

Abstract
Following the peak of the T-cell response, most of the activated effector T cells die by apoptosis driven by the proapoptotic Bcl-2 family member Bim (Bcl-2-interacting mediator of death). Whether the absence of Bim-mediated T-cell apoptosis can affect protective immunity remains unclear. Here, we used a mouse model of Leishmania major infection, in which parasite persistence and protective immunity are controlled by an equilibrium reached between parasite-specific gamma interferon (IFN-γ)-producing effector T cells and interleukin-10 (IL-10)-producing CD4+ CD25+ T regulatory cells. To further understand the role of Bim-mediated apoptosis in persistent infection and protective immunity, we infected Bim−/− mice with L. major. We found that the initial parasite growth and lesion development were similar in Bim−/− and wild-type mice after primary L. major infection. However, at later times after infection, Bim−/− mice had significantly increased L. major-specific CD4+ T-cell responses and were resistant to persistent infection. Interestingly, despite their resistance to primary L. major infection, Bim−/− mice displayed significantly enhanced protection against challenge with L. major. Increased resistance to challenge in Bim−/− mice was associated with a significant increase in the number of L. major-specific IFN-γ-producing CD4+ T cells and a lack of IL-10 production at the challenge site. Taken together, these data suggest that Bim limits protective immunity and that the absence of Bim allows the host to bypass antigen persistence for maintenance of immunity against reinfection.
Antigen-driven activation of naïve T cells during the course of an immune response to an infection results in massive expansion of antigen-specific T cells (9, 18). In general, after the peak of the response, the majority of activated T cells die by apoptosis, while some of them go on to become memory T cells. This decision between death and survival is crucial for avoiding autoimmunity and for promoting the development of immunological memory and protective immunity. We and others have recently shown that Bcl-2 family members play a significant role in the apoptotic demise of activated effector T cells during acute and chronic viral infections (12, 20, 27). However, it remains unclear whether the failure to eliminate effector T cells can result in functionally increased protective immunity in vivo. Such knowledge is critical to our understanding of the metamorphosis of activated effector T cells to memory T cells.
We and others have recently shown that the proapoptotic Bcl-2 family member Bim plays a critical role in the apoptotic demise of activated T cells in vivo (12, 14, 20, 27). The proapoptotic activity of Bim is kept in check by the antiapoptotic molecule Bcl-2 (7, 28); just prior to the apoptotic demise of T cells in vivo, Bcl-2 levels are significantly decreased, and this allows Bim to trigger apoptosis (13, 14, 17). In Bim−/− mice, the antigen-specific CD4+ and CD8+ T cells persist for a significantly longer period of time than their wild-type counterparts (14, 20). Using a viral infection model, we showed that the numbers of memory phenotype CD4+ and CD8+ T cells were increased at late times after infection in Bim−/− mice (27); however, we did not examine the functionality of memory T cells in Bim−/− mice in vivo. Thus, it remains unclear whether cells that would normally be eliminated during an immune response can, in the absence of the apoptotic signals, result in increased T-cell memory and protective immunity.
Recently, we developed a model of chronic Leishmania major infection of mice (3). This model involves intradermal inoculation of mice with a low dose of L. major, a procedure that mimics infection in the wild by the intermediate vector, phlebotomine sand flies (3). Normally resistant C57BL/6 mice inoculated in the ear dermis with a low dose of L. major develop a chronic infection in which low levels of parasites persist, which are exquisitely controlled by a population of interleukin-10 (IL-10)-producing, CD4+ CD25+ T regulatory cells (4, 5). Notably, mice inoculated with a low dose of L. major in one ear are protected from rechallenge in the second ear, despite the presence of a low-level chronic infection in the primary ear, a phenomenon known as “concomitant immunity” (16). Interestingly, in this model, elimination of L. major either through removal of T regulatory cells by anti-CD25 depletion in vivo or via ablation of IL-10 in vivo results in loss of protective immunity against rechallenge with L. major (5, 16). Thus, in this natural model of L. major infection, parasite persistence is controlled by the presence of T regulatory cells and is linked to the maintenance of protective immunity against reinfection.
In this study, we took advantage of this well-defined model to investigate the role of Bim in the control of T-cell responses to primary and secondary L. major infections. We found that although Bim−/− and wild-type mice exhibited similar parasite expansion, Bim−/− mice had a significantly increased ability to control primary infection. Taken together, our results suggest that Bim is required for the apoptotic termination of T-cell responses and that interference with this process can improve protective immunity in vivo and allow the host to bypass the requirement for antigen persistence in maintaining protective T-cell responses. These findings are relevant for vaccination strategies aimed at improving responses by limiting apoptotic cell death during the T-cell response.
MATERIALS AND METHODS
Mice and injections.
Bim-deficient mice have been described previously, were backcrossed 15 times to C57BL/6 mice (8, 14, 27), and were a generous gift from Andreas Strasser and Philippe Bouillet (WEHI, Sydney, Australia). The BL/6 mice used for experiments either were purchased from Jackson Labs, Taconic Farms, or were Bim+/+ mice from our Bim colony. Bim+/− and Bim−/− mice were identified by PCR using tail DNA. All mice used were between 8 and 18 weeks old. All animals were kept under specific-pathogen-free conditions at an American Association for the Accreditation of Laboratory Animal Care-accredited animal facility at the National Institute of Allergy and Infectious Diseases or in the Animal Facility at Cincinnati Children's Hospital Research Foundation and were housed in accordance with the procedures outlined in the Guide for the Care and Use of Laboratory Animals under animal study proposals approved by both the National Institute of Allergy and Infectious Diseases and Cincinnati Children's Hospital Research Foundation animal care and use committees.
Parasites and infection.
L. major clone V1 (MHOM/IL/80/Friedlin) promastigotes were grown at 26°C in medium 199 supplemented with 20% Hi-FCS (HyClone, Logan, UT), 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM l-glutamine, 40 mM HEPES, 0.1 mM adenine (in 50 mM HEPES), 5 μg/ml hemin (in 50% triethanolamine), and 1 μg/ml 6-biotin (M199/S). Infective-stage promastigotes (metacyclic promastigotes) of L. major were isolated from stationary-phase cultures (4 to 5 days old) by negative selection of infective forms using peanut agglutinin (Vector Laboratories Inc., Burlingame, CA). Mice were infected in the ear dermis with 3 × 103 L. major metacyclic promastigotes in 10 μl using a 27-gauge needle. In some experiments, mice were infected in one ear and 16 weeks after infection were challenged with 3 × 103 L. major promastigotes in the other ear.
Parasite quantitation.
Parasite loads in the ears were determined as previously described (3). Briefly, the ventral and dorsal sheets of the infected ears were separated and deposited dermal side down in RPMI containing 100 U/ml penicillin, 100 μg/ml streptomycin, and the liberase CI enzyme blend (50 μg/ml; Boehringer Mannheim). Ears were incubated for 40 min at 37°C. The sheets were dissociated in RPMI with 10% serum and 0.05% DNase I (Sigma) using a medimachine (BD Bioscience) according to the manufacturer's protocol. The tissue homogenates were filtered using a 70-μm cell strainer (Falcon Products Inc., St. Louis, MO) and serially diluted in a 96-well flat-bottom microtiter plate containing biphasic medium prepared using 50 μl of NNN medium containing 20% defibrinated rabbit blood overlaid with 100 μl of M199/S. The number of viable parasites in each ear was determined from the highest dilution at which promastigotes could be grown out after 7 days of incubation at 26°C. The number of parasites in the local draining lymph nodes (LNs) (retromaxilar) was also determined. The LNs were mechanically dissociated, and the parasite load in LN cells was determined by the limiting dilution method as described above.
Analysis of dermal lymphocytes.
Single-cell suspensions were obtained from the ear dermis as described above. For analysis of surface markers and intracytoplasmic staining for gamma interferon (IFN-γ), cells were stimulated with L. major-infected bone marrow-derived dendritic cells as a source of antigen for 16 h. The cells were cultured for an additional 6 h with brefeldin A (10 μg/ml) and then stained for the surface markers CD3 (145-2 C11), T-cell receptor beta (TCR-β) (H57-597), CD4 (GK1.5), and CD8 (RM4-5 and 53-6.7). Prior to staining, cells were incubated with an anti-Fcγ III/II receptor and 10% normal mouse serum in phosphate-buffered saline containing 0.1% bovine serum albumin and 0.01% NaN3. After surface staining, cells were permeabilized and incubated with antibodies against either IFN-γ or IL-10 (clones XMG1.2 and JE56-5H4). Incubation was carried out for 30 min on ice. The isotype controls used were rat immunoglobulin G2b (A95-1) and rat immunoglobulin G2a (R35-95). All antibodies were purchased from BD Pharmingen. The frequency of IFN-γ-producing CD4+ and CD8+ T cells was determined by gating on CD3+ or TCR-β+ cells. For each sample, at least 100,000 cells were analyzed. The data were collected and analyzed using either CELLQuest or FlowJo software with either a FACSCalibur or an LSRII flow cytometer (Becton Dickinson, San Jose, CA).
Cytokine measurement.
For cytokine measurement in culture supernatants, pooled cells from draining LNs were resuspended in RPMI containing fetal bovine serum, penicillin, and streptomycin at a concentration of 6 × 106 cells/ml, and 0.1 ml was plated in U-bottom 96-well plates. Cells were incubated at 37°C in the presence of 5% CO2 with uninfected or L. major-infected bone marrow-derived dendritic cells for 48 h as described previously (23). Cytokine IFN-γ and IL-10 production was analyzed by an enzyme-linked immunosorbent assay (Endogen kits) performed according to the manufacturer's protocol.
RESULTS
Lesion development and early parasite growth after L. major infection of Bim−/− mice.
Low-dose intradermal infection of C57BL/6 mice with L. major results in lesions that develop over several weeks and resolve with the onset of the T-cell response (3, 4). Following resolution of cutaneous lesions, low levels of parasites persist in BL/6 mice (3, 4). To test whether Bim−/− mice were similarly susceptible to primary L. major infection, we inoculated groups of Bim+/+ and Bim−/− mice with 3 × 103 L. major promastigotes in the left ear pinnae and monitored the development of lesions for 6 months. The lesion sizes in Bim+/+ and Bim−/− mice were not different for the first 2 months of infection, suggesting that the expansion and control of primary L. major infection were similar in Bim+/+ and Bim−/− mice (Fig. 1A). Additionally, 6 weeks after infection, there were no significant differences in parasite burden between Bim+/+ and Bim−/− mice (Fig. 1B). These data suggested that the early primary T-cell response and the initial growth of the parasite were similar in the two strains of mice.
FIG. 1.
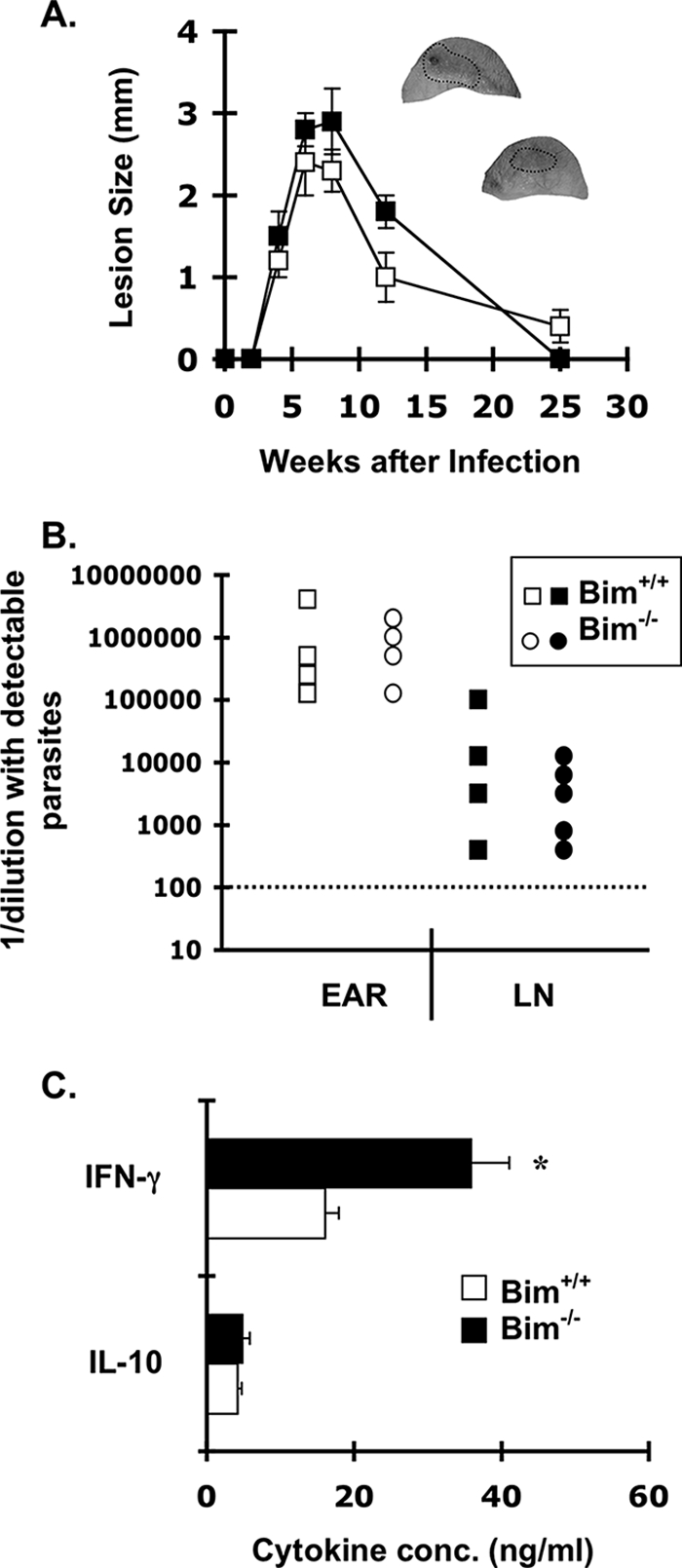
Lesion size and early parasite growth after intradermal L. major infection of Bim−/− mice. (A) Groups of either Bim+/+ or Bim−/− mice (four mice/group) were infected intradermally in the left ear with 3 × 103 L. major promastigotes. The lesion size in infected ears was determined using a vernier caliper as described previously (2); ears of representative Bim+/+ (top ear) and Bim−/− (bottom ear) are shown, and the lesion area is circled. The data are the mean lesion sizes ± standard deviations in Bim+/+ (□) and Bim−/− (▪) mice. (B) In a second experiment, groups of either Bim+/+ or Bim−/− mice (five mice/group) were infected intradermally in the left ear with 3 × 103 L. major promastigotes. At 6 weeks mice were sacrificed, and parasite loads in the ears and draining LNs of infected mice were determined by the limiting dilution method. The data are the total numbers of parasites (expressed as 1/dilution with detectable parasites) in the ears and draining LNs of Bim+/+ and Bim−/− mice. The dotted line indicates the limit of detection. (C) Single-cell suspensions from draining LNs from groups of Bim+/+ or Bim−/− mice were cultured for 48 h with soluble Leishmania antigen (25 μg/ml). IFN-γ and IL-10 levels in the supernatant were determined by an enzyme-linked immunosorbent assay. The bars indicate the mean concentrations of cytokines produced by cells isolated from either the chronic site or the challenged site on individual mice (four mice/group); the error bars indicate the standard errors of the means. The asterisk indicates that there was a statistically significant difference between the IFN-γ levels in supernatants from Bim−/− and Bim+/+ LN cells (P < 0.01, Student's t test). The data are representative of two independent experiments in which similar results were obtained.
Enhanced effector T-cell responses in L. major-infected Bim−/− mice.
Our previous work showed that during acute immune responses the peak expansion of antigen-specific T cells was similar in Bim−/− and Bim+/+ mice but that afterwards, as T cell numbers waned in Bim+/+ mice, they were maintained significantly longer in Bim−/− mice (14, 27). As the peak response in the L. major model occurs between 6 and 8 weeks after infection (2, 4, 16), we next examined the role of Bim in controlling T-cell responses during L. major infection. We first tested IFN-γ production in draining LN cells isolated from L. major-infected Bim+/+ and Bim−/− mice. At 6 weeks after infection, LN cells from Bim−/− mice produced significantly more IFN-γ than Bim+/+ mice but identical levels of IL-10 (Fig. 1C). At 8 weeks after infection, LN cells from Bim−/− mice still produced more IFN-γ but less IL-10 than Bim+/+ mice (data not shown). By 15 weeks after infection, both the percentage and the total number of L. major-specific CD4+ T cells able to produce IFN-γ were significantly greater in Bim−/− mice than in Bim+/+ mice (Fig. 2A and B). Thus, the accumulation of IFN-γ-producing cells in Bim−/− mice suggests that Bim drives the death of significant numbers of L. major-specific effector T cells in vivo. Although increased T-cell proliferation or altered trafficking of T cells in Bim−/− mice could explain this T-cell accumulation, our data obtained previously with other models systems showed that the absence of Bim does not increase T -cell proliferation or alter T-cell trafficking (14, 27).
FIG. 2.

Bim−/− mice have increased L. major-specific T-cell responses. Groups of either Bim+/+ or Bim−/− mice (six mice/group) were infected intradermally in the left ear with 3 × 103 L. major promastigotes. At 15 weeks the T-cell responses in the infected ear and draining LNs were assessed using intracellular cytokine staining and flow cytometry. The graphs show (A) the percentages and (B) the total numbers of L. major-specific CD4+ T cells able to produce IFN-γ; the error bars in panel B indicate the standard errors of the means. An asterisk indicates that there was a significant increase in the total numbers of IFN-γ-producing T cells in Bim−/− mice compared to Bim+/+ mice (P < 0.022 for ears and P < 0.012 for LNs, Student's t test).
Bim−/− mice are resistant to chronic L. major infection.
The increased numbers of Bim−/− effector T cells at the primary site of infection with L. major suggested that Bim−/− mice may have an increased capacity to control L. major infection. Indeed, while Bim+/+ mice harbored a low number of persisting parasites in the dermis and regional LNs, the number of parasites in Bim−/− mice was below the level of detection (data not shown). To reveal potential low-level infection, we administered anti-IFN-γ neutralizing antibody to groups of L. major-infected Bim+/+ or Bim−/− mice at 25 weeks and then treated them again at 27 weeks after infection. We have previously shown that neutralization of IFN-γ in vivo results in disease reactivation and parasite expansion (2). Four weeks after anti-IFN-γ treatment, mice were sacrificed and the parasites in the ears and draining LNs were quantified. While Bim+/+ mice had roughly 105 parasites/ear and 4 × 105 parasites/LN, one of three Bim−/− mice had detectable but very low levels of parasites, while the levels of parasites in the LNs were still below the limit of detection in the other two Bim−/− mice and no parasites persisted in the ears of any of the Bim−/− mice (Fig. 3). Although we did not include an isotype control-treated group in this experiment, the anti-IFN-γ antibody was effective in Bim+/+ mice because they showed evidence of disease reactivation upon macroscopic evaluation and there were high numbers of parasites in their ears and draining LNs (Fig. 3). At this late time point, the parasite burdens in untreated, L. major-infected Bim+/+ mice are generally 2 to 3 logs lower (2). Thus, Bim−/− mice had an enhanced capacity to control L. major infection. Taken together, these data suggest that Bim is critical for limiting the effector T-cell response and for promoting persistent L. major infection.
FIG. 3.

Bim−/− mice are resistant to chronic L. major infection. Groups of either Bim+/+ or Bim−/− mice were infected intradermally with 3 × 103 L. major promastigotes. At 25 weeks after infection mice were inoculated intraperitoneally with 0.5 mg anti-IFN-γ monoclonal antibody (XMG1.2). One week later mice received a second dose of anti-IFN-γ antibody. At 29 weeks after infection, mice were sacrificed, infected ears and draining LNs were removed, and L. major parasites were quantified by limiting dilution. The dotted line indicates the limit of detection. The number of parasites (expressed as 1/dilution with detectable parasites) was significantly decreased in Bim−/− mice compared to Bim+/+ mice (P < 0.0001 for ears and P < 0.0009 for LNs, Student's t test).
Enhanced protective immunity in vivo in the absence of Bim.
In persistent L. major infection of wild-type mice, the ability to mount protective immunity to secondary infection is linked to persisting antigen (5, 16). As Bim−/− mice were resistant to chronic infection, we next tested whether Bim−/− mice were able to maintain protective immunity after rechallenge. Groups of either Bim+/+ or Bim−/− mice were inoculated with 3 × 103 L. major promasitgotes in the left ear pinnae and 4 months later were challenged in the other ear with 3 × 103 promastigotes (Fig. 4A). Three weeks after the secondary challenge, mice were sacrificed and the parasites in their ears were quantified (Fig. 4A). In this model, the secondary response is also controlled by T regulatory cells, and control of parasites in wild-type mice occurs only after the fourth week after a secondary challenge (16). Similar to our results described above (Fig. 3), parasites were undetectable at the primary site in Bim−/− mice but not in Bim+/+ mice (Fig. 4B). As expected, at 3 weeks after the secondary challenge, Bim+/+ mice did not exhibit concomitant immunity; however, Bim−/− mice exhibited significant resistance to the secondary challenge, as shown by a roughly 1,000-fold reduction in the number of parasites at the challenge site (Fig. 4B). In fact, parasites were detectable in only one of three Bim−/− mice at the challenge site. Similarly, parasites were undetectable in the draining LNs of Bim−/− mice (Fig. 4C). These data demonstrate that the absence of Bim significantly enhances protective immunity in vivo, despite the apparent clearance of parasites in the primary infection.
FIG. 4.

Increased protective immunity to L. major in Bim−/− mice. (A) Experimental design of infection, challenge, and analysis. Briefly, groups of either Bim+/+ or Bim−/− mice (three mice/group) were infected intradermally in the left ear with 3 × 103 L. major promastigotes. Four months after this primary infection, mice from each group were challenged in the right ear with 3 × 103 L. major promastigotes. As a control, naïve Bim+/+ and Bim−/− mice (no primary infection) were challenged at the same time. Three weeks after this challenge, mice were sacrificed and the numbers of parasites at the chronic site (left ear) and the challenge site (right ear) and in the LNs draining either site were quantified. (B and C) Numbers of parasites (expressed as 1/dilution with detectable parasites) in either (B) the ear or (C) the draining LNs. The symbols indicate values for individual mice as follows: naïve mice, Bim+/+ (squares) and Bim−/− (circles); chronic and challenged mice, Bim+/+ (open squares and circles) and Bim−/− (filled squares and circles). The dotted lines indicate the limit of detection. The Bim−/− mice exhibited a significant reduction in the parasite burden at the challenge site (indicated by an asterisk; P < 0.037 for ears and P < 0.025 for LNs, Student's t test) compared to the naïve Bim−/− controls. In addition, in Bim−/− mice there were significant reductions in the numbers of parasites at the chronic site (P < 0.005 for ears and P < 0.033 for LNs) and at the challenge site (P < 0.032 for ears and P < 0.043 for LNs) compared to Bim+/+ controls. The numbers of parasites in challenged Bim+/+ mice were not significantly different from the numbers in naïve Bim+/+ controls (P < 0.160 for ears and P < 0.29 for LNs). n.s., not significant. (D) Representative dot plots showing IFN-γ production by CD4+ T cells of naïve mice (3 weeks after infection) or at the chronic site (19 weeks after infection) and challenge site (3 weeks after challenge) of either Bim+/+ (WT) or Bim−/− mice. (E) Total numbers of IFN-γ+ CD4+ T cells in Bim+/+ mice (open bars) and Bim−/− mice (filled bars) at either the chronic site or the challenged site. The error bars indicate the standard errors of the means. A significant increase in the number of IFN-γ+ cells at the challenge site was observed in Bim−/− mice compared to Bim+/+ mice (P < 0.03, Student's t test). The data are representative of two independent experiments in which similar results were obtained.
Elimination of L. major requires IFN-γ, which is antagonized during persistent infection by IL-10-dependent counterregulatory mechanisms (2, 5). Because Bim−/− mice displayed increased immunity to challenge with L. major, we next quantified T-cell production of IFN-γ following brief stimulation in vitro in response to L. major-infected dendritic cells. Three weeks after infection of naïve mice, the percentages of L. major-specific, IFN-γ-producing CD4+ T cells were similar in Bim−/− and Bim+/+ mice (Fig. 4D). However, at both the chronic site and the challenge site the percentage of L. major-specific, IFN-γ-producing CD4+ T cells was significantly greater in Bim−/− mice than in Bim+/+ mice (Fig. 4D). Further, while the numbers of L. major-specific, IFN-γ producing T cells were slightly increased at the chronic site, they were 100-fold greater at the challenge site of Bim−/− mice than at the challenge site of Bim+/+ mice (Fig. 4E). These data suggest that Bim is critical for limiting the numbers of L. major-specific memory T cells.
LN cells that drain either the chronic site or the challenge site were also stimulated with soluble L. major antigen, and cytokine production was measured after 48 h. Similar to what we observed in intracellular flow cytometric analyses, the IFN-γ production by cells isolated from the chronic site of infection in Bim+/+ mice was similar to the IFN-γ production in Bim−/− mice, while IFN-γ production by cells isolated from the challenge site was significantly increased in Bim−/− mice (Table 1). In contrast, cells isolated from both the chronic and challenge sites of Bim+/+ mice secreted significantly more IL-10 than draining LN cells from Bim−/− mice (Table 1). The increased IFN-γ production and the lack of IL-10 production in Bim−/− mice resulted in a dramatic skewing of the IFN-γ/IL-10 ratio for both the challenge and chronic sites (Table 1). These results are consistent with the observed in vivo protection against secondary challenge, because protection against L. major is IFN-γ dependent (6). Thus, the loss of Bim enhances protective immunity in vivo, likely by increasing the number of IFN-γ-producing memory T cells.
TABLE 1.
Increased level of IFN-γ and lack of IL-10 production in L. major-infected Bim−/− micea
| Mice | Site | IFN-γ concn (pg/ml) | IL-10 concn (pg/ml) | IFN-γ/IL-10 ratio |
|---|---|---|---|---|
| Bim+/+ | Naïve | 151 ± 48 | 257 ± 48 | 0.58 |
| Challenged | 87.7 ± 15.3 | 201 ± 18.5 | 0.44 | |
| Chronic | 5,377 ± 1,186 | 702 ± 83 | 7.66 | |
| Bim−/− | Naïve | 228 ± 80 | 276 ± 123 | 0.83 |
| Challenged | 15,270 ± 1,570b | 26.3 ± 27.9b | 580 | |
| Chronic | 5,870 ± 1,477 | 51.7 ± 29.9 | 114 |
Single-cell suspensions from draining LNs from either naïve Bim+/+ or Bim−/− mice 3 weeks after primary L. major infection or from the chronic site or the challenge site for groups of Bim+/+ or Bim−/− mice 3 weeks after secondary L. major infection were cultured for 48 h with soluble Leishmania antigen (25 μg/ml). IFN-γ and IL-10 levels in the supernatant were determined by an enzyme-linked immunosorbent assay.
The value is statistically significantly different from the value for Bim+/+ mice (P ≤ 0.005, Student's t test).
DISCUSSION
Previous work by us and others showed that Bim was required for apoptotic contraction of superantigen-reactive and virus-specific T cells in vivo (12, 14, 20, 27). We showed that following infection with lymphocytic choriomeningitis virus (LCMV), the absence of apoptotic contraction in Bim−/− mice resulted in significantly increased numbers of memory phenotype T cells based on their ability to produce IL-2 after brief stimulation in vitro (27). However, neither the superantigen model system nor the LCMV model system is an ideal system to ask questions about protective immunity. Superantigen-reactive T cells are tolerized after an encounter with superantigen in vivo (21). In addition, LCMV induces robust memory in wild-type mice (18), making it difficult to observe increased protective immunity in this model. In this study, we took advantage of a well-characterized infection model, intradermal L. major infection of mice, to demonstrate that an absence of Bim leads to increased protective immunity in vivo.
Following challenge, Bim−/− mice had decreased numbers of parasites at both the chronic site and the challenge site, which correlated with increased L. major-specific production of IFN-γ and a lack of production of IL-10. Notably, IL-10 production is normal early after infection in Bim−/− mice, suggesting that the generation of IL-10-producing T cells is not impaired in Bim−/− mice. Two possible mechanisms may explain the loss of IL-10 production in Bim−/− mice. First, it is possible that the decreased parasite burden in Bim−/− mice may result in the loss of T regulatory cells, as our recent data showed that parasite persistence was required for the maintenance of L. major-specific T regulatory cells (24). Second, at later time points, when IFN-γ was neutralized in vivo, the L. major-specific IL-10 production in Bim−/− mice was similar to that in Bim+/+ mice (data not shown), suggesting that the failure to eliminate IFN-γ-producing effector T cells results in functional silencing of IL-10 production by T regulatory cells. With either mechanism, infection of Bim−/− mice with L. major results in substantial imbalances between effector and T regulatory cells that result in resistance to parasite persistence and preservation of protective immunity.
Although some experiments have shown that major histocompatibility complex-TCR interactions and/or persisting antigen is required for the maintenance of T-cell memory (15, 16, 22), other experiments have shown that memory T cells survive in the absence of major histocompatibility complex and/or antigen (10, 19, 25). In this study, we found that even in the seeming absence of persistent parasites, protective CD4+ T-cell memory is maintained. However, we cannot be certain that the protective responses that result in “concomitant immunity” in wild-type mice are mediated by memory T cells or by a population of effector T cells that is continuously stimulated by antigen in vivo. In other model systems, persisting antigen may promote the accumulation of chronically activated effector T cells and limit their development into “true” memory T cells (11, 26). In addition, recent work has shown that an auxotrophic mutant of L. major that cannot persist in vivo is capable of stimulating a population of CD62Lhi “central” memory T cells which appear to be critical for protection from reinfection (29). Thus, it is possible that when antigen is eliminated, effector T cells die off and are replaced by CD62Lhi central memory T cells. In the absence of Bim, effector T cells are maintained at a level sufficient to drive parasite elimination, but further work is required to determine whether the protective T cells in Bim−/− mice after parasite elimination are “effector memory” or “central memory” in nature.
The implication of these data is that a decrease in the contraction of the T-cell response can lead to increased protective immunity. Experiments are under way to determine whether interference with contraction of T-cell responses in vivo could be exploited to increase vaccine efficiency. Because a major driving force of the evolution of T-cell immunity has likely been pathogen epidemics and the absence of contraction provides stronger protection against pathogens, we assume that contraction of T-cell responses is critical for the avoidance of other potentially lethal situations. For example, it was recently shown that in mice that have very high levels of persistent virus-specific memory T cells, secondary exposure to the virus results in a lethal shocklike reaction with >85% mortality by 8 days after challenge (1). Such data suggest that contraction has evolved to avoid such lethal reactions. At least three other forces that are not mutually exclusive probably favor contraction. (i) Elimination of effector T cells avoids an increased metabolic cost to the organism to keep the cells alive. (ii) Increased numbers of effector memory T cells could compete with naïve T cells for survival cytokines, such as IL-7 or IL-15; such competition could result in the loss of naïve T cells and the inability to fight off other infections. (iii) Increased numbers of effector T cells could result in increased autoimmune disease through their potential cross-reactivity with self-antigens and/or through the functional silencing of T regulatory cells. As the function of T regulatory cells is likely suppressed to allow development of T-cell responses to infection, it is tempting to speculate that contraction of T-cell responses has evolved to allow reestablishment of functional homeostasis of T regulatory cells.
Finally, our data have implications for the ability to manipulate antigen-specific T-cell responses in vivo. Strategies targeted at inhibiting Bim may be particularly relevant in the context of vaccines against pathogens known to elicit “poor-quality” immune responses. Further, some parasitic pathogens usually have few, if any, dominant antigens; thus, limiting Bim-mediated apoptosis may enhance T-cell responses to such subdominant epitopes. Altogether, our results suggest that targeting Bim may be a valid strategy to enhance the efficiency of vaccines against infectious diseases. However, we also note that inhibition of Bim may lead to other potential consequences, including increased numbers of effector T cells that could cause deleterious hyperinflammatory responses, as well as functional inactivation of T regulatory cells.
Acknowledgments
We thank Michael Jordan for providing the anti-IFN-γ neutralizing antibody and Chip Caldwell and George Deepe for contributing some Bim−/− mice.
This work was supported by generous start-up funds from the Divisions of Immunobiology and Molecular Immunology, Cincinnati Children's Hospital Medical Center and University of Cincinnati, and by Public Health Service grants AI057753 (D.A.H.) and AI057992 (C.L.K.).
Editor: J. L. Flynn
Footnotes
Published ahead of print on 17 December 2007.
REFERENCES
- 1.Badovinac, V. P., S. E. Hamilton, and J. T. Harty. 2003. Viral infection results in massive CD8+ T cell expansion and mortality in vaccinated perforin-deficient mice. Immunity 18463-474. [DOI] [PubMed] [Google Scholar]
- 2.Belkaid, Y., K. F. Hoffmann, S. Mendez, S. Kamhawi, M. C. Udey, T. A. Wynn, and D. L. Sacks. 2001. The role of interleukin (IL)-10 in the persistence of Leishmania major in the skin after healing and the therapeutic potential of anti-IL-10 receptor antibody for sterile cure. J. Exp. Med. 1941497-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Belkaid, Y., S. Kamhawi, G. Modi, J. Valenzuela, N. Noben-Trauth, E. Rowton, J. Ribeiro, and D. L. Sacks. 1998. Development of a natural model of cutaneous leishmaniasis: powerful effects of vector saliva and saliva preexposure on the long-term outcome of Leishmania major infection in the mouse ear dermis. J. Exp. Med. 1881941-1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belkaid, Y., S. Mendez, R. Lira, N. Kadambi, G. Milon, and D. Sacks. 2000. A natural model of Leishmania major infection reveals a prolonged “silent” phase of parasite amplification in the skin before the onset of lesion formation and immunity. J. Immunol. 165969-977. [DOI] [PubMed] [Google Scholar]
- 5.Belkaid, Y., C. A. Piccirillo, S. Mendez, E. M. Shevach, and D. L. Sacks. 2002. CD4+ CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature 420502-507. [DOI] [PubMed] [Google Scholar]
- 6.Belosevic, M., D. S. Finbloom, P. H. Van Der Meide, M. V. Slayter, and C. A. Nacy. 1989. Administration of monoclonal anti-IFN-gamma antibodies in vivo abrogates natural resistance of C3H/HeN mice to infection with Leishmania major. J. Immunol. 143266-274. [PubMed] [Google Scholar]
- 7.Bouillet, P., S. Cory, L. C. Zhang, A. Strasser, and J. M. Adams. 2001. Degenerative disorders caused by Bcl-2 deficiency prevented by loss of its BH3-only antagonist Bim. Dev. Cell 1645-653. [DOI] [PubMed] [Google Scholar]
- 8.Bouillet, P., D. Metcalf, D. C. Huang, D. M. Tarlinton, T. W. Kay, F. Kontgen, J. M. Adams, and A. Strasser. 1999. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science 2861735-1738. [DOI] [PubMed] [Google Scholar]
- 9.Butz, E. A., and M. J. Bevan. 1998. Massive expansion of antigen-specific CD8+ T cells during an acute virus infection. Immunity 8167-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dorfman, J. R., I. Stefanova, K. Yasutomo, and R. N. Germain. 2000. CD4+ T cell survival is not directly linked to self-MHC-induced TCR signaling. Nat. Immunol. 1329-335. [DOI] [PubMed] [Google Scholar]
- 11.Fuller, M. J., D. A. Hildeman, S. Sabbaj, D. E. Gaddis, A. E. Tebo, L. Shang, P. A. Goepfert, and A. J. Zajac. 2005. Cutting edge: emergence of CD127 high functionally competent memory T cells is compromised by high viral loads and inadequate T cell help. J. Immunol. 1745926-5930. [DOI] [PubMed] [Google Scholar]
- 12.Grayson, J. M., A. E. Weant, B. C. Holbrook, and D. Hildeman. 2006. Role of Bim in regulating CD8+ T-cell responses during chronic viral infection. J. Virol. 808627-8638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grayson, J. M., A. J. Zajac, J. D. Altman, and R. Ahmed. 2000. Cutting edge: increased expression of Bcl-2 in antigen-specific memory CD8+ T cells. J. Immunol. 1643950-3954. [DOI] [PubMed] [Google Scholar]
- 14.Hildeman, D. A., Y. Zhu, T. C. Mitchell, P. Bouillet, A. Strasser, J. Kappler, and P. Marrack. 2002. Activated T cell death in vivo mediated by proapoptotic Bcl-2 family member Bim. Immunity 16759-767. [DOI] [PubMed] [Google Scholar]
- 15.Kundig, T. M., M. F. Bachmann, S. Oehen, U. W. Hoffmann, J. J. Simard, C. P. Kalberer, H. Pircher, P. S. Ohashi, H. Hengartner, and R. M. Zinkernagel. 1996. On the role of antigen in maintaining cytotoxic T-cell memory. Proc. Natl. Acad. Sci. USA 939716-9723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mendez, S., S. K. Reckling, C. A. Piccirillo, D. Sacks, and Y. Belkaid. 2004. Role for CD4+ CD25+ regulatory T cells in reactivation of persistent leishmaniasis and control of concomitant immunity. J. Exp. Med. 200201-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mitchell, T., J. Kappler, and P. Marrack. 1999. Bystander virus infection prolongs activated T cell survival. J. Immunol. 1624527-4535. [PubMed] [Google Scholar]
- 18.Murali-Krishna, K., J. D. Altman, M. Suresh, D. J. Sourdive, A. J. Zajac, J. D. Miller, J. Slansky, and R. Ahmed. 1998. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity 8177-187. [DOI] [PubMed] [Google Scholar]
- 19.Murali-Krishna, K., L. L. Lau, S. Sambhara, F. Lemonnier, J. Altman, and R. Ahmed. 1999. Persistence of memory CD8 T cells in MHC class I-deficient mice. Science 2861377-1381. [DOI] [PubMed] [Google Scholar]
- 20.Pellegrini, M., G. Belz, P. Bouillet, and A. Strasser. 2003. Shutdown of an acute T cell immune response to viral infection is mediated by the proapoptotic Bcl-2 homology 3-only protein Bim. Proc. Natl. Acad. Sci. USA 10014175-14180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rellahan, B. L., L. A. Jones, A. M. Kruisbeek, A. M. Fry, and L. A. Matis. 1990. In vivo induction of anergy in peripheral V beta 8+ T cells by staphylococcal enterotoxin B. J. Exp. Med. 1721091-1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seddon, B., P. Tomlinson, and R. Zamoyska. 2003. Interleukin 7 and T cell receptor signals regulate homeostasis of CD4 memory cells. Nat. Immunol. 4680-686. [DOI] [PubMed] [Google Scholar]
- 23.Suffia, I., S. K. Reckling, G. Salay, and Y. Belkaid. 2005. A role for CD103 in the retention of CD4+ CD25+ Treg and control of Leishmania major infection. J. Immunol. 1745444-5455. [DOI] [PubMed] [Google Scholar]
- 24.Suffia, I. J., S. K. Reckling, C. A. Piccirillo, R. S. Goldszmid, and Y. Belkaid. 2006. Infected site-restricted Foxp3+ natural regulatory T cells are specific for microbial antigens. J. Exp. Med. 203777-788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swain, S. L., H. Hu, and G. Huston. 1999. Class II-independent generation of CD4 memory T cells from effectors. Science 2861381-1383. [DOI] [PubMed] [Google Scholar]
- 26.Wherry, E. J., D. L. Barber, S. M. Kaech, J. N. Blattman, and R. Ahmed. 2004. Antigen-independent memory CD8 T cells do not develop during chronic viral infection. Proc. Natl. Acad. Sci. USA 10116004-16009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wojciechowski, S., M. B. Jordan, Y. Zhu, J. White, A. J. Zajac, and D. A. Hildeman. 2006. Bim mediates apoptosis of CD127(lo) effector T cells and limits T cell memory. Eur. J. Immunol. 361694-1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wojciechowski, S., P. Tripathi, T. Bourdeau, L. Acero, H. L. Grimes, J. D. Katz, F. D. Finkelman, and D. A. Hildeman. 2007. Bim/Bcl-2 balance is critical for maintaining naive and memory T cell homeostasis. J. Exp. Med. [DOI] [PMC free article] [PubMed]
- 29.Zaph, C., J. Uzonna, S. M. Beverley, and P. Scott. 2004. Central memory T cells mediate long-term immunity to Leishmania major in the absence of persistent parasites. Nat. Med. 101104-1110. [DOI] [PubMed] [Google Scholar]