

Abstract
Metabotropic glutamate receptors (mGluRs) share a common molecular morphology with other G protein–linked receptors, but there expression throughout the mammalian nervous system places these receptors as essential mediators not only for the initial development of an organism, but also for the vital determination of a cell’s fate during many disorders in the nervous system that include amyotrophic lateral sclerosis, Parkinson’s disease, Alzheimer’s disease, Huntington’s disease, Multiple Sclerosis, epilepsy, trauma, and stroke. Given the ubiquitous distribution of these receptors, the mGluR system impacts upon neuronal, vascular, and glial cell function and is activated by a wide variety of stimuli that includes neurotransmitters, peptides, hormones, growth factors, ions, lipids, and light. Employing signal transduction pathways that can modulate both excitatory and inhibitory responses, the mGluR system drives a spectrum of cellular pathways that involve protein kinases, endonucleases, cellular acidity, energy metabolism, mitochondrial membrane potential, caspases, and specific mitogen-activated protein kinases. Ultimately these pathways can converge to regulate genomic DNA degradation, membrane phosphatidylserine (PS) residue exposure, and inflammatory microglial activation. As we continue to push the envelope for our understanding of this complex and critical family of metabotropic receptors, we should be able to reap enormous benefits for both clinical disease as well as our understanding of basic biology in the nervous system.
Keywords: Akt, Alzheimer’s disease, amyotrophic lateral sclerosis, apoptosis, caspases, endonucleases, epilepsy, erythropoietin, Huntington’s disease, microglia, mitogen-activated protein kinase, multiple sclerosis, Parkinson’s disease, PKA, PKC, stroke, trauma
THE G-PROTEIN mGluR FAMILY
As one of the primary excitatory neurotransmitters in the mammalian central nervous system (CNS), glutamate plays an important role during both cellular function and cellular injury. Until the mid 1980s, the actions of glutamate in mammalian brain were thought to be mediated exclusively through the activation of glutamate-gated channels named ionotropic glutamate receptors. Yet, further studies provided evidence for the existence of another family of glutamate receptors which was directly coupled to GTP-binding regulatory proteins. Early work, such as the observation of glutamate induced phospholipase C generation in neurons, indicated that glutamate had more complex roles that could not be accounted for by only N-methyl-D-aspartate (NMDA), alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), or kainate receptor families (Sladeczek, F et al., 1985). Subsequently, it became evident that a new class of glutamate receptors, termed metabotropic glutamate receptors (mGluRs), was coupled to effector systems through GTP-proteins (G-proteins) (Houamed, KM et al., 1991, Prezeau, L et al., 1992, Yuzaki, M and Mikoshiba, K, 1992). The first mGluR, now generally termed mGluR1a, was cloned in1991 by a functional expression screening procedure (Houamed, KM et al., 1991, Masu, M et al., 1991). Since molecular cloning has preceded pharmacological characterization in the identification of novel mGluRs, the mGluRs are numbered following the order in which their cDNAs have been cloned.
G-proteins consist of heterotrimeric proteins that contain three subunits termed α, β, and γ. Since a total of forty-six G-proteins have been identified with twenty-seven classified as Gα, five classified as Gβ, and fourteen classified as the Gγ, a variety of heterotrimeric combinations can be formed that may produce a broad spectrum of G-protein signaling (Albert, PR and Robillard, L, 2002). Activation of G-protein-coupled receptors results in the dissociation of the heterotrimer of the G-protein into its α and βγ subunits, which can then bind to a variety of effector molecules. A particular G-protein may be responsible for the modulation of a series of signal transduction pathways. The G-protein βγ has been associated with many effector molecules including adenylate cyclase (AC), phospholipase C-β (PLC-β), mitogen-activated protein kinases (MAPKs), and phosphoinositide 3 kinase (PI 3-K) (Hur, EM and Kim, KT, 2002).
G-protein-coupled receptors can be divided into three major subfamilies based on nucleotide and amino acid sequence similarity. Family A consists of the rhodopsin/adrenergic receptors and is characterized by the presence of a restricted number of conserved residues (Asp-Arg-Tyr). Family B consists of peptide hormone and neuropeptide receptors that are characterized by a large extracellular NH2 terminus containing six cysteine residues. Metabotropic glutamate receptors share a common molecular morphology with other G protein–linked receptors. The mGluRs are part of family C of G-protein-coupled receptors, which also includes gamma aminobutyric acid (GABA) receptors and ionotropic calcium receptor transmission. Unlike the other G-protein-coupled receptors families, mGluRs contain a long NH2 terminal chain and couple to G-proteins through their second intracellular loop rather than the third intracellular loop of the receptor.
The mGluRs are classified into three major groups based on sequence homology, G-protein coupling specificity, and agonist selectivity. Group I mGluRs (including mGluR1 and 5) couple preferentially to Gq to stimulate PLC-β. Activation of PLC-β results in the generation of two second messengers, inositol-1, 4, 5-triphosphate (IP3) and diacylglycerol (DAG), to mobilize intracellular calcium and activate protein kinase C (PKC). Group I mGluRs also can activate AC via coupling to Gs to result in an increase in cAMP (Francesconi, A and Duvoisin, RM, 1998). In contrast to group I mGluRs, group II mGluRs (including mGluR2 and 3) and group III mGluRs (including mGluR4, 6, 7, and 8) are negatively coupled to AC to reduce the amount of intracellular cAMP. In addition, activation of group II/III can modulate activity of extracellular signal-regulated protein kinases (ERKs) and PI 3-K (Ferraguti, F et al., 1999).
CELLULAR EXPRESSION AND FUNCTION OF mGluRs
Metabotropic glutamate receptors are expressed throughout the mammalian CNS (Table 1). In neuronal populations, the mGluR system participates in the processing of cognition, sensory, motor, and olfactory information. For example, mGluRs are present in the cerebral cortex (Lopez-Bendito, G et al., 2002), cerebellar neurons (Berthele, A et al., 1999), striatal neurons and in the spinal cord (Aronica, E et al., 2001). In the hippocampus, a more restricted expression of the receptor subtypes mGluR1b, mGluR2/3, mGluR4a and mGluR5 has been demonstrated (Blumcke, I et al., 1996). The receptor subtype mGluR4 also has a distinct distribution in the thalamus, hypothalamus, and caudate nucleus (Makoff, A et al., 1996a). In the retina, mGluR6 is expressed (Vardi, N et al., 2000). In contrast, mGluR3 is expressed throughout the brain with dense expression in neurons of the cerebral cortex, caudate-putamen, thalamus, and cerebellum (Makoff, A et al., 1996b).
Table 1.
Expression of mGluRs in Mammalian Brains
| mGluR Group | Tissue Expression | Cellular Expression | |
|---|---|---|---|
| Group I | mGluR1 | Cerebral cortex, striatum, medulla oblongata, basal ganglia, substantia nigra, and spinal cord hippocampus, thalamus, cerebellum | Neurons (postsynaptic membrane)
Microglia (mGluR5) Astrocytes (mGluR5) Endothelial cell |
| mGluR5 | Striatum (caudate nuclei), medulla oblongata (autonomic nuclei), basal ganglia, substantia nigra hippocampus, thalamus, cerebellum | ||
| Group II | mGluR 2/3 | Cerebral cortex, striatum, medulla oblongata (autonomic nuclei), olfactory bulb, corpus striatum, basal ganglia, caudate-putamen, thalamus, cerebellum, hippocampus | Neurons (pre and postsynaptic membrane)
Microglia (mGluR5) Astrocytes (mGluR5) |
| mGluR4 | Cerebral cortex, striatum, basal ganglia, hippocampus (CA2), thalamus, and cerebellum | Neurons (pre and postsynaptic membrane)
Microglia Astrocytes |
|
| Group III | MGluR6 | Retina | |
| mGluR7 | Medulla oblongata, thalamus, and cerebellum | ||
| mGluR8 | Olfactory bulb, pontine gray, thalamus, piriform cortex (strong expression), cerebral cortex, hippocampus, cerebellum, mammillary body (weak expression) | ||
The mGluR receptors are distributed in specific subcellular regions and alter their expression during development of the nervous system. Group I mGluRs including mGluR1a and mGluR5 predominantly exist on the post-synaptic membranes of the glutamatergic synapse junctions (Lujan, R et al., 1997). Yet, in the initial postnatal period, mGluR1a and mGluR5 can be found in proximal dendrites and the cell somata. With age, these receptors become densely distributed in the distal part of dendrites to participate in synaptic function (Liu, XB et al., 1998). Group II mGluRs (mGluR2/3) are present primarily in astrocytes surrounding the neuronal somata and synapses. A less dense population of group II mGluRs is also located in presynaptic axon terminals. The distribution pattern of mGluR2/3 is believed to be consistently maintained during postnatal development (Liu, XB et al., 1998). Of the group III mGluRs, mGluR6 is initially distributed in both the neuronal soma and dendrites in rat retinal bipolar cells, but later redistributes to postsynaptic sites (Nomura, A et al., 1994). Presynaptic expression is more common for the mGluR7 subtypes (Shigemoto, R et al., 1996). In group III mGluRs, mGluR4, mGluR6, and mGluR8 also have been identified in microglia (Taylor, DL et al., 2003) and astrocytes (Geurts, JJ et al., 2005). The presence of these receptors in a variety of cell types may be responsible for protection of neuronal cell populations (Yao, HH et al., 2005).
Little is known of the role of the mGluR system in the vascular system, but new investigations are beginning to provide evidence for a vital function for the mGluR system in brain endothelial cells. Initial work has outlined the expression of mGluRs in cultured rat cerebrovascular endothelial cells (Krizbai, IA et al., 1998) and in cardiac cells (Gill, SS et al., 1999). Further studies have now demonstrated not only the expression of specific group I mGluRs in cerebral endothelial cells, but also the potential for the mGluR system to protect against apoptotic injury (Chong, ZZ et al., 2003d, Lin, SH and Maiese, K, 2001a, Maiese, K et al., 2003b). In addition, mGluR1. mGluR2/3, mGluR4a, mGluR5, and mGluR7 have been demonstrated in the meningeal microvasculature (Gillard, SE et al., 2003). Interestingly, agents such as nicotine can inhibit the expression of mGluRs in cardiac tissue (Hu, D et al., 2002).
The G-protein-coupled receptor family is the largest family of cell-surface molecules. These receptors are activated by a wide variety of stimuli, including neurotransmitters, peptides, hormones, growth factors, ions, lipids, and light. The mGluR system is one of the principal members in this receptor family and provides an important function as presynaptic auto-receptors that mediate feedback inhibition of glutamate release in a wide variety of brain regions. One of the mechanisms of glutamate inhibition is thought to result from the down-regulation of voltage-activated calcium channels which are necessary for synaptic vesicle exocytosis (Anwyl, R, 1999). The mGluR system also is a critical mediator for the modulation of intracellular signal cascades and physiological function. Interaction among each G-protein subunit, such as -α, -β and -γ subunits, can stimulate effector molecules including adenylyl/guanylyl cyclases, phosphodiesterases, phospholipases, and phosphoinositide 3-kinase (PI 3-K) resulting in the modulation of other second messengers. Several second messenger systems, such as cAMP, cGMP, inositol (3, 4, 5)- trisphosphate (Ins(1,4,5)P3), arachidonic acid, phosphatidic acid, and calcium are active participants in these signal transduction cascades (Marinissen, MJ and Gutkind, JS, 2001).
ACTIVE INTEGRATION AND TRANSITION OF THE mGluR SIGNAL TRANSDUCTION PATHWAYS DURING DEVELOPMENT AND CELLULAR FUNCTION
During the development of the nervous system, mGluRs serve to modulate neuronal transmission at excitatory and inhibitory synapses. In addition, the mGluR system is required for the modulation of intracellular calcium homeostasis. Immature and developing neurons require higher intracellular calcium concentrations than their mature counterparts to facilitate neuronal survival, synapse formation, dendrite growth, and other cellular functions (Spitzer, NC et al., 1995). As a result, the modulation of intracellular calcium by the mGluR system has proven to be necessary for neuronal development, such in the cochlear nucleus magnocellular neurons (Zirpel, L et al., 2000) and in maturing hippocampal neurons (Maiese, K et al., 1999a). Furthermore, group I mGluR1 facilitates L-type voltage-dependent calcium channels currents through PKC (Endoh, T, 2004) and group II mGluRs can control calcium flux in the suprachiasmatic nucleus that may oversee circadian function (Haak, LL, 1999). In astrocytes, both group I and group II mGluRs have been associated with the generation of calcium oscillations (Zur Nieden, R and Deitmer, JW, 2005).
Redistribution of the expression of mGluRs also appears to be necessary for proper nervous system development. For example, redistribution of mGluR6 in rat retinal bipolar cells occurs from somatic and dendritic sites to restricted localization at postsynaptic sites (Nomura, A et al., 1994). In the mouse thalamus, subcellular relocalization of group I mGluRs also occurs during postnatal development (Liu, XB et al., 1998). In addition to redistribution of receptors, functional changes in the mGluR system also may occur during development. The generation of second messengers, such as cAMP, has been reported to vary under mGluR control during critical periods of ocular dominance and plasticity (Reid, SN et al., 1996).
In the adult mammalian, electrophysiological studies of mGluRs have shown that activation of mGluRs may lead to a range of cellular changes such as the inhibition of calcium and potassium currents, mediation of slow excitatory postsynaptic potential, and the presynaptic inhibition of transmitter release (Anwyl, R, 1999, Losonczy, A et al., 2003, White, AM et al., 2003). In particular, activation of group I mGluRs can contribute to slow-onset potentiation in the hippocampal region of CA1 (Manahan-Vaughan, D, 1997). Activation of postsynaptic group I mGluRs also suppresses transmission at excitatory synapses onto CA1 pyramidal cells (Watabe, AM et al., 2002). In addition, group I mGluRs have been shown to alter calcium homeostasis and trigger calcium-sensitive gene transcription in striatal neurons (Mao, L and Wang, JQ, 2003). Group II mGluRs have a significant role in the modulation of GABA afferent inhibition in the ventrobasal thalamus that controls functions of sleep, arousal, and sensation (Salt, TE and Turner, JP, 1998). Both group I and II receptors may be required for the activity-dependent regulation of ribosomes during auditory function (Nicholas, AH and Hyson, RL, 2004). High-frequency stimulation appears to be particularly dependent upon group I and group II metabotropic glutamate receptors (Ene, FA et al., 2003). Group III mGluRs have a greater role in motor function through the inhibition of GABA and glutamate transmission in the substantia nigra, pars reticulata (Wittmann, M et al., 2001), and the periaqueductal grey area (Marabese, I et al., 2005).
The ubiquitous distribution of glutamatergic synapses in the brain offers a great potential for mGluRs to modulate global CNS function. Behavioral and physiological studies have demonstrated that mGluRs can regulate fast synaptic transmission, changes in synaptic plasticity, and the modification of the calcium currents (Dietrich, D et al., 1997, Maiese, K et al., 1999a). During memory imprinting, group I mGluRs which are juxtaposition to NMDA receptors can modulate the potentiation of NMDA receptor activity to influence both long-term potentiation and long-term depression (Chen, J et al., 2000, Manahan-Vaughan, D, 1997). Yet, multiple members of the mGluR system may be required for memory formation (Holscher, C et al., 1999) or memory restoration following an ischemic insult (Wisniewski, K and Car, H, 2002). Activation of mGluRs also can lead to depolarization-induced synapsin I phosphorylation, a process that may be involved in synaptic vesicle exocytosis in visceral sensory neurons (Hay, M et al., 2000).
THE mGluR SYSTEM INFLUENCES A WIDE SPECTRUM OF BRAIN DISORDERS
Significant attention has focused on the protective role of the mGluR system in the CNS. Several observations support a prominent role for the mGluR system for normal physiology as well as during a variety of disease states. If one examines the diverse role of endocannabinoids, a class of lipids which includes amides, esters and ethers of long chain poly-unsaturated fatty acids, in neurovascular biology (Battista, N et al., 2004), recent work has shown that mGluRs are intimately associated with the production of endocannabinoids (Jung, KM et al., 2005). In regards to disease states, the modulation of the mGluR system has been proposed for the treatment of bipolar disease since these receptors may modulate signal transduction during affective disorders (Quiroz, JA et al., 2004). In other diseases that involve neurodegeneration, such as trisomy 21, a congenital disorder with mental impairment, enhanced expression of the mGluR subtype 5 has been reported (Oka, A and Takashima, S, 1999). Furthermore, dysfunctional signaling in the mGluR system may be responsible for other types of abnormal cognitive development found in disorders such as fragile X mental retardation (Bear, MF, 2005).
In terminal disorders such as amyotrophic lateral sclerosis (ALS), the mGluR1a receptor may be offer endogenous cellular protection, since surviving motor neurons from the spinal cord of ALS patients maintain mGluR1a at levels comparable to that from controls (Valerio, A et al., 2002). In epilepsy, group I metabotropic receptors can lead to prolonged epileptiform discharges (Wong, RK et al., 2005) and enhanced expression of group II and III metabotropic receptors in the hippocampus of patients with epilepsy has been observed (Tang, FR and Lee, WL, 2001a, Tang, FR et al., 2001b). Yet, other studies support a protective role for mGluRs in epilepsy, since activation of group II mGluRs has been shown to prevent seizures in experimental animal models (Folbergrova, J et al., 2005). Reported abnormal expression of mGluR1 has been reported in Pick’s disease (Dalfo, E et al., 2005). Modulation of synaptic activity by mGluRs during other chronic neuronal disorders, such as in Huntington’s disease, has been suggested to alter cellular susceptibility to injury (Calabresi, P et al., 1999). In particular, huntingtin protein, which is associated with excitotoxic death of striatal neurons, can lead to the desensitization of mGluR1 that may promote cell death (Anborgh, PH et al., 2005). Changes in expression patterns of group III mGluRs also have been observed during inflammatory disorders, such as multiple sclerosis (Geurts, JJ et al., 2005, Mouzaki, A et al., 2004), and during acute central (Mills, CD et al., 2001) or peripheral nerve trauma (Anneser, JM et al., 2000).
In regards to Parkinson’s disease (PD), group I mGluRs have been associated with the basal ganglia synaptic transmission through several mechanisms. Activation of group I mGluRs can invoke excitatory postsynaptic current in dopaminergic neurons (Shen, KZ and Johnson, SW, 1997), facilitate dopamine release from nigrostriatal terminals (Campusano, JM et al., 2002, Shimazoe, T et al., 2002), and modulate GABA release (Saransaari, P and Oja, SS, 2001). These results suggest that group I mGluRs agonists may have a therapeutic potential in PD. Yet, other functions of the group in the subthalamic nucleus appear to increase the excitation output in the basal ganglia. Group I mGluRs (mGluR5) can lead to an excitatory drive in subthalamic nucleus neurons (Awad, H et al., 2000). Stimulation of striatal group I mGluRs inhibits striatal projection neuronal activity, while stimulation of subthalamic metabotropic glutamate receptors increases subthalamic nucleus activity. As a result, group I mGluR antagonists also have been proposed for the treatment of PD (Golembiowska, K et al., 2002).
Group II mGluRs are presynaptically localized on subthalamic nucleus terminals. Activation of these receptors inhibits excitatory transmission at subthalamic nucleus synapses (Bradley, SR et al., 2000). As a result, selective agonists of group II mGluRs can reduce excitatory drive through the indirect pathway, which is enhanced in PD, and provide an entirely new approach to the treatment of PD. For example, in a haloperidol-induced rat model of PD, a selective agonist of group II mGluRs, LY354740 ((+)-2-aminobicyclo[3.1.0.]hexane-2,6,-dicarboxylic acid), has been demonstrated to reverse parkinsonian muscle rigidity and catalepsy (Bradley, SR et al., 2000, Wolfarth, S et al., 2000). In another model of PD through application of reserpine in rats, the parkinsonian akinesia was relieved by injection of the group II mGluR receptor agonist, (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine (Dawson, L et al., 2000). In addition, group II mGluR receptor agonists may protect through the release of trophic factors (Matarredona, ER et al., 2001) or potentially modulate extracellular glutamate uptake (Yang, YL et al., 2005). Taken together, agonists for group II mGluRs may be promising agents in the management of PD.
Group III mGluRs also play an important role in the synaptic transmission in the basal ganglia circuits. Of these, mGluR7 is presynaptically localized in the striatum, the globus pallidus, and the substantia nigra pars reticulata (Kosinski et al., 1999) and modulates synaptic transmission in both the direct and indirect pathways. Activation of mGluR7 inhibits GABA as well as glutamate transmission in the substantia nigra pars reticulata (Wittmann, M et al., 2001). As a result, modulation of excitatory and inhibitory synaptic transmission by mGluR7 may yield no alteration in the output of the substantia nigra pars reticulata. In contrast to mGluR7, mGluR4 appears to be more selectively localized in striatopallidal synapses and inhibits synaptic activity through the indirect pathway (Bradley, SR et al., 1999). Consequently, the selective agonists for mGluR4 may provide an alternate therapy for the treatment of PD.
Alzheimer’s disease (AD) is characterized by two pathologic hallmarks that consist of extracellular plaques of amyloid-β peptide aggregates and intracellular neurofibrillary tangles composed of hyperphosphorylated microtubular protein tau (Chong, ZZ et al., 2005d, Panchal, M et al., 2004). The β-amyloid deposition that constitutes the plaques is composed of a 39–42 amino acid peptide (Aβ), which is the proteolytic product of the amyloid precursor protein (APP) (Chong, ZZ et al., 2005d). Large soluble fragments (APPs) that are the result from the cleavage of APP within its Aβ domain are secreted into the extracellular medium. Overexpression of APP can accelerate Aβ secretion which can form insoluble amyloid aggregates contributing to the development of AD (Selkoe, DJ, 2001).
In AD, a down regulation of mGluR binding sites occurs (Dewar, D et al., 1991). In addition, group I mGluRs are desensitized in the front cortex in AD patients and these modifications have been correlated with the progression of AD (Albasanz, JL et al., 2005). The mGluRs also have been coupled to acceleration of APP processing. Activation of mGluR1 in human glioma and neuroblastoma cells favor the processing of APP into nonamyloidogenic APPs resulting in the reduction of Aβ formation (Lee, RK et al., 1995) as well as modulation of APP secretion (Croucher, MJ et al., 2003). In hippocampal neurons, the APPs release is also accelerated by stimulation of mGluRs, but not with ionotropic glutamate receptors (Lee, RK et al., 1995). In brain cortical and hippocampal slices, the stimulation of group I and group II mGluRs by trans-(1S,3R)-1-amino-1,3-cyclopentane dicarboxylic acid (ACPD) can increase the release of APPs. The process can be blocked by the administration of (±)-α-methyl-4-carboxyphenylglycine, a non-selective antagonist of group I and group II mGluRs (Ulus, IH and Wurtman, RJ, 1997). The regulation of APP processing by mGluRs appears to be dependent on the activation of PKC, since inhibition of PKC activity can block alterations in secretion of APPs in response to the activation of mGluRs (Lee, RK et al., 1995, Ulus, IH et al., 1997).
Given the ability of mGluRs to mediate the APP metabolism, agents that modulate the activation of mGluRs may be potentially useful in the therapy of AD. Furthermore, the involvement of mGluRs in synaptic plasticity and in the induction of long term potentiation and depression is believed to be necessary for the processing of learning and memory (Riedel, G, 1996). This implies that activation of mGluRs may improve the cognitive functions in AD patients and may spur future clinical trials for mGluR agonists in the treatment of AD.
A BALANCE BETWEEN CELLULAR PROTECTION AND TOXICITY IN THE mGluR SYSTEM
In experimental models, activation of mGluR subtypes usually can protect cells against several types of insults (Baskys, A and Blaabjerg, M, 2005). During traumatic brain injury models, activation of group II mGluRs can reduce neuronal loss (Zwienenberg, M et al., 2001). Stimulation of mGluRs also efficiently prevents motor neuron degeneration during kainate toxicity (Pizzi, M et al., 2000). A number of studies have shown that NMDA excitotoxicity can be prevented or significantly reduced by mGluR activation (Aarts, MM and Tymianski, M, 2003, Blaabjerg, M et al., 2003, Lafon-Cazal, M et al., 1999, Leker, RR and Shohami, E, 2002). Activation of group II receptors during both excitotoxic in vitro and ischemic in vivo conditions can prevent neuronal degeneration and limit infarct size (Cai, Z et al., 1999, Kingston, AE et al., 1999). Neuronal protection through the activation of the mGluR has been extended to several other models of neuronal injury such as glucose deprivation (Sagara, Y and Schubert, D, 1998), anoxia (Lin, SH and Maiese, K, 2001b, Maiese, K et al., 1996, Vincent, AM and Maiese, K, 2000), hypoxia (Cai, Z et al., 1999) hypoxia/hypoglycemia (Sabelhaus, CF et al., 2000), oxygen glucose deprivation (Kalda, A et al., 2000), nitric oxide (NO) exposure (Chong, ZZ et al., 2005a, Maiese, K et al., 1996, Vincent, AM et al., 1997), hydrogen peroxide (Zhu, P et al., 2004), hypokalemia (Borodezt, K and D’Mello, SR, 1998), and hyperglycemia (Berent-Spillson, A et al., 2004, Spillson, AB and Russell, JW, 2003), and oxidative stress (Deng, W et al., 2004). Further analysis has illustrated that protection by the mGluR system is mediated through more downstream pathways of cellular injury. For example, mGluR activation prevents, and in some cases, reverses genomic DNA degradation (Baskys, A et al., 2005, Blandini, F et al., 2004, Chong, ZZ et al., 2005c, Lin, SH et al., 2001b), modulates endonuclease activation (Vincent, AM et al., 1999), and maintains cellular membrane asymmetry (Chong, ZZ et al., 2005a, Vincent, AM et al., 2000). Cytoprotection by the mGluR system is believed to act at or below the level of free radical generation and oxidative stress (Maiese, K et al., 1996, Sagara, Y et al., 1998, Vincent, AM et al., 1997). More current work has suggested that the mGluR offers similar protective capacity to the vascular system by preventing endothelial cell DNA degradation and inhibiting a thrombotic state through the maintenance of membrane asymmetry (Lin, SH et al., 2001a, Lin, SH et al., 2001b).
Although activation of the mGluR system usually provides a supportive environment for cell survival, both the nature and extent of the cellular injury as well as the state of the cell itself may determine whether activation or inhibition of the mGluR system is ultimately required for cellular protection. As a result, some studies provide evidence that antagonism of mGluRs may be beneficial (Agrawal, SK et al., 1998, Caruso, C et al., 2004, Faden, AI et al., 2001, Henrich-Noack, P and Reymann, KG, 1999, Lu, J et al., 2003). For example, inhibition of mGluR activity during the progression of a toxic insult in some experimental models may subsequently improve neuronal survival (Pellegrini-Giampietro, DE et al., 1999, Shuaib, A and Kanthan, R, 1997). Moreover, the acute and chronic application of mGluR antagonists may have different therapeutic efficacy. For example, in a rat model of PD, it is chronic rather than acute treatment with a mGluR5 antagonist that can reverse akinetic deficits (Breysse, N et al., 2002). Furthermore, changes in the cellular environment, such as decreased intracellular calcium release, may allow antagonism of the mGluR system to exert cytoprotection (Maiese, K et al., 1999a).
Yet, the role of the mGluR system during cellular compromise is not always clear and can differ among cell systems. It should be noted that mGluRs in the CNS is complex in nature and may, at times, have adverse consequences or ineffective results (Kermer, P et al., 2001). Down-regulation of the mGluR system has been suggested to lead to the generation of post-injury pain following spinal cord injury during both pharmacological (Abraham, KE et al., 2001) and knockdown studies (Fundytus, ME et al., 2001). In other scenarios, activation of the mGluR system may potentiate activity of the capsaicin receptor and contribute to hyperalgesia (Tominaga, M et al., 2001).
TARGETING INJURY MECHANISMS TO MAINTAIN NEURONAL AND VASCULAR SURVIVAL
Several mechanisms for the cytoprotective effects of the mGluR system in both neuronal and vascular systems have been proposed. Initial work focused on the potential of the mGluR system to modulate the release of intracellular calcium in a variety of animal models (Lachica, EA et al., 1995, Stefani, A et al., 1994, Yoshino, M and Kamiya, H, 1995). Subsequent investigations examined the ability of the mGluR system to directly inhibit excitatory transmission (Bonci, A et al., 1997) and offer protection through the inhibition of NMDA activity (Lafon-Cazal, M et al., 1999) and free radical generation (Maiese, K et al., 1995, Maiese, K et al., 1996, Vincent, AM et al., 2000). These investigations have matured to identify the ability of the mGluR system to regulate specific cellular and molecular targets that may ultimately determine the fate of a cell (Chong, ZZ et al., 2003d). These pathways include apoptosis or programmed cell death (PCD) induction, protein kinase activity, intracellular pH, endonuclease activity, mitochondrial membrane potential, cysteine protease generation, and mitogen activated protein kinase activity.
The mGluR System and Programmed Cell Death
Cellular self-destruction known as PCD or apoptosis plays a significant role in both neuronal and vascular degeneration. Following one of the original morphological descriptions of PCD (Kerr, JF et al., 1972), several general biochemical and physiologic features of PCD have been identified. These processes include the loss of plasma membrane asymmetry, nuclear chromatin condensation, and DNA fragmentation. This active process is recognized as a central pathway that can lead to a cell’s demise in a variety of tissues and has recently been identified in organisms as diverse as plants (Hatsugai, N et al., 2004). PCD consists of two independent processes that involve membrane phosphatidylserine (PS) exposure and DNA fragmentation (Maiese, K et al., 2004b). Apoptotic injury is believed to contribute significantly to a variety of disease states that especially involve the nervous system such as ischemic stroke, AD, PD, and spinal cord injury (Chong, ZZ and Maiese, K, 2004b, Li, F et al., 2004). Outside of the nervous system, such as during cardiovascular injury, PCD also may be a significant precipitant of cell death. Ischemic-reperfusion injury can lead to apoptosis in cardiomyocytes (Cai, Z et al., 2003).
As an early event in the dynamics of cellular apoptosis, the biological role of membrane PS externalization can vary in different cell populations (Chong, ZZ et al., 2005c, Doonan, F and Cotter, TG, 2004). In some cell systems, PS may be required for embryogenesis (Bose, J et al., 2004). Yet, in mature tissues, membrane PS externalization is a signal for the phagocytosis of cells (Hong, JR et al., 2004). In the nervous system, cells expressing externalized PS may be removed by microglia (Chong, ZZ et al., 2003c, Li, F et al., 2004). An additional role for membrane PS externalization in the vascular cell system is the activation of coagulation cascades. The externalization of membrane PS residues in endothelial cells (ECs) can promote the formation of a procoagulant surface (Chong, ZZ et al., 2004a). Loss of vascular endothelial cells can lead to thrombosis, immune dysfunction, and inflammation throughout the vascular system (Maiese, K et al., 2005b).
The cleavage of genomic DNA into fragments is considered to be a delayed event that occurs late during apoptosis (Dombroski, D et al., 2000, Jessel, R et al., 2002, Kang, JQ et al., 2003b, Maiese, K and Vincent, AM, 2000b). Several enzymes responsible for DNA degradation have been differentiated based on their ionic sensitivities to zinc (Torriglia, A et al., 1997) and magnesium (Sun, XM and Cohen, GM, 1994). Calcium, a critical independent component that can determine cell survival (Weber, J, 2004), also may determine endonuclease activity through calcium/magnesium - dependent endonucleases such as DNase I (Madaio, MP et al., 1996). Other enzymes that may disassemble DNA include the acidic, cation independent endonuclease (DNase II) (Torriglia, A et al., 1995), cyclophilins (Montague, JW et al., 1997), and the 97 kDa magnesium -dependent endonuclease (Pandey, S et al., 1997). In the nervous system, three separate endonuclease activities are present that include a constitutive acidic cation-independent endonuclease, a constitutive calcium/magnesium-dependent endonuclease, and an inducible magnesium dependent endonuclease (Vincent, AM and Maiese, K, 1999). The physiologic characteristics of the magnesium dependent endonuclease, such as a pH range of 7.4–8.0, a dependence on magnesium, and a molecular weight of 95–108 kDa, are consistent with a recently described constitutive 97 kDa endonuclease in non-neuronal tissues.
Exposure to reactive oxygen species can lead to PCD through multiple cellular pathways. Oxidative stress, such as NO or hydrogen peroxide, results in nuclei condensation and DNA fragmentation (Chong, ZZ et al., 2003b, Goldshmit, Y et al., 2001, Pugazhenthi, S et al., 2003, Vincent, AM et al., 1999). NO produces apoptotic death in hippocampal and dopaminergic neurons (Chong, ZZ et al., 2003a, Sharma, SK and Ebadi, M, 2003, Vincent, AM and Maiese, K, 1999, Witting, A et al., 2000). Injury during NO exposure can result not only from a loss of balance between itself and the superoxide ion (Blandini, F et al., 2004), but also NO can become synergistic with hydrogen peroxide to render neurons more sensitive to oxidative injury (de la Monte, SM et al., 2003, Wang, JY et al., 2003). Hydrogen peroxide also results in neuronal injury through impaired mitochondrial function and increased levels of pro-apoptotic gene products, such as CD95/Fas (de la Monte, SM et al., 2000, Pugazhenthi, S et al., 2003, Vaudry, D et al., 2002). Externalization of membrane PS residues also occurs in neurons during anoxia (Chong, ZZ et al., 2002a), NO exposure (Chong, ZZ et al., 2003f), or during the administration of agents that induce the production of reactive oxygen species, such as 6-hydroxydopamine (Salinas, M et al., 2003).
Since DNA fragmentation and PS externalization can each lead to cellular injury, the mechanisms that induce these individual processes may be important targets for neuroprotective strategies. The mGluR system can provide cytoprotection at two distinct levels during PCD. Protection against PCD during activation of the mGluR system is broad in nature by addressing the separate components of PCD. Agonism of each group of the mGluR system can prevent the early exposure of membrane PS residues and also inhibit the later stages of genomic DNA destruction (Lin, SH et al., 2001a, Lin, SH et al., 2001b, Maiese, K et al., 2000a, Vincent, AM et al., 2000). Post-treatment paradigms also have demonstrated a “window of opportunity” to prevent the further progression of membrane PS residue exposure once an injury has been initiated. Interestingly, the maintenance of membrane PS asymmetry also provides more long-term protection by inhibiting the destruction of cells by phagocytes (Savill, J, 1997) and maintaining a normal anticoagulant state in endothelial cells (Bombeli, T et al., 1997, Lin, SH et al., 2001b).
The mGluR System Modulates Microglial Proliferation and Activation
Activation of mGluRs not only can directly preserve neuronal or vascular function, but also may prevent microglial neurotoxicity, such as during Aβ application (Taylor, DL et al., 2003). In addition, during condition that involves microglial activation, diminished activity of mGluRs may prove useful for cellular protection. For example, inhibition of group II mGluRs can attenuate microglial activation and subsequent neurotoxicity during toxic stimuli such as chromogranin A (Taylor, DL et al., 2003), a protein up-regulated in Alzheimer disease.
Although usually maintained in a quiescent state, microglia can become activated during a variety of pathological insults (Chong, ZZ et al., 2004b). Activated microglia may lead to cellular damage during oxidative stress (Sankarapandi, S et al., 1998). The secretion of cytokines by microglia also may represent another source of cytotoxicity for this cell population. Microglia produce a variety of cytokines in response to toxic stimulation, such as interleukins and tumor necrosis factor-α (TNF-α). TNF-α production by microglia may be linked to neurodegeneration by increasing the sensitivity of neurons to free radical exposure. For example, Aβ induced microglial secretion of TNF-α during Aβ deposition lead to the neuronal expression of inducible nitric oxide synthase, peroxinitrite production, and neuronal apoptosis (Combs, CK et al., 2001).
Once activated, microglia function to remove cellular debri and apoptotic cells through phagocytosis. Several potential mechanisms may regulate the phagocytosis of cells that have entered the apoptotic pathway. Generation of annexin I and membrane PS exposure appears to be necessary to tether an apoptotic cell with a phagocyte (Arur, S et al., 2003). Secreted factors by either apoptotic or phagocytic cells, such as milk fat globule EGF8 (Hanayama, R et al., 2002), fractalkine (Hatori, K et al., 2002), and lipid lysophosphosphatidylcholine (Lauber, K et al., 2003) also have been shown to assist with the phagocytic removal of injured cells.
Yet, a common denominator that appears to be critical for the removal of apoptotic cells by phagocytic sentries is the translocation of membrane PS residues from the inner cellular membrane to the outer surface (Fadok, VA et al., 2001, Kang, JQ et al., 2003b, Maiese, K et al., 2000b). In cells that are without injury, the phospholipids of the plasma membrane are distributed asymmetrically with the outer leaflet of the plasma membrane consisting primarily of choline-containing lipids, such as phosphatidylcholine and sphingomyelin, and the inner leaflets consisting of aminophospholipids that include phosphatidylethanolamine and PS. The disruption of membrane phospholipid asymmetry leads to the externalization of membrane PS residues and serves to identify cells for phagocytosis (Chong, ZZ et al., 2003d, Hoffmann, PR et al., 2001, Kang, JQ et al., 2003b, Maiese, K and Chong, ZZ, 2003a).
Protein kinase B, also known as Akt, may provide another clue to the regulation of microglial activation. Akt can modulate the spatial regulation of actin assembly, suggesting a relationship between Akt and the coordination of cytoskeletal organization (Lemmon, MA et al., 2002). Furthermore, through a series of investigations, Akt has recently been shown to be a necessary component for the modulation of membrane PS externalization and prevent microglial activation (Kang, JQ et al., 2003a, Kang, JQ et al., 2003b). Initially, microglial activation and proliferation have been shown to occur during oxidative stress that includes free radical exposure (Chong, ZZ et al., 2003b). In addition, through the use of an antibody to the PS receptor, it has been demonstrated that membrane PS residue exposure is both necessary and sufficient to induce microglial activation and proliferation (Chong, ZZ et al., 2003b, Kang, JQ et al., 2003a, Kang, JQ et al., 2003b). Furthermore, media taken from cells that overexpress active, phosphorylated Akt during cellular injury leads to a significant reduction in microglial activation and proliferation (Kang, JQ et al., 2003a, Kang, JQ et al., 2003b). Taken together, this series of studies illustrate that Akt can directly modulate microglial activation and proliferation through the modulation of membrane PS exposure on cells and conceivably prevent the shedding of membrane PS residues that is known to occur during apoptosis (Simak, J et al., 2002).
Given the evidence that loss of cellular membrane asymmetry and exposure of membrane PS residues is necessary and sufficient to result in microglial activation, we have investigated the effect of mGluRs on microglial activation and its activity of phagocytosis. Activation of group I mGluRs can not only prevent neuronal membrane PS exposure, but also inhibit microglial activation by decreasing the expression of proliferating cell nuclear antigen (PCNA) and uptake of bromodeoxyuridine (BrdU) that results through conditioned neuronal media following NO exposure (Fig. (1) and (Fig. (3)). Through the continuous assessment of individual neurons in real time, activation of mGluRs was documented to block neuronal PS exposure and prevented subsequent neuronal cell engulfment by microglia seeking “PS tagged” neurons. Furthermore, maintenance of cellular integrity and inhibition of microglial activity by mGluR activation was dependent upon the activation of Akt1 (Chong, ZZ et al., 2005a).
Fig. (1).
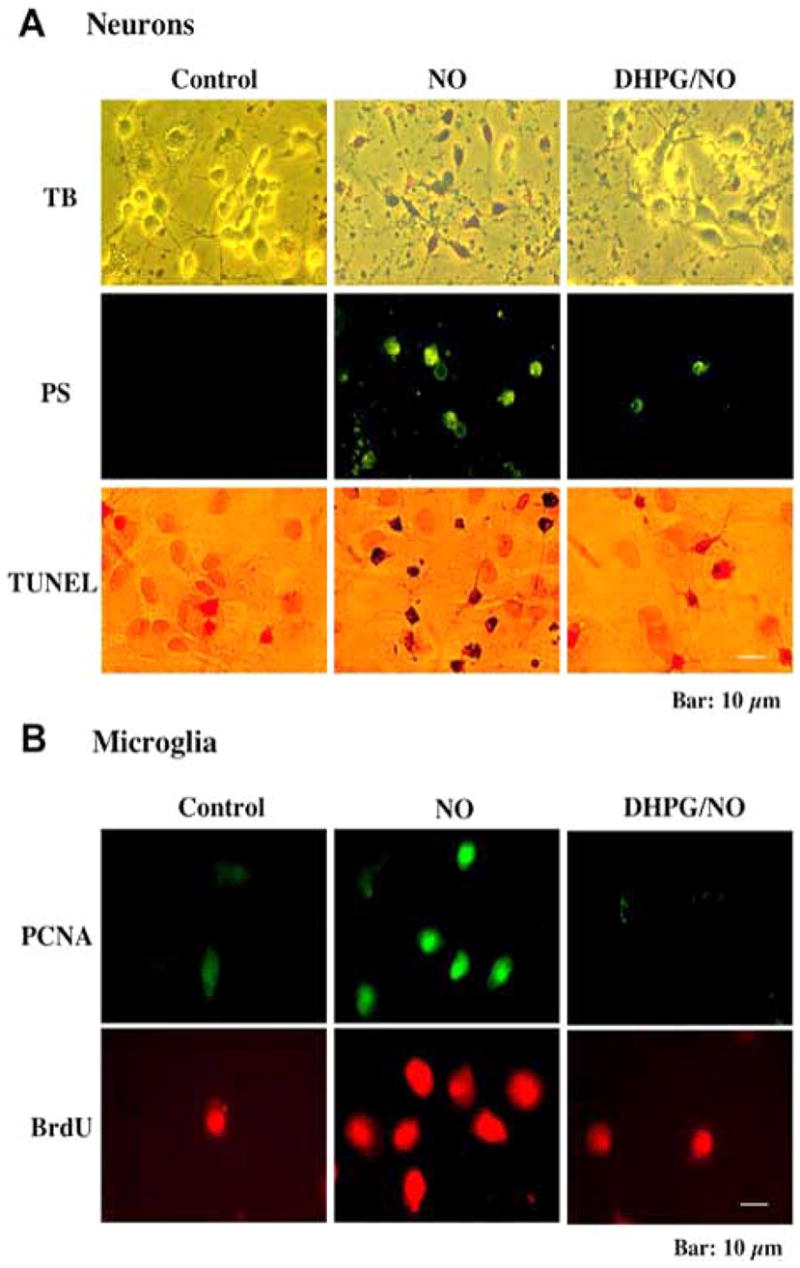
Activation of group I mGluRs by 3,5-dihydroxyphenylglycine (DHPG) maintains genomic DNA integrity and membrane phosphatidylserine (PS) asymmetry and prevents microglial activation. (A) Representative images illustrate cell membrane disruption with trypan blue staining, DNA fragmentation with terminal deoxynucleotidyl transferase nick end labeling (TUNEL), and phosphatidylserine (PS) exposure with annexin V phycoerythrin labeling in hippocampal neurons 24 hours following nitric oxide (NO) exposure (NOC-9, 300 μM). Pretreatment with the group I agonist DHPG (750 μM) 1 hour prior to the NO insult results in a significant reduction in trypan blue staining, DNA fragmentation, and membrane PS exposure. (B) Representative images of microglia are shown that illustrate increased micoglial activity assessed by proliferating cell nuclear antigen (PCNA) expression or the uptake of bromodeoxyuridine (BrdU) following the application of neuronal media that was exposed to either NO (NOC-9, 300 μM) or NO (NOC-9, 300 μM) plus DHPG. Administration of DHPG (750 μg/ml) one hour prior to neurons exposed to NO subsequently prevented PCNA expression and BrdU uptake in microglia illustrating that mGluR1 activation can block inflammatory microglial activation.
Fig. (3).
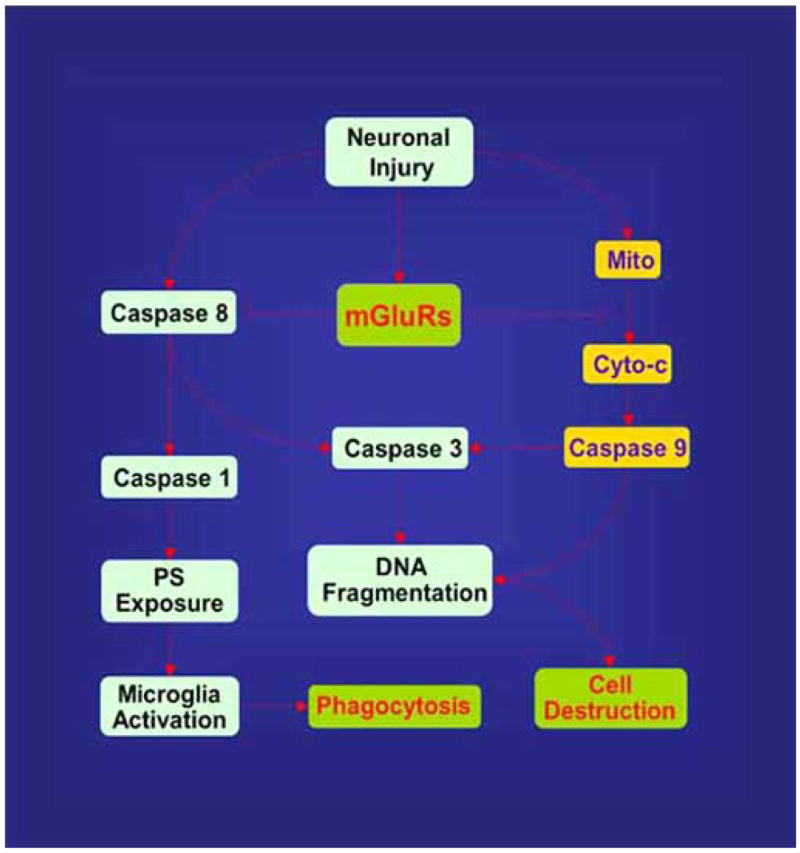
Metabotropic glutamate receptors (mGluRs) prevent apoptotic neuronal injury, caspase induction, and microglial activation. Activation of mGluRs maintains mitochondrial membrane potential (Mito) to prevent the release of cytochrome c (Cyto c) and subsequent caspase activation. With the blockade of caspase activity, apoptotic DNA fragmentation and membrane phosphatidylserine (PS) exposure is prevented. The ability of mGluRs to block microglia activation relies upon the prevention of membrane phosphatidylserine (PS) exposure.
The “ABCs” of the mGluR System
Modulation of protein kinase A (PKA) by the mGluR system is one potential pathway that may improve synaptic plasticity (Yamamoto, M et al., 2005) and offer cytoprotection during toxic cellular insults (Maiese, K et al., 1993a, Maiese, K et al., 1993b) (Fig. (2)). The mGluR system employs PKA activation for the regulation of memory retrieval (Szapiro, G et al., 2000, Vianna, MR et al., 2004) and long-term depression (Huang, LQ et al., 1999). During paradigms of cellular injury, activation of PKA can prevent the progression of PCD in a number of cell types, including neurons, neutrophils, and smooth muscle cells (Maiese, K et al., 1993b, Orlov, SN et al., 1999, Rossi, AG et al., 1995). In addition, loss of PKA activity during toxic insults can promote the progression of PCD (Findik, D et al., 1995, Maiese, K et al., 1993b, Nishio, E and Watanabe, Y, 1997). Protection by PKA is believed to reside upstream from the inhibition of caspase 3 - like activity (Parvathenani, LK et al., 1998). Furthermore, PKA has been shown to phosphorylate Bad, a member of Bcl-2 protein family, which can prevent the induction of cell injury (Lizcano, JM et al., 2000). In the mGluR system, the subtype mGluR4 requires the activation of PKA to prevent cellular injury following acute neurodegenerative insults (Maiese, K et al., 1996).
Fig. (2).
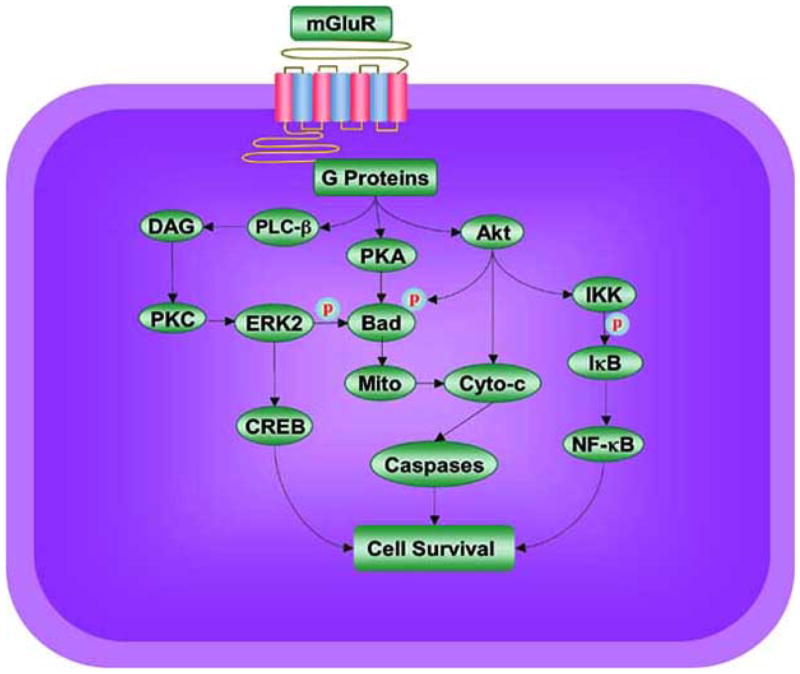
Potential signal transduction pathways of metabotropic glutamate receptors (mGluRs) that may foster cellular protection. mGluRs employ G-protein βγ to activate phospholipase β (PLC-β), diacylglycerol (DAG), and phosphoinositide 3 kinase (PI 3-K). These pathways lead to the activation of protein kinases A (PKA), B (Akt), and C (PKC). PKA has been shown to phosphorylate (p) Bad, a member of Bcl-2 protein family, which can prevent the induction of cell injury. Akt provides an anti-apoptotic survival signal through the phosphorylation and inactivation of Bad and stimulation of NF-kappaB (NF-κB) activity. Akt can activate IκB kinase (IKK) that can precipitate the phosphorylation (p) and degradation of IκB. This is followed by liberation of free NF-κB to promote cell survival. In regards to PKC, mGluRs can activate ERK2 through PKC. ERKs also may employ phosphorylation (p) of the pro-apoptotic protein Bad and induction of pro-survival gene expression via the cAMP responsive element-binding (CREB) protein dependent pathway to lead to cellular protection. mGluRs also preserve mitochondrial membrane potential (Mito) to block cytochrome c (Cyto-c) release and caspase activation that ultimately will lead to cellular demise.
Protein kinase B (Akt) is a common mediator of cell survival in a variety of circumstances via its anti-apoptotic effects (Chong, ZZ et al., 2005b). Increased activity of Akt can provide protection against neuronal and vascular injury. Maximal activity of Akt is achieved through phosphorylation by phosphoinositide-dependent kinase 1 at Ser473 to confer protection against genomic DNA degradation (Chong, ZZ et al., 2002a, Wick, A et al., 2002, Yamaguchi, A et al., 2001) and membrane PS exposure (Chong, ZZ et al., 2002a, Chong, ZZ et al., 2003b, Kang, JQ et al., 2003b). Akt activity also can be facilitated by a 90 kDa heat shock protein (Hsp90). Hsps are characterized by their mass in kilodaltons, are induced in response to heat in essentially all organisms, and are highly conserved between different species. Hsps, such as Hsp90 can be cytoprotective, such as preventing cell injury against heat thermal stress (Beere, HM et al., 2000, Kalwy, SA et al., 2003, Latchman, D, 2004).
Initial work has demonstrated that overexpression of Akt in neurons prevents apoptosis during growth factor withdrawal (Datta, SR et al., 1997). Similar investigations that employed superior cervical ganglion neurons also illustrated that Akt was necessary to prevent cell death during nerve growth factor withdraw (Philpott, KL et al., 1997). Additional studies have shown that Akt can be both necessary and sufficient for the survival of neurons, since expression of a dominant-negative Akt or inhibition of PI 3-K yields apoptotic cell death during trophic factor administration (Crowder, RJ and Freeman, RS, 1998) and precipitates cell death during oxidative stress (Kang, JQ et al., 2003a, Kang, JQ et al., 2003b). Akt also impacts upon the function and survival of cerebral vascular ECs. Recent investigations have shown that Akt modulates cerebral blood flow and vasomotor tone (Luo, Z et al., 2000) and prevents apoptotic injury during compromises in mitochondrial function and caspase regulation (Chong, ZZ et al., 2002a Chong, ZZ et al., 2004a).
Further work has illustrated an important role for Akt for the survival of cells during a number of injury paradigms. Enhanced Akt activity can foster cell survival during free radical exposure (Chong, ZZ et al., 2003b, Matsuzaki, H et al., 1999), matrix detachment (Rytomaa, M et al., 2000), neuronal axotomy (Namikawa, K et al., 2000), DNA damage (Chong, ZZ et al., 2002a, Chong, ZZ et al., 2004a, Henry, MK et al., 2001, Kang, JQ et al., 2003a), anti-Fas antibody administration (Suhara, T et al., 2001), oxidative stress (Chong, ZZ et al., 2003b, Kang, JQ et al., 2003a, Kang, JQ et al., 2003b, Yamaguchi, H and Wang, HG, 2001), hypoxic preconditioning (Wick, A et al., 2002), Aβ exposure (Martin, D et al., 2001), and transforming growth factor-β (TGF-β) application (Conery, AR et al., 2004).
Interestingly, several trophic factors and cytokines, such as erythropoietin (EPO), may depend upon Akt to offer cellular protection (Maiese, K et al., 2003b). EPO can phosphorylate Akt and is dependent upon the activation of PI 3-K and Janus Kinase 2 (Jak2) (Chong, ZZ et al., 2002a, Witthuhn, BA et al., 1993). Yet, central to the ability of EPO to prevent cellular apoptosis is the activation of Akt by EPO (Maiese, K et al., 2004b). During anoxia or free radical exposure, expression of the active form of Akt (phospho-Akt) is increased (Kang, JQ et al., 2003a, Kang, JQ et al., 2003b). EPO can significantly enhance the activity of Akt during oxidative stress and prevent inflammatory activation of microglia (Maiese, K et al., 2005b). This up-regulation of Akt activity during injury paradigms appears to be vital for EPO protection, since prevention of Akt phosphorylation blocks cellular protection by EPO (Chong, ZZ et al., 2003a, Chong, ZZ et al., 2003b, Chong, ZZ et al., 2003e). Yet, this same modulation of Akt by EPO may not always be desirable, such as during increased neoplastic growth (Maiese, K et al., 2005a).
Akt provides an anti-apoptotic survival signal by several mechanisms, including the phosphorylation and inactivation of Bad and stimulation of NF-kappaB (NF-κB) activity (Barber, AJ et al., 2001, Khwaja, A, 1999) (Fig. (2)). In regards to NF-κB, Akt uses IκB kinase (IKK) and the IKKα catalytic subunit to efficiently stimulate the transactivation domain of the p65 subunit of NF-κB. Once activated, NF-κB results in the induction of several anti-apoptotic genes (Chong, ZZ et al., 2005c). Akt also inhibits PCD through its ability to prevent caspase activation that is initiated at either a pre- or a post-mitochondrial level. Recent studies have illustrated that Akt acts to prevent the release of cytochrome c from mitochondria (Kennedy, SG et al., 1999) and functions to inhibit the activation of cysteine proteases following the release of cytochrome c (Rytomaa, M et al., 2000, Zhou, H et al., 2000). Yet, feedback systems exist that can modulate the half-life of Akt. Activity of Akt can be eliminated by caspase 3 induction, since caspase 3 has been shown to cleave Akt leading to the inhibition of Akt kinase activity (Bachelder, RE et al., 2001). In some cellular systems, work has suggested that mGluRs can increase cellular survival during injury paradigms through the activation of the PI 3-K/Akt pathways (Chong, ZZ et al., 2003d). In rat hippocampal neuronal cultures, application of the group I mGluR agonist DHPG prevented neuronal injury during NO toxicity through increased Akt activity (Chong, ZZ et al., 2005a). This enhancement of Akt activity by mGluR1 may proceed through the formation of a complex that includes Homer, an adaptor protein, and PI 3-K to prevent neuronal apoptosis (Rong, R et al., 2003). As a second possible protective mechanism, mGluR inhibition of caspase 3 - like activity may serve to prevent the caspase 3 mediated cleavage of Akt and foster increased cellular survival through a prolonged half-life of Akt (Lin, SH et al., 2001a, Lin, SH et al., 2001b, Maiese, K and Vincent, AM, 1999b).
Protein kinase C (PKC) represents a family of serine-threonine kinases that are physiologically activated by a number of lipid cofactors and are considered to be important transducers in several agonist-induced signaling cascades (Fig. (2)). To date, PKC comprises at least 12 distinct serine/threonine kinase isoenzymes that have important actions in transmembrane signal transduction pathways regulating cell proliferation, differentiation, cytoskeletal functions, gene transcription, PCD, and drug resistance (Musashi, M et al., 2000). Studies have been shown that activation of PKC may be either pro-apoptotic or anti-apoptotic depending on the cell type (Maiese, K et al., 1993b, Maiese, K et al., 1996, Musashi, M et al., 2000). Studies have begun to define isoform-specific functions of PKC in the apoptotic pathway and the alterations of specific PKC isoforms during injury (Selvatici, R et al., 2003). For example, PKC isoforms that appear to be anti-apoptotic include PKC-α, PKC-βII, and PKC-ε and the atypical isoforms PKC-λ and PKC-ζ (Lin, WW et al., 1997). During free radical NO exposure and anoxia, activation of mGluRs has been shown to protect neurons through pathways which modulate PKC. Neuroprotection by the subtypes mGluR1a, mGluR2, and mGluR5 appears to be dependent on the direct modulation of PKC activity (Maiese, K et al., 1996).
The mGluR System and its Control of Cellular Acidity and Endonuclease Activity
Changes in intracellular pH can significantly impact on cellular survival (Franco-Cea, A et al., 2004), since cellular proteins such as enzymes, ion channels, ion transporters, and ions (Sensi, SL and Jeng, JM, 2004) are especially sensitive to alterations in intracellular pH (Willoughby, D et al., 2001). During cellular injury, reactive oxygen species have been postulated as a potential mechanism for the induction of acidosis-induced cellular toxicity (Shen, H et al., 1995). Other experimental models of cell injury, including hypercapnia (Ritucci, NA et al., 1997), hypoxia (Roberts, E, Jr. and Chih, CP, 1997), and glutamate toxicity (Zhan, RZ et al., 1997), also have been linked to disturbances in intracellular pH.
Under some cellular conditions, intracellular acidification has been demonstrated to be both necessary and sufficient for the induction of PCD (Vincent, AM et al., 1999). Free radical generation results in a biphasic, transient intracellular acidification that can occur within 30 minutes and directly precipitate cellular degeneration (Lin, SH et al., 2000, Vincent, AM et al., 1999). Prevention of intracellular acidification markedly improves cell survival, suggesting that intracellular acidification is required for free radical induced cellular degeneration.
Cellular injury mediated by intracellular pH also is tied to the activation of acidic-dependent endonucleases (Villalba, M et al., 1995, Vincent, AM et al., 1999). Cleavage of chromosomal DNA into oligonucleosomal size fragments is an integral part of cell injury that involves hydrolysis of genomic DNA catalyzed by a number of endonucleases. Endonucleases can be divided into several groups according to their ionic sensitivities to zinc (Torriglia, A et al., 1997) and magnesium (Sun, XM et al., 1994). Endonucleases associated with PCD induction may be classified as calcium-magnesium dependent endonucleases, magnesium dependent endonucleases, and cation-independent endonucleases. DNase I is a secreted digestive enzyme that is a calcium-magnesium dependent endonuclease and consists of a single polypeptide with a molecular weight of 31 kDa (Madaio, MP et al., 1996). DNase II, a lysosomal, acidic pH activated enzyme, also is involved in DNA cleavage during injury (Torriglia, A et al., 1995). Activation of other endonucleases, such as a magnesium dependent endonuclease and a caspase-activated DNase, can result in the induction of PCD (Pandey, S et al., 1997).
Within the nervous system, modulation of endonuclease activity directly influences cell survival (Vincent, AM et al., 1999, Vincent, AM et al., 1999). Three separate endonuclease activities are present in neurons. They are a constitutive acidic cation independent endonuclease, a constitutive calcium/magnesium-dependent endonuclease, and an inducible magnesium dependent endonuclease (Vincent, AM et al., 1999). The inducible magnesium dependent endonuclease may be unique for the nervous system (Vincent, AM et al., 1999). The physiologic characteristics of the magnesium dependent endonuclease, such as a pH range of 7.4–8.0, a dependence on magnesium, and a molecular weight of 95–108 kDa, are consistent with a recently described constitutive 97 kDa endonuclease in non-neuronal tissues, but in contrast the endonuclease in the nervous system is inducible rather than constitutive in nature.
The mGluR system may offer protection against PCD through the regulation of intracellular acidification, since activation of the mGluRs directly modulates cellular pH (Vincent, AM et al., 1999). In addition, activation of mGluRs prevents cellular injury through the modulation of endonuclease activity that is linked to changes in intracellular pH (Vincent, AM et al., 1999). Prior work has demonstrated that specific mGluRs subtypes can modulate neuronal endonuclease activity during PCD. For example, activation of group III mGluRs inhibits calcium-magnesium dependent endonuclease activity. Yet, it is the activation of group I mGluRs that inhibits magnesium dependent endonuclease activity (Vincent, AM et al., 1999). Thus, the ability of mGluRs to protect genomic DNA integrity is closely linked to the modulation of both intracellular pH and endonuclease activity by the mGluR system.
The “Extrinsic and Intrinsic” Pathways of the mGluR System
Cytoprotection through the mGluR system can occur at several levels, but ultimate protection against genomic DNA degradation and membrane PS exposure may be dependent upon the modulation of cysteine protease activity that involves caspases (Fig. (3)). Caspases are usually synthesized as inactive zymogens that are proteolytically cleaved into subunits at the onset of apoptosis and function as active caspases after reconstitution to molecular heterodimers (Chong, ZZ et al., 2005c). Caspases are composed of three domains including an N-terminal prodomain, a large subunit, and a small subunit (Earnshaw, WC et al., 1999). As a result of their activation sequence, they are classified as either initiator caspases (also known as apical caspases) or effector caspases (Shi, Y, 2004). An initiator caspase cleaves and subsequently activates an effector caspase. The apoptotic-associated caspases include initiator caspases, such as caspase 2, 8, 9, and 10, that activate downstream effector caspases, resulting in an amplification of cascade activity. The initiator caspases consist of long N-terminal prodomains that contain caspase recruitment domains (CARDs) in caspase 2 and caspase 9, or death effector domains (DEDs) in caspase 8 and caspase 10 (Hofmann, K et al., 1997). The effector caspases have short or absent prodomains and consist of caspase 3, 6, and 7 that function to directly cleave crucial cellular protein substrates that result in cell destruction.
Activation of caspases proceeds through extrinsic and intrinsic pathways (Chong, ZZ et al., 2005d, Ekshyyan, O and Aw, TY, 2004). The extrinsic pathway is initiated by death receptor activation at the cell surface, resulting in the recruitment and activation of the initiator caspase 8 upon apoptotic stimuli (Ashkenazi, A and Dixit, VM, 1998). The intracellular death domain of death receptors, such as the TNF superfamily, CD95/Fas/Apo-1, and the death receptor 3, undergoes conformational change upon binding to extracellular ligands and forms an intracellular death-inducing signaling complex following recruitment of adaptor molecules, such as the Fas associated death domain (FADD). FADD recruits caspase 8 through its DED domain and this leads to caspase 8 activation (Juo, P et al., 1998, Varfolomeev, EE et al., 1998). Caspase 8 can subsequently activate caspase 3. In addition, caspase 8 activation also may result in the cleavage of Bid, a pro-apoptotic member of Bcl-2 family, allowing the truncated Bid (tBid) to translocate to the mitochondria (Li, H et al., 1998). This leads to cytochrome c release through Bax resulting in the subsequent activation of executioner caspases (Yin, XM et al., 2002).
The intrinsic caspase pathway involves mitochondrial dysfunction (Doonan, F et al., 2004). The mitochondrial pathway is associated with the release of cytochrome c and subsequent activation of caspase 9 followed by activation of caspase 3 (Liu, X et al., 1996). The process is regulated by the Bcl-2 subfamily BH3-only proteins, which are normally located in cellular compartments other than mitochondria, but translocate to the mitochondria in response to apoptotic stimuli (Cosulich, SC et al., 1997). The translocation of these proteins delivers an apoptotic signal to mitochondria through the interaction with Bax to induce the release of cytochrome c that then binds to apoptotic protease-activating factor-1 (Apaf-1). Apaf-1 consists of three different domains that include CARDs, repeats of tryptophan and aspartate residues (WD-40 repeats), and a nucleotide-binding domain CED-4. Binding of cytochrome c to Apaf-1 results in the removal of the WD-40 domain, masking the CED-4 and CARDs, and leads to the oligomerization of Apaf-1 with the requirement of dATP/ATP (Hu, Y et al., 1999). The oligomerization of Apaf-1 promotes the allosteric activation of caspase 9 by forming the Apaf-1 apoptosome (Li, P et al., 1997). Caspase 9 can subsequently activate caspase 3 (Li, P et al., 1997) as well as caspase 1 through the intermediary caspase 8 (Takahashi, H et al., 1999). Together, caspase 1 and caspase 3 lead to both DNA fragmentation and membrane PS exposure (Chong, ZZ et al., 2002a, Li, P et al., 1997, Maiese, K et al., 2000b).
Control of the caspase pathway during oxidative stress may be vital to provide protection against PCD (Figueroa, S et al., 2005, Paucard, A et al., 2004). The caspases 1 and 3 have each been linked to the independent apoptotic pathways of genomic DNA cleavage and cellular membrane PS exposure (Chong, ZZ et al., 2003a, Chong, ZZ et al., 2003e, Takahashi, H et al., 1999). These caspases, in addition to caspase 8 and 9, are also tied to the direct activation and proliferation of microglia (Chong, ZZ et al., 2003b, Kang, JQ et al., 2003a, Kang, JQ et al., 2003b). Caspase 1 is believed to be principally responsible for the externalization of membrane PS residues in several cell systems that can subsequently activate microglial phagocytosis (Maiese, K et al., 2000b, Vanags, DM et al., 1996). Furthermore, caspase 9 is activated through a process that involves the cytochrome c -Apaf-1 complex (Chong, ZZ et al., 2002b, Li, P et al., 1997). In addition, caspase 8 serves as an upstream initiator of executioner caspases, such as caspase 3, and also leads to the mitochondrial release of cytochrome c (Engels, IH et al., 2000, Stegh, AH et al., 2002). Following caspase 8 and caspase 9 activation, caspase 3 directly leads to genomic DNA degradation.
Caspase activation has been closely associated to the pathogenesis of neurodegenerative disorders. Caspase activation can be a significant factor for cellular injury during acute oxidative stress exposure, such as during cerebral ischemia (Benchoua, A et al., 2004), trauma (Clausen, F et al., 2004), free radical exposure (Chong, ZZ et al., 2002c, Maiese, K et al., 2000a), and early retinal degenerative disease (Haynes, T and Del Rio-Tsonis, K, 2004). Yet, what may be more interesting is the potential contribution of caspase activity during chronic neurodegeneration. Several studies support the belief that caspase activation is involved in the pathological process of chronic neurodegenerative diseases, such as AD (Chong, ZZ et al., 2005e). The elevation of caspase genes including caspase 1, 2, 3, 5, 6, 7, 8, and 9 has been observed in human postmortem brains from AD patients (Pompl, PN et al., 2003). In addition, single neurons with DNA fragmentation have been shown to contain cytoplasmic immunoreactivity for active caspase 3, implying that apoptotic injury results during AD. Caspase 3 immunoreactivity also was co-localized with paired helical filaments in neurons, suggesting that caspase 3 activation may contribute to the formation of neurofibrillary tangles (Gastard, MC et al., 2003). Additional work in cell culture experiments has demonstrated that treatment with amyloid directly results in the activation of caspase 1 (Jordan, J et al., 1997), caspase 2, and caspase 3 (Troy, CM et al., 2000).
Caspase activation also is necessary for the processing of APP. Caspases cleave APP at three major caspase recognition sites, one at the C-terminus, D720, and two at the N-terminus, D197 and D219. Caspase activation results in the increased production of Aβ. Yet, in some cases, Aβ generation may not be entirely dependent upon the cleavage of APP at its C-terminal (D720) and/or N-terminal caspase sites. During etoposide-induced apoptosis ablation of caspase-dependent cleavage at D720, D197 and D219 (by site-directed mutagenesis) does not prevent enhanced Aβ production (Tesco, G et al., 2003). It is conceivable that APP may lead to cell injury through a more direct route that involves the generation of the C-terminal fragment C31. Production of C31 is a result of APP cleavage at the caspase site D720 of the C-terminus. Following caspase 3 activation, caspase 3 generates the carboxyl-terminally truncated fragment C31 from APP, which has been shown to be capable of apoptotic injury independent of caspase 3 (Nishimura, I et al., 2002). Furthermore, caspase-dependent APP cleavage at D720 also has been observed in brains of AD patients through demonstration of C31 expression (Lu, DC et al., 2000).
Interestingly, protection by the mGluR system against cellular injury appears to be linked to the modulation of caspase activity (Fig. (3)). Activation of mGluRs attenuates the induction of caspase 9, caspase 1, and caspase 3 activities (Lin, SH et al., 2001b, Maiese, K et al., 1999a, Maiese, K et al., 2000a, Vincent, AM et al., 2000). Protection by the mGluR system appears to function at two distinct levels. Activation of the mGluRs directly prevents induction of caspase activity (Berent-Spillson, A et al., 2004, Chong, ZZ et al., 2003d, Kajta, M et al., 2005). In addition, mGluRs employ more upstream mechanisms that preserve mitochondrial membrane potential and prevent the release of cytochrome c (Baskys, A et al., 2005, Blandini, F et al., 2004, Chong, ZZ et al., 2005c, Lin, SH et al., 2001b).
Poly(ADP-ribose) polymerase (PARP) functions within a variety of cellular processes, including the repair of DNA breaks (Maiese, K et al., 2003a, Maruta, H et al., 1997). Inhibition of PARP activity leads to the activation of PCD and to subsequent DNA fragmentation, suggesting that loss of PARP activity can significantly mediate apoptotic DNA degradation (Lin, SH et al., 2001b, Lin, SH et al., 2000, Thies, RL and Autor, AP, 1991). In order to successfully utilize PARP for DNA repair, a fine modulation of PARP activity must take place. Although activation of PARP can be beneficial for the repair of DNA, excessive activation of PARP may lead to the depletion of intracellular nicotinamide adenine dinucleotide (NAD+) and limit cellular ATP reserves (Wielckens, K et al., 1982).
The maintenance of cellular energy reserves is critical for cellular survival and new evidence suggests that the mGluR system may regulate body weight and metabolism (Duvoisin, RM et al., 2005). NAD+ is closely tied to cellular metabolism and genomic DNA repair (Li, F et al., 2004, Maiese, K et al., 2001). During a cellular insult that affects DNA integrity, PARP catalyses the synthesis of poly(ADP-ribose) from its substrate NAD+, which stimulates the process of DNA repair (Satoh, MS and Lindahl, T, 1992). Increased activation of PARP leads to an extensive turnover of NAD+ and a significant reduction in NAD+ levels. This can trigger the loss of NAD+ and ATP, leading to the death of a cell. Furthermore, oxidative stress can trigger the opening of mitochondrial membrane permeability transition pore (Chong, ZZ et al., 2003a, Di Lisa, F et al., 2001, Kang, JQ et al., 2003b, Lin, SH et al., 2000) and subsequently result in the release of NAD+ from mitochondria (Di Lisa, F et al., 2001). During conditions of oxidative stress and energy depletion in neurons, poly(ADP-ribosylation) activation and loss of NAD+ stores in mitochondria have been shown to lead to apoptotic injury. Restoration of NAD+ content in mitochondria with liposomal NAD+ prevents neuronal injury (Du, L et al., 2003). To complicate this scenario, caspase 3 -like activity leads to the specific cleavage of PARP (Lin, SH et al., 2001b, Lin, SH et al., 2000, Maiese, K et al., 2000a). The mGluR system appears to closely interface with PARP (Aarts, MM and Tymianski, M, 2004). In particular, mGluR prevents PARP degradation and allow for DNA repair through the either the maintenance of mitochondrial membrane potential or through the direct inhibition of caspase 3-like activity (Chong, ZZ et al., 2003d). Preserving mitochondrial integrity not only inhibits caspase 3 activation through the prevention of cytochrome c release, but also maintains intracellular metabolic homeostasis preventing energy depletion during the DNA repair process (Chong, ZZ et al., 2005f).
The mGluR System and Regulation of Mitochondrial Membrane Potential
Given the detrimental effects of caspase activation, preservation of mitochondrial integrity can be a key determinant for the maintenance of normal cellular physiology as well as for cellular recovery from toxic insults. In both neuronal and vascular populations, loss of mitochondrial membrane permeability represents a significant determinant for cell injury and the induction of the apoptotic cascade (Chong, ZZ et al., 2003a, Lin, SH et al., 2000, Ueda, S et al., 2002). Mitochondria are a significant source of superoxide radicals that can generate oxidative stress. Impairment of the electron transfer chain at the flavin mononucleotide group of complex I (NADPH ubiquinone oxidoreductase) or at ubiquinone site of complex III (ubiquinone-cytochrome c reductase) results in the active generation of reactive oxygen species (Bosca, L and Hortelano, S, 1999, Liu, D et al., 2003). Once generated, reactive oxygen species further impair mitochondrial electron transport and enhance reactive oxygen species production (Smeitink, JAM et al., 2004).
A subsequent drop in mitochondrial membrane potential has been suggested to be an important trigger for the release of cytochrome c, a critical determinant of cell survival (Bosca, L et al., 1999, Liu, D et al., 2003, Maiese, K and Chong, ZZ, 2004a). Once released into the cytosol, cytochrome c binds to Apaf-1 leading to the caspase cascade activation. In addition to cytochrome c, other mitochondrial proteins, such as endonuclease G (Li, LY et al., 2001), Smac/Diablo (Verhagen, AM et al., 2000), and apoptosis-inducing factor (AIF) (Susin, SA et al., 1999) also can be released in response to injury. Endonuclease G is recognized as a DNase responsible for DNA fragmentation during apoptosis. The caspase co-activator Smac/Diablo competes with caspase 9 for binding to the X-chromosome-linked inhibitor of apoptosis proteins (XIAPs) to block activities of these proteins. AIF translocates from the mitochondria to the nucleus to promote chromatin condensation and large-scale DNA fragmentation.
With the delicate function of the mitochondria and the potential release of cytochrome c during cell injury, it is conceivable that maintenance of mitochondrial membrane potential by the mGluR system may offer one avenue for cytoprotection (Fig. (2) and Fig. (3)). Recent work has demonstrated the ability of group I mGluRs to preserve mitochondrial membrane potential in endothelial cells (Chong, ZZ et al., 2003d, Lin, SH et al., 2002, Lin, SH et al., 2001b) and neurons (Chong, ZZ et al., 2005a, Lin, SH et al., 2001b). Free radical exposure with NO can result in the loss of mitochondrial membrane potential when compared to untreated control cells. Yet, pretreatment of endothelial cells or neurons one hour prior to NO exposure with DHPG (a group I mGluR agonist), but not AIDA (a group I mGluR antagonist) prevents the loss of membrane potential in mitochondria (Chong, ZZ et al., 2005a, Lin, SH et al., 2001b). The precise mechanisms by which the mGluRs employ to preserve mitochondrial integrity are not clear, but may involve the modulation of Akt1 activity, intracellular calcium stores, and reactive oxygen species. Reduction in mitochondrial intracellular calcium stores and free radical levels has been suggested to promote the maintenance of mitochondrial membrane potential and integrity (Sullivan, PG et al., 1999). Activation of group I mGluRs has been demonstrated to regulate the release of intracellular calcium from both Ins (1,4,5)P3-sensitive and ryanodine-sensitive calcium stores (Maiese, K et al., 1999a). In addition, group I mGluRs can modulate free radical signal transduction cascades in both neuronal and endothelial cell populations (Lin, SH et al., 2001b, Maiese, K et al., 2000a, Vincent, AM et al., 2000).
The Extracellular Signal-Related Kinases, c-Jun-Amino Terminal Kinases, and the p38 Kinases of the mGluR System
MAPKs consist of ERKs, the c-Jun-amino terminal kinases (JNKs), and the p38 kinase. Activation or phosphorylation of MAPKs can trigger a number of downstream signal transduction pathways and modulate cell differentiation, growth, and death. Phosphorylated MAPKs also can translocate to the cell nucleus to regulate both the activation of transcription factors and the subsequent expression of genes.
ERK is pro-survival factor and the activation of ERKs is usually associated with inhibition of apoptosis (Zablocka, B et al., 2003). In many cellular injury models, such as serum deprivation (Desire, L et al., 2000), hypoxic-ischemia (Han, BH and Holtzman, DM, 2000) and oxidative stress (Wang, X et al., 1998), the activation of ERKs also has been associated with neuronal protection. In contrast to ERKs, enhanced activity of the MAPKs p38 and JNK can sometimes lead to cell injury. The MAPKs p38 and JNK have been shown to increase the activity of both caspase 1 and caspase 3 (Ko, HW et al., 2000, MacFarlane, M et al., 2000). Yet, the relationship of p38 and JNK during injury paradigms in the neuronal and vascular systems is not entirely evident and may require combined signal transduction systems (Lee, SR and Lo, EH, 2003). The MAPK p38 and possibly JNK are believed to have a role during cellular stress and during neurodegenerative diseases (Hensley, K et al., 1999). Work has linked p38 activation to Bax translocation during free radical exposure (Ghatan, S et al., 2000) and to endothelial injury during cytokine administration (Yue, TL et al., 1999).
The mGluR system may employ several downstream signal transduction pathways of the ERKs that mediate anti-apoptotic signaling (Fig. (2)). For example, group I mGluRs can increase the phosphorylation of ERK2. This induction of ERK2 phosphorylation is inhibited by PKC inhibitor, suggesting that group I mGluRs leads to ERK2 activation through PKC (Voulalas, PJ et al., 2005). In addition, ERKs may employ phosphorylation of the pro-apoptotic protein Bad and induction of pro-survival gene expression via the cAMP responsive element-binding (CREB) protein dependent pathway to lead to cellular protection (Choe, ES and Wang, JQ, 2001, Choe, ES and Wang, JQ, 2002). It appears not so evident whether the protective mechanisms utilized by mGluRs also involve the cellular pathways of the MAPKs. Work has suggested that activation of p38 and JNK may contribute to either neuronal or endothelial degeneration. Within 10 minutes following an ischemic insult, significant activation of p38 and JNK is present in both neurons and endothelial cells (Lin, SH et al., 2001a, Lin, SH et al., 2001b). Yet, this activation of p38 and JNK does not appear to be linked to the protective effect of mGluRs. Activation of mGluRs does not alter the activity of either p38 or JNK, suggesting that protection by mGluRs is independent or below the level of p38 activation (Lin, SH et al., 2001a, Lin, SH et al., 2001b).
FUTURE PERSPECTIVES
The mGluR system is involved in a number of brain functions and disorders that range from memory imprinting to cognitive loss with AD. Activation of specific mGluR subtypes can block the progression of distinct pathways of apoptosis that involve genomic DNA degradation, the exposure of membrane PS residues, and the induction of inflammatory pathways through microglial activation. Upstream pathways may involve the protein kinases PKA, PKB, and PKC. Subsequently, modulation of mitochondrial membrane potential, intracellular pH, and intracellular calcium can ultimately regulate specific caspase activity. Interestingly, these cytoprotective mechanisms offered by the mGluR system can rely upon ERKs, but may remain independent from the MAPKs p38 and JNK. As our knowledge of the broad implications of the mGluR system continues to unfold, we will be able to identify specific cellular mGluR targets that can not only enhance the clinical therapeutic utility of the mGluR system, but also foster greater understanding of the cellular pathways that initially lead to disorders of the nervous system.
Acknowledgments
This research was supported by the following grants (KM): American Heart Association (National), Bugher Foundation Award, Janssen Neuroscience Award, LEARN Foundation Award, MI Life Sciences Challenge Award, and NIH NIEHS (P30 ES06639).
References
- Aarts MM, Tymianski M. Novel treatment of excitotoxicity: targeted disruption of intracellular signalling from glutamate receptors. Biochem Pharmacol. 2003;66(6):877–86. doi: 10.1016/s0006-2952(03)00297-1. [DOI] [PubMed] [Google Scholar]
- Aarts MM, Tymianski M. Molecular mechanisms underlying specificity of excitotoxic signaling in neurons. Curr Mol Med. 2004;4(2):137–47. doi: 10.2174/1566524043479202. [DOI] [PubMed] [Google Scholar]
- Abraham KE, McGinty JF, Brewer KL. The role of kainic acid/AMPA and metabotropic glutamate receptors in the regulation of opioid mRNA expression and the onset of pain-related behavior following excitotoxic spinal cord injury. Neuroscience. 2001;104(3):863–74. doi: 10.1016/s0306-4522(01)00134-8. [DOI] [PubMed] [Google Scholar]
- Agrawal SK, Theriault E, Fehlings MG. Role of group I metabotropic glutamate receptors in traumatic spinal cord white matter injury. J Neurotrauma. 1998;15(11):929–41. doi: 10.1089/neu.1998.15.929. [DOI] [PubMed] [Google Scholar]
- Albasanz JL, Dalfo E, Ferrer I, Martin M. Impaired metabotropic glutamate receptor/phospholipase C signaling pathway in the cerebral cortex in Alzheimer’s disease and dementia with Lewy bodies correlates with stage of Alzheimer’s-disease-related changes. Neurobiol Dis. 2005 doi: 10.1016/j.nbd.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Albert PR, Robillard L. G protein specificity. Traffic direction required. Cell Signal. 2002;14(5):407–18. doi: 10.1016/s0898-6568(01)00259-5. [DOI] [PubMed] [Google Scholar]
- Anborgh PH, Godin C, Pampillo M, Dhami GK, Dale LB, Cregan SP, Truant R, Ferguson SS. Inhibition of metabotropic glutamate receptor signalling by the huntingtin binding protein optineurin. J Biol Chem. 2005 doi: 10.1074/jbc.M504508200. [DOI] [PubMed] [Google Scholar]
- Anneser JM, Berthele A, Borasio GD, Castro-Lopes JM, Zieglgansberger W, Tolle TR. Axotomy of the sciatic nerve differentially affects expression of metabotropic glutamate receptor mRNA in adult rat motoneurons. Brain Res. 2000;868(2):215–21. doi: 10.1016/s0006-8993(00)02332-5. [DOI] [PubMed] [Google Scholar]
- Anwyl R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Brain Res Rev. 1999;29(1):83–120. doi: 10.1016/s0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- Aronica E, Catania MV, Geurts J, Yankaya B, Troost D. Immunohistochemical localization of group I and II metabotropic glutamate receptors in control and amyotrophic lateral sclerosis human spinal cord: upregulation in reactive astrocytes. Neuroscience. 2001;105(2):509–20. doi: 10.1016/s0306-4522(01)00181-6. [DOI] [PubMed] [Google Scholar]
- Arur S, Uche UE, Rezaul K, Fong M, Scranton V, Cowan AE, Mohler W, Han DK. Annexin I is an endogenous ligand that mediates apoptotic cell engulfment. Dev Cell. 2003;4(4):587–98. doi: 10.1016/s1534-5807(03)00090-x. [DOI] [PubMed] [Google Scholar]
- Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281(5381):1305–8. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- Awad H, Hubert GW, Smith Y, Levey AI, Conn PJ. Activation of metabotropic glutamate receptor 5 has direct excitatory effects and potentiates NMDA receptor currents in neurons of the subthalamic nucleus. J Neurosci. 2000;20(21):7871–9. doi: 10.1523/JNEUROSCI.20-21-07871.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachelder RE, Wendt MA, Fujita N, Tsuruo T, Mercurio AM. The cleavage of Akt/protein kinase B by death receptor signaling is an important event in detachment-induced apoptosis. J Biol Chem. 2001;276(37):34702–7. doi: 10.1074/jbc.M102806200. [DOI] [PubMed] [Google Scholar]
- Barber AJ, Nakamura M, Wolpert EB, Reiter CE, Seigel GM, Antonetti DA, Gardner TW. Insulin rescues retinal neurons from apoptosis by a phosphatidylinositol 3-kinase/Akt-mediated mechanism that reduces the activation of caspase-3. J Biol Chem. 2001;276(35):32814–21. doi: 10.1074/jbc.M104738200. [DOI] [PubMed] [Google Scholar]
- Baskys A, Blaabjerg M. Understanding regulation of nerve cell death by mGluRs as a method for development of successful neuroprotective strategies. J Neurol Sci. 2005;229–230:201–9. doi: 10.1016/j.jns.2004.11.028. [DOI] [PubMed] [Google Scholar]
- Battista N, Fezza F, Maccarrone M. Endocannabinoids and their involvement in the neurovascular system. Curr Neurovasc Res. 2004;1(2):129–140. doi: 10.2174/1567202043480107. [DOI] [PubMed] [Google Scholar]
- Bear MF. Therapeutic implications of the mGluR theory of fragile X mental retardation. Genes Brain Behav. 2005;4(6):393–8. doi: 10.1111/j.1601-183X.2005.00135.x. [DOI] [PubMed] [Google Scholar]
- Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of pro-caspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2000;2(8):469–75. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- Benchoua A, Braudeau J, Reis A, Couriaud C, Onteniente B. Activation of proinflammatory caspases by cathepsin B in focal cerebral ischemia. J Cereb Blood Flow Metab. 2004;24(11):1272–9. doi: 10.1097/01.WCB.0000140272.54583.FB. [DOI] [PubMed] [Google Scholar]
- Berent-Spillson A, Robinson AM, Golovoy D, Slusher B, Rojas C, Russell JW. Protection against glucose-induced neuronal death by NAAG and GCP II inhibition is regulated by mGluR3. J Neurochem. 2004;89(1):90–9. doi: 10.1111/j.1471-4159.2003.02321.x. [DOI] [PubMed] [Google Scholar]
- Berthele A, Platzer S, Laurie DJ, Weis S, Sommer B, Zieglgansberger W, Conrad B, Tolle TR. Expression of metabotropic glutamate receptor subtype mRNA (mGluR1-8) in human cerebellum. Neuroreport. 1999;10(18):3861–7. doi: 10.1097/00001756-199912160-00026. [DOI] [PubMed] [Google Scholar]
- Blaabjerg M, Fang L, Zimmer J, Baskys A. Neuroprotection against NMDA excitotoxicity by group I metabotropic glutamate receptors is associated with reduction of NMDA stimulated currents. Exp Neurol. 2003;183(2):573–80. doi: 10.1016/s0014-4886(03)00204-8. [DOI] [PubMed] [Google Scholar]
- Blandini F, Braunewell KH, Manahan-Vaughan D, Orzi F, Sarti P. Neurodegeneration and energy metabolism: from chemistry to clinics. Cell Death Differ. 2004;11(4):479–84. doi: 10.1038/sj.cdd.4401323. [DOI] [PubMed] [Google Scholar]
- Blumcke I, Behle K, Malitschek B, Kuhn R, Knopfel T, Wolf HK, Wiestler OD. Immunohistochemical distribution of metabotropic glutamate receptor subtypes mGluR1b, mGluR2/3, mGluR4a and mGluR5 in human hippocampus. Brain Res. 1996;736(1–2):217–26. doi: 10.1016/0006-8993(96)00697-x. [DOI] [PubMed] [Google Scholar]
- Bombeli T, Karsan A, Tait JF, Harlan JM. Apoptotic vascular endothelial cells become procoagulant. Blood. 1997;89(7):2429–42. [PubMed] [Google Scholar]
- Bonci A, Grillner P, Siniscalchi A, Mercuri NB, Bernardi G. Glutamate metabotropic receptor agonists depress excitatory and inhibitory transmission on rat mesencephalic principal neurons. Eur J Neurosci. 1997;9(11):2359–69. doi: 10.1111/j.1460-9568.1997.tb01653.x. [DOI] [PubMed] [Google Scholar]
- Borodezt K, D’Mello SR. Decreased expression of the metabotropic glutamate receptor-4 gene is associated with neuronal apoptosis. J Neurosci Res. 1998;53(5):531–41. doi: 10.1002/(SICI)1097-4547(19980901)53:5<531::AID-JNR3>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Bosca L, Hortelano S. Mechanisms of nitric oxide-dependent apoptosis: involvement of mitochondrial mediators. Cell Signal. 1999;11(4):239–44. doi: 10.1016/s0898-6568(98)00064-3. [DOI] [PubMed] [Google Scholar]
- Bose J, Gruber AD, Helming L, Schiebe S, Wegener I, Hafner M, Beales M, Kontgen F, Lengeling A. The phosphatidylserine receptor has essential functions during embryogenesis but not in apoptotic cell removal. J Biol. 2004;3(4):15. doi: 10.1186/jbiol10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley SR, Marino MJ, Wittmann M, Rouse ST, Awad H, Levey AI, Conn PJ. Activation of group II metabotropic glutamate receptors inhibits synaptic excitation of the substantia Nigra pars reticulata. J Neurosci. 2000;20(9):3085–94. doi: 10.1523/JNEUROSCI.20-09-03085.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley SR, Standaert DG, Rhodes KJ, Rees HD, Testa CM, Levey AI, Conn PJ. Immunohistochemical localization of subtype 4a metabotropic glutamate receptors in the rat and mouse basal ganglia. J Comp Neurol. 1999;407(1):33–46. [PubMed] [Google Scholar]
- Breysse N, Baunez C, Spooren W, Gasparini F, Amalric M. Chronic but not acute treatment with a metabotropic glutamate 5 receptor antagonist reverses the akinetic deficits in a rat model of park-insonism. J Neurosci. 2002;22(13):5669–78. doi: 10.1523/JNEUROSCI.22-13-05669.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Manalo DJ, Wei G, Rodriguez ER, Fox-Talbot K, Lu H, Zweier JL, Semenza GL. Hearts from rodents exposed to intermittent hypoxia or erythropoietin are protected against ischemia-reperfusion injury. Circulation. 2003;108(1):79–85. doi: 10.1161/01.CIR.0000078635.89229.8A. [DOI] [PubMed] [Google Scholar]
- Cai Z, Xiao F, Fratkin JD, Rhodes PG. Protection of neonatal rat brain from hypoxic-ischemic injury by LY379268, a Group II metabotropic glutamate receptor agonist. Neuroreport. 1999;10(18):3927–31. doi: 10.1097/00001756-199912160-00037. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Pisani A, Bernardi G. Metabotropic glutamate receptors and cell-type-specific vulnerability in the striatum: implication for ischemia and Huntington’s disease. Exp Neurol. 1999;158(1):97–108. doi: 10.1006/exnr.1999.7092. [DOI] [PubMed] [Google Scholar]
- Campusano JM, Abarca J, Forray MI, Gysling K, Bustos G. Modulation of dendritic release of dopamine by metabotropic glutamate receptors in rat substantia nigra. Biochem Pharmacol. 2002;63(7):1343–52. doi: 10.1016/s0006-2952(02)00870-5. [DOI] [PubMed] [Google Scholar]
- Caruso C, Bottino MC, Pampillo M, Pisera D, Jaita G, Duvilanski B, Seilicovich A, Lasaga M. Glutamate induces apoptosis in anterior pituitary cells through group II metabotropic glutamate receptor activation. Endocrinology. 2004;145(10):4677–84. doi: 10.1210/en.2004-0550. [DOI] [PubMed] [Google Scholar]
- Chen J, Heinke B, Sandkuhler J. Activation of group I metabotropic glutamate receptors induces long- term depression at sensory synapses in superficial spinal dorsal horn. Neuropharmacology. 2000;39(12):2231–43. doi: 10.1016/s0028-3908(00)00084-8. [DOI] [PubMed] [Google Scholar]
- Choe ES, Wang JQ. Group I metabotropic glutamate receptor activation increases phosphorylation of cAMP response element-binding protein, Elk-1, and extracellular signal-regulated kinases in rat dorsal striatum. Brain Res Mol Brain Res. 2001;94(1–2):75–84. doi: 10.1016/s0169-328x(01)00217-0. [DOI] [PubMed] [Google Scholar]
- Choe ES, Wang JQ. CREB and Elk-1 phosphorylation by metabotropic glutamate receptors in striatal neurons. Int J Mol Med. 2002;9(1):3–10. [PubMed] [Google Scholar]
- Chong ZZ, Kang J-Q, Li F, Maiese K. mGluRI targets microglial activation and selectively prevents neuronal cell engulfment through Akt and caspase dependent pathways. Curr Neurovasc Res. 2005a;2(3):197–211. doi: 10.2174/1567202054368317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Erythropoietin is a novel vascular protectant through activation of Akt1 and mitochondrial modulation of cysteine proteases. Circulation. 2002a;106(23):2973–9. doi: 10.1161/01.cir.0000039103.58920.1f. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Hematopoietic Factor Erythropoietin Fosters Neuroprotection Through Novel Signal Transduction Cascades. J Cereb Blood Flow Metab. 2005b;22(5):503–514. doi: 10.1097/00004647-200205000-00001. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Apaf-1, Bcl-xL, Cytochrome c, and Caspase-9 Form the Critical Elements for Cerebral Vascular Protection by Erythropoietin. J Cereb Blood Flow Metab. 2003a;23(3):320–30. doi: 10.1097/01.WCB.0000050061.57184.AE. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Erythropoietin fosters both intrinsic and extrinsic neuronal protection through modulation of microglia, Akt1, Bad, and caspase-mediated pathways. Br J Pharmacol. 2003b;138(6):1107–1118. doi: 10.1038/sj.bjp.0705161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Erythropoietin: cytoprotection in vascular and neuronal cells. Curr Drug Targets Cardiovasc Haematol Disord. 2003c;3(2):141–54. doi: 10.2174/1568006033481483. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Metabotropic glutamate receptors promote neuronal and vascular plasticity through novel intracellular pathways. Histol Histopathol. 2003d;18(1):173–89. doi: 10.14670/HH-18.173. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Akt1 drives endothelial cell membrane asymmetry and microglial activation through Bcl-x(L) and caspase 1, 3, and 9. Exp Cell Res. 2004a;296(2):196–207. doi: 10.1016/j.yexcr.2004.01.021. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Activating Akt and the brain’s resources to drive cellular survival and prevent inflammatory injury. Histol Histopathol. 2005b;20(1):299–315. doi: 10.14670/hh-20.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Oxidative stress in the brain: Novel cellular targets that govern survival during neurodegenerative disease. Prog Neurobiol. 2005c;75(3):207–46. doi: 10.1016/j.pneurobio.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Stress in the brain: novel cellular mechanisms of injury linked to Alzheimer’s disease. Brain Res Brain Res Rev. 2005d;49(1):1–21. doi: 10.1016/j.brainresrev.2004.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Li FQ, Maiese K. Employing new cellular therapeutic targets for Alzheimer’s disease: A change for the better? Curr Neurovasc Res. 2005e;2(1):55–72. doi: 10.2174/1567202052773508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Lin S-H, Li F, Maiese K. The sirtuin inhibitor nicotinamide enhances neuronal cell survival during acute anoxic injury through Akt, Bad, PARP, and mitochondrial associated “anti-apoptotic” pathways. Curr Neurovasc Res. 2005f doi: 10.2174/156720205774322584. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Lin SH, Kang JQ, Maiese K. Erythropoietin prevents early and late neuronal demise through modulation of Akt1 and induction of caspase 1, 3, and 8. J Neurosci Res. 2003e;71(5):659–69. doi: 10.1002/jnr.10528. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Lin SH, Kang JQ, Maiese K. The tyrosine phosphatase SHP2 modulates MAP kinase p38 and caspase 1 and 3 to foster neuronal survival. Cell Mol Neurobiol. 2003f;23(4–5):561–78. doi: 10.1023/A:1025158314016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Lin SH, Maiese K. Nicotinamide Modulates Mitochondrial Membrane Potential and Cysteine Protease Activity during Cerebral Vascular Endothelial Cell Injury. J Vasc Res. 2002c;39(2):131–47. doi: 10.1159/000057762. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Maiese K. Targeting WNT, protein kinase B, and mitochondrial membrane integrity to foster cellular survival in the nervous system. Histol Histopathol. 2004b;19(2):495–504. doi: 10.14670/hh-19.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen F, Lundqvist H, Ekmark S, Lewen A, Ebendal T, Hillered L. Oxygen free radical-dependent activation of extracellular signal-regulated kinase mediates apoptosis-like cell death after traumatic brain injury. J Neurotrauma. 2004;21(9):1168–82. doi: 10.1089/neu.2004.21.1168. [DOI] [PubMed] [Google Scholar]
- Combs CK, Karlo JC, Kao SC, Landreth GE. beta-Amyloid stimulation of microglia and monocytes results in TNFalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci. 2001;21(4):1179–88. doi: 10.1523/JNEUROSCI.21-04-01179.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conery AR, Cao Y, Thompson EA, Townsend CM, Jr, Ko TC, Luo K. Akt interacts directly with Smad3 to regulate the sensitivity to TGF-beta induced apoptosis. Nat Cell Biol. 2004;6(4):366–72. doi: 10.1038/ncb1117. [DOI] [PubMed] [Google Scholar]
- Cosulich SC, Worrall V, Hedge PJ, Green S, Clarke PR. Regulation of apoptosis by BH3 domains in a cell-free system. Curr Biol. 1997;7(12):913–20. doi: 10.1016/s0960-9822(06)00410-6. [DOI] [PubMed] [Google Scholar]
- Croucher MJ, Patel H, Walsh DT, Moncaster JA, Gentleman SM, Fazal A, Jen LS. Up-regulation of soluble amyloid precursor protein fragment secretion in the rat retina in vivo by metabotropic glutamate receptor stimulation. Neuroreport. 2003;14(17):2271–4. doi: 10.1097/00001756-200312020-00027. [DOI] [PubMed] [Google Scholar]
- Crowder RJ, Freeman RS. Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J Neurosci. 1998;18(8):2933–43. doi: 10.1523/JNEUROSCI.18-08-02933.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalfo E, Albasanz JL, Rodriguez A, Martin M, Ferrer I. Abnormal group I metabotropic glutamate receptor expression and signaling in the frontal cortex in Pick disease. J Neuropathol Exp Neurol. 2005;64(7):638–47. doi: 10.1097/01.jnen.0000171649.86718.f2. [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91(2):231–41. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Dawson L, Chadha A, Megalou M, Duty S. The group II metabotropic glutamate receptor agonist, DCG-IV, alleviates akinesia following intranigral or intraventricular administration in the reserpine-treated rat. Br J Pharmacol. 2000;129(3):541–6. doi: 10.1038/sj.bjp.0703105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Monte SM, Chiche J, von dem Bussche A, Sanyal S, Lahousse SA, Janssens SP, Bloch KD. Nitric oxide synthase-3 overexpression causes apoptosis and impairs neuronal mitochondrial function: relevance to Alzheimer’s-type neurodegeneration. Lab Invest. 2003;83(2):287–98. doi: 10.1097/01.lab.0000056995.07053.c0. [DOI] [PubMed] [Google Scholar]
- de la Monte SM, Lu BX, Sohn YK, Etienne D, Kraft J, Ganju N, Wands JR. Aberrant expression of nitric oxide synthase III in Alzheimer’s disease: relevance to cerebral vasculopathy and neurodegeneration. Neurobiol Aging. 2000;21(2):309–19. doi: 10.1016/s0197-4580(99)00108-6. [DOI] [PubMed] [Google Scholar]
- Deng W, Wang H, Rosenberg PA, Volpe JJ, Jensen FE. Role of metabotropic glutamate receptors in oligodendrocyte excitotoxicity and oxidative stress. Proc Natl Acad Sci USA. 2004;101(20):7751–6. doi: 10.1073/pnas.0307850101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desire L, Courtois Y, Jeanny JC. Endogenous and exogenous fibroblast growth factor 2 support survival of chick retinal neurons by control of neuronal neuronal bcl-x(L) and bcl- 2 expression through a fibroblast berowth factor receptor 1- and ERK- dependent pathway. J Neurochem. 2000;75(1):151–63. doi: 10.1046/j.1471-4159.2000.0750151.x. [DOI] [PubMed] [Google Scholar]
- Dewar D, Chalmers DT, Graham DI, McCulloch J. Glutamate metabotropic and AMPA binding sites are reduced in Alzheimer’s disease: an autoradiographic study of the hippocampus. Brain Res. 1991;553(1):58–64. doi: 10.1016/0006-8993(91)90230-s. [DOI] [PubMed] [Google Scholar]
- Di Lisa F, Menabo R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem. 2001;276(4):2571–5. doi: 10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- Dietrich D, Beck H, Kral T, Clusmann H, Elger CE, Schramm J. Metabotropic glutamate receptors modulate synaptic transmission in the perforant path: pharmacology and localization of two distinct receptors. Brain Res. 1997;767(2):220–7. doi: 10.1016/s0006-8993(97)00579-9. [DOI] [PubMed] [Google Scholar]
- Dombroski D, Balasubramanian K, Schroit AJ. Phosphatidylserine expression on cell surfaces promotes antibody- dependent aggregation and thrombosis in beta2-glycoprotein I-immune mice. J Autoimmun. 2000;14(3):221–9. doi: 10.1006/jaut.2000.0365. [DOI] [PubMed] [Google Scholar]
- Doonan F, Cotter TG. Apoptosis: A potential therapeutic target for retinal degenerations. Curr Neurovasc Res. 2004;1(1):41–53. doi: 10.2174/1567202043480215. [DOI] [PubMed] [Google Scholar]
- Du L, Zhang X, Han YY, Burke NA, Kochanek PM, Watkins SC, Graham SH, Carcillo JA, Szabo C, Clark RS. Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J Biol Chem. 2003;278(20):18426–33. doi: 10.1074/jbc.M301295200. [DOI] [PubMed] [Google Scholar]
- Duvoisin RM, Zhang C, Pfankuch TF, O’Connor H, Gayet-Primo J, Quraishi S, Raber J. Increased measures of anxiety and weight gain in mice lacking the group III metabotropic glutamate receptor mGluR8. Eur J Neurosci. 2005;22(2):425–36. doi: 10.1111/j.1460-9568.2005.04210.x. [DOI] [PubMed] [Google Scholar]
- Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- Ekshyyan O, Aw TY. Apoptosis: A key in Neurodegenerative disorders. Curr Neurovasc Res. 2004;1(4):355–371. doi: 10.2174/1567202043362018. [DOI] [PubMed] [Google Scholar]
- Endoh T. Characterization of modulatory effects of postsynaptic metabotropic glutamate receptors on calcium currents in rat nucleus tractus solitarius. Brain Res. 2004;1024(1–2):212–24. doi: 10.1016/j.brainres.2004.07.074. [DOI] [PubMed] [Google Scholar]
- Ene FA, Kullmann PH, Gillespie DC, Kandler K. Glutamatergic calcium responses in the developing lateral superior olive: receptor types and their specific activation by synaptic activity patterns. J Neurophysiol. 2003;90(4):2581–91. doi: 10.1152/jn.00238.2003. [DOI] [PubMed] [Google Scholar]
- Engels IH, Stepczynska A, Stroh C, Lauber K, Berg C, Schwenzer R, Wajant H, Janicke RU, Porter AG, Belka C, Gregor M, Schulze-Osthoff K, Wesselborg S. Caspase-8/FLICE functions as an executioner caspase in anticancer drug-induced apoptosis. Oncogene. 2000;19(40):4563–73. doi: 10.1038/sj.onc.1203824. [DOI] [PubMed] [Google Scholar]
- Faden AI, O’Leary DM, Fan L, Bao W, Mullins PG, Movsesyan VA. Selective blockade of the mGluR1 receptor reduces traumatic neuronal injury in vitro and improves outcome after brain trauma. Exp Neurol. 2001;167(2):435–44. doi: 10.1006/exnr.2000.7577. [DOI] [PubMed] [Google Scholar]
- Fadok VA, de Cathelineau A, Daleke DL, Henson PM, Bratton DL. Loss of phospholipid asymmetry and surface exposure of phosphatidylserine is required for phagocytosis of apoptotic cells by macrophages and fibroblasts. J Biol Chem. 2001;276(2):1071–7. doi: 10.1074/jbc.M003649200. [DOI] [PubMed] [Google Scholar]
- Ferraguti F, Baldani-Guerra B, Corsi M, Nakanishi S, Corti C. Activation of the extracellular signal-regulated kinase 2 by metabotropic glutamate receptors. Eur J Neurosci. 1999;11(6):2073–2082. doi: 10.1046/j.1460-9568.1999.00626.x. [DOI] [PubMed] [Google Scholar]
- Figueroa S, Canadas S, Arce C, Oset-Gasque MJ, Gonzalez MP. SNAP, a NO donor, induces cortical neuron death by a mechanism in which the caspase pathway is implicated. Brain Res. 2005;1047(2):168–76. doi: 10.1016/j.brainres.2005.04.044. [DOI] [PubMed] [Google Scholar]
- Findik D, Song Q, Hidaka H, Lavin M. Protein kinase A inhibitors enhance radiation-induced apoptosis. J Cell Biochem. 1995;57(1):12–21. doi: 10.1002/jcb.240570103. [DOI] [PubMed] [Google Scholar]
- Folbergrova J, Druga R, Otahal J, Haugvicova R, Mares P, Kubova H. Seizures induced in immature rats by homocysteic acid and the associated brain damage are prevented by group II metabotropic glutamate receptor agonist (2R,4R)-4-aminopyrrolidine-2,4-dicarboxylate. Exp Neurol. 2005;192(2):420–36. doi: 10.1016/j.expneurol.2004.12.019. [DOI] [PubMed] [Google Scholar]
- Francesconi A, Duvoisin RM. Role of the second and third intracellular loops of metabotropic glutamate receptors in mediating dual signal transduction activation. J Biol Chem. 1998;273(10):5615–24. doi: 10.1074/jbc.273.10.5615. [DOI] [PubMed] [Google Scholar]
- Franco-Cea A, Valencia A, Sanchez-Armass S, Dominguez G, Moran J. Role of ionic fluxes in the apoptotic cell death of cultured cerebellar granule neurons. Neurochem Res. 2004;29(1):227–38. doi: 10.1023/b:nere.0000010501.25627.0f. [DOI] [PubMed] [Google Scholar]
- Fundytus ME, Yashpal K, Chabot JG, Osborne MG, Lefebvre CD, Dray A, Henry JL, Coderre TJ. Knockdown of spinal metabotropic glutamate receptor 1 (mGluR(1)) alleviates pain and restores opioid efficacy after nerve injury in rats. Br J Pharmacol. 2001;132(1):354–67. doi: 10.1038/sj.bjp.0703810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gastard MC, Troncoso JC, Koliatsos VE. Caspase activation in the limbic cortex of subjects with early Alzheimer’s disease. Ann Neurol. 2003;54(3):393–8. doi: 10.1002/ana.10680. [DOI] [PubMed] [Google Scholar]
- Geurts JJ, Wolswijk G, Bo L, Redeker S, Ramkema M, Troost D, Aronica E. Expression patterns of Group III metabotropic glutamate receptors mGluR4 and mGluR8 in multiple sclerosis lesions. J Neuroimmunol. 2005;158(1–2):182–90. doi: 10.1016/j.jneuroim.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Ghatan S, Larner S, Kinoshita Y, Hetman M, Patel L, Xia Z, Youle RJ, Morrison RS. p38 MAP kinase mediates bax translocation in nitric oxide-induced apoptosis in neurons. J Cell Biol. 2000;150(2):335–47. doi: 10.1083/jcb.150.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill SS, Pulido OM, Mueller RW, McGuire PF. Immunochemical localization of the metabotropic glutamate receptors in the rat heart. Brain Res Bull. 1999;48(2):143–6. doi: 10.1016/s0361-9230(98)00154-3. [DOI] [PubMed] [Google Scholar]
- Gillard SE, Tzaferis J, Tsui HC, Kingston AE. Expression of metabotropic glutamate receptors in rat meningeal and brain microvasculature and choroid plexus. J Comp Neurol. 2003;461(3):317–32. doi: 10.1002/cne.10671. [DOI] [PubMed] [Google Scholar]
- Goldshmit Y, Erlich S, Pinkas-Kramarski R. Neuregulin rescues PC12-ErbB4 cells from cell death induced by H(2)O(2). Regulation of reactive oxygen species levels by phosphatidylinositol 3-kinase. J Biol Chem. 2001;276(49):46379–85. doi: 10.1074/jbc.M105637200. [DOI] [PubMed] [Google Scholar]
- Golembiowska K, Konieczny J, Ossowska K, Wolfarth S. The role of striatal metabotropic glutamate receptors in degeneration of dopamine neurons: review article. Amino Acids. 2002;23(1–3):199–205. doi: 10.1007/s00726-001-0129-z. [DOI] [PubMed] [Google Scholar]
- Haak LL. Metabotropic glutamate receptor modulation of glutamate responses in the suprachiasmatic nucleus. J Neurophysiol. 1999;81(3):1308–17. doi: 10.1152/jn.1999.81.3.1308. [DOI] [PubMed] [Google Scholar]
- Han BH, Holtzman DM. BDNF protects the neonatal brain from hypoxic-ischemic injury in vivo via the ERK pathway. J Neurosci. 2000;20(15):5775–81. doi: 10.1523/JNEUROSCI.20-15-05775.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002;417(6885):182–7. doi: 10.1038/417182a. [DOI] [PubMed] [Google Scholar]
- Hatori K, Nagai A, Heisel R, Ryu JK, Kim SU. Fractalkine and fractalkine receptors in human neurons and glial cells. J Neurosci Res. 2002;69(3):418–26. doi: 10.1002/jnr.10304. [DOI] [PubMed] [Google Scholar]
- Hatsugai N, Kuroyanagi M, Yamada K, Meshi T, Tsuda S, Kondo M, Nishimura M, Hara-Nishimura I. A plant vacuolar protease, VPE, mediates virus-induced hypersensitive cell death. Science. 2004;305(5685):855–8. doi: 10.1126/science.1099859. [DOI] [PubMed] [Google Scholar]
- Hay M, Hoang CJ, Hasser EM, Price EM. Activation of metabotropic glutamate receptors inhibits synapsin I phosphorylation in visceral sensory neurons. J Membr Biol. 2000;178(3):195–204. doi: 10.1007/s002320010027. [DOI] [PubMed] [Google Scholar]
- Haynes T, Del Rio-Tsonis K. Retina repair, stem cells and beyond. Curr Neurovasc Res. 2004;1(3):231–239. doi: 10.2174/1567202043362216. [DOI] [PubMed] [Google Scholar]
- Henrich-Noack P, Reymann KG. (1S,3R)-ACPD, a metabotropic glutamate receptor agonist, enhances damage after global ischaemia. Eur J Pharmacol. 1999;365(1):55–8. doi: 10.1016/s0014-2999(98)00865-6. [DOI] [PubMed] [Google Scholar]
- Henry MK, Lynch JT, Eapen AK, Quelle FW. DNA damage-induced cell-cycle arrest of hematopoietic cells is overridden by activation of the PI-3 kinase/Akt signaling pathway. Blood. 2001;98(3):834–41. doi: 10.1182/blood.v98.3.834. [DOI] [PubMed] [Google Scholar]
- Hensley K, Floyd RA, Zheng NY, Nael R, Robinson KA, Nguyen X, Pye QN, Stewart CA, Geddes J, Markesbery WR, Patel E, Johnson GV, Bing G. p38 kinase is activated in the Alzheimer’s disease brain. J Neurochem. 1999;72(5):2053–8. doi: 10.1046/j.1471-4159.1999.0722053.x. [DOI] [PubMed] [Google Scholar]
- Hoffmann PR, deCathelineau AM, Ogden CA, Leverrier Y, Bratton DL, Daleke DL, Ridley AJ, Fadok VA, Henson PM. Phosphatidylserine (PS) induces PS receptor-mediated macropinocytosis and promotes clearance of apoptotic cells. J Cell Biol. 2001;155(4):649–59. doi: 10.1083/jcb.200108080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann K, Bucher P, Tschopp J. The CARD domain: a new apoptotic signalling motif. Trends Biochem Sci. 1997;22(5):155–6. doi: 10.1016/s0968-0004(97)01043-8. [DOI] [PubMed] [Google Scholar]
- Holscher C, Gigg J, O’Mara SM. Metabotropic glutamate receptor activation and blockade: their role in long-term potentiation, learning and neurotoxicity. Neurosci Biobehav Rev. 1999;23(3):399–410. doi: 10.1016/s0149-7634(98)00045-1. [DOI] [PubMed] [Google Scholar]
- Hong JR, Lin GH, Lin CJ, Wang WP, Lee CC, Lin TL, Wu JL. Phosphatidylserine receptor is required for the engulfment of dead apoptotic cells and for normal embryonic development in zebrafish. Development. 2004;131(21):5417–27. doi: 10.1242/dev.01409. [DOI] [PubMed] [Google Scholar]
- Houamed KM, Kuijper JL, Gilbert TL, Haldeman BA, O’Hara PJ, Mulvihill ER, Almers W, Hagen FS. Cloning, expression, and gene structure of a G protein-coupled glutamate receptor from rat brain. Science. 1991;252(5010):1318–21. doi: 10.1126/science.1656524. [DOI] [PubMed] [Google Scholar]
- Hu D, Cao K, Peterson-Wakeman R, Wang R. Altered profile of gene expression in rat hearts induced by chronic nicotine consumption. Biochem Biophys Res Commun. 2002;297(4):729–36. doi: 10.1016/s0006-291x(02)02280-5. [DOI] [PubMed] [Google Scholar]
- Hu Y, Benedict MA, Ding L, Nunez G. Role of cytochrome c and dATP/ATP hydrolysis in Apaf-1-mediated caspase- 9 activation and apoptosis. EMBO J. 1999;18(13):3586–95. doi: 10.1093/emboj/18.13.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LQ, Rowan MJ, Anwyl R. Role of protein kinases A and C in the induction of mGluR-dependent long-term depression in the medial perforant path of the rat dentate gyrus in vitro. Neurosci Lett. 1999;274(2):71–4. doi: 10.1016/s0304-3940(99)00561-3. [DOI] [PubMed] [Google Scholar]
- Hur EM, Kim KT. G protein-coupled receptor signalling and cross-talk. Achieving rapidity and specificity. Cell Signal. 2002;14(5):397–405. doi: 10.1016/s0898-6568(01)00258-3. [DOI] [PubMed] [Google Scholar]
- Jessel R, Haertel S, Socaciu C, Tykhonova S, Diehl HA. Kinetics of apoptotic markers in exogeneously induced apoptosis of EL4 cells. J Cell Mol Med. 2002;6(1):82–92. doi: 10.1111/j.1582-4934.2002.tb00313.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan J, Galindo MF, Miller RJ. Role of calpain- and interleukin-1 beta converting enzyme-like proteases in the beta-amyloid-induced death of rat hippocampal neurons in culture. J Neurochem. 1997;68(4):1612–21. doi: 10.1046/j.1471-4159.1997.68041612.x. [DOI] [PubMed] [Google Scholar]
- Jung KM, Mangieri R, Stapleton C, Kim J, Fegley D, Wallace M, Mackie K, Piomelli D. Stimulation of endocannabinoid formation in brain slice cultures through activation of group I metabotropic glutamate receptors. Mol Pharmacol. 2005 doi: 10.1124/mol.105.013961. [DOI] [PubMed] [Google Scholar]
- Juo P, Kuo CJ, Yuan J, Blenis J. Essential requirement for caspase-8/FLICE in the initiation of the Fas-induced apoptotic cascade. Curr Biol. 1998;8(18):1001–8. doi: 10.1016/s0960-9822(07)00420-4. [DOI] [PubMed] [Google Scholar]
- Kajta M, Marszal M, Kubera M, Lason W. Effects of estrone on quisqualate-induced toxicity in primary cultures of rat cortical neurons. J Physiol Pharmacol. 2005;56(2):233–45. [PubMed] [Google Scholar]
- Kalda A, Kaasik A, Vassiljev V, Pokk P, Zharkovsky A. Neuroprotective action of group I metabotropic glutamate receptor agonists against oxygen-glucose deprivation-induced neuronal death. Brain Res. 2000;853(2):370–3. doi: 10.1016/s0006-8993(99)02166-6. [DOI] [PubMed] [Google Scholar]
- Kalwy SA, Akbar MT, Coffin RS, de Belleroche J, Latchman DS. Heat shock protein 27 delivered via a herpes simplex virus vector can protect neurons of the hippocampus against kainic-acid-induced cell loss. Brain Res Mol Brain Res. 2003;111(1–2):91–103. doi: 10.1016/s0169-328x(02)00692-7. [DOI] [PubMed] [Google Scholar]
- Kang JQ, Chong ZZ, Maiese K. Akt1 protects against inflammatory microglial activation through maintenance of membrane asymmetry and modulation of cysteine protease activity. J Neurosci Res. 2003a;74(1):37–51. doi: 10.1002/jnr.10740. [DOI] [PubMed] [Google Scholar]
- Kang JQ, Chong ZZ, Maiese K. Critical role for Akt1 in the modulation of apoptotic phosphatidylserine exposure and microglial activation. Mol Pharmacol. 2003b;64(3):557–69. doi: 10.1124/mol.64.3.557. [DOI] [PubMed] [Google Scholar]
- Kennedy SG, Kandel ES, Cross TK, Hay N. Akt/Protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol Cell Biol. 1999;19(8):5800–10. doi: 10.1128/mcb.19.8.5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kermer P, Klocker N, Bahr M. Modulation of metabotropic glutamate receptors fails to prevent the loss of adult rat retinal ganglion cells following axotomy or N-methyl-D-aspartate lesion in vivo. Neurosci Lett. 2001;315(3):117–20. doi: 10.1016/s0304-3940(01)02318-7. [DOI] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239–57. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khwaja A. Akt is more than just a Bad kinase. Nature. 1999;401(6748):33–4. doi: 10.1038/43354. [DOI] [PubMed] [Google Scholar]
- Kingston AE, O’Neill MJ, Lam A, Bales KR, Monn JA, Schoepp DD. Neuroprotection by metabotropic glutamate receptor glutamate receptor agonists: LY354740, LY379268 and LY389795. Eur J Pharmacol. 1999;377(2–3):155–65. doi: 10.1016/s0014-2999(99)00397-0. [DOI] [PubMed] [Google Scholar]
- Ko HW, Han KS, Kim EY, Ryu BR, Yoon WJ, Jung YK, Kim SU, Gwag BJ. Synergetic activation of p38 mitogen-activated protein kinase and caspase-3-like proteases for execution of calyculin A-induced apoptosis but not N-methyl-d-aspartate-induced necrosis in mouse cortical neurons. J Neurochem. 2000;74(6):2455–61. doi: 10.1046/j.1471-4159.2000.0742455.x. [DOI] [PubMed] [Google Scholar]
- Krizbai IA, Deli MA, Pestenacz A, Siklos L, Szabo CA, Andras I, Joo F. Expression of glutamate receptors on cultured cerebral endothelial cells. J Neurosci Res. 1998;54(6):814–9. doi: 10.1002/(SICI)1097-4547(19981215)54:6<814::AID-JNR9>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Lachica EA, Rubsamen R, Zirpel L, Rubel EW. Glutamatergic inhibition of voltage-operated calcium channels in the avian cochlear nucleus. J Neurosci. 1995;15(3 Pt 1):1724–34. doi: 10.1523/JNEUROSCI.15-03-01724.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafon-Cazal M, Fagni L, Guiraud MJ, Mary S, Lerner-Natoli M, Pin JP, Shigemoto R, Bockaert J. mGluR7-like metabotropic glutamate receptors inhibit NMDA-mediated excitotoxicity in cultured mouse cerebellar granule neurons. Eur J Neurosci. 1999;11(2):663–72. doi: 10.1046/j.1460-9568.1999.00475.x. [DOI] [PubMed] [Google Scholar]
- Latchman D. Protective Effect of Heat Shock Proteins in the Nervous System. Curr Neurovasc Res. 2004;1(1):21–27. doi: 10.2174/1567202043480206. [DOI] [PubMed] [Google Scholar]
- Lauber K, Bohn E, Krober SM, Xiao YJ, Blumenthal SG, Lindemann RK, Marini P, Wiedig C, Zobywalski A, Baksh S, Xu Y, Autenrieth IB, Schulze-Osthoff K, Belka C, Stuhler G, Wesselborg S. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction Signal. Cell. 2003;113(6):717–30. doi: 10.1016/s0092-8674(03)00422-7. [DOI] [PubMed] [Google Scholar]
- Lee RK, Wurtman RJ, Cox AJ, Nitsch RM. Amyloid precursor protein processing is stimulated by metabotropic glutamate receptors. Proc Natl Acad Sci USA. 1995;92(17):8083–7. doi: 10.1073/pnas.92.17.8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SR, Lo EH. Interactions between p38 mitogen-activated protein kinase and caspase-3 in cerebral endothelial cell death after hypoxia-reoxygenation. Stroke. 2003;34(11):2704–9. doi: 10.1161/01.STR.0000096540.40826.BA. [DOI] [PubMed] [Google Scholar]
- Leker RR, Shohami E. Cerebral ischemia and trauma-different etiologies yet similar mechanisms: neuroprotective opportunities. Brain Res Brain Res Rev. 2002;39(1):55–73. doi: 10.1016/s0165-0173(02)00157-1. [DOI] [PubMed] [Google Scholar]
- Lemmon MA, Ferguson KM, Abrams CS. Pleckstrin homology domains and the cytoskeleton. FEBS Lett. 2002;513(1):71–6. doi: 10.1016/s0014-5793(01)03243-4. [DOI] [PubMed] [Google Scholar]
- Li F, Chong ZZ, Maiese K. Navigating novel mechanisms of cellular plasticity with the NAD+ precursor and nutrient nicotinamide. Front Biosci. 2004;9:2500–2520. doi: 10.2741/1412. [DOI] [PubMed] [Google Scholar]
- Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94(4):491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412(6842):95–9. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91(4):479–89. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Lin SH, Chong ZZ, Maiese K. The metabotropic glutamate receptor system: G-Protein mediated pathways that modulate neuronal and vascular cellular injury. Curr Med Chem -Central Nervous System Agents. 2002;2(1):17–28. [Google Scholar]
- Lin SH, Maiese K. Group I metabotropic glutamate receptors prevent endothelial programmed cell death independent from MAP kinase p38 activation in rat. Neurosci Lett. 2001a;298(3):207–11. doi: 10.1016/s0304-3940(00)01766-3. [DOI] [PubMed] [Google Scholar]
- Lin SH, Maiese K. The metabotropic glutamate receptor system protects against ischemic free radical programmed cell death in rat brain endothelial cells. J Cereb Blood Flow Metab. 2001b;21(3):262–75. doi: 10.1097/00004647-200103000-00010. [DOI] [PubMed] [Google Scholar]
- Lin SH, Vincent A, Shaw T, Maynard KI, Maiese K. Prevention of nitric oxide-induced neuronal injury through the modulation of independent pathways of programmed cell death. J Cereb Blood Flow Metab. 2000;20(9):1380–91. doi: 10.1097/00004647-200009000-00013. [DOI] [PubMed] [Google Scholar]
- Lin WW, Wang CW, Chuang DM. Effects of depolarization and NMDA antagonists on the role survival of cerebellar granule cells: a pivotal role for protein kinase C isoforms. J Neurochem. 1997;68(6):2577–86. doi: 10.1046/j.1471-4159.1997.68062577.x. [DOI] [PubMed] [Google Scholar]
- Liu D, Slevin JR, Lu C, Chan SL, Hansson M, Elmer E, Mattson MP. Involvement of mitochondrial K+ release and cellular efflux in ischemic and apoptotic neuronal death. J Neurochem. 2003;86(4):966–79. doi: 10.1046/j.1471-4159.2003.01913.x. [DOI] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86(1):147–57. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Liu XB, Munoz A, Jones EG. Changes in subcellular localization of metabotropic glutamate receptor subtypes during postnatal development of mouse thalamus. J Comp Neurol. 1998;395(4):450–65. doi: 10.1002/(sici)1096-9861(19980615)395:4<450::aid-cne3>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Lizcano JM, Morrice N, Cohen P. Regulation of BAD by cAMP-dependent protein kinase is mediated via phosphorylation of a novel site, Ser155. Biochem J. 2000;349(Pt 2):547–57. doi: 10.1042/0264-6021:3490547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Bendito G, Shigemoto R, Fairen A, Lujan R. Differential distribution of group I metabotropic glutamate receptors during rat cortical Development. Cereb Cortex. 2002;12(6):625–38. doi: 10.1093/cercor/12.6.625. [DOI] [PubMed] [Google Scholar]
- Losonczy A, Somogyi P, Nusser Z. Reduction of excitatory postsynaptic responses by persistently active metabotropic glutamate receptors in the hippocampus. J Neurophysiol. 2003;89(4):1910–9. doi: 10.1152/jn.00842.2002. [DOI] [PubMed] [Google Scholar]
- Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, Salvesen GS, Koo EH, Bredesen DE. A second cytotoxic proteolytic peptide derived from amyloid beta-protein precursor. Nat Med. 2000;6(4):397–404. doi: 10.1038/74656. [DOI] [PubMed] [Google Scholar]
- Lu J, Goula D, Sousa N, Almeida OF. Ionotropic and metabotropic glutamate receptor mediation of glucocorticoid-induced apoptosis in hippocampal cells and the neuroprotective role of synaptic N-methyl-D-aspartate receptors. Neuroscience. 2003;121(1):123–31. doi: 10.1016/s0306-4522(03)00421-4. [DOI] [PubMed] [Google Scholar]
- Lujan R, Roberts JD, Shigemoto R, Ohishi H, Somogyi P. Differential plasma membrane distribution of metabotropic glutamate receptors mGluR1 alpha, mGluR2 and mGluR5, relative to neurotransmitter release sites. J Chem Neuroanat. 1997;13(4):219–41. doi: 10.1016/s0891-0618(97)00051-3. [DOI] [PubMed] [Google Scholar]
- Luo Z, Fujio Y, Kureishi Y, Rudic RD, Daumerie G, Fulton D, Sessa WC, Walsh K. Acute modulation of endothelial Akt/PKB activity alters nitric oxide-dependent vasomotor activity in vivo. J Clin Invest. 2000;106(4):493–9. doi: 10.1172/JCI9419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane M, Cohen GM, Dickens M. JNK (c-Jun N-terminal kinase) and p38 activation in receptor-mediated and chemically-induced apoptosis of T-cells: differential requirements for caspase activation. Biochem J. 2000;348(Pt 1):93–101. [PMC free article] [PubMed] [Google Scholar]
- Madaio MP, Fabbi M, Tiso M, Daga A, Puccetti A. Spontaneously produced anti-DNA/DNase I autoantibodies modulate nuclear apoptosis in living cells. Eur J Immunol. 1996;26(12):3035–41. doi: 10.1002/eji.1830261232. [DOI] [PubMed] [Google Scholar]
- Maiese K, Ahmad I, TenBroeke M, Gallant J. Metabotropic glutamate receptor subtypes independently modulate neuronal intracellular calcium. J Neurosci Res. 1999a;55:472–485. doi: 10.1002/(SICI)1097-4547(19990215)55:4<472::AID-JNR7>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Maiese K, Boniece I, DeMeo D, Wagner JA. Peptide growth factors protect against ischemia in culture by preventing nitric oxide toxicity. J Neurosci. 1993a;13(7):3034–40. doi: 10.1523/JNEUROSCI.13-07-03034.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiese K, Boniece IR, Skurat K, Wagner JA. Protein kinases modulate the sensitivity of hippocampal neurons to nitric oxide toxicity and anoxia. J Neurosci Res. 1993b;36(1):77–87. doi: 10.1002/jnr.490360109. [DOI] [PubMed] [Google Scholar]
- Maiese K, Chong ZZ. Nicotinamide: necessary nutrient emerges as a novel cytoprotectant for the brain. Trends Pharmacol Sci. 2003a;24(5):228–32. doi: 10.1016/S0165-6147(03)00078-6. [DOI] [PubMed] [Google Scholar]
- Maiese K, Chong ZZ. Insights into oxidative stress and potential novel therapeutic targets for Alzheimer disease. Restor Neurol Neurosci. 2004a;22(2):87–104. [PubMed] [Google Scholar]
- Maiese K, Chong ZZ, Kang J. Transformation into treatment: Novel therapeutics that begin within the Cell. In: Maiese K, editor. Neuronal and Vascular Plasticity: Elucidating Basic Cellular Mechanisms for Future Therapeutic Discovery. Kluwer Academic Publishers; Norwell, MA: 2003b. pp. 1–26. [Google Scholar]
- Maiese K, Greenberg R, Boccone L, Swiriduk M. Activation of the metabotropic glutamate receptor is neuroprotective during nitric oxide toxicity in primary hippocampal neurons of rats. Neurosci Lett. 1995;194(3):173–6. doi: 10.1016/0304-3940(95)11753-j. [DOI] [PubMed] [Google Scholar]
- Maiese K, Li F, Chong ZZ. Erythropoietin in the brain: can the promise to protect be fulfilled? Trends Pharmacol Sci. 2004b;25(11):577–583. doi: 10.1016/j.tips.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Maiese K, Li F, Chong ZZ. Erythropoietin and cancer. JAMA. 2005a;293(15):1858–1859. doi: 10.1001/jama.293.15.1858-a. [DOI] [PubMed] [Google Scholar]
- Maiese K, Li F, Chong ZZ. New avenues of exploration for erythropoietin. JAMA. 2005b;293(1):90–5. doi: 10.1001/jama.293.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiese K, Lin S, Chong ZZ. Elucidating neuronal and vascular injury through the cytoprotective agent nicotinamide. Curr Med Chem-Imm Endoc Metab Agents. 2001;1(3):257–267. [Google Scholar]
- Maiese K, Swiriduk M, TenBroeke M. Cellular mechanisms of protection by metabotropic glutamate receptors during anoxia and nitric oxide toxicity. J Neurochem. 1996;66(6):2419–28. doi: 10.1046/j.1471-4159.1996.66062419.x. [DOI] [PubMed] [Google Scholar]
- Maiese K, Vincent A, Lin SH, Shaw T. Group I and Group III metabotropic glutamate receptor subtypes provide enhanced neuroprotection. J Neurosci Res. 2000a;62(2):257–272. doi: 10.1002/1097-4547(20001015)62:2<257::AID-JNR10>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Maiese K, Vincent AM. Group I metabotropic receptors down-regulate nitric oxide induced caspase-3 activity in rat hippocampal neurons. Neurosci Lett. 1999b;264(1–3):17–20. doi: 10.1016/s0304-3940(99)00199-8. [DOI] [PubMed] [Google Scholar]
- Maiese K, Vincent AM. Membrane asymmetry and DNA degradation: functionally distinct determinants of neuronal programmed cell death. J Neurosci Res. 2000b;59(4):568–80. doi: 10.1002/(SICI)1097-4547(20000215)59:4<568::AID-JNR13>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Makoff A, Lelchuk R, Oxer M, Harrington K, Emson P. Molecular characterization and localization of human metabotropic glutamate receptor type 4. Brain Res Mol Brain Res. 1996a;37(1–2):239–48. doi: 10.1016/0169-328x(95)00321-i. [DOI] [PubMed] [Google Scholar]
- Makoff A, Volpe F, Lelchuk R, Harrington K, Emson P. Molecular characterization and localization of human metabotropic glutamate receptor type 3. Brain Res Mol Brain Res. 1996b;40(1):55–63. doi: 10.1016/0169-328x(96)00037-x. [DOI] [PubMed] [Google Scholar]
- Manahan-Vaughan D. Group 1 and 2 metabotropic glutamate receptors play differential roles in hippocampal long-term depression and long-term potentiation in freely moving rats. J Neurosci. 1997;17(9):3303–11. doi: 10.1523/JNEUROSCI.17-09-03303.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao L, Wang JQ. Group I metabotropic glutamate receptor-mediated calcium signalling and immediate early gene expression in cultured rat striatal neurons. Eur J Neurosci. 2003;17(4):741–50. doi: 10.1046/j.1460-9568.2003.02495.x. [DOI] [PubMed] [Google Scholar]
- Marabese I, de Novellis V, Palazzo E, Mariani L, Siniscalco D, Rodella L, Rossi F, Maione S. Differential roles of mGlu8 receptors in the regulation of glutamate and gamma-aminobutyric acid release at periaqueductal grey level. Neuropharmacology. 2005;49(Suppl):157–66. doi: 10.1016/j.neuropharm.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Marinissen MJ, Gutkind JS. G-protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol Sci. 2001;22(7):368–76. doi: 10.1016/s0165-6147(00)01678-3. [DOI] [PubMed] [Google Scholar]
- Martin D, Salinas M, Lopez-Valdaliso R, Serrano E, Recuero M, Cuadrado A. Effect of the Alzheimer amyloid fragment Abeta(25–35) on Akt/PKB kinase and survival of PC12 cells. J Neurochem. 2001;78(5):1000–8. doi: 10.1046/j.1471-4159.2001.00472.x. [DOI] [PubMed] [Google Scholar]
- Maruta H, Matsumura N, Tanuma S. Role of (ADP-ribose)n catabolism in DNA repair. Biochem Biophys Res Commun. 1997;236(2):265–9. doi: 10.1006/bbrc.1997.6910. [DOI] [PubMed] [Google Scholar]
- Masu M, Tanabe Y, Tsuchida K, Shigemoto R, Nakanishi S. Sequence and expression of a metabotropic glutamate receptor. Nature. 1991;349(6312):760–5. doi: 10.1038/349760a0. [DOI] [PubMed] [Google Scholar]
- Matarredona ER, Santiago M, Venero JL, Cano J, Machado A. Group II metabotropic glutamate receptor activation protects striatal dopaminergic nerve terminals against MPP+-induced neurotoxicity along with brain-derived neurotrophic factor induction. J Neurochem. 2001;76(2):351–60. doi: 10.1046/j.1471-4159.2001.00056.x. [DOI] [PubMed] [Google Scholar]
- Matsuzaki H, Tamatani M, Mitsuda N, Namikawa K, Kiyama H, Miyake S, Tohyama M. Activation of Akt kinase inhibits apoptosis and changes in Bcl-2 and Bax expression induced by nitric oxide in primary hippocampal neurons. J Neurochem. 1999;73(5):2037–46. [PubMed] [Google Scholar]
- Mills CD, Fullwood SD, Hulsebosch CE. Changes in metabotropic glutamate receptor expression following spinal cord injury. Exp Neurol. 2001;170(2):244–57. doi: 10.1006/exnr.2001.7721. [DOI] [PubMed] [Google Scholar]
- Montague JW, Hughes F, Jr, Cidlowski JA. Native recombinant cyclophilins A, B, and C degrade DNA independently of peptidylprolyl cis-trans-isomerase activity. Potential roles of cyclophilins in apoptosis. J Biol Chem. 1997;272(10):6677–84. doi: 10.1074/jbc.272.10.6677. [DOI] [PubMed] [Google Scholar]
- Mouzaki A, Tselios T, Papathanassopoulos P, Matsoukas I, Chatzantoni K. Immunotherapy for Multiple Sclerosis: Basic insights for new clinical strategies. Curr Neurovasc Res. 2004;1(4):325–340. doi: 10.2174/1567202043362180. [DOI] [PubMed] [Google Scholar]
- Musashi M, Ota S, Shiroshita N. The role of protein kinase C isoforms in cell proliferation and apoptosis. Int J Hematol. 2000;72(1):12–9. [PubMed] [Google Scholar]
- Namikawa K, Honma M, Abe K, Takeda M, Mansur K, Obata T, Miwa A, Okado H, Kiyama H. Akt/protein kinase B prevents injury-induced motoneuron death and accelerates axonal regeneration. J Neurosci. 2000;20(8):2875–86. doi: 10.1523/JNEUROSCI.20-08-02875.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas AH, Hyson RL. Group I and II metabotropic glutamate receptors are necessary for the activity-dependent regulation of ribosomes in chick auditory neurons. Brain Res. 2004;1014(1–2):110–9. doi: 10.1016/j.brainres.2004.03.066. [DOI] [PubMed] [Google Scholar]
- Nishimura I, Uetsuki T, Kuwako K, Hara T, Kawakami T, Aimoto S, Yoshikawa K. Cell death induced by a caspase-cleaved transmembrane fragment of the Alzheimer amyloid precursor protein. Cell Death Differ. 2002;9(2):199–208. doi: 10.1038/sj.cdd.4400931. [DOI] [PubMed] [Google Scholar]
- Nishio E, Watanabe Y. Nitric oxide donor-induced apoptosis in smooth muscle cells is modulated by protein kinase C and protein kinase A. Eur J Pharmacol. 1997;339(2–3):245–51. doi: 10.1016/s0014-2999(97)01368-x. [DOI] [PubMed] [Google Scholar]
- Nomura A, Shigemoto R, Nakamura Y, Okamoto N, Mizuno N, Nakanishi S. Developmentally regulated postsynaptic localization of a metabotropic glutamate receptor in rat rod bipolar cells. Cell. 1994;77(3):361–9. doi: 10.1016/0092-8674(94)90151-1. [DOI] [PubMed] [Google Scholar]
- Oka A, Takashima S. The up-regulation of metabotropic glutamate receptor 5 (mGluR5) in Down’s syndrome brains. Acta Neuropathol (Berl) 1999;97(3):275–8. doi: 10.1007/s004010050985. [DOI] [PubMed] [Google Scholar]
- Orlov SN, Thorin-Trescases N, Dulin NO, Dam TV, Fortuno MA, Tremblay J, Hamet P. Activation of cAMP signaling transiently inhibits apoptosis in vascular smooth muscle cells in a site upstream of caspase-3. Cell Death Differ. 1999;6(7):661–72. doi: 10.1038/sj.cdd.4400539. [DOI] [PubMed] [Google Scholar]
- Panchal M, Rholam M, Brakch N. Abnormalities of peptide metabolism in Alzheimer disease. Curr Neurovasc Res. 2004;1(4):317–323. doi: 10.2174/1567202043362117. [DOI] [PubMed] [Google Scholar]
- Pandey S, Walker PR, Sikorska M. Identification of a novel 97 kDa endonuclease capable of internucleosomal DNA cleavage. Biochemistry. 1997;36(4):711–20. doi: 10.1021/bi962387h. [DOI] [PubMed] [Google Scholar]
- Parvathenani LK, Buescher ES, Chacon-Cruz E, Beebe SJ. Type I cAMP-dependent protein kinase delays apoptosis in human neutrophils at a site upstream of caspase-3. J Biol Chem. 1998;273(12):6736–43. doi: 10.1074/jbc.273.12.6736. [DOI] [PubMed] [Google Scholar]
- Paucard A, Palmier B, Croci N, Taillieu F, Plotkine M, Margaill I. Biphasic modulation by nitric oxide of caspase activation due to malonate injection in rat striatum. Eur J Pharmacol. 2004;483(2–3):259–65. doi: 10.1016/j.ejphar.2003.10.038. [DOI] [PubMed] [Google Scholar]
- Pellegrini-Giampietro DE, Peruginelli F, Meli E, Cozzi A, Albani-Torregrossa S, Pellicciari R, Moroni F. Protection with metabotropic glutamate 1 receptor antagonists in models of ischemic neuronal death: time-course and mechanisms. Neuropharmacology. 1999;38(10):1607–19. doi: 10.1016/s0028-3908(99)00097-0. [DOI] [PubMed] [Google Scholar]
- Philpott KL, McCarthy MJ, Klippel A, Rubin LL. Activated phosphatidylinositol 3-kinase and Akt kinase promote survival of superior cervical neurons. J Cell Biol. 1997;139(3):809–15. doi: 10.1083/jcb.139.3.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzi M, Benarese M, Boroni F, Goffi F, Valerio A, Spano PF. Neuroprotection by metabotropic glutamate receptor agonists on kainate-induced degeneration of motor neurons in spinal cord slices from adult rat. Neuropharmacology. 2000;39(5):903–10. doi: 10.1016/s0028-3908(99)00257-9. [DOI] [PubMed] [Google Scholar]
- Pompl PN, Yemul S, Xiang Z, Ho L, Haroutunian V, Purohit D, Mohs R, Pasinetti GM. Caspase gene expression in the brain as a function of the clinical progression of Alzheimer disease. Arch Neurol. 2003;60(3):369–76. doi: 10.1001/archneur.60.3.369. [DOI] [PubMed] [Google Scholar]
- Prezeau L, Manzoni O, Homburger V, Sladeczek F, Curry K, Bockaert J. Characterization of a metabotropic glutamate receptor: direct negative coupling to adenylyl cyclase and involvement of a pertussis toxin-sensitive G protein. Proc Natl Acad Sci USA. 1992;89(17):8040–4. doi: 10.1073/pnas.89.17.8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugazhenthi S, Nesterova A, Jambal P, Audesirk G, Kern M, Cabell L, Eves E, Rosner MR, Boxer LM, Reusch JE. Oxidative stress-mediated down-regulation of bcl-2 promoter in hippocampal neurons. J Neurochem. 2003;84(5):982–96. doi: 10.1046/j.1471-4159.2003.01606.x. [DOI] [PubMed] [Google Scholar]
- Quiroz JA, Singh J, Gould TD, Denicoff KD, Zarate CA, Manji HK. Emerging experimental therapeutics for bipolar disorder: clues from the molecular pathophysiology. Mol Psychiatry. 2004;9(8):756–76. doi: 10.1038/sj.mp.4001521. [DOI] [PubMed] [Google Scholar]
- Reid SN, Daw NW, Gregory DS, Flavin H. cAMP levels increased by activation of metabotropic glutamate receptors correlate with visual plasticity. J Neurosci. 1996;16(23):7619–26. doi: 10.1523/JNEUROSCI.16-23-07619.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel G. Function of metabotropic glutamate receptors in learning and memory. Trends Neurosci. 1996;19(6):219–24. doi: 10.1016/0166-2236(96)20012-8. [DOI] [PubMed] [Google Scholar]
- Ritucci NA, Dean JB, Putnam RW. Intracellular pH response to hypercapnia in neurons from chemosensitive areas of the medulla. Am J Physiol. 1997;273(1 Pt 2):R433–41. doi: 10.1152/ajpregu.1997.273.1.R433. [DOI] [PubMed] [Google Scholar]
- Roberts E, Jr, Chih CP. The influence of age of pH regulation in hippocampal slices before, during, and after anoxia. J Cereb Blood Flow Metab. 1997;17(5):560–6. doi: 10.1097/00004647-199705000-00010. [DOI] [PubMed] [Google Scholar]
- Rong R, Ahn JY, Huang H, Nagata E, Kalman D, Kapp JA, Tu J, Worley PF, Snyder SH, Ye K. PI3 kinase enhancer-Homer complex couples mGluRI to PI3 kinase, preventing neuronal apoptosis. Nat Neurosci. 2003;6(11):1153–61. doi: 10.1038/nn1134. [DOI] [PubMed] [Google Scholar]
- Rossi AG, Cousin JM, Dransfield I, Lawson MF, Chilvers ER, Haslett C. Agents that elevate cAMP inhibit human neutrophil apoptosis. Biochem Biophys Res Commun. 1995;217(3):892–9. doi: 10.1006/bbrc.1995.2855. [DOI] [PubMed] [Google Scholar]
- Rytomaa M, Lehmann K, Downward J. Matrix detachment induces caspase-dependent cytochrome c release from mitochondria: inhibition by PKB/Akt but not Raf signalling. Oncogene. 2000;19(39):4461–8. doi: 10.1038/sj.onc.1203805. [DOI] [PubMed] [Google Scholar]
- Sabelhaus CF, Schroder UH, Breder J, Henrich-Noack P, Reymann KG. Neuroprotection against hypoxic/hypoglycaemic injury after the insult by the group III metabotropic glutamate receptor agonist (R, S)-4- phosphonophenylglycine. Br J Pharmacol. 2000;131(4):655–8. doi: 10.1038/sj.bjp.0703646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagara Y, Schubert D. The activation of metabotropic glutamate receptors protects nerve cells from oxidative stress. J Neurosci. 1998;18(17):6662–71. doi: 10.1523/JNEUROSCI.18-17-06662.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas M, Diaz R, Abraham NG, Ruiz de Galarreta CM, Cuadrado A. Nerve growth factor protects against 6-hydroxydopamine-induced oxidative stress by increasing expression of heme oxygenase-1 in a phosphatidylinositol 3-kinase-dependent manner. J Biol Chem. 2003;278(16):13898–904. doi: 10.1074/jbc.M209164200. [DOI] [PubMed] [Google Scholar]
- Salt TE, Turner JP. Modulation of sensory inhibition in the ventrobasal thalamus via activation of group II metabotropic glutamate receptors by 2R,4R- aminopyrrolidine-2,4-dicarboxylate. Exp Brain Res. 1998;121(2):181–5. doi: 10.1007/s002210050450. [DOI] [PubMed] [Google Scholar]
- Sankarapandi S, Zweier JL, Mukherjee G, Quinn MT, Huso DL. Measurement and characterization of superoxide generation in microglial cells: evidence for an NADPH oxidase-dependent pathway. Arch Biochem Biophys. 1998;353(2):312–21. doi: 10.1006/abbi.1998.0658. [DOI] [PubMed] [Google Scholar]
- Saransaari P, Oja SS. Metabotropic glutamate receptors modulate GABA release from mouse hippocampal slices. Neurochem Res. 2001;26(2):175–80. doi: 10.1023/a:1011055014357. [DOI] [PubMed] [Google Scholar]
- Satoh MS, Lindahl T. Role of poly(ADP-ribose) formation in DNA repair. Nature. 1992;356(6367):356–8. doi: 10.1038/356356a0. [DOI] [PubMed] [Google Scholar]
- Savill J. Recognition and phagocytosis of cells undergoing apoptosis. Br Med Bull. 1997;53(3):491–508. doi: 10.1093/oxfordjournals.bmb.a011626. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81(2):741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Selvatici R, Marino S, Piubello C, Rodi D, Beani L, Gandini E, Siniscalchi A. Protein kinase C activity, translocation, and selective isoform subcellular redistribution in the rat cerebral cortex after in vitro ischemia. J Neurosci Res. 2003;71(1):64–71. doi: 10.1002/jnr.10464. [DOI] [PubMed] [Google Scholar]
- Sensi SL, Jeng JM. Rethinking the excitotoxic ionic milieu: the emerging role of zn(2+) in ischemic neuronal injury. Curr Mol Med. 2004;4(2):87–111. doi: 10.2174/1566524043479211. [DOI] [PubMed] [Google Scholar]
- Sharma SK, Ebadi M. Metallothionein attenuates 3-morpholinosydnonimine (SIN-1)-induced oxidative stress in dopaminergic neurons. Antioxid Redox Signal. 2003;5(3):251–64. doi: 10.1089/152308603322110832. [DOI] [PubMed] [Google Scholar]
- Shen H, Chan J, Kass IS, Bergold PJ. Transient acidosis induces delayed neurotoxicity in cultured hippocampal slices. Neurosci Lett. 1995;185(2):115–8. doi: 10.1016/0304-3940(94)11238-e. [DOI] [PubMed] [Google Scholar]
- Shen KZ, Johnson SW. A slow excitatory postsynaptic current mediated by G-protein-coupled metabotropic glutamate receptors in rat ventral tegmental dopamine neurons. Eur J Neurosci. 1997;9(1):48–54. doi: 10.1111/j.1460-9568.1997.tb01352.x. [DOI] [PubMed] [Google Scholar]
- Shi Y. Caspase activation: revisiting the induced proximity model. Cell. 2004;117(7):855–8. doi: 10.1016/j.cell.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Kulik A, Roberts JD, Ohishi H, Nusser Z, Kaneko T, Somogyi P. Target-cell-specific concentration of a metabotropic glutamate receptor in the presynaptic active zone. Nature. 1996;381(6582):523–5. doi: 10.1038/381523a0. [DOI] [PubMed] [Google Scholar]
- Shimazoe T, Doi Y, Arai I, Yoshimatsu A, Fukumoto T, Watanabe S. Both metabotropic glutamate I and II receptors mediate augmentation of dopamine release from the striatum in methamphetamine-sensitized rats. Jpn J Pharmacol. 2002;89(1):85–8. doi: 10.1254/jjp.89.85. [DOI] [PubMed] [Google Scholar]
- Shuaib A, Kanthan R. Amplification of inhibitory mechanisms in cerebral ischemia: an alternative approach to neuronal protection. Histol Histopathol. 1997;12(1):185–94. [PubMed] [Google Scholar]
- Simak J, Holada K, Vostal JG. Release of annexin V-binding membrane microparticles from cultured human umbilical vein endothelial cells after treatment with camptothecin. BMC Cell Biol. 2002;3(1):11. doi: 10.1186/1471-2121-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladeczek F, Pin J-P, Recasens M, Bockaert J, Weiss S. Glutamate stimulates inositol phosphate formation in striatal neurones. Nature (Lond) 1985;317:717–719. doi: 10.1038/317717a0. [DOI] [PubMed] [Google Scholar]
- Smeitink JAM, van den Heuvel L, Koopman WJH, Nijtmans LGJ, Ugalde C, Willems P. Cell biological consequences of mitochondrial NADH: Ubiquinone oxidoreductase deficiency. Curr Neurovasc Res. 2004;1(1):29–40. doi: 10.2174/1567202043480224. [DOI] [PubMed] [Google Scholar]
- Spillson AB, Russell JW. Metabotropic glutamate receptor regulation of neuronal cell death. Exp Neurol. 2003;184(Suppl 1):S97–105. doi: 10.1016/j.expneurol.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Spitzer NC, Olson E, Gu X. Spontaneous calcium transients regulate neuronal plasticity in developing neurons. J Neurobiol. 1995;26(3):316–24. doi: 10.1002/neu.480260304. [DOI] [PubMed] [Google Scholar]
- Stefani A, Pisani A, Mercuri NB, Bernardi G, Calabresi P. Activation of metabotropic glutamate receptors inhibits calcium currents and GABA-mediated synaptic potentials in striatal neurons. J Neurosci. 1994;14(11 Pt 1):6734–43. doi: 10.1523/JNEUROSCI.14-11-06734.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegh AH, Barnhart BC, Volkland J, Algeciras-Schimnich A, Ke N, Reed JC, Peter ME. Inactivation of caspase-8 on mitochondria of Bcl-xL-expressing MCF7-Fas cells: role for the bifunctional apoptosis regulator protein. J Biol Chem. 2002;277(6):4351–60. doi: 10.1074/jbc.M108947200. [DOI] [PubMed] [Google Scholar]
- Suhara T, Mano T, Oliveira BE, Walsh K. Phosphatidylinositol 3-kinase/Akt signaling controls endothelial cell sensitivity to Fas-mediated apoptosis via regulation of FLICE- inhibitory protein (FLIP) Circ Res. 2001;89(1):13–9. doi: 10.1161/hh1301.092506. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Thompson MB, Scheff SW. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp Neurol. 1999;160(1):226–34. doi: 10.1006/exnr.1999.7197. [DOI] [PubMed] [Google Scholar]
- Sun XM, Cohen GM. Mg(2+)-dependent cleavage of DNA into kilobase pair fragments is responsible for the initial degradation of DNA in apoptosis. J Biol Chem. 1994;269(21):14857–60. [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397(6718):441–6. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Szapiro G, Izquierdo LA, Alonso M, Barros D, Paratcha G, Ardenghi P, Pereira P, Medina JH, Izquierdo I. Participation of hippocampal metabotropic glutamate receptors, protein kinase A and mitogen-activated protein kinases in memory retrieval. Neuroscience. 2000;99(1):1–5. doi: 10.1016/s0306-4522(00)00236-0. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Nakamura S, Asano K, Kinouchi M, Ishida-Yamamoto A, Iizuka H. Fas antigen modulates ultraviolet B-induced apoptosis of SVHK cells: sequential activation of caspases 8, 3, and 1 in the apoptotic process. Exp Cell Res. 1999;249(2):291–8. doi: 10.1006/excr.1999.4476. [DOI] [PubMed] [Google Scholar]
- Tang FR, Lee WL. Expression of the group II and III metabotropic glutamate receptors in the hippocampus of patients with mesial temporal lobe epilepsy. J Neurocytol. 2001a;30(2):137–43. doi: 10.1023/a:1011939223872. [DOI] [PubMed] [Google Scholar]
- Tang FR, Lee WL, Yang J, Sim MK, Ling EA. Metabotropic glutamate receptor 8 in the rat hippocampus after pilocarpine induced status epilepticus. Neurosci Lett. 2001b;300(3):137–40. doi: 10.1016/s0304-3940(01)01579-8. [DOI] [PubMed] [Google Scholar]
- Taylor DL, Diemel LT, Pocock JM. Activation of microglial group III metabotropic glutamate receptors protects neurons against microglial neurotoxicity. J Neurosci. 2003;23(6):2150–60. doi: 10.1523/JNEUROSCI.23-06-02150.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesco G, Koh YH, Tanzi RE. Caspase activation increases Abeta generation independently of caspase cleavage of APP. J Biol Chem. 2003;5:5. doi: 10.1074/jbc.M307809200. [DOI] [PubMed] [Google Scholar]
- Thies RL, Autor AP. Reactive oxygen injury to cultured pulmonary artery endothelial cells: mediation by poly(ADP-ribose) polymerase activation causing NAD depletion and altered energy balance. Arch Biochem Biophys. 1991;286(2):353–63. doi: 10.1016/0003-9861(91)90051-j. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Wada M, Masu M. Potentiation of capsaicin receptor activity by metabotropic ATP receptors as a possible mechanism for ATP-evoked pain and hyperalgesia. Proc Natl Acad Sci USA. 2001;98(12):6951–6. doi: 10.1073/pnas.111025298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torriglia A, Chaudun E, Chany-Fournier F, Jeanny JC, Courtois Y, Counis MF. Involvement of DNase II in nuclear degeneration during lens cell differentiation. J Biol Chem. 1995;270(48):28579–85. doi: 10.1074/jbc.270.48.28579. [DOI] [PubMed] [Google Scholar]
- Torriglia A, Chaudun E, Courtois Y, Counis MF. On the use of Zn2+ to discriminate endonucleases activated during apoptosis. Biochimie. 1997;79(7):435–8. doi: 10.1016/s0300-9084(97)86153-6. [DOI] [PubMed] [Google Scholar]
- Troy CM, Rabacchi SA, Friedman WJ, Frappier TF, Brown K, Shelanski ML. Caspase-2 mediates neuronal cell death induced by beta-amyloid. J Neurosci. 2000;20(4):1386–92. doi: 10.1523/JNEUROSCI.20-04-01386.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda S, Masutani H, Nakamura H, Tanaka T, Ueno M, Yodoi J. Redox control of cell death. Antioxid Redox Signal. 2002;4(3):405–14. doi: 10.1089/15230860260196209. [DOI] [PubMed] [Google Scholar]
- Ulus IH, Wurtman RJ. Metabotropic glutamate receptor agonists increase release of soluble amyloid precursor protein derivatives from rat brain cortical and hippocampal slices. J Pharmacol Exp Ther. 1997;281(1):149–54. [PubMed] [Google Scholar]
- Valerio A, Ferrario M, Paterlini M, Liberini P, Moretto G, Cairns NJ, Pizzi M, Spano P. Spinal cord mGlu1a receptors: possible target for amyotrophic lateral sclerosis therapy. Pharmacol Biochem Behav. 2002;73(2):447–54. doi: 10.1016/s0091-3057(02)00835-3. [DOI] [PubMed] [Google Scholar]
- Vanags DM, Porn-Ares MI, Coppola S, Burgess DH, Orrenius S. Protease involvement in fodrin cleavage and phosphatidylserine exposure in apoptosis. J Biol Chem. 1996;271(49):31075–85. doi: 10.1074/jbc.271.49.31075. [DOI] [PubMed] [Google Scholar]
- Vardi N, Duvoisin R, Wu G, Sterling P. Localization of mGluR6 to dendrites of ON bipolar cells in primate retina. J Comp Neurol. 2000;423(3):402–12. doi: 10.1002/1096-9861(20000731)423:3<402::aid-cne4>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, Rebrikov D, Brodianski VM, Kemper OC, Kollet O, Lapidot T, Soffer D, Sobe T, Avraham KB, Goncharov T, Holtmann H, Lonai P, Wallach D. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9(2):267–76. doi: 10.1016/s1074-7613(00)80609-3. [DOI] [PubMed] [Google Scholar]
- Vaudry D, Pamantung TF, Basille M, Rousselle C, Fournier A, Vaudry H, Beauvillain JC, Gonzalez BJ. PACAP protects cerebellar granule neurons against oxidative stress-induced apoptosis. Eur J Neurosci. 2002;15(9):1451–60. doi: 10.1046/j.1460-9568.2002.01981.x. [DOI] [PubMed] [Google Scholar]
- Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102(1):43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- Vianna MR, Coitinho AS, Izquierdo I. Role of the hippocampus and amygdala in the extinction of fear-motivated learning. Curr Neurovasc Res. 2004;1(1):55–60. doi: 10.2174/1567202043480170. [DOI] [PubMed] [Google Scholar]
- Villalba M, Ferrari D, Bozza A, Del Senno L, Di Virgilio F. Ionic regulation of endonuclease activity in PC12 cells. Biochem J. 1995;311(Pt 3):1033–8. doi: 10.1042/bj3111033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent AM, Maiese K. Direct temporal analysis of apoptosis induction in living adherent neurons. J Histochem Cytochem. 1999;47(5):661–72. doi: 10.1177/002215549904700508. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Maiese K. Nitric oxide induction of neuronal endonuclease activity in programmed cell death. Exp Cell Res. 1999;246(2):290–300. doi: 10.1006/excr.1998.4282. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Maiese K. The metabotropic glutamate system promotes neuronal survival through distinct pathways of programmed cell death. Exp Neurol. 2000;166(1):65–82. doi: 10.1006/exnr.2000.7487. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Mohammad Y, Ahmad I, Greenberg R, Maiese K. Metabotropic glutamate receptors prevent nitric oxide-induced programmed cell death. J Neurosci Res. 1997;50(4):549–64. doi: 10.1002/(SICI)1097-4547(19971115)50:4<549::AID-JNR6>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Vincent AM, TenBroeke M, Maiese K. Metabotropic glutamate receptors prevent programmed cell death through the modulation of neuronal endonuclease activity and intracellular pH. Exp Neurol. 1999;155(1):79–94. doi: 10.1006/exnr.1998.6966. [DOI] [PubMed] [Google Scholar]
- Vincent AM, TenBroeke M, Maiese K. Neuronal intracellular pH directly mediates nitric oxide-induced programmed cell death. J Neurobiol. 1999;40(2):171–84. doi: 10.1002/(sici)1097-4695(199908)40:2<171::aid-neu4>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Voulalas PJ, Holtzclaw L, Wolstenholme J, Russell JT, Hyman SE. Metabotropic glutamate receptors and dopamine receptors cooperate to enhance extracellular signal-regulated kinase phosphorylation in striatal neurons. J Neurosci. 2005;25(15):3763–73. doi: 10.1523/JNEUROSCI.4574-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JY, Shum AY, Ho YJ. Oxidative neurotoxicity in rat cerebral cortex neurons: synergistic effects of H2O2 and NO on apoptosis involving activation of p38 mitogen-activated protein kinase and caspase-3. J Neurosci Res. 2003;72(4):508–19. doi: 10.1002/jnr.10597. [DOI] [PubMed] [Google Scholar]
- Wang X, Martindale JL, Liu Y, Holbrook NJ. The cellular response to oxidative stress: influences of mitogen- activated protein kinase signalling pathways on cell survival. Biochem J. 1998;333(Pt 2):291–300. doi: 10.1042/bj3330291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe AM, Carlisle HJ, O’Dell TJ. Postsynaptic Induction and Presynaptic Expression of Group 1 mGluR- Dependent LTD in the Hippocampal CA1 Region. J Neurophysiol. 2002;87(3):1395–403. doi: 10.1152/jn.00723.2001. [DOI] [PubMed] [Google Scholar]
- Weber J. Calcium Homeostasis Following Traumatic Neuronal Injury. Curr Neurovasc Res. 2004;1(2):151–171. doi: 10.2174/1567202043480134. [DOI] [PubMed] [Google Scholar]
- White AM, Kylanpaa RA, Christie LA, McIntosh SJ, Irving AJ, Platt B. Presynaptic group I metabotropic glutamate receptors modulate synaptic transmission in the rat superior colliculus via 4-AP sensitive K+ channels. Br J Pharmacol. 2003 doi: 10.1038/sj.bjp.0705570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick A, Wick W, Waltenberger J, Weller M, Dichgans J, Schulz JB. Neuroprotection by hypoxic preconditioning requires sequential activation of vascular endothelial growth factor receptor and Akt. J Neurosci. 2002;22(15):6401–7. doi: 10.1523/JNEUROSCI.22-15-06401.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wielckens K, Schmidt A, George E, Bredehorst R, Hilz H. DNA fragmentation and NAD depletion. Their relation to the turnover of endogenous mono(ADP-ribosyl) and poly(ADP-ribosyl) proteins. J Biol Chem. 1982;257(21):12872–7. [PubMed] [Google Scholar]
- Willoughby D, Thomas R, Schwiening C. The effects of intracellular pH changes on resting cytosolic calcium in voltage-clamped snail neurones. J Physiol. 2001;530(Pt 3):405–16. doi: 10.1111/j.1469-7793.2001.0405k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski K, Car H. (S)-3,5-DHPG: a review. CNS Drug Rev. 2002;8(1):101–16. doi: 10.1111/j.1527-3458.2002.tb00218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witthuhn BA, Quelle FW, Silvennoinen O, Yi T, Tang B, Miura O, Ihle JN. JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin. Cell. 1993;74(2):227–36. doi: 10.1016/0092-8674(93)90414-l. [DOI] [PubMed] [Google Scholar]
- Witting A, Muller P, Herrmann A, Kettenmann H, Nolte C. Phagocytic clearance of apoptotic neurons by Microglia/Brain macrophages in vitro: involvement of lectin-, integrin-, and phosphatidylserine-mediated recognition. J Neurochem. 2000;75(3):1060–70. doi: 10.1046/j.1471-4159.2000.0751060.x. [DOI] [PubMed] [Google Scholar]
- Wittmann M, Marino MJ, Bradley SR, Conn PJ. Activation of Group III mGluRs Inhibits GABAergic and Glutamatergic Transmission in the Substantia Nigra Pars Reticulata. J Neurophysiol. 2001;85(5):1960–8. doi: 10.1152/jn.2001.85.5.1960. [DOI] [PubMed] [Google Scholar]
- Wolfarth S, Konieczny J, Lorenc-Koci E, Ossowska K, Pilc A. The role of metabotropic glutamate receptor (mGluR) ligands in parkinsonian muscle rigidity. Amino Acids. 2000;19(1):95–101. doi: 10.1007/s007260070038. [DOI] [PubMed] [Google Scholar]
- Wong RK, Bianchi R, Chuang SC, Merlin LR. Group I mGluR-induced Epileptogenesis: Distinct and Overlapping Roles of mGluR1 and mGluR5 and Implications for Antiepileptic Drug Design. Epilepsy Curr. 2005;5(2):63–8. doi: 10.1111/j.1535-7597.2005.05207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi A, Tamatani M, Matsuzaki H, Namikawa K, Kiyama H, Vitek MP, Mitsuda N, Tohyama M. Akt activation protects hippocampal neurons from apoptosis by inhibiting transcriptional activity of p53. J Biol Chem. 2001;276:5256–5264. doi: 10.1074/jbc.M008552200. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Wang HG. The protein kinase PKB/Akt regulates cell survival and apoptosis by inhibiting Bax conformational change. Oncogene. 2001;20(53):7779–86. doi: 10.1038/sj.onc.1204984. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Urakubo T, Tominaga-Yoshino K, Ogura A. Long-lasting synapse formation in cultured rat hippocampal neurons after repeated PKA activation. Brain Res. 2005;1042(1):6–16. doi: 10.1016/j.brainres.2005.01.102. [DOI] [PubMed] [Google Scholar]
- Yang YL, Meng CH, Ding JH, He HR, Ellsworth K, Wu J, Hu G. Iptakalim hydrochloride protects cells against neurotoxin-induced glutamate transporter dysfunction in in vitro and in vivo models. Brain Res. 2005;1049(1):80–8. doi: 10.1016/j.brainres.2005.04.073. [DOI] [PubMed] [Google Scholar]
- Yao HH, Ding JH, Zhou F, Wang F, Hu LF, Sun T, Hu G. Enhancement of glutamate uptake mediates the neuroprotection exerted by activating group II or III metabotropic glutamate receptors on astrocytes. J Neurochem. 2005;92(4):948–61. doi: 10.1111/j.1471-4159.2004.02937.x. [DOI] [PubMed] [Google Scholar]
- Yin XM, Luo Y, Cao G, Bai L, Pei W, Kuharsky DK, Chen J. Bid-mediated mitochondrial pathway is critical to ischemic neuronal apoptosis and focal cerebral ischemia. J Biol Chem. 2002;277(44):42074–81. doi: 10.1074/jbc.M204991200. [DOI] [PubMed] [Google Scholar]
- Yoshino M, Kamiya H. Suppression of presynaptic calcium influx by metabotropic glutamate receptor agonists in neonatal rat hippocampus. Brain Res. 1995;695(2):179–85. doi: 10.1016/0006-8993(95)00743-a. [DOI] [PubMed] [Google Scholar]
- Yue TL, Ni J, Romanic AM, Gu JL, Keller P, Wang C, Kumar S, Yu GL, Hart TK, Wang X, Xia Z, DeWolf WE, Jr, Feuerstein GZ. TL1, a novel tumor necrosis factor-like cytokine, induces apoptosis in endothelial cells. Involvement of activation of stress protein kinases (stress-activated protein kinase and p38 mitogen-activated protein kinase) and caspase-3-like protease. J Biol Chem. 1999;274(3):1479–86. doi: 10.1074/jbc.274.3.1479. [DOI] [PubMed] [Google Scholar]
- Yuzaki M, Mikoshiba K. Pharmacological and immunocytochemical characterization of metabotropic glutamate receptors in cultured Purkinje cells. J Neurosci. 1992;12(11):4253–63. doi: 10.1523/JNEUROSCI.12-11-04253.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zablocka B, Dluzniewska J, Zajac H, Domanska-Janik K. Opposite reaction of ERK and JNK in ischemia vulnerable and resistant regions of hippocampus: involvement of mitochondria. Brain Res Mol Brain Res. 2003;110(2):245–52. doi: 10.1016/s0169-328x(02)00653-8. [DOI] [PubMed] [Google Scholar]
- Zhan RZ, Fujiwara N, Yamakura T, Taga K, Fukuda S, Endoh H, Shimoji K. NMDA induces a biphasic change in intracellular pH in rat hippocampal slices. Brain Res. 1997;760(1–2):179–86. doi: 10.1016/s0006-8993(97)00278-3. [DOI] [PubMed] [Google Scholar]
- Zhou H, Li XM, Meinkoth J, Pittman RN. Akt regulates cell survival and apoptosis at a postmitochondrial level. J Cell Biol. 2000;151(3):483–94. doi: 10.1083/jcb.151.3.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P, DeCoster MA, Bazan NG. Interplay among platelet-activating factor, oxidative stress, and group I metabotropic glutamate receptors modulates neuronal survival. J Neurosci Res. 2004;77(4):525–31. doi: 10.1002/jnr.20175. [DOI] [PubMed] [Google Scholar]
- Zirpel L, Janowiak MA, Taylor DA, Parks TN. Developmental changes in metabotropic glutamate receptor-mediated calcium homeostasis. J Comp Neurol. 2000;421(1):95–106. [PubMed] [Google Scholar]
- Zur Nieden R, Deitmer JW. The Role of Metabotropic Glutamate Receptors for the Generation of Calcium Oscillations in Rat Hippocampal Astrocytes In Situ. Cereb Cortex. 2005 August 3; doi: 10.1093/cercor/bhj013. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Zwienenberg M, Gong QZ, Berman RF, Muizelaar JP, Lyeth BG. The effect of groups II and III metabotropic glutamate receptor activation on neuronal injury in a rodent model of traumatic brain injury. Neurosurgery. 2001;48(5):1119–26. doi: 10.1097/00006123-200105000-00031. discussion 1126–7. [DOI] [PubMed] [Google Scholar]