 0.071 (Table 1). An Arrhenius plot (not shown) of log(knr) versus 1/T (where knr is the nonradiative rate constant of the phototautomer) gives an activation energy to radiationless channels of 1.8 (±0.2) kcal/mol.
Table 1.
Photophysical parameters of the proton-transfer fluorescence band of HPIP in different media and temperatures
| Solvent | T, °C | λem, nm ±2 | <r>, ±0.003 | α1, ±0.05 | τ1, ns ±0.3 | α2, ±0.05 | τ2, ns ±0.2 | <τ>/ns | φ, ±0.007 |
|---|---|---|---|---|---|---|---|---|---|
| Cyclohexane | |||||||||
| 10 | 1 | 2.0* | 1.8 | 0.081 | |||||
| 15 | 1 | 1.9* | 1.7 | 0.076 | |||||
| 20 | 590 | 0 | 1 | 1.8 | 1.6 | 0.071 | |||
| 30 | 1 | 1.7* | 1.5 | 0.067 | |||||
| DMPC | |||||||||
| 15 | 560 | 0.243 | 0.64 | 1.1 | 0.36 | 4.0 | 3.0 | ||
| 18 | 563 | 0.235 | 0.65 | 1.2 | 0.35 | 3.6 | 2.7 | ||
| 29 | 587 | 0.158 | 0.70 | 0.8 | 0.30 | 1.4 | 1.0 | ||
| 39 | 590 | 0.82 | 0.6 | 0.18 | 1.1 | 0.7 | |||
| PMMA | |||||||||
| 20 | 535 | 0.335 |
λem, <r>, αi, τi, and φ are the wavelength of the emission maximum, the steady-state anisotropy, the preexponential factors, the fluorescence lifetimes, and the fluorescence quantum yield, respectively. <τ> = (τ12 + τ12)/(τ1 + τ2).
Estimated from the quantum yield.
In paraffin oil at 6°C (η = 4.6 poise), the absorption spectrum is similar to that obtained in cyclohexane but the emission band is centered at 578 nm instead of 590 nm. When the temperature is increased from 6 to 30°C, (the viscosity decreases from 4.6 to 1 poise), the emission maximum shifts to 586 nm, close to the value observed in cyclohexane at room temperature (Fig. 2A). For both media the spectral shift is linear in η/T and extrapolates to 590 nm as η → 0 (Fig. 2B). In the same interval of temperature, the spectral change in paraffin oil is much more important than that observed in cyclohexane. Changes in viscosity therefore are more influential than temperature in their effects on the spectral shift. The green fluorescence of HPIP in a polymeric film of poly(methyl methacrylate) (535 nm) at room temperature (17) agrees well with this conclusion.
Figure 2.

(A) Normalized absorption and emission spectra of 10−5 M HPIP in neutral water (—) and in paraffin oil (—) at 20°C. Excitation wavelength, 320 nm. (B) Plot of the wavelength of the maximum proton-transfer emission intensity of HPIP in paraffin oil (○) and in cyclohexane (•) as a function of η/T, and in DMPC at 30°C (▪) and 15°C (∗).
As mentioned, the intramolecular proton transfer of HPIP is prevented in aqueous solution, and absorption and emission spectra arise from 4 (Figs. 1 and 2A). The molar absorption coefficient in water at 320 nm is ≈15,000 M−1 ⋅ cm−1, and the estimated fluorescence quantum yield at room temperature is 0.05. The calculated activation energy to radiationless states is about 1.5 (±0.1) kcal/mol, a value lower than that determined for 3 in cyclohexane [1.8 (±0.2) kcal/mol].
HPIP in Lipid Vesicles.
Upon addition of DMPC vesicles to an aqueous solution of HPIP at 23°C, an emission band centered in the 550- to 600-nm region appears, whereas the 390-nm fluorescence band intensity decreases (Fig. 3). A similar behavior has been observed when adding CDx to an aqueous solution of HPIP (17), and the phenomenon was explained as the incorporation and subsequent conformational change (formation of O—H … N internal H bond) of the dye inside the CDx hydrophobic pocket. We conclude that in the presence of DMPC vesicles, part of 4 is incorporated into the lipid membrane and converted to 1. The excited state of structure 1 experiences an intramolecular ESPT reaction and produces an emission in the green-red region.
Figure 3.

Fluorescence excitation and emission spectra of HPIP in water containing large unilamellar vesicles of DMPC at 23°C. The excitation spectra were recorded observing at 400 (—) and 570 nm (—), and the emission spectrum was recorded exciting at 300 nm. The emission anisotropy, <r> (•), at 23°C also is displayed.
The phase-transition temperature of DMPC from the gel to the fluid phase is ≈23.5°C (23, 24). The incorporation of 4 into fluid-phase bilayers of this lipid is fast (a few minutes). To determine the distribution of the probe between the aqueous and the lipid phases, large unilamellar vesicles of DMPC were used. Because the fluorescence bands of HPIP in aqueous (390 nm) and lipid (590 nm) phases are well separated, the penetration of the probe into DMPC vesicles was quantified by the fluorescence technique of Sklar (22). The lipid concentration was varied whereas that of HPIP was kept constant (3 × 10−5 M). A plot of 1/IF versus mole H2O/mole lipid, where IF is the fluorescence intensity at 580 nm, gives a straight line (data not shown). The partition coefficient (Kp′) of the probe between the lipid and aqueous phases is 4 (±2) × 104 at 30°C. Kp′ = RL/RA, where RL is the ratio between the number of moles of HPIP in lipid phase and that of total moles of lipids, and RA is the ratio between the number of moles of HPIP in the aqueous phase and that of moles of water.
The degree of penetration of HPIP into lipid bilayers was studied by using the relative quenching of the proton-transfer fluorescence by the lipophilic spin probes 5NS and 16NS. The nitroxide groups are located at 12 Å (5NS) and 3 Å (16NS) from the center (25). Proton-transfer fluorescence is quenched by both 5NS and 16NS. The Stern–Volmer (SV) plot of the fluorescence intensity in large unilamellar vesicles of DMPC at 30°C is linear (Fig. 4) and gives the SV constant, KSV. The ratio KSV(16NS)/KSV(5NS) ∼ 5, indicating a deep penetration of the dye, species 1, within the membrane and away from the lipid/water interface. Precise information about the position and orientation of the buried HPIP cannot be extracted from the present experiments because the complex mechanism of quenching of 16NS (20, 25, 26) requires additional experiments (time-resolved) and theoretical considerations. The purpose of our steady-state data using 5NS and 16NS is to get a global picture of HPIP in DMPC. Based on the above ratio and from the molecular structure of the probe, we suggest that the long axis of the dye tends to be oriented parallel to the acyl chains. A population of 1 at the center of the vesicles, oriented perpendicular to the acyl chains, is not excluded, however.
Figure 4.

Stern–Volmer plots of the intramolecular proton-transfer fluorescence intensity in DMPC upon addition of 5NS and 16NS at 30°C; I0 is the intensity without the spin label.
Probing the Structural Change of DMPC and DPPC Bilayers.
When the temperature changes from 5 to 35°C, through the phase transition of DMPC (23.5°C), the emission band maximum shifts from 558 to 590 nm (Fig. 5), showing an abrupt change at ≈23°C (λmax = 580 nm). In the gel phase, HPIP presumably is entrapped between the rigid lipid chains, so the fluorescence at 558 nm is mainly a result of the photoproduct of the proton-transfer reaction. The local viscosities of the DMPC bilayer (related to the rate of chains motion) at 15, 25, and 31°C are 2.9, 0.46, and 0.34 poise, respectively (3). In the fluid phases, the fluorescence band (structure 3) exhibits maxima at 587 nm (25°C) and 589 nm (31°C). Both values agree with the linear behavior of Fig. 2B. Therefore, the plot in Fig. 2B could in principle be used as a calibration curve to determine viscosities in fluid-phase lipid membranes. However, at 15°C, the value of the viscosity extracted from this curve (0.9 poise) is lower than that reported (2.9 poise) (3). This might be explained by the fact that in gel phase not only the viscosity is responsible for the emission behavior of HPIP, but also the acyl chain order (related to the extent of chains motion) plays a role by hindering the internal rotation of the phototautomer, 2, around the C2—C1′ single bond.
Figure 5.

Spectral shift (wavelength of the maximum intensity) of the proton-transfer fluorescence band in DMPC vesicles as a function of temperature.
The steady-state anisotropy (<r>) of HPIP was recorded in large unilamellar vesicles of DMPC and DPPC (transition temperature, TT = 41°C). As Fig. 6 shows, the value of <r> decreases significantly when the temperature increases, with a sharp change at TT. It is remarkable that the pretransition (transition associated with a change from the tilted chains to a lamellar structure distorted by a ripple) temperature of DPPC (34°C) also is detected by the small change of the anisotropy around this temperature, showing that HPIP does not disrupt the overall structure of the lipid bilayers. The data were compared with the <r> values recorded in paraffin oil (Fig. 6). Under the viscous conditions of paraffin oil, the effect of temperature is similar to that observed for fluid phases of DMPC and DPPC. For instance, in DMPC at 23°C, where gel and fluid phases coexist, <r> decreases from 0.225 at 510 nm to 0.196 at 600 nm (Fig. 3). This indicates that excited 2 and 3 coexist at this temperature, and most probably emit from different environments, with different anisotropies. Structure 2 will emit from the gel phase, whereas 3 will fluoresce from the fluid environment. In the fluid phase of DMPC, <r> is constant with the emission wavelength (data not shown). In polymeric films of poly(methyl methacrylate) where reorientation of the fluorophore is prevented during the lifetime of the excited phototautomer, the steady-state anisotropy of the probe is 0.335, indicating that the direction of the absorption and emission transition moments of HPIP are not very different. The <r> values in the gel phase are lower and much more temperature-dependent than those obtained by using other fluorescent probes. This property makes HPIP a potential probe for following structural changes in ordered phases.
Figure 6.

Steady-state anisotropy (<r>) of the proton-transfer fluorescence in large unilamellar vesicles of DPPC (•) and DMPC (○) and in paraffin oil (∗) as a function of temperature.
The decay of the proton-transfer fluorescence intensity of HPIP in large unilamellar vesicles of DMPC at temperatures from 15 to 39°C (both gel and fluid phases) is biexponential (Table 1). The fluorescence lifetimes slowly decrease when the temperature increases, with an abrupt change at the transition temperature of the lipid. Assuming that the radiative lifetimes of 2 and 3 are similar, we can relate the preexponential factors (α1 and α2 in Table 1) to the populations of these fluorophores. Taking into account that lowering the temperature increases the viscosity and thus decreases the conversion of 2 to 3, the trend of the variation of the preexponential factors with the temperature suggests that the longest lifetime, ι2 (1–4 ns), could be associated with the emission of 2, whereas ι1 (0.6–1 ns) should be assigned to the fraction of 3. In lipid bilayers both excited species coexist even below the phase transition, although the fraction of excited 2 significantly increases in the gel phase. This accords with the spectral position of the emission band in gel phase (≈560 nm), which is intermediate between the wavelength for cyclohexane (590 nm) and that for polymeric films of poly(methyl methacrylate) (535 nm). Therefore, the rigid chains of the gel phase allow a twisting motion of 2 around the C2—C1′ bond and the partial reorientation of the molecule during the lifetime in the excited state. The relatively high value of <r> supports this conclusion (Table 1).
HPIP in Mixed DMPC/Cholesterol Bilayers.
HPIP was incorporated into large unilamellar vesicles of DMPC at different cholesterol concentrations ranging from 0 to 40 mol%. The steady-state fluorescence spectra and anisotropy measurements were carried out in these lipid mixtures at 15°C (gel phase) and at 30°C (fluid phase). In both phases, upon addition of cholesterol up to 30 mol%, the proton-transfer emission band shifts to short wavelengths (Fig. 7A). Further additions of cholesterol do not modify the spectral position. The effect is larger in the fluid phase than in the gel phase: with 30 mol% of cholesterol, the shifts are about 40 nm (fluid phase) and 20 nm (gel phase).
Figure 7.

Spectral shift (A) and <r> (B) measured at the wavelength of the maximum intensity of the proton-transfer fluorescence band in DMPC vesicles upon addition of cholesterol. χchol is the molar fraction of cholesterol.
The observed shift at 30°C is expected because it has been shown (27) that the presence of cholesterol in a fluid phase reduces the angular range for rotational motion of the acyl chains, without a substantial change of the lateral and rotational mobility of these chains. Internal rotation of excited 2 around the C2—C1′ bond might be prevented at an increased cholesterol concentration. At 15°C, the phenomenon is much more interesting. It is known that the presence of cholesterol in a gel phase induces local disorder and increases the rotational motion of the lipid chains (27), and thus emission from 3 mostly is expected. However, we observed that upon addition of cholesterol, the maximum of the proton-transfer band shifts from 570 to 548 nm, indicating that the fluorescence is mainly a result of the emission of 2.
Several physical techniques have provided strong evidence for phase separation in model lipid membranes containing cholesterol concentrations above 6–8% in both gel and fluid phases (28). Thus, HPIP could preferentially migrate into cholesterol-rich regions and be located in immediate contact with the rigid steroid nucleus where its mobility should be prevented as in rigid media. Then 3 would not be formed and emission would occur mostly from 2 in both fluid and gel phases. Therefore, the steady-state anisotropy of the probe was measured (Fig. 7B). In the fluid phase, the anisotropy increases with the concentration of cholesterol, tending to plateau at 30 mol%. In the gel phase, addition of 10 mol% of cholesterol induces an increase of <r> from 0.245 to 0.275. Further addition above 10 mol% is less effective to change the steady-state anisotropy. This result accords with the behavior of the spectral shift of the proton-transfer fluorescence band and agrees with the explanation regarding the proximity of the dye to the steroid nucleus.
In conclusion, we have shown that the photoinduced intramolecular proton-transfer reaction coupled to the internal twisting motion of HPIP can be used to follow the structural changes of lipid bilayers. The highly Stokes-shifted fluorescence band, coming solely from the dye inside the vesicle, the use of static emission parameters (spectral shift and anisotropy) as well as dynamic measurements (fluorescence lifetime), and the photostability of the dye and its sensitivity to the space offered by the environment make this combination of proton transfer and twisting motion suitable for exploring the structure/dynamics of microdomains in lipid media. We are continuing such studies by using HPIP and other similar dyes.
Acknowledgments
Helpful discussion with Dr. A. U. Acuña is gratefully acknowledged. We also thank Dr. F. Amat-Guerri for his help in the synthesis of HPIP. We also thank the referees for their suggestions and editing of the manuscript. This work was supported by the University of Castilla–La Mancha and the Dirección General de Investigación Cientifica y Ténica (Spain), project PB93–0126. C.R.M. was supported by a Dirección General de Investigación Cientifica y Ténica research contract.
ABBREVIATIONS
- ESPT
excited-state proton transfer
- CDx
cyclodextrins
- HPIP
2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridine
- DMPC
dimyristoyl-phosphatidylcholine
- DPPC
dipalmitoyl-phosphatidylcholine
- 5NS
5-doxyl-stearic acid
- 16NS
16-doxyl-stearic acid
References
- 1.Sklar L A, Hudson B S, Simoni R D. Biochemistry. 1977;16:819–828. doi: 10.1021/bi00624a002. [DOI] [PubMed] [Google Scholar]
- 2.Lentz B R. Chem Phys Lipids. 1989;50:171–190. [Google Scholar]
- 3.Mateo C R, Souto A A, Amat-Guerri F, Acuña A U. Biophys J. 1996;71:2177–2191. doi: 10.1016/S0006-3495(96)79419-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holmes A S, Birch D J S, Sanderson A, Aloisi G G. Chem Phys Lett. 1997;266:309–316. [Google Scholar]
- 5.Neyroz P, Franzoni L, Menna C, Spisni A, Masotti L. J Fluorescence Chem. 1996;6:127–138. doi: 10.1007/BF00732052. [DOI] [PubMed] [Google Scholar]
- 6.Sytnik A, Gormin D, Kasha M. Proc Natl Acad Sci USA. 1994;91:11968–11972. doi: 10.1073/pnas.91.25.11968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sytnik A, Kasha M. Proc Natl Acad Sci USA. 1994;91:8627–8630. doi: 10.1073/pnas.91.18.8627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sytnik A, Litvinyuk Y. Proc Natl Acad Sci USA. 1996;93:12959–12963. doi: 10.1073/pnas.93.23.12959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Douhal A, Lahmani F, Zewail A H. Chem Phys. 1996;207:477–498. [Google Scholar]
- 10.Douhal A, Kim S K, Zewail A H. Nature (London) 1995;378:260–263. doi: 10.1038/378260a0. [DOI] [PubMed] [Google Scholar]
- 11.Douhal A. Science. 1997;276:221–222. [Google Scholar]
- 12.Pimentel G C, McClellan A L, editors. The Hydrogen Bonds. San Francisco: Freeman; 1960. [Google Scholar]
- 13.Schowen R L. Angew Chem Int Ed Engl. 1997;36:1434–1438. [Google Scholar]
- 14.Watson J D H, Crick F H C. Nature (London) 1953;171:337–738. [Google Scholar]
- 15.Guallar V, Moreno M, Lluch J M, Amat-Guerri F, Douhal A. J Phys Chem. 1996;100:19789–19794. [Google Scholar]
- 16.Douhal A, Amat-Guerri F, Acuña A U. J Phys Chem. 1995;99:76–80. [Google Scholar]
- 17.Douhal A, Amat-Guerri F, Acuña A U. Angew Chem Int Ed Engl. 1997;36:1514–1516. [Google Scholar]
- 18.Douhal A, Amat-Guerri F, Lillo M P, Acuña A U. J Photochem Photobiol A. 1994;79:127–138. [Google Scholar]
- 19.Mateo C R, Lillo M P, González-Rodriguez J, Acuña A U. Eur Biophys J. 1991;20:41–52. doi: 10.1007/BF00183278. [DOI] [PubMed] [Google Scholar]
- 20.Castanho M, Prieto M E. Biophys J. 1995;69:155–168. doi: 10.1016/S0006-3495(95)79886-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wardlaw J R, Sawyer W H, Ghiggino K P. FEBS Lett. 1987;231:20–24. doi: 10.1016/0014-5793(87)80502-1. [DOI] [PubMed] [Google Scholar]
- 22.Sklar L A. Mol Cell Biochem. 1980;32:169–177. doi: 10.1007/BF00227444. [DOI] [PubMed] [Google Scholar]
- 23.Ladbrooke B D, Chapman D. Chem Phys Lipids. 1969;3:304–356. doi: 10.1016/0009-3084(69)90040-1. [DOI] [PubMed] [Google Scholar]
- 24.Mabrey S, Sturtevant J M. Proc Natl Acad Sci USA. 1976;73:3862–3866. doi: 10.1073/pnas.73.11.3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chattopadhyay A, London E. Biochemistry. 1987;26:39–45. doi: 10.1021/bi00375a006. [DOI] [PubMed] [Google Scholar]
- 26.Castanho M, Prieto M, Acuña A U. Biochim Biophys Acta. 1996;1279:164–168. doi: 10.1016/0005-2736(95)00251-0. [DOI] [PubMed] [Google Scholar]
- 27.Yeagle P L. Biochim Biophys Acta. 1985;822:267–287. doi: 10.1016/0304-4157(85)90011-5. [DOI] [PubMed] [Google Scholar]
- 28.Almeida P F F, Vaz W L C, Thompson T E. Biochemistry. 1992;31:6739–6747. doi: 10.1021/bi00144a013. [DOI] [PubMed] [Google Scholar]