

Abstract
This perspective article provides an assessment of the state-of-the-art in the molecular-resolution analysis of complex organic materials. These materials can be divided into biomolecules in complex mixtures (which are amenable to successful separation into unambiguously defined molecular fractions) and complex nonrepetitive materials (which cannot be purified in the conventional sense because they are even more intricate). Molecular-level analyses of these complex systems critically depend on the integrated use of high-performance separation, high-resolution organic structural spectroscopy and mathematical data treatment. At present, only high-precision frequency-derived data exhibit sufficient resolution to overcome the otherwise common and detrimental effects of intrinsic averaging, which deteriorate spectral resolution to the degree of bulk-level rather than molecular-resolution analysis. High-precision frequency measurements are integral to the two most influential organic structural spectroscopic methods for the investigation of complex materials—NMR spectroscopy (which provides unsurpassed detail on close-range molecular order) and FTICR mass spectrometry (which provides unrivalled resolution)—and they can be translated into isotope-specific molecular-resolution data of unprecedented significance and richness. The quality of this standalone de novo molecular-level resolution data is of unparalleled mechanistic relevance and is sufficient to fundamentally advance our understanding of the structures and functions of complex biomolecular mixtures and nonrepetitive complex materials, such as natural organic matter (NOM), aerosols, and soil, plant and microbial extracts, all of which are currently poorly amenable to meaningful target analysis. The discrete analytical volumetric pixel space that is presently available to describe complex systems (defined by NMR, FT mass spectrometry and separation technologies) is in the range of 108–14 voxels, and is therefore capable of providing the necessary detail for a meaningful molecular-level analysis of very complex mixtures. Nonrepetitive complex materials exhibit mass spectral signatures in which the signal intensity often follows the number of chemically feasible isomers. This suggests that even the most strongly resolved FTICR mass spectra of complex materials represent simplified (e.g. isomer-filtered) projections of structural space.
Keywords: NMR, FT mass spectrometry, Compositional space, Separation, Intrinsic averaging, Isomers, Resolution, Complexity, Complex systems
Introduction
Status of the molecular-level analysis of complex materials
Natural molecules can be classified into two main groups according to their functions and how they are synthesized (Fig. 1). In living organisms, a genetic code initiates and controls the synthesis of functional, discrete molecules, which range in size from multienzyme complexes and molecular machines through mid-sized natural products to small molecules (e.g. NO). These molecules are often assigned to well-established distinct classes, such as proteins/peptides, carbohydrates, lipids, and (less structurally related), natural products and metabolites. Groups of these molecules frequently act together in regulatory networks [1–4] so as to enable critical life functions. Typical biofluids and tissues are very complex mixtures which can be resolved into defined molecular fractions using current high-performance separation technologies.
Fig. 1.

Natural complex organic materials divide into either functional biomolecules which eventually derive from a genetic code or complex biogeochemical nonrepetitive materials which are formed according to the general constraints of thermodynamics and kinetics from geochemical or ultimately biogenic molecules. While biomaterials are amenable to successful separation into unambiguously defined molecular fractions, complex nonrepetitive materials cannot be purified in the conventional meaning of purity due to their extreme intricacy; in fact, the molecular signatures of these supermixtures often approach the limitations imposed by the laws of chemical binding. Improvements in the resolution and sensitivity of analytical techniques combined with the use of minimal (non)invasive sampling techniques have enabled environmental and living systems to be observed to a degree of molecular resolution that was considered unthinkable only a few years ago. A molecular-level understanding of biogeochemical and life processes implies a key role for de novo structural analysis, which depends on the combined use of separation technology hyphenated to organic structural spectroscopy and integrated mathematical data analysis. Analyses of supermixtures depend even more on the mathematical analysis of correlated data obtained from complementary molecular-level precision analytical methods. The formation of NOM on the Earth preceded the evolution of life; the binding of NOM-derived prebiotic molecules to borate contributed to the synthesis of ribose, a crucial precursor of nucleotides, in good yield [5]. Later on in the Earth’s history, coevolution occurred between prebiotic/abiotic molecules, NOM and primitive and higher forms of life. The near-continuum of binding sites available to ions and organic molecules acts to buffer against environmental and chemical extremes in the geo- and biosphere, which could damage life because of their potent reactivity. This key supportive role of (for example) natural organic matter (NOM) in life processes is sustained by strong interactions between biological and geochemical cycles (Fig. 2). Hence, plant and animal residues are key ingredients of NOM synthesis, while NOM itself, which defines the bioavailability of crucial organic and inorganic nutrients, is indispensable for the sustenance of the microbial life at the bottom of the food chain
The fate of the second vast group of molecules in the bio- and geosphere is governed by the rather fundamental restraints of thermodynamics and kinetics (Figs. 1 and 2). In these intricate materials, the “classical” signatures of the (geogenic or ultimately biogenic) precursor molecules, like lipids, glycans and proteins, have been attenuated [26, 27], often beyond recognition, during a succession of biotic and abiotic (e.g. photo- and redox chemistry) reactions. Because of this loss of a biochemical signature, these materials can be designated nonrepetitive complex systems. The quantity of molecules in the Earth’s crust that can be attributed to these nonrepetitive complex materials, in the form of kerogens and natural organic matter (NOM) alone, exceeds the quantity of functional biomolecules by several orders of magnitude [28, 29]. Examples include freshwater, marine, and soil organic matter, kerogens and aerosols, among others. These materials typically exhibit an extremely complex array of chemical structures and interactions across a large range of size- and timescales, resulting in molecular signatures that reflect the fundamentals of chemical binding rather than those of their precursors. These novel signatures may in fact cover a sizable proportion of the theoretically feasible molecular composition space (Fig. 9). This extraordinary heterogeneity of molecularly diverse species renders these materials refractory and also implies a limited probability of detecting identical molecules [18]. This contrasts sharply with even the most complex mixtures of biomolecules extracted from any living organism, from which molecularly pure fractions can be readily obtained.
Fig. 2.

Natural organic matter (NOM) continuously interacts with a broad range of terrestrial, limnic and marine ecosystems. Common to all of these environments are the fundamental molecular aspects of life, and an availability of extended mineral surfaces for interactions with and binding of NOM. The dynamic equilibrium of NOM generation and decomposition spans timescales of many different orders of magnitude (from microseconds to hundreds of thousands of years), and it results from a combined action of biotic and abiotic reactions. NOM may be intrinsically recalcitrant because of the chemical structures of its organic molecules; alternatively, strong NOM–mineral interactions could alter the reactivity of these organic molecules towards increased resistance to degradation. The physical protection of organic matter at interior mineral surfaces provides alternative pathways that enable the recalcitrance of NOM. Photochemical degradation, one of the most significant abiotic reactions of NOM, often results in small molecules like CO2, which are mobile and are easily distributed within various ecosystems. Biomolecules derived from photosynthesis or otherwise originating from a genetic code are eventually decomposed according to the general laws and constraints of thermodynamics and kinetics (Fig. 1). Over very long timescales, the interactions of NOM with minerals at elevated temperatures result in the formation of geopolymers, like kerogen, coal, and oil shales. These ancient materials participate in bio- and geochemical cycling through natural and anthropogenic combustion and through weathering [6]
Fig. 9.

Van Krevelen diagram (which illustrates the C,H,O-compositional space) for consolidated ESI, APPI, APCI positive and negative ions of Suwannee river fulvic acid (SuwFA) in the mass range 200–700 Da (see the main text and Fig. 8)
Given these unique features, nonrepetitive complex systems epitomize supermixtures. The purification of a supermixture would, in the ultimate sense, approach a molecule-by-molecule separation—a feat beyond our reach, both conceptually and practically. Therefore, these complex nonrepetitive systems are operationally defined according to their properties rather than according to their chemical structures, and their purification (in the conventional sense of the word) remains elusive [18].
While the analysis of complex biomolecules has advanced to the degree that it is possible to obtain well-resolved three-dimensional molecular structures and even meaningful descriptions of dynamics and interactions [30–35], the molecular-level precision analysis of complex nonrepetitive materials remains rather rudimentary in comparison [20, 36–40]. First of all, theoretically well-founded approaches to numerically describe the complex, polydisperse and nonstoichiometric characteristics of nonrepetitive unknowns are missing at present, limiting our understanding of molecular structures and any application of quantitative structure–activity relationships (QSAR) when modelling their properties. Novel approaches suitable for a quantitative description of various hierarchical levels of molecular organisation (e.g. elements, fragments, molecules) must be developed. Secondly, a meaningful molecular-level analysis of nonrepetitive systems—such as aerosols, natural organic matter and native cell extracts—obviously cannot rely on target analysis, as most of the chemical environments and linkages present are simply not known (Fig. 3).
Fig. 3.

Hierarchical order of intricacy associated with the structural analysis of materials, in terms of polydispersity and molecular heterogeneity (see the main text). The structures (connectivities and stereochemistry) of monodisperse molecules are readily accessible (provided that sufficient amounts of the material are available) by organic structural spectroscopy [7–9]. Supramolecular structures [10–12] require an adequate definition of the covalently bonded molecules and of their noncovalent interactions [13–16]. The structural analysis of nonrepetitive complex unknowns, which feature substantial levels of both polydispersity and molecular heterogeneity [17, 18], is most demanding in terms of methodology and concepts [19–22]. Any highly resolved three-dimensional structure of a monodisperse biomolecule is based on a precise description of the unique chemical environment of any single atom [23, 24]. Currently, the molecular-level structural analysis of complex systems is primarily focused on covalent bond definition. Future high-quality structural analyses of these materials will have to assess both the (classes of) individual molecules and their interaction mechanisms [25]
Consequently, any comparative analysis of nonrepetitive unknowns with reference materials is very unlikely to provide satisfactory molecular resolution, because rather tiny variations in chemical binding may strongly and often unpredictably affect the properties commonly used for detection, such as retention times and spectral signatures. These fundamental restrictions that are intrinsic to comparative and target analysis are not easily circumvented and they necessitate an independent, spectroscopic “bottom-up” approach to the molecular-resolution characterisation of these complex unknowns.
Information transfer in organic structural spectroscopy and separation technologies
Interestingly, this “bottom-up” approach to the molecular characterisation of complex systems and materials necessarily relies upon spectroscopic methods that translate high-precision frequency measurements into important molecular-level information. Frequencies can be measured to an accuracy of 15 digits, and recently (2005) a Nobel Prize in Physics was awarded for improvements of laser-based precision spectroscopy that enable even more exact frequency measurements to be obtained [41]. This high accuracy of frequency measurement translates directly into high resolution, itself a very useful and even indispensable feature when producing information-rich data. Well-resolved signatures are less susceptible to the detrimental consequences of intrinsic averaging, which is an often overlooked key feature of any low-resolution (spectroscopic and separation) method, when they are employed in the characterisation of complex systems (Fig. 4).
Fig. 4.

Information transfer in organic structural spectroscopy. These images define a three-dimensional space composed of the area of the image (pixel resolution along the x- and y-axes) and the depth of the color space. This three-dimensional space offers n options for depicting dissimilarity (n = x × y × color depth). Significant resolution is only attained if there is sufficient information to enable a meaningful assessment of data [here, the apparent differentiation of (a) a fish, (b) a mountain, (c) a human, and (d) a beetle is only possible at panels C and D]. In analogy with these considerations, a three-dimensional analytical volumetric pixel space comprising NMR spectroscopy, mass spectrometry and separation is developed in Fig. 12. The current expansion of this analytical volumetric pixel space (log 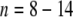 ) is sufficient to elaborate meaningful detail at molecular resolution from the most complex biological and biogeochemical mixtures
) is sufficient to elaborate meaningful detail at molecular resolution from the most complex biological and biogeochemical mixtures
Bulk data of complex systems, like physical parameters, total acidity and elemental analyses, seem to be more precisely defined [42], but exhibit limited resolution. However, any sound structural model of these materials must conform to the constraints defined by these “hard” bulk data. High-energy methods of organic structural organic spectroscopy, like XANES, UV/VIS and infrared spectroscopy, exhibit intermediate structural resolution, which is sufficient, for example, for the characterisation of specific chemical environments [43]; for instance, functional group analysis (carbonyl derivatives, aromatics, heterocycles) in intricate materials.
In general, the degree of significant detail generated by a certain analytical technique will depend on both the intrinsic resolution of the respective method and the characteristics of the analysed material. Any inadequate relationship between the resolving power of the technique and intrinsic analyte properties will be wasteful. Investigations of near-featureless materials with methods of supreme resolution could result in unnecessary effort and expenditure. Insufficient resolution of any analytical method with respect to the properties of the analyte will inevitably result in intrinsic averaging, which typically results in poorly resolved properties (which affect the separation) and/or poorly resolved chemical environments (which affect the spectra). Intrinsic averaging is visualized in Fig. 4 in the form of images of ever-degrading resolution. Similarly, insufficient resolution deteriorates detail in spectra and chromatograms of complex nonrepetitive materials, producing low-resolution signatures and limited bandwidths of variance in bulk and spectral properties.
Hence, any organic structural spectroscopy with a limited peak capacity (Fig. 5) will inevitably lead to a summary bulk-type description of complex materials and considerable averaging, rather than to a meaningful molecular-level resolution analysis. In the case of NOM, this inevitable relationship has been observed in many spectroscopic, separation and chemical experiments, resulting in data with a remarkably limited bandwidth of variance, even when advanced techniques (e.g. at the level of one-dimensional solid-state 13C NMR spectroscopy) are used [44, 45].
Fig. 5.

Characteristic resolutions (peak capacity: total range / half width [57]) of various separation technologies and organic structural spectroscopic methods (see the main text). This diagram represents a two-dimensional projection of the analytical volumetric pixel space—comprising NMR spectroscopy, mass spectrometry and separation technologies—that defines our current capacity to depict variance in complex systems with molecular resolution (see Fig. 12)
Analogously, the widespread use of the idiom HULIS (or humic-like substances) in the fields of, for instance, aerosol and remediation research [46–53] reflects the operational definition of humic materials as well as our current inability to perform a meaningful molecular-level analysis of complex unknowns, as materials currently denoted HULIS or humic-like substances undoubtedly encompass a wide range of very different species.
Due to the huge peak capacity of FTICR mass spectrometry, FT mass spectra provide the most convincing direct experimental evidence for the extraordinary molecular diversity of complex materials at present. In these, the molecular-level intricacy of the complex unknowns is most adequately converted into very highly resolved and, consequently, extremely information-rich signatures.
Analogous considerations to those given here for spectroscopic characterisation also apply to the separation of complex materials [54, 55, 56].
Instrumentation and methods
The 13C NMR spectrum of Suwannee river fulvic acid (SuwFA) shown in Fig. 7 was acquired at the GSF with a Bruker (Bremen, Germany) AC 400 NMR spectrometer, operating at 100 MHz for 13C. FTICR mass spectra were acquired at Bruker’s facilities with a 9.4-T APEXq FT mass spectrometer (data in Fig. 9) and a 12-T APEXq FT mass spectrometer at the GSF (Fig. 8). Here, FTMS spectra were acquired with a time domain size of 1 MWord (Fig. 8a; Fig. 9, typical resolution 3 × 105) or 4 MWord (Fig. 8b, typical resolution 7 × 105). For Figs. 8a and 8b, elemental compositions were computed with the DataAnalysis software, version 3.4 (Bruker), using the following restrictions: C, H, N, O, unlimited; S, P, 0–5; H/C ratio < 3, mass error ≤ 0.5 ppm; observance of the nitrogen rule. Exactly one elemental formula was obtained for each peak. The elemental formulae of Fig. 9 were batch-calculated using a software tool written in-house, as described elsewhere [36].
Fig. 7.

Molecular-level resolution spectroscopic data represent projections of the vast total structural space of molecules, for which count estimates range from 1060 to 10200 [72]. The complementarity of NMR spectroscopy and mass spectrometry for the spectral characterisation of intricate materials is caused by the entirely different atomic and molecular processes that these methods rely upon (Fig. 6). Mass spectra reflect the isomer-filtered complement of the entire space of molecular structures. The compositional space of molecules can be probed with ultrahigh-resolution FTICR mass spectroscopy, resulting in single peaks for molecules (in the absence of fragmentation). NMR spectra represent site- and isotope-specific projections of the molecular environments (the projected NMR spectrum given here shows a 13C NMR spectrum for an aquatic NOM). Accordingly, typical organic molecules exhibit single mass peaks (molecular ions given here: C9H9NO3) and more elaborate NMR signatures (13C NMR data are shown here for C9H9NO3; see also Fig. 6). Because these atomic and molecular signatures are not entirely orthogonal, the data provided by NMR and MS show correlations that can be used to reconstruct chemical structures by empirical and mathematical back-projection. In mixture analysis, separation offers a (near-)orthogonal means of expanding the two-dimensional spectroscopic projection area into a three-dimensional analytical volumetric pixel space (see Fig. 12). Following separation, molecules that often exhibit wide concentration variances in mixtures can be investigated by combined NMR/MS, with acquisition parameters specifically adapted in order to maintain the optimum dynamic range, which is necessary for good-quality spectra
Fig. 8.

FT mass spectral resolution and the C,H,O-compositional space (see the main text and Fig. 9)
The numbers of isomers, as displayed in Fig. 11, were calculated by constructive enumeration using the software MOLGEN (Department of Mathematics, University of Bayreuth, Bayreuth, Germany). For this computation [58], certain restrictions were applied in order to exclude structures that are mathematically possible but are not likely to occur in materials of biogeochemical origin; this means that there are (1) no peroxides; i.e. no –O–O– connectivities; (2) no triple bonds (–C≡C–); (3) no three- or four-membered rings; and (4) no carbon with cumulative double bonds (=C=). The 1H and 13C NMR spectra of cholesterolacetate (section of 1H NMR spectrum in Fig. 6, top panels) and 2-carboxypyrene (Fig. 6, middle panels) were computed with ACD software (ACD/HNMR and ACD/CNMR Predictor, v. 5.0); the mass spectral isotope peaks of Ciguatoxin C60H86O19 (Fig. 6, middle panels) were computed with Bruker Compass FTMS software.
Fig. 11.

The relationship between the FTICR mass spectral intensity and the number of feasible isomers in complex materials. The distribution of (chemically relevant) C,H,O-isomer counts within the C,H,O-compositional space is visualized here in a van Krevelen diagram (left) for the eleven feasible molecular compositions CnHmOq that have an IUPAC nominal mass of 178 Da (see the main text). These molecules are arranged into three series (series 1a–1c with three members; series 2a–2f with six members; series 3a–3b with two members) of isobaric molecules, which are related by a formal exchange of CH4 against oxygen. For any molecular composition, the number of chemically relevant isomers is given in units of 104. Carboxyl-rich alicyclic molecules (CRAM), which represent a complex mixture of molecules with near-absent olefinic and aromatic unsaturation, have recently been identified in marine organic matter [18], and they occupy an area of C,H,O-compositional space for which the largest number of feasible C,H,O-isomers is expected (see the main text), suggesting that the actual number of different molecules in the mass spectra of CRAM for any given mass may correlate with mass spectral intensity patterns. The availability of aromatic moieties in Suwannee river fulvic acid (SuwFA) also allows chemically relevant structures at lower H/C ratios (see the main text). The green circumfenced area denotes the van Krevelen compositional space of SuwFA, as provided in Fig. 9 (note that the SuwFA here represents consolidated positive and negative ion APCI+APPI+ESI FTICR mass spectra, as opposed to the negative ion ESI FTICR mass spectra given in the case of CRAM; see the main text). The panel on the right shows the CnHmOq isomers computed for an IUPAC nominal mass of 178 Da in an intensity versus mass display (analogous to a mass spectrum), denoted according to series 1–3 (see the main text). This pattern resembles the clusters of peaks observed in good-quality FT mass spectra of NOM (see Fig. 6, bottom panels, and Fig. 8), suggesting that the intensities of the C,H,O-derived mass spectral peaks of NOM follow the number of chemically relevant C,H,O-isomers computed
Fig. 6.

The two most significant methods of organic structural spectroscopy, nuclear magnetic resonance (NMR) and FTICR mass spectrometry, are based on high-precision frequency measurements. The top panels illustrate NMR and FTICR MS for atomic and molecular processes. The precessions of atomic magnetic moments in molecules are defined by the chemical environment, and this means that NMR yields unsurpassed resolution of short-range molecular order (in noncrystalline materials, for which X-ray crystallography is not available). In FT mass spectrometry, the orbital frequencies of ions depend on their molecular masses. Mid-size molecules (see middle panels) provide information-rich signatures in NMR and only single peaks in mass spectra (under conditions of non-fragmentation), while complex non-repetitive materials (see bottom panels) produce low-resolution signatures in NMR because of extensive peak overlap. High FT mass spectra resolution is retained for complex molecules, however, because of the extensive peak capacity of the technique (see Fig. 5)
High-precision frequency-derived organic structural spectroscopy
The two most influential organic structural spectroscopic methods for the investigation of complex materials, which depend upon high-precision frequency measurements, are NMR spectroscopy and FTICR mass spectrometry (Table 1, Figs. 6, 7). In NMR, the precession frequencies of individual atomic nuclei in an external magnetic field B0 are influenced by their respective chemical surroundings; in FTICR mass spectrometry, the orbital frequencies of ions in an ion trap cell depend on the mass and charge of the molecule of interest [59]. Both methods are isotope-specific, and the combination of NMR and FTICR mass spectral data provides more useful spectral information on complex unknowns at the molecular level than any other spectroscopic method at present.
Table 1.
Characteristics and significance of key molecular-level resolution techniques
| Molecular-level resolution technique | Advantages | Current weaknesses and future developments |
|---|---|---|
| NMR spectroscopy | NMR spectroscopy provides isotope-specific information, in unsurpassed detail, on short-range molecular order (the arrangement of chemical bonds, including connectivities, stereochemistry and spatial proximity), dynamics [33, 74] and reactivity [35] | Relative insensitivity compared with other analytical techniques |
| Nondestructive and isotope-specific [32] analysis across almost the entire periodic table combines with the most accurately defined near-quantitative relationship between the spin number and the area of the NMR signal. This key feature of NMR when applied to the analysis of complex systems implies the use of NMR spectrometry as a quantitative reference for other, complementary analytical methods [75–81] | Intricate physics and chemistry of intra- and intermolecular interactions in complex mixtures may interfere with the direct relationship between chemical shift and molecular structure and, because of relaxation-induced variable line widths, quantification | |
| The unique ability to generate and analyse data from multiple 1,2,3-D NMR experiments performed on a single sample enables the significance and authenticity of individual spectra to be assessed [82–84] | Near-identical chemical shifts do not necessarily imply similar chemical structures [85] | |
| Extensive and far-reaching information can be obtained, even from ill-resolved NMR spectra, for “small” nuclei (e.g. 1H, 13C, 15N, 31P) because of the plausible correspondence between chemical shift and extended substructures | Sensitivity and resolution increased by high-field magnets [86, 87], cryogenic [88] and micro- [89–92] probes, and by changing (to nanoliter) sample size [93, 94] | |
| Throughput increased by using fast higher dimensional spectroscopy with superior sensitivity and through the parallel acquisition of NMR spectra [95–99] | ||
| FTICR mass spectrometry | Best combination of spectral resolution and sensitivity, which allows miniaturisation [100] and hyphenation of mass spectrometry with high-performance separation techniques like capillary electrophoresis and UPLC (CE/UPLC with mass-selective detection) | Molecular-level structural information is mainly restricted to ionizable compounds |
| Due to its supreme mass accuracy and resolution, molecular formulae from thousands of compounds can be obtained in a single experiment directly from mixtures [63, 69, 101–104] | Isomer differentiation is a nontrivial task [71] | |
| Fragmentation provides further molecular-level structural information beyond molecular composition | Quantification is difficult, even for identical molecules in mixtures, because of the variable ionization efficiencies of individual compounds, which strongly depend on the experimental conditions and mixture composition | |
| Column adsorption and fractionation as well as electrochemical and redox reactions associated with the spray conditions may interfere with authentic sample representation | ||
| A wide range of ionization techniques (electrospray, ESI; chemical ionization, CI; photoionization, PI; desorption ionization, DESI; field ionization, FI, among others, all performed in either positive or/and negative modes) for mixtures is available under specifically adapted conditions [105–110] | ||
| Mass-selective imaging is feasible with high spatial and mass resolution; qTOF mass spectrometry allows for very fast scan rates, and is perfectly suited for hyphenation with high-performance separation techniques (CE and UPLC) as well as mass-selective imaging | Further miniaturisation of separation and detection devices in conjunction with ultrahigh-resolution FTICR mass spectrometry will permit highly resolved and information-rich data to be obtained from tiny amounts of sample (chip-MS) [100] | |
| High-performance separation techniques (UPLC/HPLC and capillary electrophoresis) | Large separation capacity and extensive miniaturisation; is cost-effective; can be highly automated | Gives only limited structure-specific information about the short-range molecular order [111, 112] |
| Electrophoretic mobility and chromatographic retention time carry structure-specific information, which can be adapted to a wide range of experimental conditions in order to probe size, shape, charge characteristics and reactivity | ||
| Sensitive and versatile suite of separation methods and of structure-specific (and nondestructive) detection systems, such as (laser-induced) fluorescence, UV/VIS, radioisotope or mass-selective detection | ||
| CE complements NMR information about primary chemical structures (covalent bonds) by providing data on the corresponding secondary and tertiary structure | ||
| Feasibility of up-scaling from capillary zone electrophoresis (CZE) to a preparative level by means of free flow electrophoresis (FFE) and from UPLC to any preparative LC method | Further miniaturisation offers hyphenation options down to single-cell analysis and compartments within |
When studying typical organic molecules, NMR spectra provide more information-rich data than mass spectra, because any single atom within can produce an individual NMR signature (Figs. 6 and 7) [7, 60–62]. In the absence of fragmentation, the mass spectrum of any particular molecule will contain only a single peak (in conjunction with its corresponding isotopic pattern [63, 64]). NMR spectra of ever more complex materials will eventually become near featureless because of the extensive overlap between individual NMR resonances [65]; however, the significance of the information is maintained because of the quantification reliability and insightful relationships between NMR chemical shift and extended substructures (at least for the “small” NMR-active nuclei, e.g. 1H, 13C, 15N, 31P, which represent the key players in organic structural spectroscopy) [66]. Mass spectrometry retains its supreme resolution for extremely complex systems [63, 67–70], but will eventually become limited by the inability of mass spectrometry to (easily) discriminate between isomers [71]. Hence, mass spectra will at best represent isomer-filtered projections of the entire structural space of molecules (see Figs. 7, 8, 9, 10, 11).
Fig. 10.

Elucidating mass spectra of complex materials requires advanced means of data analysis, such as van Krevelen diagrams, which are based upon assigned molecular formulae, and fragment- or molecule-specific Kendrick mass defect analyses [73]. Any dot in the van Krevelen diagram of a complex material represents a projection of the elemental ratios derived from assigned molecular formulae, irrespective of molecular mass. Accordingly, any of these dots could represent an intrinsic superposition of all feasible isomers from possibly different molecular compositions, sharing only their respective elemental ratios. The numbers of chemically reasonable isomers easily account for many millions of isomers seen for moderately sized molecules (a few hundred Daltons), even when only a few double bond equivalents and heteroatoms (e.g. oxygen) are present (see Fig. 11)
Molecular-level resolution spectroscopic data represent projections of the vast total structural space of molecules, for which count estimates range from 1060 to 10200 [72]. The complementarity of NMR and mass spectrometry for the spectral characterisation of intricate materials is caused by the entirely different atomic and molecular processes these methods rely upon (Fig. 6).
Mass spectra reflect the isomer-filtered complement of the entire space of molecular structures. The compositional space of molecules can be probed with ultrahigh-resolution FTICR mass spectroscopy, resulting in single peaks for molecules (in the absence of fragmentation). Two-dimensional projections of the structural space, like van Krevelen diagrams and Kendrick mass defect analyses, are indispensable tools for the evaluation of mass spectra of complex materials (Figs. 8, 9, 10, 11) [73, 113, 114]. NMR spectra represent site- and isotope-specific projections of the molecular environments. Therefore, typical organic molecules exhibit single mass peaks (molecule ions) in mass spectra and more elaborate NMR signatures (Figs. 6 and 7). Because these atomic and molecular signatures are not entirely orthogonal, the data provided by NMR and MS exhibit correlations that can be used to reconstruct chemical structures by empirical and mathematical back-projection.
The NMR and mass spectral data can be acquired via direct hyphenation or in separate experiments [115–117]. In mixture analysis, separation offers a (near-)orthogonal means of expanding the two-dimensional spectroscopic projection area into a three-dimensional analytical volumetric pixel space (see Fig. 12). Following separation, molecules that often exhibit wide concentration variances in mixtures can be investigated by combined NMR/MS, with acquisition parameters specifically adapted to maintain the optimum dynamic range necessary for good quality spectra. Particularly in the case of mixture analysis, any joint mathematical evaluation of these correlated data will reveal hidden detail and will considerably enhance the resolution as well as the significance of molecular-level information (see also Figs. 4 and 12).
Fig. 12.

The currently accessible discrete volumetric pixel (voxel) space for the characterisation of complex materials is in the range of 108–14 voxels. Its expansion is defined by the significant resolution of the complementary techniques of nuclear magnetic resonance (102–5 buckets, depicting the short-range order of molecules), ultrahigh-resolution FTICR mass spectrometry (104–5 buckets, depicting molecular masses and formulae of gas-phase ions) and high-performance separation (102–4 buckets, depicting both ions and molecules; this provides a way to validate NMR against MS data); see also Fig. 5 for even wider expansion. The various projections of this voxel space, like separation/MS, separation/NMR and NMR/MS, can be realised in the form of direct hyphenation [104, 115, 117–120] and via mathematical analysis [121, 122]
FTICR mass spectra show supreme resolution, as indicated by the 12-T negative ionization ESI FT mass spectra of a barley extract (Fig. 8a) and IHSS Suwannee River Natural Organic Matter (International Humic Substances Society NOM; Fig. 8b). Here, CnHmOq molecules contribute most to the total ion count. These molecules can be arranged into series, which are related by the formal exchange of CH4 against oxygen. Figure 8c denotes the mass peaks corresponding to the 37 theoretically possible and chemically reasonable C,H,O-compositions depicted in Fig. 8d that have a nominal mass of 301 Da. Note that negative M–H+ ions (i.e. [M−H+e]−) are observed in the FTICR mass spectra (Fig. 8b), and the C,H,O-compositions of molecules M are denoted in Fig. 8c. M and M–H+ differ in mass by one hydrogen (1.007825032 Da) minus an electron (0.000548625 Da); in Fig. 8c this difference is decomposed into a mass shift of one (see the shift between the mass axes) and an additional small mass spacing Δm = 0.000233878 Da. The molecules in the barley extract exhibit mass peaks outside of the range accessible for any C,H,O-composition (dotted purple box in Fig. 8a), indicating the presence of additional heteroatoms (e.g. N, P, S) in these ions.
Figure 8d denotes a van Krevelen diagram of the 37 chemically reasonable CnHmOq molecules, in which the 16 C,H,O-ions observed in Fig. 8b are highlighted. The number of peaks identified corresponds to a coverage of 43% of the entire C,H,O-compositional space. These ions occupy an area for which the largest number of feasible C,H,O-isomers is expected (see Fig. 11).
Molecularly intricate materials, like natural organic matter (NOM), exhibit molecular signatures approaching the theoretical limits defined by the laws of chemical binding. In Fig. 9, a van Krevelen diagram of Suwannee River fulvic acid (SuwFA) depicts the elemental ratios of CnHmOq ions (the ions shown represent a consolidation of the ions obtained by ESI, APCI and APPI positive and negative ionization from 9.4-T FTICR mass spectra; unpublished data). The peaks observed in the negative/positive ionization mode only are coloured green/orange; peaks observed in both positive and negative modes are depicted in black. The lack of signatures from biochemical precursor molecules [123] indicates the considerable level of processing typical of NOM. Within a mass range of 200–700 Da and the given limits of the H/C and O/C ratios, the minimum consolidated number of individual C,H,O-molecular compositions (4270) represents a sizable fraction (23%) of the entire feasible compositional space of CnHmOq molecules (18414 in total; small grey dots). To further appreciate the remarkable intricacy of natural organic matter, it should be noted that any dot in the van Krevelen diagrams of these complex materials represents a projection of the elemental ratios derived from assigned molecular formulae, irrespective of molecular mass. Hence, the dots in the van Krevelen diagrams can represent multiple molecular formulae (Figs. 10 and 11), while any identified molecular composition reflects an intrinsic superposition of all feasible isomers (Fig. 11). Considering typical molecular weights of several hundreds of Daltons in the mass spectra of NOM (Fig. 6, bottom panels), it is readily anticipated that the mass spectra of such systems represent simplified (e.g. isomer-filtered) projections of a still hugely more expansive structural space (Fig. 7).
Key trends relating molecular composition to the number of feasible isomers
For any exceedingly complex material, it is logical to postulate that many isomers will contribute to any given molecular formula. Analogously, the intensities of the mass spectral peaks, which superimpose all of the isomers present, will be a function of the abundances of these isomers in these materials and the ionization efficiency of each isomer under the given experimental conditions.
For molecules of a given mass composed of carbon, hydrogen, and oxygen, two major and independent trends are expected to define the number of feasible isomers. First, decreasing the H/C ratio from fully saturated molecules (CnH2n+2) means removing hydrogen atoms, which is equivalent to introducing double bonds or (ali)cyclic structures (double bond equivalents, DBEs). Molecules with large H/C ratios are structurally fairly uniform, consisting mainly of various branched chains of single bonds. Introducing large numbers of DBEs will lead to many new structures with double bonds and or (ali)cyclic structures in various positions. For an H/C ratio of close to one, on average two carbons carry one DBE, and the introduction of further DBEs will lead to a lack of single bonds. Hence, the maximum number of feasible C,H,O-isomers is expected to occur for intermediate numbers of DBEs in a molecule, because the occurrence of a DBE (which solely depends on the H/C ratio) enables double-bond displacement and the formation of (ali)cyclic structures, both of which greatly enlarge the number of feasible isomers. In contrast, only highly condensed structures can be assembled at very low H/C ratios [37], and this constraint severely diminishes the number of feasible C,H,O-isomers (if mathematically possible but chemically unlikely isomers are excluded; see Fig. 11).
Second, the insertion of oxygen into potentially any carbon–carbon (creating C–O–C units) or carbon–hydrogen bond (creating C–OH functionalities) will result in many more feasible isomers at low O/C ratios; in the presence of DBEs, “terminal” carbonyl derivatives (C=O) can also be constructed. At higher O/C ratios, however, further insertion of oxygen decreases the number of feasible isomers for two reasons: oxygen provides fewer (two) options for forming (single) bonds with other partners than carbon (four); in addition, the higher mass of oxygen (16 Da) compared with that of carbon (12 Da) decreases the total number of “heavy” atoms available for the construction of CnHmOq molecules of a given mass.
These considerations imply that the number of feasible C,H,O-isomers for a given mass will reach maximum values at intermediate H/C and O/C ratios, and that these numbers will (sharply) decline at extreme (high and low) H/C and O/C ratios, respectively.
These dependencies are displayed in a van Krevelen diagram (Fig. 11), in which the numbers of chemically relevant isomers for any given molecular composition CnHmOq of a single nominal IUPAC mass are provided. For any given nominal mass, the mathematically possible and chemically relevant structures composed solely of carbon, hydrogen and oxygen atoms can be constructively enumerated for each composition (molecular formula) [58]. By “chemically relevant isomers”, we mean all mathematically possible isomers (not counting stereoisomers) except for those containing O–O bonds, C≡C bonds, three- or four-membered rings, or =C= fragments (cumulated double bonds), which are not assumed to occur in the materials of interest (natural organic matter here). These data are displayed in the right panel of Fig. 11, where they are arranged according to actual mass.
For practical reasons, we have selected CnHmOq compositions with a nominal IUPAC mass of 178, for which the number of isomers can be computed within a reasonable time on a desktop computer; within the given limits of H/C and O/C elemental ratios, eleven feasible C,H,O-molecules are found, which are grouped into three series of isobaric molecules, related by a formal exchange of CH4 for oxygen (Fig. 11).
Series 1 represents highly unsaturated molecules in which the number of isomers declines sharply with decreasing H/C ratio. Series 2 presents the maximum number of isomers at intermediate H/C (and O/C) ratios and the decline in the number of isomers at both high and low H/C (and O/C) ratios, as anticipated (see above).
The maximum H/C ratio found for a series 2 molecule amounts to almost 1.7, and the corresponding molecule C13H22 (2a) features three DBEs, thereby allowing for a much larger array of unsaturation-related isomers than obtained for a fully saturated parent molecule. This is demonstrated by the variance in the isomer count when the fully saturated analogue C13H28 (184 Da, 802 isomers) is compared with the series 2 “endmember” C13H22 (2a; 178 Da, 1.7 × 105 isomers); analogous relationships are found for the series 1 “endmember” C14H10 (1a; 178 Da, 5.3 × 106 isomers) in comparison with its fully saturated parent molecule C14H30 (198 Da, 1858 isomers).
A considerable fraction of the 16.6-fold increase in the isomer count observed when comparing C13H22 (2a; three DBEs) and C12H18O (2b; four DBEs) results from the ability to produce novel isomers with singly bonded oxygen and those with a C=O bond (carbonyl derivative). The maximum number of isomers (~1.1 × 107 each) is attained for the molecules C11H14O2 (2c; five DBEs) and C10H10O3 (2d; six DBEs), respectively. Further exchange of CH4 against oxygen again sharply decreases the number of feasible isomers [by a factor of 5.5 when proceeding from C10H10O3 (2d) to C9H6O4 (2e), and by a factor of 78 when changing from C9H6O4 (2e) to C8H2O5 (2f)].
The series 3 molecules C7H14O5 (3a) and C6H10O6 (3b) feature rather limited numbers of isomers because of their large O/C ratios (see above). A comparison of C8H2O5 (2f) and C7H14O5 (3a) indicates that extreme hydrogen deficiency restricts the feasible number of isomers more severely than almost full saturation. Molecules C14H10 (1a; ten DBEs, 5.3 × 106 isomers; series 1), C10H10O3 (3b; six DBEs, 1.1 × 107 isomers; series 2) and C6H10O6 (3b; two DBEs, 6 × 104 isomers; series 3) are all related by a formal exchange of four carbons for three oxygen atoms. The introduction of oxygen initially outweighs the decrease in the number of carbon atoms and DBEs available because of (i) the reduced severity of unsaturation and (ii) the availability of oxygen to construct isomers (see above). Upon the transition from C10H10O3 (2d) to C6H10O6 (3b), however, both the lesser ability of oxygen to participate in chemical bonding (two bonds for any oxygen instead of four for any carbon) and the decline in available DBEs lead to a drastic decrease in the number of accessible isomers.
Mass spectral intensities and number of feasible isomers in marine and terrestrial NOM
In a highly processed and supposedly exceedingly complex material such as deep sea marine organic matter, most of the molecules of formula CnHmOq will contain an intermediate amount of unsaturation and numerous oxygen atoms [18, 20]. This flexibility to generate a potentially huge number of isomers implies that (in the absence of severe ion suppression) mass spectral intensities should correlate roughly with the number of feasible isomers for any given molecular composition. Recently, carboxyl-rich alicyclic molecules (CRAM) have been identified as prominent constituents of marine (and possibly freshwater and terrestrial) organic matter [18]. CRAM likely represent highly processed products of ultimately terpenoid origin and are expected to represent an extremely complex mixture of molecules. Based on the molecules of formulae CnHmOq and a recognition of FT mass spectral intensities, the CRAM that occur in deep ocean marine ultrafiltered organic matter comform mainly to the region inside the dotted ellipsoid in the van Krevelen diagram of Fig. 11, which appears to coincide with the maximum number of feasible C,H,O-isomers.
The availability of aromatic structures in terrestrially and freshwater-derived NOM, such as that in Suwannee river fulvic acid (SuwFA; Fig. 9), opens up the compositional space of chemically relevant NOM molecules (see above) to significantly lower H/C ratios than accessible solely on the basis of open-chain unsaturation (e.g. olefinic and carbonyl) and alicyclic double-bond equivalents (DBE). These dependencies are nicely illustrated by comparing the van Krevelen diagrams of marine ultrafiltered dissolved organic matter (UDOM) [18], a blackwater NOM [18, 124], and that of SuwFA (Figs. 9 and 11). While mass spectra of marine UDOM are dominated by carboxyl-rich alicyclic molecules (CRAM), composed mainly of carboxylic groups and alicyclic rings with only negligible aromatic and olefinic unsaturation [18], the significant terrestrial, aromatic-rich signature present in both blackwater NOM and SuwFA populates the compositional space with notably lower elemental H/C ratios than feasible in marine UDOM [18].
It should be noted that oxygen-depleted molecules of formula CnHm are less likely to be ionized in standard ESI FTICR mass spectra in comparison with oxygenated molecules of formula CnHmOq. Carbohydrates, which are oxygen-rich, also are less efficiently ionized under standard ESI-FTICR mass spectral conditions than carboxyl-rich molecules like CRAM. CRAM therefore represent the most likely constituents of NOM to produce strong signals in ESI-FTICR mass spectra.
The compositional space of Suwannee river fulvic acid (SuwFA) given in Figs. 9 and 11 is derived from consolidated positive and negative ion FTICR mass spectra, obtained via APCI+APPI+ESI ionization modes, thereby facilitating the observation of oxygen-depleted molecules (Fig. 9).
The discrete analytical volumetric pixel space defines our current capacity to depict molecular-level variance in complex systems
The current capacity to describe complex materials at molecular resolution can be visualized in the form of an analytical space comprising individual volumetric pixels (voxels). The range of this discrete and quantized space is 108–14 voxels, as defined by the significant resolution of the complementary techniques of nuclear magnetic resonance (102–5 buckets, depicting the short-range order of molecules), ultrahigh-resolution FTICR mass spectrometry (104–5 buckets, depicting molecular masses and formulae of gas-phase ions) and high-performance separation (102–4 buckets, has the capacity to investigate both ions and molecules, and so provides a way to validate NMR against MS data).
An investigation of these correlated data is feasible at the level of the direct hyphenation of separation and spectroscopy [e.g. LC/NMR and LC or CE/MS; corresponding to the rear faces of the voxel space [119, 125–128]) and by means of statistical heterospectroscopy (SHY) [129, 130]; corresponding to the top face (or any two faces) of the voxel space]. Any joint mathematical analysis of these correlated data will enhance the effective resolution of the data and the significance of the molecular-level analysis of complex unknowns [119, 121, 129].
This voxel space can be readily expanded to higher dimensions by including complementary data, like those derived from genomic and proteomic analyses [84, 131–134] or by recognising selective chemical reaction products [135–140]. Degradative approaches to the characterisation of complex systems produce limited amounts of unambiguously identifiable small molecules but lose crucial linkage information. Soft and selective biochemical and chemical reactions like mild hydrolysis, reduction, oxidation and derivatisation [141, 142] of complex systems will often result in larger fragments with valuable positional and stereochemical information for the assessment of synthesis and degradation pathways.
The chemical transformation of functional groups with NMR- and MS-recognisable labels enables isotope-specific functional group analysis based on structural rather than behavioural characteristics [143, 144]. Information concerning stereochemistry and stable isotope composition will become more important when assessing the origins and diagenesis of complex natural materials. Any progress in the determination of position-specific stable isotope composition (e.g. by NMR and MS methods) will be useful for advancing this field. Physical and chemical fractionation will greatly assist in these studies; further miniaturisation will enhance separation capacity and thereby improve the resolution of the analytical voxel space (Figs. 5 and 12).
Integrated biomarker profiling approaches [145–147] with higher resolutions, significances and accuracies will substantially improve the quality and relevance of current systems biology approaches in the health and environmental sciences. The great progress made in the molecular-level characterisation of complex systems over the last few years and foreseeable improvements in nascent technology and concepts will lead to strong synergetic effects that will further advance our understanding of any complex natural and living system whose properties and functioning depend on both strong (covalent) and weak (noncovalent) interactions.
Electronic supplementary material
Below is the link to the electronic supplementary material.
(PDF 12.9 MB)
References
- 1.Duarte NC, Becker SA, Jamshidi N, Thiele I, Mo ML, Vo TD, Srivas R, Palsson BO (2007) Proc Natl Acad Sci USA 104:1777–1782 [DOI] [PMC free article] [PubMed]
- 2.Ewing RM, Chu P, Elisma F, Li H, Taylor P, Climie S, McBroom-Cerajewski L, Robinson MD, O’Connor L, Li M, Taylor R, Dharsee M, Ho Y, Heilbut A, Moore L, Zhang S, Ornatsky O, Bukhman YV, Ethier M, Sheng Y, Vasilescu J, Abu-Farha M, Lambert JP, Duewel HS, Stewart II, Kuehl B, Hogue K, Colwill K, Gladwish K, Muskat B, Kinach R, Adams SL, Moran MF, Morin GB, Topaloglou T, Figeys D (2007) Mol Syst Biol 3:89 [DOI] [PMC free article] [PubMed]
- 3.Patil KR, Nielsen J (2005) Proc Natl Acad Sci USA 102:2685–2689 [DOI] [PMC free article] [PubMed]
- 4.Ravasz E, Somera AL, Mongru DA, Oltvai ZN, Barabasi AL (2002) Science 297:1551–1555 [DOI] [PubMed]
- 5.Ricardo A, Carrigan MA, Olcott AN, Benner SA (2004) Science 303:196–196 [DOI] [PubMed]
- 6.Dickens AF, Gelinas Y, Masiello CA, Wakeham S, Hedges JI (2004) Nature 427:336–339 [DOI] [PubMed]
- 7.Cook LR, Oinuma H, Semones MA, Kishi Y (1997) J Am Chem Soc 119:7928–7937
- 8.Duus JO, Gotfredsen CH, Bock K (2000) Chem Rev 100:4589 [DOI] [PubMed]
- 9.Barrientos LG, Louis JM, Ratner DM, Seeberger PH, Gronenborn AM (2003) J Mol Biol 325:211–223 [DOI] [PubMed]
- 10.Boyer JL, Kuhlman ML, Rauchfuss TB (2007) Acc Chem Res 40:233–242 [DOI] [PubMed]
- 11.Shuai XT, Merdan T, Unger F, Kissel T (2005) Bioconjug Chem 16:322–329 [DOI] [PubMed]
- 12.Takahashi H, Nakanishi T, Kami K, Arata Y, Shimada I (2000) Nat Struct Biol 7:220–223 [DOI] [PubMed]
- 13.Badjic JD, Nelson A, Cantrill SJ, Turnbull WB, Stoddart JF (2005) Acc Chem Res 38:723–732 [DOI] [PubMed]
- 14.Guillot-Nieckowski M, Joester D, Stohr M, Losson M, Adrian M, Wagner B, Kansy M, Heinzelmann H, Pugin R, Diederich F, Gallani JL (2007) Langmuir 23:737–746 [DOI] [PubMed]
- 15.Paulini R, Muller K, Diederich F (2005) Angew Chem Int Edit 44:1788–1805 [DOI] [PubMed]
- 16.Sprangers R, Kay LE (2007) Nature 445:618–622 [DOI] [PubMed]
- 17.Burdon J (2001) Soil Sci 166:752–769
- 18.MacCarthy P (2001) Soil Sci 166:738–751
- 19.Hertkorn N, Perdue EM, Kettrup A (2004) Anal Chem 76:6327–6341 [DOI] [PubMed]
- 20.Kujawinski EB (2002) Environ Forensics 3:207–216
- 21.Perdue EM, Hertkorn N, Kettrup A (2007) Anal Chem 79:1010–1021 [DOI] [PubMed]
- 22.Presciutti F, Capitani D, Sgamellotti A, Brunetti BG, Costantino F, Viel S, Segre A (2005) J Phys Chem B 109:22147–22158 [DOI] [PubMed]
- 23.Gratias R, Konat R, Kessler H, Crisma M, Valle G, Polese A, Formaggio F, Toniolo C, Broxterman QB, Kamphuis J (1998) J Am Chem Soc 120:4763–4770
- 24.Hirsch AKH, Fischer FR, Diederich F (2007) Angew Chem Int Edit 46:338–352 [DOI] [PubMed]
- 25.Harper JK, Grant DM, Zhang YG, Lee PL, Von Dreele R (2006) J Am Chem Soc 128:1547–1552 [DOI] [PubMed]
- 26.Reemtsma T, These A, Springer A, Linscheid M (2006) Environ Sci Technol 40:5839–5845 [DOI] [PubMed]
- 27.Cabaniss SE, Madey G, Leff L, Maurice PA, Wetzel R (2005) Biogeochemistry 76:319–347
- 28.Hedges JI, Oades JM (1997) Org Geochem 27:319–361
- 29.Vandenbroucke M, Largeau C (2007) Org Geochem 38:719–833
- 30.Hermann J, Marti-Arbona R, Fedorov AA, Fedorov E, Almo SC, Shoichet BK, Raushel FM (2007) Nature 448(7155):775–779 [DOI] [PMC free article] [PubMed]
- 31.Homans SW (2004) Angew Chem Int Edit 43:290–300 [DOI] [PubMed]
- 32.Iwahara J, Zweckstetter M, Clore GM (2006) Proc Natl Acad Sci USA 103:15062–15067 [DOI] [PMC free article] [PubMed]
- 33.Mittermaier A, Kay LE (2006) Science 312:224–228 [DOI] [PubMed]
- 34.Pellecchia M, Sem DS, Wuthrich K (2002) Nature Rev Drug Discov 1:211–219 [DOI] [PubMed]
- 35.Shuker SB, Hajduk PJ, Meadows RP, Fesik SW (1996) Science 274:1531–1534 [DOI] [PubMed]
- 36.Hertkorn N, Benner R, Frommberger M, Schmitt-Kopplin P, Witt M, Kaiser K, Kettrup A, Hedges JI (2006) Geochim Cosmochim Acta 70:2990–3010
- 37.Koch BP, Dittmar T (2006) Rapid Commun Mass Spectrom 20:926–932
- 38.Koch BP, Witt MR, Engbrodt R, Dittmar T, Kattner G (2005) Geochim Cosmochim Acta 69:3299–3308
- 39.Mopper K, Stubbins A, Ritchie JD, Bialk HM, Hatcher PG (2007) Chem Rev 107:419–442 [DOI] [PubMed]
- 40.Stenson AC, Marshall AG, Cooper WT (2003) Anal Chem 75:1275–1284 [DOI] [PubMed]
- 41.Haensch TW (2006) Chemphyschem 7:1170–1187 [DOI] [PubMed]
- 42.Gelinas Y, Baldock JA, Hedges JI (2001) Org Geochem 32:677–693
- 43.Solomon D, Lehmann J, Kinyangi J, Liang BQ, Schafer T (2005) Soil Sci Soc Am J 69:107–119
- 44.Mahieu N, Powlson DS, Randall EW (1999) Soil Sci Soc Am J 63:307–319
- 45.Ritchie JD, Perdue EM (2003) Geochim Cosmochim Acta 67:85–96
- 46.Decesari S, Facchini MC, Matta E, Mircea M, Fuzzi S, Chughtai AR, Smith DM (2002) Atmos Environ 36:1827–1832
- 47.Emmenegger C, Reinhardt A, Hueglin C, Zenobi R, Kalberer M (2007) Environ Sci Technol 41:2473–2478 [DOI] [PubMed]
- 48.Hopkins RJ, Tivanski AV, Marten BD, Gilles MK (2007) Aerosol Sci 38:573–591
- 49.Kiss G, Tombacz E, Varga B, Alsberg T, Persson L (2003) Atmos Environ 37:3783–3794
- 50.Krivacsy Z, Kiss G, Varga B, Galambos I, Sarvari Z, Gelencser A, Molnar A, Fuzzi S, Facchini MC, Zappoli S, Andracchio A, Alsberg T, Hansson HC, Persson L (2000) Atmos Environ 34:4273–4281
- 51.Perminova IV, Kovalenko AN, Schmitt-Kopplin P, Hatfield K, Hertkorn N, Belyaeva EY, Petrosyan VS (2005) Environ Sci Technol 39:8518–8524 [DOI] [PubMed]
- 52.Quagliotto P, Montoneri E, Tambone F, Adani F, Gobetto R, Viscardi G (2006) Environ Sci Technol 40:1686–1692 [DOI] [PubMed]
- 53.Samburova V, Didenko T, Kunenkov E, Emmenegger C, Zenobi R, Kalberer M (2007) Atmos Environ 41:4703–4710
- 54.Gao MX, Zhang J, Deng CH, Yang PY, Zhang XM (2006) J Proteom Res 5:2853–2860 [DOI] [PubMed]
- 55.Yu WJ, Li Y, Deng CH, Zhang XM (2006) Electrophoresis 27:2100–2110 [DOI] [PubMed]
- 56.Zhang J, Xu XQ, Gao MX, Yang PY, Zhang XM (2007) Proteomics 7:500–512 [DOI] [PubMed]
- 57.Shen YF, Lee ML (1998) Anal Chem 70:3853–3856
- 58.Gugisch R, Kerber A, Laue R, Meringer C, Ruecker C (2007) MATCH Commun Math Comput Chem 58:239–280
- 59.Marshall AG, Hendrickson CL, Jackson GS (1998) Mass Spectrom Rev 17:1–35 [DOI] [PubMed]
- 60.Murata M, Naoki H, Matsunaga S, Satake M, Yasumoto T (1994) J Am Chem Soc 116:7098–7107
- 61.Nicolaou K, Frederick MO (2007) Angew Chem Int Edit 46:5278–5282 [DOI] [PubMed]
- 62.Satake M, Ishida S, Yasumoto T (1995) J Am Chem Soc 117:7019–7020
- 63.Kind T, Fiehn O (2007) BMC Bioinform 8:105 [DOI] [PMC free article] [PubMed]
- 64.Roussis SG, Proulx R (2003) Anal Chem 75:1470–1482 [DOI] [PubMed]
- 65.Keeler C, Kelly EF, Maciel GE (2006) Geoderma 130:124–140
- 66.Hertkorn N, Kettrup A (2005) Molecular level structural analysis of natural organic matter and of humic substances by multinuclear and higher dimensional NMR spectroscopy. In: Perminova IV, Hatfield K, Hertkorn N (eds) Use of Humic Substances to Remediate Polluted Environments: From Theory to Practice. Kluver Academic Publishers, Dordrecht, pp 391–435
- 67.Bristow AWT (2006) Mass Spectrom Rev 25:99–111 [DOI] [PubMed]
- 68.He F, Hendrickson CL, Marshall AG (2001) Anal Chem 73:647–650 [DOI] [PubMed]
- 69.Kim S, Rodgers RP, Marshall AG (2006) Int J Mass Spectrom 251:260–265
- 70.Marshall AG, Rodgers RP (2004) Acc Chem Re 37:53–59 [DOI] [PubMed]
- 71.Baker CB, Li F, Tang K, Tolmachev AV, Prior DC, Belov ME, Smith RD (2007) J Am Soc Mass Spectrom 18:1176 [DOI] [PMC free article] [PubMed]
- 72.Petit-Zeman S (2004) Exploring Biological Space. Horizon Symposium, Black Point Inn, Maine, USA: Charting chemical space: Finding New Tools to Explore Biology. Nature Publishing Group, 1–3
- 73.Kendrick E (1963) Anal Chem 35:2146–2154
- 74.Seco JM, Quinoa E, Riguera R (2004) Chem Rev 104:17–117 [DOI] [PubMed]
- 75.Xia Z, Akim LG, Argyropoulos DS (2001) J Agric Food Chem 49:3573–3578 [DOI] [PubMed]
- 76.Argyropoulos DS, Li HY, Gaspar AR, Smith K, Lucia LA, Rojas OJ (2006) Bioorg Med Chem 14:4017–4028 [DOI] [PubMed]
- 77.Craig A, Cloarec O, Holmes E, Nicholson JK, Lindon JC (2006) Anal Chem 78:2262–2267 [DOI] [PubMed]
- 78.Crockford DJ, Keun HC, Smith LM, Holmes E, Nicholson JK (2005) Anal Chem 77:4556–4562 [DOI] [PubMed]
- 79.Emeis D, Anker W, Wittern KP (2007) Anal Chem 79:2096–2100 [DOI] [PubMed]
- 80.Pauli GF, Jaki BU, Lankin DC (2005) J Nat Prod 68:133–149 [DOI] [PubMed]
- 81.Teahan O, Gamble S, Holmes E, Waxman J, Nicholson JK, Bevan C, Keun HC (2006) Anal Chem 78:4307–4318 [DOI] [PubMed]
- 82.Cloarec O, Dumas ME, Craig A, Barton RH, Trygg J, Hudson J, Blancher C, Gauguier D, Lindon JC, Holmes E, Nicholson J (2005) Anal Chem 77:1282–1289 [DOI] [PubMed]
- 83.Schroeder FC, Gibson DM, Churchill ACL, Sojikul P, Wursthorn EJ, Krasnoff SB, Clardy J (2007) Angew Chem Int Edit 46:901–904 [DOI] [PubMed]
- 84.Tian C, Chikayama E, Tsuboi Y, Kuromori T, Shinozaki K, Kikuchi J, Hirayama T (2007) J Biol Chem 282:18532–18541 [DOI] [PubMed]
- 85.Hertkorn N, Permin A, Perminova I, Kovalevskii D, Yudov M, Petrosyan V, Kettrup A (2002) J Environ Qual 31:375–387 [PubMed]
- 86.Chung J, Kroon G (2003) J Magn Reson 163:360–368 [DOI] [PubMed]
- 87.Nagai H, Sato A, Kiyoshi T, Matsumoto F, Wada H, Ito S, Miki T, Yoshikawa M, Kawate Y, Fukui S (2001) Cryogenics 41:623–630
- 88.Hinse C, Richter C, Provenzani A, Stockigt J (2003) Bioorg Med Chem 11:3913–3919 [DOI] [PubMed]
- 89.Sakellariou D, Le Goff G, Jacquinot JF (2007) Nature 447:694 [DOI] [PubMed]
- 90.Schlotterbeck G, Ross A, Hochstrasser R, Senn H, Kuhn T, Marek D, Schett O (2002) Anal Chem 74:4464–4471 [DOI] [PubMed]
- 91.van Bentum PJM, Janssen JWG, Kentgens PM (2004) Analyst 129:793–803 [DOI] [PubMed]
- 92.Walton JH, de Ropp JS, Shutov MV, Goloshevsky AG, McCarthy MJ, Smith RL, Collins SD (2003) Anal Chem 75:5030–5036
- 93.Lacey ME, Subramanian R, Olson DL, Webb AG, Sweedler JV (1999) Chem Rev 99:3133 [DOI] [PubMed]
- 94.Wu N, Peck TL, Webb AG, Magin RL, Sweedler JV (1994) Anal Chem 66:3849–3857
- 95.Frydman L, Scherf T, Lupulescu A (2002) Proc Natl Acad Sci USA 99:15858–15862 [DOI] [PMC free article] [PubMed]
- 96.Gal M, Schanda P, Brutscher B, Frydman L (2007) J Am Chem Soc 129:1372–1377 [DOI] [PubMed]
- 97.Kim S, Szyperski T (2003) J Am Chem Soc 125:1385–1393 [DOI] [PubMed]
- 98.Perez-Trujillo M, Nolis P, Bermel W, Parella T (2007) Magn Reson Chem 45:325–329 [DOI] [PubMed]
- 99.Ross A, Schlotterbeck G, Senn H, von Kienlin M (2001) Angew Chem Int Edit 40:3243 [DOI] [PubMed]
- 100.Rubakhin SS, Churchill JD, Greenough WT, Sweedler JV (2006) Anal Chem 78:7267–7272 [DOI] [PMC free article] [PubMed]
- 101.Koch BP, Dittmar T, Witt M, Kattner G (2007) Anal Chem 79:1758–1763 [DOI] [PubMed]
- 102.Kujawinski EB, Behn MD (2006) Anal Chem 78:4363–4373 [DOI] [PubMed]
- 103.Meija J (2006) Anal Bioanal Chem 385:486–499 [DOI] [PubMed]
- 104.Zimmer JSD, Monroe ME, Qian WJ, Smith RD (2006) Mass Spectrom Rev 25:450–482 [DOI] [PMC free article] [PubMed]
- 105.Cai SS, Syage JA (2006) Anal Chem 78:1191–1199 [DOI] [PubMed]
- 106.Cooks RG, Ouyang Z, Takats Z, Wiseman JM (2006) Science 311:1566–1570 [DOI] [PubMed]
- 107.Hanold KA, Fischer SM, Cormia PH, Miller CE, Syage JA (2004) Anal Chem 76:2842–2851 [DOI] [PubMed]
- 108.Keski-Hynnila H, Kurkela M, Elovaara E, Antonio L, Magdalou J, Luukkanen L, Taskinen J, Kostiainen R (2002) Anal Chem 74:3449–3457 [DOI] [PubMed]
- 109.Purcell JM, Hendrickson CL, Rodgers RP, Marshall AG (2006) Anal Chem 78:5906–5912 [DOI] [PubMed]
- 110.Schaub TM, Hendrickson CL, Quinn JP, Rodgers RP, Marshall AG (2005) Anal Chem 77:1317–1324 [DOI] [PubMed]
- 111.Plenert ML, Shear JB (2003) Proc Natl Acad Sci USA 100:3853–3857 [DOI] [PMC free article] [PubMed]
- 112.Trapp O (2007) Angew Chem Int Edit 46:5609–5613 [DOI] [PubMed]
- 113.Hughey CA, Hendrickson CL, Rodgers RP, Marshall AG, Qian KN (2001) Anal Chem 73:4676–4681 [DOI] [PubMed]
- 114.Xiong Y, Schroeder K, Greenbaum NL, Hendrickson CL, Marshall AG (2004) Anal Chem 76:1804–1809 [DOI] [PubMed]
- 115.Lindon JC, Nicholson JK, Wilson ID (2000) J Chromatogr B 748:233–258 [DOI] [PubMed]
- 116.Pan ZZ, Raftery D (2007) Anal Bioanal Chem 387:525–527 [DOI] [PubMed]
- 117.Spraul M, Freund AS, Nast RE, Withers RS, Maas WE, Corcoran O (2003) Anal Chem 75:1536–1541 [DOI] [PubMed]
- 118.Chalmers MJ, Mackay CL, Hendrickson CL, Wittke S, Walden M, Mischak H, Fliser D, Just I, Marshall AG (2005) Anal Chem 77:7163–7171 [DOI] [PubMed]
- 119.Smith CA, Want EJ, O’Maille G, Abagyan R, Siuzdak G (2006) Anal Chem 78:779–787 [DOI] [PubMed]
- 120.Want EJ, O’Maille G, Smith CA, Brandon TR, Uritboonthai W, Qin C, Trauger SA, Siuzdak G (2006) Anal Chem 78:743–752 [DOI] [PubMed]
- 121.Cloarec O, Campbell A, Tseng LH, Braumann U, Spraul M, Scarfe G, Weaver R, Nicholson JK (2007) Anal Chem 79:3304–3311 [DOI] [PubMed]
- 122.Cloarec O, Dumas ME, Trygg J, Craig A, Barton RH, Lindon JC, Nicholson JK, Holmes E (2005) Anal Chem 77:517–526 [DOI] [PubMed]
- 123.Hockaday WC, Grannas AM, Kim S, Hatcher PG (2007) Geochim Cosmochima Acta 71:3432–3445
- 124.Kim S, Kramer RW, Hatcher PG (2003) Anal Chem 75:5336–5344 [DOI] [PubMed]
- 125.Godejohann M (2007) J Chromatogr A 1156:87–93 [DOI] [PubMed]
- 126.Gu X, Deng CH, Yan GQ, Zhang XM (2006) J Proteom Res 5:3186–3196 [DOI] [PubMed]
- 127.Seger C, Godejohann M, Tseng LH, Spraul M, Girtler A, Sturm S, Stuppner H (2005) Anal Chem 77:878–885 [DOI] [PubMed]
- 128.Want EJ, Nordstrom A, Morita H, Siuzdak G (2007) J Proteom Res 6:459–468 [DOI] [PubMed]
- 129.Crockford DJ, Holmes E, Lindon JC, Plumb RS, Zirah S, Bruce SJ, Rainville P, Stumpf CL, Nicholson JK (2006) Anal Chem 78:363–371 [DOI] [PubMed]
- 130.Smith LM, Maher AD, Cloarec O, Rantalainen M, Tang H, Elliott P, Stamler J, Lindon JC, Holmes E, Nicholson JK (2007) Anal Chem 79:5682–5689 [DOI] [PubMed]
- 131.Coen M, Ruepp SU, Lindon JC, Nicholson JK, Pognan F, Lenz EM, Wilson ID (2004) J Pharm Biomed Anal 35:93–105 [DOI] [PubMed]
- 132.Craig A, Sidaway J, Holmes E, Orton T, Jackson D, Rowlinson R, Nickson J, Tonge R, Wilson I, Nicholson J (2006) J Proteom Res 5:1586–1601 [DOI] [PubMed]
- 133.Rantalainen M, Cloarec O, Beckonert O, Wilson ID, Jackson D, Tonge R, Rowlinson R, Rayner S, Nickson J, Wilkinson RW, Mills JD, Trygg J, Nicholson JK, Holmes E (2006) J Proteom Res 5:2642–2655 [DOI] [PubMed]
- 134.Saito N, Robert M, Kitamura S, Baran R, Soga T, Mori H, Nishioka T, Tomita M (2006) J Proteom Res 5:1979–1987 [DOI] [PubMed]
- 135.Desando MA, Ripmeester JA (2002) Fuel 81:1305–1319
- 136.Hall MP, Ashrafi S, Obegi I, Petesch R, Peterson JN, Schneider LV (2003) J Mass Spectrom 38:809–816 [DOI] [PubMed]
- 137.Lopez SN, Ramallo IA, Sierra MG, Zacchino SA, Furlan RLE (2007) Proc Natl Acad Sci USA 104:441–444 [DOI] [PMC free article] [PubMed]
- 138.Macnaughtan MA, Kane AM, Prestegard JH (2005) J Am Chem Soc 127:17626–17627 [DOI] [PMC free article] [PubMed]
- 139.Sforza S, Dall’Asta C, Moseritti A, Galaverna G, Dossena A, Marchelli R (2005) Angew Chem Int Edit 44:5126–5130 [DOI] [PubMed]
- 140.Shanaia N, Desilva MA, Gowda GN, Raftery MA, Hainline BE (2007) Proc Natl Acad Sci USA 104:11540–11544 [DOI] [PMC free article] [PubMed]
- 141.Frimmel FH (1998) Acta Hydrochim Hydrobiol 26:129–129
- 142.Hertkorn N, Guenzl A, Freitag D, Kettrup A (2002) Nuclear Magnetic Resonance Spectroscopic Investigations of Silylated Refractory Organic Substances. In: Frimmel FH, Abbt-Braun G, Heumann KG, Hock B, Luedemann HD, Spiteller M (eds) Refractory Organic Substances in the Environment. Wiley-VCH, Weinheim, pp 129–145
- 143.Go EP, Uritboonthai W, Apon JV, Trauger SA, Nordstrom A, O’Maille G, Brittain SM, Peters EC, Siuzdak G (2007) J Proteom Res 6:1492–1499 [DOI] [PMC free article] [PubMed]
- 144.Thorn KA, Arterburn JB, Mikita MA (1992) Env Sci Technol 26:107–116
- 145.Lindon JC, Holmes E, Nicholson JK (2007) FEBS J 274:1140–1151 [DOI] [PubMed]
- 146.Martin FPJ, Dumas ME, Wang YL, Legido-Quigley C, Yap IKS, Tang HR, Zirah S, Murphy GM, Cloarec O, Lindon JC, Sprenger N, Fay LB, Kochhar S, van Bladeren P, Holmes E, Nicholson JK (2007) Mol Syst Biol 3 [DOI] [PMC free article] [PubMed]
- 147.Oresic M, Clish CB, Davidov EJ, Verheij E, Vogels J, Havekes LM, Neumann E, Adourian A, Naylor S, van der Greef J, Plasterer T (2004) Appl Bioinform 3:205–217 [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Below is the link to the electronic supplementary material.
(PDF 12.9 MB)