

Abstract
Interactions between subunit a and oligomeric subunit c are essential for the coupling of proton translocation to rotary motion in the ATP synthase. A pair of previously described mutants, R210Q/Q252R and P204T/R210Q/Q252R (Hatch et al., J. Biol. Chem. (1995) 270:29417-29412) have been constructed and further analyzed. These mutants, in which the essential arginine of subunit a, R210, was switched with a conserved glutamine residue, Q252, are shown here to be capable of both ATP synthesis by oxidative phosphorylation, and ATP-driven proton translocation. In addition, lysine can replace the arginine at position 252 with partial retention of both activities. The pH dependence of ATP-driven proton translocation was determined after purification of mutant enzymes, and reconstitution into liposomes. Proton translocation by the lysine mutant, and to a lesser extent the arginine mutant, dropped off sharply above pH 7.5, consistent with the requirement for a positive charge during function. Finally, the rates of ATP synthesis and of ATP-driven proton translocation were completely inhibited by treatment with DCCD (N, N’-dicyclohexyl carbodiimide), while rates of ATP hydrolysis by the mutants were not significantly affected, indicating that DCCD modification disrupts the F1-Fo interface. The results suggest that minimal requirements for proton translocation by the ATP synthase include a positive charge in subunit a and a weak interface between subunit a and oligomeric subunit c.
Keywords: ATP synthase, F1Fo, subunit a, proton translocation, ATP synthesis, rotary motor
1. Introduction
ATP synthesis is catalyzed by a rotary enzyme found in the membranes of mitochondria, chloroplasts and bacteria called the F1Fo-ATPase or ATP synthase (for reviews see [1-4]). The enzyme from Escherichia coli contains eight different types of subunits in a stoichiometry of α3β3γδεab2c10. [5, 6] This enzyme comprises two functional sectors: F1 (α3β3γδε) which contains the sites for ATP synthesis and hydrolysis, and Fo (ab2c10) which contains the proton pore through the membrane. Mechanistically, γεc10, are the rotary subunits, and the others compose the stator. In E. coli, proton movement through Fo drives rotation of oligomeric c subunits relative to subunit a, with which it forms an interface. Simultaneous rotation of the elongated and asymmetric γ subunit brings about conformational changes at the nucleotide binding sites in F1 that are necessary for ATP synthesis. In the absence of a proton gradient, ATP hydrolysis will drive proton translocation.
Two hydrophobic membrane proteins are directly involved in proton translocation: subunit a and subunit c, of which the latter is found in an oligomeric ring [7]. Subunit a is a protein of 30.3 kDa [8] and five transmembrane spans [9, 10]. Monomeric subunit c is 8.3 kDa and has two transmembrane spans. Ten monomers form a ring with a central cavity filled with lipid [11]. The interface is formed by the fourth transmembrane span, aTM41, of subunit a and the outside of the ring of c subunits [12], which is primarily the C-terminal helix, cTM2. A key interaction between these subunits involves R210 of subunit a and D61 of subunit c. In subunit a, R210 is found near the center of aTM4, while in subunit c D61 is found near the center of cTM2. An extensive study of disulfide cross-linking between these two transmembrane spans provides support for the close interaction of subunits a and c, and the two key residues [12]. Other studies [13-15] have indicated a potential proton pathway through aTM2, aTM4 and aTM5 in subunit a, from the periplasmic space to the region of R210. A second potential proton pathway is suggested to exist from the cytoplasmic face to the region of R210 at the interface of subunits a and c. Thus, during ATP synthesis, the likely path of protons starts from the periplasm through subunit a, to cD61 of one subunit c. Then rotation of the ring of c subunits takes the proton through nearly 360° of revolution until it a-c interface, where it is released and travels to the cytoplasm between the two subunits.
A key feature of the essential Arg is thought to be its positive charge. The charge might be necessary for electrostatic attraction leading to rotation, or it might be necessary for modulation of the pKa of cD61 leading to proton release. However, since the E. coli ATP synthase does not tolerate the conservative substitution of Lys for aR210 [16], its role might be more complex.
A mutant was described over ten years ago [17] in which aR210 was switched with another conserved residue aQ252. These residues are expected to be near the center of aTM4 and aTM5, respectively. This double mutant was shown to be capable of growth on succinate minimal medium, an indicator of oxidative phosphorylation. A spontaneous third mutation, aP204T, was discovered which improved the growth on succinate. However, these mutants were found to have no ATP-driven proton translocation in a fluorescence quenching assay using membrane vesicles. No results regarding rates of ATP synthesis were provided.
This double mutant is similar to several described for subunit c, in which the essential aspartic acid, cD61, was moved from cTM2 to position 24 in cTM1, normally Ala [18, 19]. The double mutant with the highest activity was cA24D/D61N. The functionality of such mutants is apparently due to the close proximity of the two residues, and the features of a rotary mechanism. The current study was undertaken to examine the enzymatic function of subunit a mutants in which the essential Arg was moved to position 252, and to verify that ATP synthesis actually occurs.
2. Materials and Methods
2.1 Materials
Restriction endonucleases were obtained from New England Biolabs, (Beverly, MA). Synthetic oligonucleotides were obtained from Operon Technologies, (Huntsville, AL). LDAO and DCCD were purchased from Sigma (St. Louis, MO). Materials for purification of plasmid DNA were obtained from Qiagen (Chatsworth, CA). Reagents for electrophoresis and immunoblotting were obtained from Bio-Rad (Hercules, CA). Rabbit polyclonal anti-a antibodies and monoclonal anti-c antibodies were a generous gift from Dr. Karlheinz Altendorf (Universität Osnabrück, Germany). Monoclonal anti-b antibodies were a generous gift of Dr. Roderick Capaldi (Univeristy of Oregon, Eugene, OR, USA). DNA sequencing was done by Lone Star Labs, (Houston, TX).
2.2 Plasmids, mutagenesis, growth and expression
Mutations were constructed in subunit a by cassette mutagenesis using plasmid pTW1-HisHA [9], which encodes an HA-epitope tag and a hexahistidine tag at the carboxy-terminus of subunit a. Saturation mutagenesis of position 252 was carried out using cassette mutagenesis at unique sites AseI and PvuI in pTW1-HisHA/R210Q, using the following oligonucleotides: TAATCATTACGCTGNNSGCCTTCATCTTCATGGTGCTGACGAT and CGTCAGCACCATGAAGATGAAGGCSNNCAGCGTAATGAT. N represents an equal mixture of all four nucleotides, while S represents an equal mixture of G and C. Thus, 32 different codons are possible and each amino acid is represented at least once. Mutations were transferred to plasmid pFV2 [20], which encodes all eight structural genes for the ATP synthase. The transfer was a two-step procedure, utilizing an intermediate plasmid pIP. The intermediate plasmid was constructed from pFV2 by first digesting it with XbaI and BsrGI, and purifying the 3.3 kb fragment. This fragment, containing the ampicillin resistance gene and the gene for subunit a, was ligated to a synthetic oligonucleotide linker that regenerated the staggered ends of the XbaI and BsrGI sites, and also contained a BlpI site for identification. Mutations from pTW1-His HA were transferred into pIP by isolating the 0.36 kb DraIII-PshAI fragment and ligating it to the 2.9 kb PshAI-DraIII fragment of pIP. To generate pFV2 with subunit a mutations, the 0.8 kb PflMI-BsrGI fragment was isolated from the intermediate plasmid and ligated to the 8.4 kb BsrGI-PflMI fragment from the wild-type pFV2. Plasmid pFV2 with mutant alleles of subunit a were used to transform strain DK8 [21], which lacks the eight structural genes for the ATP synthase. F1Fo-ATPase was purified from E. coli strain DK8 harboring plasmid pFV2, as previously described. Growth of cultures, preparation of membrane vesicles, purification of F1Fo, and reconstitution into liposomes were also carried out as described previously [20, 22]. Succinate minimal medium was made from minimal medium A [23], supplemented with 0.2% succinic acid (from a stock adjusted to pH 6.4 with KOH), and 0.2 mM L-valine, L-leucine and L-isoleucine.
2.3 Functional assays
ATP hydrolysis activity was measured either with an ATP regenerating system (isolated or reconstituted enzyme) or with the pH-indicator phenol red (membrane preparations), at 37° C, essentially as described [20]. Enzyme was reconstituted into liposomes, as described previously [20], using soybean asolectin at an initial lipid to protein ratio of 20. For measurements with an ATP regenerating system the medium (1 ml) contained 10 mM HEPES/KOH, pH 8.0, 100 mM KCl, 2.5 mM MgCl2, 0.1 mM EDTA, 1 mM ATP, 200 μM NADH, 2 mM phosphoenolpyruvate, lactate dehydrogenase (5 units/ml), pyruvate kinase (5 units/ml). In the case of reconstituted enzyme, 5 μM FCCP was added. Using the pH-indicator phenol red, the medium (2 ml) contained 10 mM HEPES/KOH, pH 8.0, 100 mM KCl, 10 mM ATP, 4 mM MgCl2, 0.1 mM EDTA, 60 μM phenol red, and 5 μM FCCP. Measurements of ATP-dependent ACMA-fluorescence quenching were performed at pH 8.0 and at 15° C, as described previously [20]. For pH dependence ACMA-fluorescence quenching, the buffer was 5mM Tris/ 5 mM maleate, supplemented with 15 μM valinomycin. ATP synthesis was carried out using a luciferase assay as described previously [22]. Prior to measurement of ATP synthesis, membrane vesicles were resuspended in 1 ml of buffer (200 mM Tricine/HCl, pH 7.8, 100 mM KCl, 5 mM MgCl2, and 2.5% glycerol), passed through a 10-ml Sephadex G-50 column, equilibrated with the same buffer. The ATP synthesis reaction was initiated by addition of 20 mM succinate to 2 ml of a medium containing 200 μg membrane protein, 10 mM Tricine/KOH, pH 8.0, 100 mM KCl, 5 mM MgCl2, 1 mM Pi, 0.1 mM ADP, 125 μM luciferin, and 100 ng luciferase. 20 nmol ATP was added for calibration after each reaction was finished. No ATP synthesis was observed without ADP or Pi, with the uncoupler FCCP, or after incubation with DCCD. DCCD inhibition was carried out as described previously [20].
2.4 Analytical methods
Native gel electrophoresis and determination of protein concentration were performed as described earlier [20]. Western blots were carried out as described earlier [24] using a dilution of 1:1000 of the anti-serum. Bands were quantified using NIH ImageJ software (http://rsb.info.nih.gov/ij/).
3. Results
A double and a triple mutant in subunit a (R210Q/Q252R and P204T/R210Q/Q252R), previously described in the literature [17], were constructed in the plasmid pTW1-HisHA, and were then used to transform the strain RH305 [25, 26], which does not produce subunit a. The mutants were tested for growth on succinate minimal medium. The positive results confirmed the original observations in a different genetic background [17]. The implication of these results is that the essential Arg of subunit a can function at position 252 in aTM5 nearly as well as it does at its normal position 210 in aTM4. No amino acid substitutions for Arg at position 210 are known to allow growth on succinate minimal medium [16, 27, 28]. To confirm that the Arg at position 252 was essential for growth on succinate, saturation mutagenesis was applied at position 252 in the background of aR210Q. A total of 48 isolates were identified as able to grow on succinate minimal medium. Twenty were analyzed further by restriction digests and DNA sequencing. Of those, eight were confirmed to be Arg, but twelve were found to be Lys. The Lys mutation, aR210Q/Q252K, was transferred to the aP204T/R210Q background, resulting in an additional mutant: aP204T/R210Q/Q252K. For further analysis all four mutants were transferred to the whole operon plasmid pFV2, and were analyzed in strain DK8, which is deleted for the structural genes of the atp (unc) operon [21]. Three additional mutations were constructed and analyzed in the same background: aP204T, aR210K, and aR210Q.
The results of a growth test of the mutants in pFV2 in the background of strain DK8 on a succinate minimal medium plate, was used as an indicator of in vivo oxidative phosphorylation. All four double and triple mutants can grow, with growth of the triple mutants (aP204T/R210Q/Q252R, aP204T/R210Q/Q252K) somewhat better than the double mutants (aR210Q/Q252R, aR210Q/Q252K). The aP204T mutant grows as well as the wild type, and was not analyzed further. Strains DK8 and both aR210K and aR210Q mutants did not grow on succinate minimal medium, confirming earlier studies [16, 28].
A previous study had not shown whether aR210Q/Q252R mutants could synthesize ATP by oxidative phosphorylation [17], although growth on succinate minimal medium is assumed to be a strong indicator of that. The six mutants described above, i.e., omitting aP204T, were tested for the rate of succinate-driven ATP synthesis in preparations of membrane vesicles. The results are shown in Fig. 1A. The double and triple mutants with aQ252R had rates of ATP synthesis that were about 20% of the wild type rate (96 nmol/min/mg protein) at 27°C. The double and triple mutants with aQ252K had somewhat lower rates, but were clearly non-zero, as shown by the expanded time scale in Fig. 1B. All rates of ATP synthesis were dependent on addition of ADP, and all were inhibited by both FCCP and DCCD (Fig. 1C). The single mutants, aR210Q and aR210K, showed no ATP synthesis. To estimate the expression level of subunit a in the mutant strains, immunoblots were prepared of membrane fractions. Using an antibody against subunit a, the results from several blots were quantified and are presented in Fig. 2, along with a representative blot. Subunit a is not expected to survive outside of an Fo complex [29, 30], but the ATP synthase is able to partially assemble in the absence of subunit a [31, 32], so these levels provide only a rough estimate of the level of fully assembled ATP synthase. For comparison, immunoblots for b and c subunits are also shown in Fig. 2, and it is clear that these subunits do not vary as much as subunit a.
Fig. 1.

ATP synthesis by membrane vesicles. The results shown are representative of four similar sets of traces, from two different preparations. (A) ATP synthesis is measured in real-time using a luciferin-luciferase system at 27°C in a 2 ml chamber. Membrane vesicles (200 μg) are energized by succinate (20 mM). The numbers in parentheses are the rates in nmol ATP/min/mg protein. All samples were prepared from DK8/pFV2 with the indicated mutations: (1) wild type. (2) P204T/R210Q/Q252R. (3) R210Q/Q252R. (4) P204T/R210Q/Q252K. (5) R210Q/Q252K. (6) R210Q. (7) R210K. (B) An expanded time scale is shown for samples 4, 5, 6, and 7 of panel A. (C) Sensitivity of ATP synthesis by (2) P204T/R210Q/Q252R to treatment by DCCD (50 μM at 27°C for 25 min).
Fig. 2.
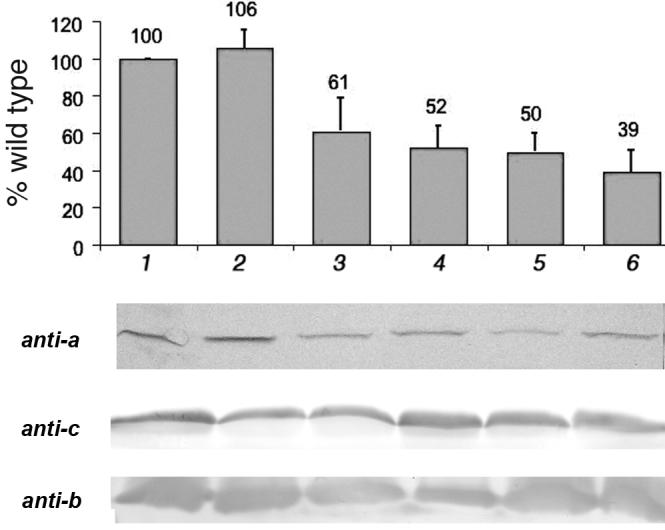
Quantification of subunit a in membrane vesicles by immunoblotting. Membrane vesicles were solubilized by dodecyl sulfate, separated by SDS-PAGE, blotted, and probed with polyclonal anti-a antibodies. The bands were measured by densitometric analysis, and the means of 3-5 experiments are shown, with standard deviations. The y-axis is the percent of the wild type value. All samples were from DK8/pFV2 with the indicated mutations: (1) wild type. (2) P204T/R210Q/Q252R. (3) P204T/R210Q/Q252K. (4) R210Q/Q252R. (5) R210Q/Q252K. (6) R210Q. Below, representative blots are shown using anti-a, anti-c and anti-b antibodies.
It was clear from the previous results that mutants lacking R210 in subunit a could synthesize ATP at significant rates. We next asked if these mutants could also use ATP hydrolysis to translocate protons, i.e., carry out the reverse reaction. Membrane vesicles were prepared and ATP-driven proton translocation was monitored with the fluorescent dye ACMA, where quenching of fluorescence indicates the generation of a proton gradient across the membrane. As shown in Fig. 3A, the two triple mutants (aP204T/R210Q/Q252R, aP204T/R210Q/Q252K) both showed modest rates of proton translocation, while the two double mutants (aR210Q/Q252R, aR210Q/Q252K) showed very slow, but non-zero rates. These rates were fully sensitive to pre-treatment by the inhibitor DCCD (Fig. 3A). The two single mutants, aR210K and aR210Q, showed essentially no proton translocation. The quenching could be rapidly eliminated by the addition of 1 μM FCCP (results not shown). The low rates were not due to general leakiness of the membranes, as was shown by the use of NADH, which generates a proton gradient due to electron transport reactions (Fig. 3B). Finally, the membranes were stripped of F1 to examine the rates of passive proton permeability due to Fo by each mutant. The results, shown in Fig. 3C, show that the wild type Fo compromises the ability of NADH to generate a proton gradient. For each mutant, the rate of proton permeability through Fo is not sufficient to significantly diminish the proton gradient generated by NADH.
Fig. 3.

Proton translocation rates by membrane vesicles. Proton translocation is measured as the quenching of fluorescence of ACMA using 40 μg of membrane protein in a 2 ml chamber at 15°C. Representative traces from 2-4 measurements are shown. (A) ATP-driven proton translocation by membrane vesicles indicates the functionality of the ATP synthase in the reverse direction. The reaction was initiated with 0.2 mM ATP. (B) NADH-driven proton translocation indicates the integrity of the membrane vesicles. The reaction was initiated with 0.25 mM NADH. Other conditions were the same as in panel A. (C) After stripping the membranes of F1, the passive-permeability of Fo to protons is measured by its effect on the rate of NADH-driven proton translocation. Same conditions were used as in panel B. All samples are DK8/pFV2 with the indicated mutations: (1) wild type. (2) P204T/R210Q/Q252R. (3) P204T/R210Q/Q252K. (4) R210Q/Q252R. (5) R210Q/Q252K. (6) R210Q. (7) R210K. In panel (A), sample 2 was pretreated with 50 μM DCCD at 25°C for 25 min.
Most models proposed for the mechanism of proton translocation by Fo require a positively charged residue supplied by subunit a, and that would normally be aR210 [33-37]. The high pKa of Arg would allow function over a broad pH range. The functionality of the two triple mutants allowed us to look for a difference in pH dependence that might be ascribed to the lower pKa of Lys, and therefore provide evidence that a positive charge is actually essential. For these experiments, ATP synthase was purified from the wild type strain, and from the two triple mutants (aP204T/R210Q/Q252R and aP204T/R210Q/Q252K), and reconstituted into liposomes. The resulting proteoliposomes were assayed for ATP-driven proton translocation over a pH range of 6.0 to 8.7, and the results are shown in Fig. 4. To enhance fluorescence quenching by the mutants, about 20-fold more protein was used in the assay chamber. All three display nearly maximal quenching at pH 7.5, but at pH values above 8.0, the quenching by the Lys mutant drops off significantly, relative to that of the wild type. The quenching of the Arg mutant is intermediate in this pH range. Analysis by native gel electrophoresis indicated that the mutant ATP synthase did not dissociate at pH 8.5 (results not shown).
Fig. 4.

Rates of proton translocation by reconstituted ATP synthase in proteoliposomes. ATP-dependent quenching was enhanced by using 15 μM valinomycin, which reduces the membrane potential as ΔpH develops. The buffer was 5 mM Tris/5 mM maleate, adjusted to the indicated pH by KOH. Reactions were initiated by addition of 0.2 mM ATP and were stopped by addition of 2 μM FCCP (not shown). Representative traces from 2-3 measurements are shown. (A) Wild type enzyme was reconstituted into liposomes and assayed at the indicated pH values, using 0.5 μg protein. (B) Mutant enzyme P204T/R210Q/Q252R was reconstituted into liposomes and assayed at the indicated pH values, using 10 μg protein. (C) Mutant enzyme P204T/R210Q/Q252K was reconstituted into liposomes and assayed at the indicated pH values, using 10 μg protein.
Finally, the rates of ATP hydrolysis by the mutants in membrane vesicles are presented in Table 1. The use of LDAO in the assay medium allows F1-ATPase activity to be uncoupled from Fo in the membrane, and therefore provides an indication of the level of F1 present in the membrane preparations. This provides a rough estimate of the level of assembled F1Fo. A pre-incubation with the reagent DCCD inhibits proton translocation through Fo, and will also inhibit ATP hydrolysis if the F1 and Fo sectors are tightly coupled, as in a wild type enzyme. The results presented here indicate that even after reaction with DCCD, the double and triple mutants are able to hydrolyze ATP at uninhibited rates. This might occur if the interactions between a and c subunits were altered in the mutants such that the DCCD-modified c subunits can rotate past subunit a. It is clear that DCCD reacts with the mutants since both ATP synthesis and ATP-driven proton translocation are inhibited normally.
Table 1.
Rates of ATP Hydrolysis by Membrane Vesicles1
| Mutation | ATP hydrolysis2 (μmol/min/mg) | + LDAO3 (fold increase4) | + DCCD5 (% inhibition) |
|---|---|---|---|
| Wild type | 0.9 | 3.9 (4.3) | 80 |
| R210Q/Q252R | 0.25 | 3.2 (13) | < 5 |
| P204T/R210Q/Q252R | 0.2 | 2.5 (12.7) | < 5 |
| R210Q/Q252K | 0.5 | 2.4 (4.8) | < 5 |
| P204T/R210Q/Q252K | 0.35 | 1.75 (5) | < 5 |
| R210Q | 0.2 | 1.45 (7.2) | 22 |
Rates shown are a typical set of values from three different experiments with similar results
ATP hydrolysis has been measured using phenol red as indicated in Materials and Methods.
ATP hydrolysis has been measured in the presence of 0.3% LDAO. Rates are given in μmol/min/mg.
The fold increase, shown in parentheses, is the ratio of the rate of ATP hydrolysis in the presence of 0.3% LDAO to the rate in the absence of LDAO.
Membrane vesicles were diluted 5-fold with buffer and incubated with 50 μM DCCD for 30 min at room temperature before assay, as described previously [20]. For the double and triple mutants it should be noted that the DCCD-inhibited rates were never significantly different from the similarly-treated controls.
4. Discussion
Early studies [16, 28] could find no amino acid substitutions for aR210 that retained the ability of the E. coli ATP synthase to function, including Lys. Only aR210A retained the ability to passively transport protons through Fo, after F1 had been removed [17, 27]. This indicated that the role of the Arg side chain was not likely to be a part of the proton translocation pathway, but rather that it might be to provide a positive charge. The findings of Hatch et al. [17] were consistent with a rotary mechanism for Fo by showing that the Arg could be moved to position 252, by switching the conserved residues aR210 and aQ252. These residues are found at a similar depth in the membrane, and are found in adjacent transmembrane spans, and so are likely to be found with only a slight lateral displacement relative to each other. In the original report of this mutant [17], no direct evidence of ATP synthesis was shown, only the ability to grow on succinate minimal medium was demonstrated. Furthermore, rates of ATP-driven proton translocation were found to be near zero.
In this report we show that both aR210Q/Q252R and the triple mutant aP204T/R210Q/Q252R have substantial rates of ATP synthesis, about 25% of the wild type rate. While it is generally assumed that growth on a non-fermentable carbon source requires a functional ATP synthase, these results eliminate the possibilities of an extremely marginal rate of synthesis, or an alternative explanation for the growth. Furthermore, we demonstrated rates of ATP-dependent proton translocation that can be estimated in the range of 15-25% of the wild type rate. Therefore we can conclude that the switch of R210 and Q252 in subunit a results in an ATP synthase than can function in both directions.
A saturation mutagenesis procedure, followed by a selection on succinate minimal plates, showed that Lys, and probably only Lys, can substitute for Arg in the double mutant aR210Q/Q252R. When compared to the corresponding double and triple mutants that contain Arg, the Lys mutants had somewhat lower rates of ATP synthesis, and similar rates of ATP-dependent proton translocation. One interpretation for the functionality of Lys at position 252, but not at position 210, is that it is facilitated by the reduced complementarity between the altered subunit a and the ring of c subunits. Altered subunit a-subunit c interactions would also account for the lack of sensitivity of ATP hydrolysis to DCCD. Since the stator is known to have flexibility in the b subunits [38], it would seem possible for a mutant subunit a to disengage from the ring of c subunits, and allow rotation of a DCCD-modified c subunit, while the remaining complex is intact. DCCD must react with c subunits in the mutant enzymes, since it inhibits both ATP synthesis and ATP-driven proton translocation. These mutants appear to be similar to the partially functional cA25T mutant described many years ago, in which it was found that DCCD treatment caused the ATP synthase to disassemble, and thereby lose its coupled function [39].
Results presented here have demonstrated that either Lys or Arg at position 252 can provide the positive charge that appears to be necessary for function. Proton translocation dropped off above pH 7.5 most sharply for the Lys mutant, and less so for the Arg mutant, but not for the wild type. Native gel analysis showed that the complex remained intact. A simple Brownian rotor would not necessarily require a positive charge near the sites of protonation. However, calculations have shown that a positive charge would be required to generate the torque necessary for ATP synthesis [36]. In studies of Fo from Rhodobacter capsulatus membranes [40], evidence for two pKa values was seen in proton conduction: at about 6 and 10, and those groups were best modeled at the membrane/electrolyte surface. The broad pH dependence seen in proton translocation by the wild type enzyme at high pH probably does not reflect the pKa of R210, which is likely to be somewhat higher. However, substitution by a Lys with a pKa lower than 10 could account for some of the change in pH dependence seen in this study. The change in position of the positive group from 210 to 252 may contribute to the altered pH dependence, since the R210Q/Q252R mutant is also different from the wild type.
What is the special role of aR210 that does not permit function after replacement with Lys? The results here indicate that the lower pKa of Lys does not prevent function at pH values below 8. One possibility is that the Lys side chain is too short to make necessary interactions with the Asp residues of subunit c. Or that, if it does interact, the more localized charge of Lys relative to Arg might hinder rotation, due to a stronger electrostatic interaction, as has been suggested in the case of the sodium-translocating ATP synthase [41]. An interesting possibility is that the unique hydrogen bonding capacity of the guanidinium group of Arg might be instrumental in the rotation mechanism. The interactions between aR210 and cD61 must be dynamic. One way to facilitate that is for alternative conformations of the side chain of aR210 to exist in which it is drawn away from cD61, such that rotation can occur. Such a conformation might be stabilized by hydrogen bonding to one or more N-H groups of the guanidinium. Rotation would then be a consequence of two events: protonation of one cD61, and movement of the aR210 side chain away from its site between two c subunits.
In the model proposed by Aksimentiev et al., based on molecular dynamics simulations [33], the simultaneous interaction of aR210 with the cD61 side chains from two adjacent c subunits is emphasized. Protonation of one cD61 causes it to rotate away from the aR210, which remains in contact with the other cD61. Such a mechanism would not seem to preclude conformational changes within the side chain of aR210 during the rotation step. In a model for the sodium-translocating ATP synthase [42], the Arg of the stator moves in response to the membrane potential. It is likely that there are subtle differences between this proton translocating enzyme and a sodium translocating one.
In conclusion, the ability of the aQ252R and aQ252K residues to function in ATP synthesis in place of aR210, seems to reflect a mechanism of proton translocation that does not require a series of conformational changes in which each drives the next, for example, the helix movement in subunit a proposed in one model [34]. Rather it reflects a requirement for a positive charge that is in position to interact with cD61. A loose interaction between subunit a and the ring of c subunits appears to be sufficient, since ATP synthesis can occur even when sensitivity of ATP hydrolysis to DCCD is lost. Not yet clear is whether a particular pathway for protons is necessary, or if it is sufficient that protons are prevented from a short-circuit path. A recent study [43] found that the double mutant R210A/N214R appeared to allow such a short circuit of protons, without coupling function. No vertical re-positoning of R210 within TM4 allowed coupled function, even when cD61 was re-positioned concomitantly.
Acknowledgments
This work was supported by grant GM40508 from the National Institutes of Health, and grant N-1378 from the Welch Foundation. We thank Drs. Karlheinz Altendorf and Gabriele Deckers-Hebestreit, Universität Osnabrück, Germany, and Dr. Roderick Capaldi, University of Oregon, Eugene OR, USA for the kind gifts of antibodies. We also acknowledge the technical assistance of Leon Bae, Jessica DeLeon-Rangel, and Qianqian Ma of Southern Methodist University.
This work was supported by grants from the NIH (GM-40508) and the Welch Foundation (N-1378) to S.B.V.
Abbreviations
- ACMA
9-amino-6-chloro-2-methoxyacridine
- DCCD
N,N’-dicyclohexylcarbodiimide
- FCCP
carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone
- LDAO
lauryldimethylamine N-oxide
- aTM4
the fourth transmembrane span of subunit a, for example
- cD61
residue 61 (aspartic acid) of subunit c, for example
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Pedersen PL. Transport ATPases: structure, motors, mechanism and medicine: a brief overview. J. Bioenerg. Biomembr. 2005;37:349–357. doi: 10.1007/s10863-005-9470-3. [DOI] [PubMed] [Google Scholar]
- [2].Dimroth P, von Ballmoos C, Meier T. Catalytic and mechanical cycles in F-ATP synthases. Fourth in the Cycles Review Series. EMBO Rep. 2006;7:276–282. doi: 10.1038/sj.embor.7400646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Nakanishi-Matsui M, Futai M. Stochastic proton pumping ATPases: from single molecules to diverse physiological roles. IUBMB Life. 2006;58:318–322. doi: 10.1080/15216540600702255. [DOI] [PubMed] [Google Scholar]
- [4].Ackerman SH, Tzagoloff A. Function, structure, and biogenesis of mitochondrial ATP synthase. Prog. Nucleic Acid Res. Mol. Biol. 2005;80:95–133. doi: 10.1016/S0079-6603(05)80003-0. [DOI] [PubMed] [Google Scholar]
- [5].Bragg PD, Hou C. Subunit composition, function, and spatial arrangement in the Ca2+-and Mg2+-activated adenosine triphosphatases of Escherichia coli and Salmonella typhimurium. Arch. Biochem. Biophys. 1975;167:311–321. doi: 10.1016/0003-9861(75)90467-1. [DOI] [PubMed] [Google Scholar]
- [6].Foster DL, Fillingame RH. Stoichiometry of subunits in the H+-ATPase complex of Escherichia coli. J. Biol. Chem. 1982;257:2009–2015. [PubMed] [Google Scholar]
- [7].Stock D, Leslie AG, Walker JE. Molecular architecture of the rotary motor in ATP synthase. Science. 1999;286:1700–1705. doi: 10.1126/science.286.5445.1700. [DOI] [PubMed] [Google Scholar]
- [8].Walker JE, Saraste M, Gay NJ. The unc operon. Nucleotide sequence, regulation and structure of ATP-synthase. Biochim. Biophys. Acta. 1984;768:164–200. doi: 10.1016/0304-4173(84)90003-x. [DOI] [PubMed] [Google Scholar]
- [9].Long JC, Wang S, Vik SB. Membrane topology of subunit a of the F1Fo ATP synthase as determined by labeling of unique cysteine residues. J. Biol. Chem. 1998;273:16235–16240. doi: 10.1074/jbc.273.26.16235. [DOI] [PubMed] [Google Scholar]
- [10].Valiyaveetil FI, Fillingame RH. Transmembrane topography of subunit a in the Escherichia coli F1Fo ATP synthase. J. Biol. Chem. 1998;273:16241–16247. doi: 10.1074/jbc.273.26.16241. [DOI] [PubMed] [Google Scholar]
- [11].Oberfeld B, Brunner J, Dimroth P. Phospholipids occupy the internal lumen of the c ring of the ATP synthase of Escherichia coli. Biochemistry. 2006;45:1841–1851. doi: 10.1021/bi052304+. [DOI] [PubMed] [Google Scholar]
- [12].Jiang W, Fillingame RH. Interacting helical faces of subunits a and c in the F1Fo ATP synthase of Escherichia coli defined by disulfide cross-linking. Proc. Natl. Acad. Sci. U. S. A. 1998;95:6607–6612. doi: 10.1073/pnas.95.12.6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Angevine CM, Fillingame RH. Aqueous access channels in subunit a of rotary ATP synthase. J. Biol. Chem. 2003;278:6066–6074. doi: 10.1074/jbc.M210199200. [DOI] [PubMed] [Google Scholar]
- [14].Angevine CM, Herold KA, Fillingame RH. Aqueous access pathways in subunit a of rotary ATP synthase extend to both sides of the membrane. Proc. Natl. Acad. Sci. U. S. A. 2003;100:13179–13183. doi: 10.1073/pnas.2234364100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Angevine CM, Herold KAG, Vincent OD, Fillingame RH. Aqueous access pathways in ATP synthase subunit a: Reactivity of cysteine substituted into transmembrane helices 1, 3, and 5. J. Biol. Chem. 2007;282:9001–9007. doi: 10.1074/jbc.M610848200. [DOI] [PubMed] [Google Scholar]
- [16].Cain BD, Simoni RD. Proton translocation by the F1Fo ATPase of Escherichia coli. Mutagenic analysis of the a subunit. J. Biol. Chem. 1989;264:3292–3300. [PubMed] [Google Scholar]
- [17].Hatch LP, Cox GB, Howitt SM. The essential arginine residue at position 210 in the a subunit of the Escherichia coli ATP synthase can be transferred to position 252 with partial retention of activity. J. Biol. Chem. 1995;270:29407–29412. doi: 10.1074/jbc.270.49.29407. [DOI] [PubMed] [Google Scholar]
- [18].Miller MJ, Oldenburg M, Fillingame RH. The essential carboxyl group in subunit c of the F1Fo ATP synthase can be moved and H+-translocating function retained. Proc. Natl. Acad. Sci. U. S. A. 1990;87:4900–4904. doi: 10.1073/pnas.87.13.4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhang Y, Fillingame RH. Essential aspartate in subunit c of F1Fo ATP synthase. Effect of position 61 substitutions in helix-2 on function of Asp24 in helix-1. J. Biol. Chem. 1994;269:5473–5479. [PubMed] [Google Scholar]
- [20].Ishmukhametov RR, Galkin MA, Vik SB. Ultrafast purification and reconstitution of His-tagged cysteine-less Escherichia coli F1Fo ATP synthase. Biochim. Biophys. Acta. 2005;1706:110–116. doi: 10.1016/j.bbabio.2004.09.012. [DOI] [PubMed] [Google Scholar]
- [21].Klionsky DJ, Brusilow WS, Simoni RD. In vivo evidence for the role of the ε subunit as an inhibitor of the proton-translocating ATPase of Escherichia coli. J. Bacteriol. 1984;160:1055–1060. doi: 10.1128/jb.160.3.1055-1060.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Galkin MA, Ishmukhametov RR, Vik SB. A functionally inactive, cold-stabilized form of the Escherichia coli F1Fo ATP synthase. Biochim. Biophys. Acta. 2006;1757:206–214. doi: 10.1016/j.bbabio.2006.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 1972. [Google Scholar]
- [24].Zhang D, Vik SB. Helix packing in subunit a of the Escherichia coli ATP synthase as determined by chemical labeling and proteolysis of the cysteine-substituted protein. Biochemistry. 2003;42:331–337. doi: 10.1021/bi026649t. [DOI] [PubMed] [Google Scholar]
- [25].Hartzog PE, Cain BD. Mutagenic analysis of the a subunit of the F1Fo ATP synthase in Escherichia coli: Gln-252 through Tyr-263. J. Bacteriol. 1993;175:1337–1343. doi: 10.1128/jb.175.5.1337-1343.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Humbert R, Brusilow WS, Gunsalus RP, Klionsky DJ, Simoni RD. Escherichia coli mutants defective in the uncH gene. J. Bacteriol. 1983;153:416–422. doi: 10.1128/jb.153.1.416-422.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Valiyaveetil FI, Fillingame RH. On the role of Arg-210 and Glu-219 of subunit a in proton translocation by the Escherichia coli FoF1-ATP synthase. J. Biol. Chem. 1997;272:32635–32641. doi: 10.1074/jbc.272.51.32635. [DOI] [PubMed] [Google Scholar]
- [28].Lightowlers RN, Howitt SM, Hatch L, Gibson F, Cox GB. The proton pore in the Escherichia coli FoF1-ATPase: a requirement for arginine at position 210 of the a-subunit. Biochim. Biophys. Acta. 1987;894:399–406. doi: 10.1016/0005-2728(87)90118-6. [DOI] [PubMed] [Google Scholar]
- [29].Hermolin J, Fillingame RH. Assembly of Fo sector of Escherichia coli H+ ATP synthase. Interdependence of subunit insertion into the membrane. J. Biol. Chem. 1995;270:2815–2817. doi: 10.1074/jbc.270.6.2815. [DOI] [PubMed] [Google Scholar]
- [30].Akiyama Y, Kihara A, Ito K. Subunit a of proton ATPase Fo sector is a substrate of the FtsH protease in Escherichia coli. FEBS Lett. 1996;399:26–28. doi: 10.1016/s0014-5793(96)01283-5. [DOI] [PubMed] [Google Scholar]
- [31].Ono S, Sone N, Yoshida M, Suzuki T. ATP synthase that lacks Foa-subunit: isolation, properties, and indication of Fob2-subunits as an anchor rail of a rotating c-ring. J. Biol. Chem. 2004;279:33409–33412. doi: 10.1074/jbc.M404993200. [DOI] [PubMed] [Google Scholar]
- [32].Vik SB, Simoni RD. F1Fo-ATPase from Escherichia coli with mutant Fo subunits. Partial purification and immunoprecipitation of F1Fo complexes. J. Biol. Chem. 1987;262:8340–8346. [PubMed] [Google Scholar]
- [33].Aksimentiev A, Balabin IA, Fillingame RH, Schulten K. Insights into the molecular mechanism of rotation in the Fo sector of ATP synthase. Biophys. J. 2004;86:1332–1344. doi: 10.1016/S0006-3495(04)74205-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Rastogi VK, Girvin ME. Structural changes linked to proton translocation by subunit c of the ATP synthase. Nature. 1999;402:263–268. doi: 10.1038/46224. [DOI] [PubMed] [Google Scholar]
- [35].Junge W, Lill H, Engelbrecht S. ATP synthase: an electrochemical transducer with rotatory mechanics. Trends Biochem. Sci. 1997;22:420–423. doi: 10.1016/s0968-0004(97)01129-8. [DOI] [PubMed] [Google Scholar]
- [36].Elston T, Wang H, Oster G. Energy transduction in ATP synthase. Nature. 1998;391:510–513. doi: 10.1038/35185. [DOI] [PubMed] [Google Scholar]
- [37].Vik SB, Antonio BJ. A mechanism of proton translocation by F1Fo ATP synthases suggested by double mutants of the a subunit. J. Biol. Chem. 1994;269:30364–30369. [PubMed] [Google Scholar]
- [38].Sorgen PL, Caviston TL, Perry RC, Cain BD. Deletions in the second stalk of F1Fo-ATP synthase in Escherichia coli. J. Biol. Chem. 1998;273:27873–27878. doi: 10.1074/jbc.273.43.27873. [DOI] [PubMed] [Google Scholar]
- [39].Fimmel AL, Jans DA, Hatch L, James LB, Gibson F, Cox GB. The F1Fo-ATPase of Escherichia coli. The substitution of alanine by threonine at position 25 in the c-subunit affects function but not assembly. Biochim. Biophys. Acta. 1985;808:252–258. doi: 10.1016/0005-2728(85)90007-6. [DOI] [PubMed] [Google Scholar]
- [40].Feniouk BA, Kozlova MA, Knorre DA, Cherepanov DA, Mulkidjanian AY, Junge W. The of ATP synthase: ohmic conductance (10 fS), and absence of voltage gating. Biophys. J. 2004;86:4094–4109. doi: 10.1529/biophysj.103.036962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wehrle F, Kaim G, Dimroth P. Molecular mechanism of the ATP synthase’s Fo motor probed by mutational analyses of subunit a. J. Mol. Biol. 2002;322:369–381. doi: 10.1016/s0022-2836(02)00731-3. [DOI] [PubMed] [Google Scholar]
- [42].Xing J, Wang H, von Ballmoos C, Dimroth P, Oster G. Torque generation by the Fo motor of the sodium ATPase. Biophys. J. 2004;87:2148–2163. doi: 10.1529/biophysj.104.042093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Langemeyer L, Engelbrecht S. Essential arginine in subunit a and aspartate in subunit c of FoF1 ATP synthase: Effect of repositioning within Helix 4 of subunit a and Helix 2 of subunit c. Biochim. Biophys. Acta. 2007;1767:998–1005. doi: 10.1016/j.bbabio.2007.05.007. [DOI] [PubMed] [Google Scholar]