

SUMMARY
Proper distribution of mitochondria within axons and at synapses is critical for neuronal function. While one-third of axonal mitochondria are mobile, a large proportion remains in a stationary phase. However, the mechanisms controlling mitochondrial docking within axons remain elusive. Here we report a role for axon-targeted syntaphilin (SNPH) in mitochondrial docking through its interaction with microtubules. Axonal mitochondria that contain exogenously or endogenously expressed SNPH lose mobility. Deletion of the mouse snph gene results in a substantially higher proportion of axonal mitochondria in the mobile state and reduces the density of mitochondria in axons. The snph mutant neurons exhibit enhanced short-term facilitation during prolonged stimulation, probably by affecting calcium signaling at presynaptic boutons. This phenotype is fully rescued by reintroducing the snph gene into the mutant neurons. These findings demonstrate a molecular mechanism for controlling mitochondrial docking in axons that has a physiological impact on synaptic function.
Introduction
The proper transport and intracellular distribution of mitochondria are critical for the normal physiology of neurons. Mitochondria accumulate in the vicinity of active growth cones of developing neurons (Morris and Hollenbeck, 1993) and are present at some synaptic terminals (Shepherd and Harris, 1998; Rowland et al., 2000). In addition to the aerobic production of ATP, mitochondria regulate Ca2+ concentrations (Werth and Thayer, 1994) and have been implicated in certain forms of short-term synaptic plasticity by buffering Ca2+ at synapses (Tang and Zucker, 1997; Billups and Forsythe, 2002; Levy et al., 2003; Yang et al., 2003). Loss of mitochondria from axon terminals in Drosophila results in defective synaptic transmission (Stowers et al., 2002; Guo et al., 2005; Verstreken et al., 2005). Mitochondria in the cell bodies of neurons are transported down neuronal processes in response to changes in the local energy state and metabolic demand. Because of their extreme polarity, neurons require specialized mechanisms to regulate the transport and retention of mitochondria at specific subcellular locations. While cytoplasmic dynein is the driving force behind retrograde movement, the kinesin family of motors and their adaptors Milton and syntabulin are responsible for anterograde transport of axonal mitochondria (Stowers et al., 2002; Gorska-Andrzejak et al., 2003; Cai et al., 2005; Glater et al., 2006). Defective transport of axonal mitochondria is implicated in human neurological disorders and neurodegenerative diseases (see reviews by Hirokawa and Takemura, 2004; Chan, 2006; Stokin and Goldstein, 2006).
Mitochondria in axons display distinct motility patterns and undergo saltatory bidirectional movements where they stop and start moving, frequently changing direction. While approximately one-third of axonal mitochondria are mobile at instantaneous velocities of 0–2.0 μm/sec in mature neurons, a large proportion remains in stationary phase. Their net movement is significantly influenced by recruitment between stationary and motile states (Hollenbeck, 1996; Ligon and Steward, 2000). Such complex mobility patterns suggest that axonal mitochondria might be coupled to two opposing motors and docking machinery. Efficient control of mitochondrial docking at particular sites of axons in response to cellular processes and synaptic stimuli is likely essential for neuronal development and synaptic function (Chada and Hollenbeck, 2004; Reynolds and Rintoul, 2004). However, the proteins mediating mitochondrial docking and retention within axons have not yet been identified.
Syntaphilin (SNPH) is a neuron-specific protein initially identified as a candidate inhibitor of presynaptic function (Lao et al., 2000). We recently generated mouse mutants with a homozygous deletion for the snph gene, leading to the discovery of a novel role for SNPH as a docking receptor of axonal mitochondria. Our findings indicate that SNPH is targeted to and required for maintaining a large portion of axonal mitochondria in stationary state through an interaction with the microtubule-based cytoskeleton. First, we show that axonal mitochondria containing exogenously expressed GFP-SNPH are nearly immobilized. Second, endogenous SNPH-tagged mitochondria are strongly correlated with stationary mitochondria. Third, deletion of the snph gene in mice dramatically increases mitochondrial motility, reduces their density in axons, and consequently influences short-term facilitation during prolonged high frequency stimulation, probably by affecting calcium dynamics at presynaptic boutons. The observed phenotype can be fully rescued with the reintroduction of the snph gene into the mutant neurons. Furthermore, the snph mutant mice show impaired motor coordination. These combined molecular, cellular, and genetic studies elucidate a mechanism underlying the docking of axonal mitochondria and provide evidence that the increased motility and/or reduced density of axonal mitochondria have a significant impact on presynaptic function.
RESULTS
SNPH is an Axon-targeted Protein Associated with Mitochondria
Previous studies using recombinant proteins suggested that SNPH is a neuron-specific protein that likely plays an inhibitory role for presynaptic function (Lao et al., 2000). To further evaluate its role, we examined the subcellular localization of SNPH in neurons. First, transfection of GFP-SNPH into cultured hippocampal neurons revealed punctate or vesicular-tubular structures preferentially enriched in axonal processes (Figure S1A). Second, co-immunostaining for SNPH with an antibody against the SNPH residues 225-428 and the dendritic marker MAP2 in mouse brain slices demonstrated that the majority of SNPH staining is not co-localized with the MAP-2 positive dendritic processes. In contrast, most axonal tracts in the CNS, including the internal capsule, cerebral and cerebellar peduncles, fimbria of the hippocampus, and almost all cranial nerves, showed relatively enriched expression of SNPH (Figure S1B). Third, immunocytochemical analysis of cultured hippocampal neurons demonstrates that endogenous SNPH is predominantly distributed in the MAP2-negative and Tau-positive axonal processes (Figure 1A) and clustered alongside but not within dendritic profiles (Figure S2A), thus indicating its axonal localization. Furthermore, 65±14% (mean±SD, 54 images) of axonal mitochondria are co-localized with SNPH (Figure 1B), whereas little SNPH is co-localized with the synaptic vesicle marker synaptophysin (Figure S2B).
Figure 1. Axonal Mitochondrial Targeting of SNPH in Cultured Hippocampal Neurons.

(A) Hippocampal neurons at DIV14 were co-immunostained with antibodies against SNPH and the dendritic marker MAP2 or the axonal marker tau.
(B) Neurons were co-immunostained for SNPH and mitochondrial marker cytochrome c. Arrows point to SNPH-associated mitochondria and arrowheads indicate SNPH-negative mitochondria within an axonal process. Scale bars, 10 μm.
(C) Schematic diagrams of GFP-tagged SNPH truncated or deleted mutants and their capability for targeting to mitochondria and axons of hippocampal neurons.
Identification of the molecular determinants for its axonal localization and mitochondrial targeting is critical to understand the role of SNPH in neurons. Localization analysis of GFP-SNPH truncated mutants reveals that its carboxyl terminal tail (TM1 and TM2) is necessary for mitochondrial association (Figure 1C and Figure S3). TM1 and TM2 have similar signal structures specific for mitochondrial outer-membrane-targeting (Rapaport, 2003), which are moderately hydrophobic and relatively short (16-20 residues) with net positive charges flanking both sides (Figure S3A). The mitochondrial targeting of SNPH is consistent with the immunogold EM observations that showed its localization to the outer mitochondrial membrane (Das et al., 2003).
We next sought to determine the sequence responsible for axonal sorting. The majority of SNPH staining is localized at axonal mitochondria (Figures 1 and S3B). In support of these observations, we identified an axon-sorting domain of SNPH (residues 381-469)(Figure 1C). Expression of the truncated mutant of SNPH lacking this sequence results in its distribution into all mitochondria including those in the soma and dendrites (Figure S3B), suggesting that SNPH may use this axon-sorting sequence for axonal localization independent of its mitochondrial targeting.
SNPH Immobilizes Axonal Mitochondria
Proper axonal transport and distribution of mitochondria are critical for neuronal activity and synaptic transmission. To characterize the role of SNPH in axonal mitochondrial trafficking, we conducted live cell time-lapse imaging using confocal microscopy in cultured hippocampal neurons co-expressing DsRed-mito and GFP-SNPH. Axonal motile and stationary mitochondria were identified using kymographs, as previously described (Miller and Sheetz, 2004). Those mitochondria not labeled with GFP-SNPH migrate dynamically and bi-directionally along the axonal process, whereas GFP-SNPH-labeled mitochondria remain stationary (Video 1). Consistent with previous reports (Morris and Hollenbeck, 1993; Ligon and Steward, 2000; Miller and Sheetz, 2004), 38±16% (mean±SD) of axonal mitochondria are motile in control neurons expressing DsRed-mito alone. Strikingly, almost no mitochondrion (0.3±1%, mean±SD) labeled with GFP-SNPH is mobile (Figure 2). This phenomenon is consistent across all axons (n=39) examined, indicating that GFP-SNPH-tagged mitochondria are stationary. In contrast, 35±9% (mean±SD) of axonal mitochondria labeled with SNPH-ΔMTB, a SNPH mutant lacking the microtubule-binding domain (130-203), are mobile (Figure 2B, Video 2, Figure S4) with no significant difference from the DsRed-mito control group (p=0.34, U test). These data suggest that exogenously expressed SNPH inhibits the motility of axonal mitochondria and its microtubule-binding domain is required for the SNPH-mediated immobilization.
Figure 2. SNPH Immobilizes Axonal Mitochondria.

Neurons were co-transfected at DIV6 with DsRed-mito and GFP-SNPH (A) or GFP-SNPH-ΔMTB mutant lacking the microtubule-binding (MTB) domain (B). Axonal mitochondrial motility was observed in live neurons one week after transfection.
(A) While GFP-SNPH-negative mitochondrion (red, pointed by arrows) migrates along the axonal process, GFP-SNPH-labeled mitochondria (yellow) remain stationary during the time-lapse observation (16 min). (A'), Motion data in (A) is presented in kymograph, in which vertical lines represent stationary mitochondria and slant line or curve indicate motile one.
(B) Kymograph showing the motility of axonal mitochondria labeled with GFP-SNPH-ΔMTB. Scale bars in all panels, 10 μm.
(C) Relative motility of the axonal mitochondria labeled with DsRed-mito alone as controls (n=298 from 16 axons) or co-labeled with DsRed-mito and GFP-SNPH (n=1294 from 39 axons) or DsRed-mito and GFP-SNPH-ΔMTB (n=226 from 9 axons). Error bars: s.e.m.
To exclude an artificial effect due to over-expression of SNPH, we next examined whether there are distinct motion patterns of axonal mitochondria in relation to the association with endogenous SNPH. Mitochondrial movement in living hippocampal neurons was recorded, followed by retrospective immunostaining for endogenous SNPH. Statistical analysis reveals a strong correlation (r=0.98, 6 paired experiments) between SNPH-positive mitochondria (green and yellow puncta shown in Figure 3C and D) and stationary mitochondria (vertical red lines shown in Figure 3B). Axonal mitochondria appear as two populations: one associated with SNPH and the other not, demonstrating a binominal B(n, p) distribution (Figure 3E). The proportion (ρ) of SNPH-tagged mitochondria is approximately 62% based on quantitative analysis (Supplementary information). The mean percentage of endogenous SNPH-tagged mitochondria (65±14%, mean±SD) is consistent with the mean percentage of stationary mitochondria (62±15%, mean±SD), and both fit well with 90% confidence intervals based on binomial statistics (Figure 3F and G). These results suggest a complementary relationship between endogenous SNPH-tagged mitochondria and motile mitochondria (38±15%, mean±SD) (Figure 3E), indicating that SNPH is associated with stationary mitochondria.
Figure 3. Motility Analysis of Axonal Mitochondria Containing Endogenous SNPH.
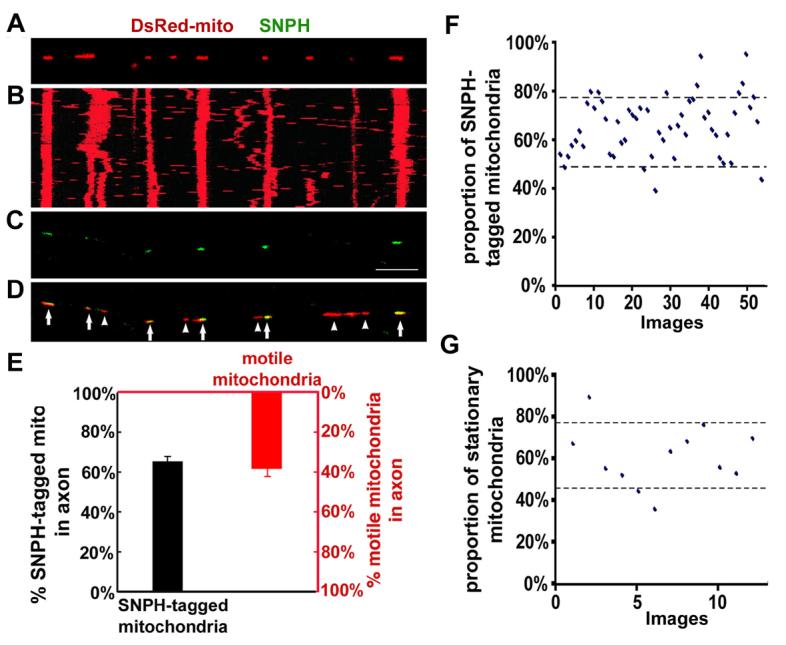
(A) Hippocampal neurons were transfected with DsRed-mito, and time-lapse imaged to monitor mitochondrial movement within axons.
(B) The kymograph for the time-lapse images of (A).
(C, D) The image taken in the same region of the axon as (A) after fixation and staining with an anti-SNPH antibody immediately following time-lapse imaging. Scale bar: 10 μm. Arrows point to the mitochondria tagged with SNPH (yellow) and arrowheads indicate the SNPH-negative mitochondria (red). Note the correlation between the endogenous SNPH-tagged mitochondria in (C) and stationary mitochondria (vertical line) in (B).
(E) A complementary relationship between the mean percentages of the SNPH-tagged mitochondria (black, 1126/1727, 54 images) and the motile mitochondria (red, 120/305, 12 time-lapse images).
(F, G) Scatter plots with 90% confidence intervals show the percentages of the SNPH-tagged mitochondria (62±14%, 54 images)(F) and the stationary mitochondria within axons (62±15%, 12 time-lapse images)(G).
SNPH Docks Axonal Mitochondria through an Interaction with Microtubule-based Cytoskeleton
Recent studies indicate that intracellular signaling stimulated by nerve growth factor can regulate axonal mitochondrial motility by influencing static interactions between mitochondria and the actin cytoskeleton through unidentified docking receptors (Chada and Hollenbeck, 2004). To identify cytoskeletal elements through which SNPH might mediate inhibition of mitochondrial movement, we performed three lines of experiments. First, we used COS7 cells instead of hippocampal neurons since axons are too thin to evaluate cytoskeletal association using confocal microscopy. While GFP-SNPH (1-469) co-localized predominantly with microtubules in transfected COS7 cells, the pharmacological disruption of microtubules with Nocadazol or deletion of the microtubule-binding domain of SNPH resulted in diffusion throughout the cytoplasm (Figure 4A). These results indicate that the microtubule-binding domain of SNPH is required for its association with the microtubule-based cytoskeleton in COS cells. Second, we found that the microtubule-binding domain is required for SNPH binding to Taxol-stabilized microtubules in an in vitro spin-down assay (Figure 4B). The majority of SNPH (1-469) was spun down with microtubules in the pellet, while the mutant SNPH lacking the microtubule-binding domain remained in the supernatant. Finally, to test whether the microtubule-binding domain alone is sufficient for mitochondrial docking, we constructed a chimeric transgene, in which the microtubule-binding domain was placed between GFP and the mitochondrial outer-membrane protein TOM22. While 35%±12% (mean±SD) of GFP-TOM22-labeled mitochondria showed dynamic trafficking along axons, only a small portion (4%±7%) of mitochondria labeled with the chimeric mutant was mobile (Figures 4C and D).
Figure 4. The Microtubule-binding Domain of SNPH Is Sufficient to Immobilize Axonal Mitochondria.

(A) SNPH associates with microtubules. COS cells were transfected with either GFPSNPH (1-469), a truncated SNPH in which the C-terminal mitochondrial targeting domain was deleted, or GFP-SNPH (1-469)ΔMTB, followed by staining for β-tubulin one day after transfection. In the middle pane, the transfected cells were treated with 10 μM of Nocadazol for 1 hr to disassemble microtubules.
(B) The microtubule-binding domain is required for SNPH to be co-pelleted with microtubules in a spin down assay. GST-SNPH (1-469) or GST-SNPH (1-469)ΔMTB was incubated in the absence or presence of Taxol-stabilized microtubules. The supernatant (S) and pellets (P) were analyzed by SDS-PAGE and visualized by Coomassie blue staining.
(C, D) Schematic diagrams and mobility of axonal mitochondria labeled with GFP-TOM22 or GFP-MTB-TOM22, a chimeric transgene in which the MTB was placed between GFP and the mitochondrial outer-membrane protein TOM22. Kymographs: 10 min. Scale bars: 10μm.
As controls, we did not observe any significant association of SNPH with F-actin in COS cells (Figure S5). In addition, the presence of the SNPH-labeled mitochondria in axons (Figure S6) and their stationary state (data not shown) are not dependent on neurofilaments. Given that the sequence of SNPH is not homologous to any known microtubule-binding protein in the database, SNPH likely acts as a docking receptor specific for axonal mitochondria through a unique interaction with the microtubule-based cytoskeleton. These findings provide a molecular explanation for the biochemical interactions between neuronal mitochondria and microtubules (Linden et al., 1989; Jung et al., 1993; Leterrier et al., 1994), and morphological observations of axonal ultra-structures, which revealed cross-bridges between mitochondria and microtubule in vivo (Smith et al., 1977; Hirokawa, 1982; Benshalom and Reese, 1985; Pannese et al., 1986; Price et al., 1991). These cross-bridges may represent both dynamic (motor proteins) and static (docking or adaptor proteins) links between axonal mitochondria and the microtubule-based cytoskeleton.
Deletion of the Mouse snph Gene Results in Dramatically Increased Motility and Decreased Density of Mitochondria in Axons
To further confirm the role of SNPH in docking axonal mitochondria, we generated snph knockout mice. Inactivation of the snph gene in TC-1 ES cells was achieved by replacing all four protein-coding exons with a PGKneo gene cassette (Figure S7A) (Deng et al., 1996). Southern blot shows correct gene targeting and Western blot analysis of brain homogenates demonstrates a dose-dependent loss of SNPH protein in heterozygous and homozygous mutant animals (Figure S7B and C). SNPH is undetectable in the slices of cortex tissue from the snph (−/−) mice (Figure S7D). In addition, the expression of SNPH in wild-type mouse brain is strictly developmentally regulated, with low expression at postnatal day 7 and peaking two weeks after birth (Figure S7E). Both homozygote and heterozygote mutant animals were viable, fertile, and morphologically normal. Hippocampal neurons dissociated from P1 snph (−/−) mice differentiated normally in culture.
A behavioral evaluation of the snph mice at 3 months of age reveals subtle but significant impairments in motor ability, especially in challenging tasks of balance and coordination, although the overall performance of the mutant mice was generally comparable to the wild-type animals. In the rotorod test, there were no significant differences between the snph (+/+) and (−/−) mice for constant speeds (p=0.782, Tukey test), but for each of the two accelerating speeds, the (−/−) mice failed to remain on the wheels as long as the (+/+) controls at both initial speeds (for 3.0-30.0 rpm, p=0.004; for 4.0-40.0 rpm, p=0.020, Tukey test) (Figure S7F). The impairment in rotorod tests at accelerating speeds was consistent across all mutant animals (n=10).
To characterize the phenotypes of the snph mutant neurons in vitro, we monitored and quantified the movement of DsRed-mito-labeled mitochondria by time-lapse imaging in living hippocampal neurons at days in vitro 12 (DIV12). Stationary and motile axonal mitochondria during the entire recording time (16 min) are shown as representative kymographs and the relative percentages of motile axonal mitochondria were calculated (Figure 5A-C). In axons of wild-type neurons, approximately one-third of mitochondria (36±15%, mean±SD, 23 axons) were in motion (Video 3), consistent with our quantitative data from rat hippocampal neurons (Figure 2C). In contrast, deletion of the snph gene resulted in a dramatic increase in the percentage of motile axonal mitochondria (76±20%, mean±SD, 26 axons, P<0.01, U test) (Video 4), a phenotype that has not yet been reported in mature hippocampal neurons in culture.
Figure 5. Increased Mobility and Reduced Density of Axonal Mitochondria in the Neurons of the snph (−/−) Mice.

(A, B) Four representative kymographs recorded from the snph (+/+) (A) and (−/−) (B) neurons. Hippocampal neurons were transfected with DsRed-mito at DIV6, followed by time-lapse imaging six days post-transfection. Density of stationary axonal mitochondria is visualized during the entire recording time (16 min). Scale bars: 10 μm.
(C, D) Quantitative analysis of the percentage of motile axonal mitochondria (C) and relative density of axonal mitochondria (D) (**p<0.01, U test).
To address whether SNPH contributes to the axonal distribution of mitochondria, we measured mitochondrial densities. Deletion of the snph gene resulted in decreased mitochondrial density within axons (1.0±0.2/10μm, mean±SD) compared to those of snph (+/+) neurons (1.7±0.4/10μm, mean±SD, p<0.01, U test) (Figure 5D). We then quantified the ratio of anterograde to retrograde movement in axons. In the snph (−/−) neurons the ratio was 1.03±0.30 (mean±SD), which is slightly lower but not significantly different from the ratio in the snph (+/+) axons (1.27±0.77, mean±SD, p=0.30, U test). The slightly decreased ratio of anterograde to retrograde movement and the significantly reduced density of mitochondria in the snph (−/−) axons suggest that the SNPH-mediated docking/retention contributes to the maintenance of proper mitochondrial density in axons. Thus, the enhanced motility and reduced density of axonal mitochondria are likely two parallel effects by the deletion of the snph gene. In contrast, the deletion of the snph gene has no apparent effect on the velocities of motile mitochondria (p>0.05, t test) averaged at 0.5±0.4 μm/sec (mean±SD) for both the snph (+/+) and (−/−) neurons and on the averaged size (1.3±0.9 μm2, mean±SD) of randomly selected axonal mitochondria (p>0.05, t test) from the snph (+/+) and (−/−) neurons.
Next, we determined the distribution of mitochondria relative to presynaptic boutons along the axonal processes of the snph (+/+) and (−/−) hippocampal neurons in culture (Figure S8). While no significant difference (P=0.25, U test) was found in the colocalization (at least 20% overlapping) of presynaptic boutons (marked by synaptophysin) with mitochondria between the snph (+/+) (18.54±6.28%, mean±SD, n=13 axons) and (−/−) neurons (15.38±6.77%, mean±SD, n=10 axons), the proportion of presynaptic boutons with proximal mitochondria (within the vicinity of 0.5 μm) is significantly reduced (P<0.001, U test) in the snph (−/−) neurons (32.80±4.15%) relative to the snph (+/+) controls (57.02±4.00%). We further quantified the ratio of cytochrome c to synaptophysin puncta within axons in cultured hippocampal neurons (Figure S8D). The ratio in the snph (−/−) axons (0.4±0.1, mean±SD) is significantly lower (p<0.001, U test) than that in the snph (+/+) axons (0.7±0.2, mean±SD). Given that the density of presynaptic boutons per 10 μm axon in length (3±1, mean±SD) is not significantly changed in the snph (−/−) neurons (P=0.25, U test) (Figure S8C) and that the majority of mitochondria are found within axons rather than in presynaptic boutons, the reduced density of axonal mitochondria reflects a reduced inter-bouton density of axonal mitochondria in the snph (−/−) neurons (0.84±0.06) relative to that of wild-type controls (1.36±0.12, P<0.001).
Previous electron microscopy analysis of rat hippocampal CA3-CA1 slices showed that 42% of presynaptic boutons contain mitochondria (Shepherd and Harris, 1998). Using electron microscopy we further examined the relative density of mitochondria at presynaptic terminals of hippocampal CA3 slices from adult (P35) snph (+/+) and (−/−) mice from the same litters (Figure S8E). Double-blind quantitative analysis reveals that the ratio of presynaptic boutons containing mitochondria in the snph (−/−) mice is 33%, significantly lower than that found in the snph (+/+) mice (42%, P=0.02, χ2 test) (Figure S8F). Considering the 10-25% shrinkage of slices during EM fixation (Shepherd and Harris, 1998), some mitochondria in the vicinity of presynaptic boutons might be counted as presynaptic mitochondria in our quantitative analysis.
The snph Mutant Neurons Exhibit Short-term Facilitation
It is well documented that mitochondria are critical in neuronal development and function, including axonal outgrowth and synapse formation, and particularly synaptic plasticity (see recent reviews by Jonas, 2006; Mattson, 2007). Synaptic mitochondria play important roles in calcium homeostasis and in the ATP-dependent mobilization of synaptic vesicles from the reserve pool (Stowers et al., 2002; Verstreken et al., 2005; Guo et al., 2005). However, whether changes in mitochondrial mobility and/or density in axons have an impact on synaptic transmission and plasticity remains elusive. By taking advantage of the observed phenotypes in the snph (−/−) neurons, we further looked at synaptic physiology by dual whole-cell patch-clamp recording on paired hippocampal neurons in culture. By delivering high frequency (10 Hz and 50 Hz) pulse trains to presynaptic neurons, we consistently observed a substantial facilitation in the snph (−/−) neurons (Figure 6A, B), a phenotype not shown in the snph (+/+) neurons under these stimulation frequencies. The facilitation could last for about 1 sec under 10 Hz stimulation and 200 ms under 50 Hz tetanus train and was followed by depression. However, no significant differences were observed in both frequency and amplitude of mini AMPA events between the snph (+/+) and (−/−) neurons (Figure 6C). Furthermore, averaged amplitude (Figure 6D) and kinetics (Table S1) of EPSCs (0.05 Hz) recorded from the paired snph (−/−) neurons was not statistically different from the snph (+/+) neurons. The results from both mini and evoked EPSC suggest that, with the deletion of the snph gene, the resultant effects on docking/retention of axonal mitochondria have no significant impact on basal synaptic transmission.
Figure 6. Deletion of the snph Gene Results in Synaptic Short-term Facilitation.
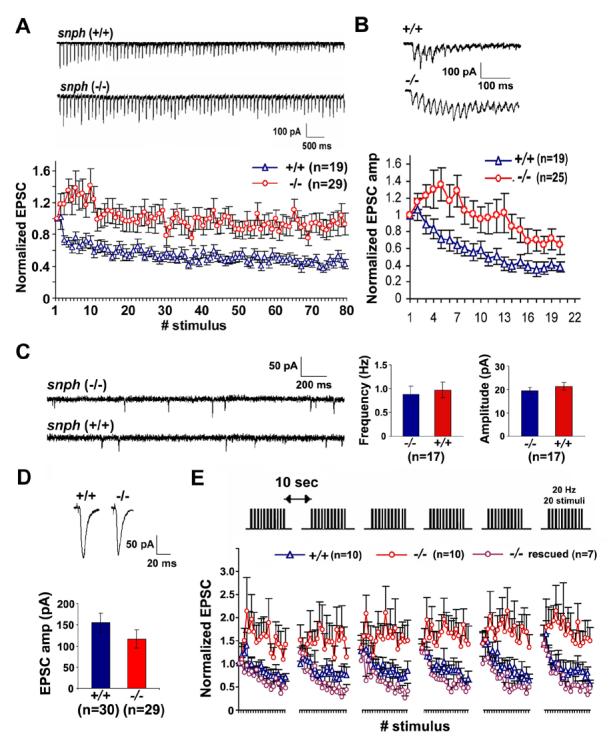
(A, B) Sample traces recorded from the snph (+/+) and (−/−) neurons evoked by 10 Hz, 8 sec (A, upper panel) and 50 Hz, 60 msec (B, upper panel) and their normalized EPSC amplitudes plotted against stimulus number (lower panels of A and B). Substantial short-term facilitation was observed in the snph (−/−) neurons (red circles).
(C) Deletion of the snph gene has no significant effect on the miniature AMPA currents. Left panel: representative miniature AMPA currents recorded from the snph (−/−) and (+/+) hippocampal neurons (DIV14). Right panel: bar graphs of averaged mini AMPA current frequency and amplitude.
(D) Representative EPSCs (upper panels) recorded from paired neurons of the snph (+/+) and (−/−) mice at 0.05 Hz and the bar graph of mean EPSC amplitude (lower panel). No statistical difference was found between the snph (+/+) and (−/−) neurons (P=0.22, t test).
(E) Deletion of the snph gene produces sustained short-term facilitation. 20 Hz, 1 sec stimulus train was delivered repetitively six times at 10-second intervals (upper panel). Normalized EPSC amplitudes were plotted against stimulus number (lower panel). Note that persistent facilitation in synaptic responses was shown only in the snph (−/−) neurons (red circles). Reintroducing the snph gene into the mutant presynaptic neuron (purple circles) eliminates the short-term facilitation and fully rescues the (+/+) phenotype (blue triangles).
To allow the recovery of synaptic vesicles, we applied short stimulus trains (20 Hz, 1 s) to the presynaptic neuron at 10-second intervals. Persistent facilitation was reproduced in the snph (−/−) neurons under repetitive pulse train stimulation (Figure 6E), suggesting a possible role of residual [Ca2+] in maintaining short-term facilitation of synaptic response. In contrast, facilitation was only observed in a very short time frame and was immediately followed by depression in the snph (+/+) neurons. The observed phenotype could be fully rescued by expressing the full-length SNPH in the mutant neurons, further confirming that the enhanced short-term facilitation was due to the deletion of the snph gene (Figure 6E). As controls, expression of SNPH mutant lacking the mitochondria-targeting signal at the carboxyl terminus is unable to rescue the phenotype (Figure S9).
The deletion of the snph gene results in enhanced short-term facilitation but does not significantly affect the basal synaptic transmission. One possible explanation for this observation is a rapid buildup of intracellular [Ca2+] at presynaptic terminals during intensive stimulation. To test our hypothesis, we conducted calcium imaging at nerve terminals of cultured hippocampal neurons using Fluo-4 NW (Experimental Procedures). Presynaptic boutons were labeled with DsRed-synaptophysin and calcium signal within the bouton was imaged every 50 ms throughout the experiment (Figure 7A). Due to limitation of the relatively low signal-to-noise ratio of calcium imaging, we are unable to resolve calcium transients at a single bouton in response to single stimulation. Instead, the train stimulation (100 pulses at 10 Hz) was applied. The change in fluorescence intensity of Fluo-4 NW over baseline (ΔF/F0) at presynaptic boutons of the snph (+/+) neurons showed an initial fast-rising phase, reached a steady state plateau after 10 stimuli (around 1 sec), and finally recovered rapidly (Figure 7B). Peak values of ΔF/F0, expressed as % increase of fluorescence intensity over baseline and averaged from the last 10 stimuli (20 frames of calcium imaging), was significantly larger in the snph (−/−) neurons (32.51%±2.02%, n=33) than that found in the snph (+/+) cells (23.70%±1.58%, n=32, p<0.001, t test). Furthermore, the initial rising phases (first 15 stimuli) of the two groups were not significantly different. By sigmoid fitting, half-maximum increase in fluorescence (ΔF/F0) is 0.67±0.02s for the snph (−/−) neurons and 0.63±0.03s for the snph (+/+) neurons (p=0.82, u test). Linear fittings of steady state plateaus revealed that the plateau in the snph (+/+) is quite flat (0.05%±0.06%, r=0.18), whereas ΔF/F0 in the snph (−/−) neurons displayed a steady increase (0.80%±0.07%, r=0.94). In addition, by single exponential fitting, the initial decay phase after stimulation in the snph (−/−) group is significantly faster than that in the snph (+/+) (time constant rate: 0.66±0.03s and 0.76±0.03s, respectively, p<0.001, U test). Hence, we propose that disrupting the SNPH-mediated mitochondrial docking/retention mechanism changes the dynamics of the global [Ca2+] at presynaptic boutons during prolonged high-frequency stimulation. To further test this hypothesis, we applied EGTA-AM in the recording buffer and found that 100 μM EGTA-AM was effective in abolishing the phenotype of the snph (−/−) neurons, and the short-term facilitation in the snph (−/−) synapses were fully reversed to the depression level observed in wild type neurons after the EGTA application (Figure S10). Altogether, these electrophysiological and calcium imaging studies provide an explanation for the SNPH-mediated modulation of synaptic plasticity by controlling axonal mitochondrial docking/retention and consequently affecting calcium dynamics at nerve terminals.
Figure 7. Presynaptic Calcium Dynamics in the snph (+/+) and snph (−/−) Neurons.

(A) Representative calcium images at presynaptic boutons of the snph (+/+) and (−/−) neurons before stimulation (1), at the beginning (2) and the end (3), and after stimulation (4). Calcium transients within presynaptic boutons labeled with DsRed-monomersynaptophysin were imaged using Fluo-4NW at 50 ms intervals upon stimulation at 10 Hz for 10 seconds. The images are pseudocolored, with blue representing low [Ca2+] concentration and red representing high [Ca2+] concentration. Scale bars: 1 μm.
(B) The time course of changes in presynaptic fluorescent intensity over baseline (ΔF/F0) from the snph (+/+) (black, n=32) and (−/−) (red, n=33) neurons. Inset: the peak values of intracellular [Ca2+] levels within boutons were averaged from last 10 stimuli (20 frames of calcium imaging), expressed as % increase of fluorescence intensity over baseline (ΔF/F0), and are significantly different (p<0.001, t test) between the snph (+/+) (24%±2%) and (−/−) (33%±2%) neurons. Error bars: s.e.m.
DISCUSSION
SNPH Controls Axonal Mitochondrial Mobility
In this study, we reveal that SNPH acts as a receptor for docking/retaining mitochondria in axons and is required for maintaining a large number of axonal mitochondria in a stationary state through an interaction with the microtubule-based cytoskeleton. Such a mechanism may enable neurons to maintain proper densities of stationary mitochondria within axons and in the proximity of synapses. Given that defective trafficking of axonal mitochondria is implicated in the pathogenesis of neurodegenerative diseases such as Alzheimer's and Huntington's (Chan, 2006), these studies will shed light on fundamental neuronal processes that may impact our understanding of human neurodegenerative disorders.
Our study elucidates the mechanism for modulating complex behavior of mitochondria within axons. The majority of SNPH puncta are not co-localized with synaptophysin in the cultured hippocampal neurons (Figure S2), and ∼65% of axonal mitochondria associate with endogenous SNPH and remain in a stationary state. In addition, few of the synaptophysin puncta (1.5±1.1%, mean±SD, 28/1577, 11 images) are co-localized with the SNPH-labeled mitochondria. Thus, it is reasonable to propose that SNPH is not a protein directly mediating docking of the stationary mitochondria at presynaptic boutons. In contrast, via docking mitochondria at microtubules, SNPH controls mitochondrial docking/retention at non-synaptic sites within the axons or in the proximity of synapses.
The coordination of mitochondrial motility with axonal physiology plays a crucial role in maintaining mitochondrial density at appropriate sites in axons. Mitochondria within nerve fibers are relatively enriched at the nodes of Ranvier (Berthold et al., 1993) where ATP production is in high demand (Aiello et al., 2000). Considering the limited diffusion of ATP in an intracellular environment (Belles et al., 1987; Hubley et al., 1996), docked mitochondria ideally serve as local energy stations, providing ATP to maintain the high activity of Na+-K+ ATPase and fast spike propagation. It is expected that defective docking machinery could affect normal neuronal functions, particularly for cells with a long axonal process such as motor neurons. Our behavioral evaluation of the snph mutant mice reveals impaired motor ability, further supporting the notion that the proper docking of axonal mitochondria is crucial for neuronal function.
The Impact of Axonal Mitochondrial Mobility on Short-term Facilitation
Mitochondria play an important role in maintaining calcium homeostasis at some synapses by buffering extra intracellular [Ca2+] during tetanic stimulation and releasing calcium after stimulation to prolong the tail of residual [Ca2+] (Jonas, 2006). The snph mutant neurons provide us with a unique model for studying the physiological impact of mitochondrial docking/retention on synaptic transmission. Our study demonstrates that disruption of the docking mechanism for axonal mitochondria changes the global [Ca2+] dynamics at presynaptic boutons during intensive and prolonged stimulation, probably through reduced [Ca2+] buffering by mitochondria or impaired ability of the terminals to pump [Ca2+] out due to an insufficient supply of ATP. Our results suggest that the inter-bouton or non-synaptic axonal mitochondria might play a comparable role in buffering calcium at nerve terminals during high frequency stimulation.
The proportion of synaptic terminals co-localized with mitochondria is between 10-20% for both cultured snph (+/+) and (−/−) neurons. By applying Monte Carlo simulation analysis, we suggest that motile mitochondria passing presynaptic boutons could account for ∼13% of the co-localization rate in snph (−/−) neurons (Supplemental Information). However, the proportion of presynaptic terminals with proximal mitochondria (within the vicinity of 0.5 μm) is significantly reduced (P<0.001, U test) in the snph (−/−) neurons (32.80±4.15%) relative to the snph (+/+) controls (57.02±4.00%). Thus, the reduced density of inter-bouton axonal mitochondria or proximal presynaptic mitochondria in snph (−/−) neurons is likely the main factor contributing to the mutant phenotypes observed in hippocampal neurons.
The results from calcium imaging (Figure 7) and application of 100 μM EGTA-AM (Figure S10) suggest that the presence of elevated residual calcium is the main cause to the phenotype of the snph (−/−) neurons which show consistent and repeated short-term facilitation during prolonged and intensive stimulation. Although the time course of calcium imaging and short-term facilitation do not synchronize, the distinctions between local [Ca2+] at release sites and global [Ca2+] transients in the terminals may account for the phenotype. Our findings are consistent with recent study in the dMiro mutant neuromuscular junctions in Drosophila, a mutation that causes defective axonal transport of mitochondria and impaired neurotransmitter release and calcium buffering during intensive stimulations (Guo et al., 2005). Unlike the loss of mitochondria at neuromuscular junctions in the drp1 mutant Drosophila, which results in faster depression of synaptic responses during prolonged pulse train stimulation (Verstreken et al., 2005), deletion of the snph gene in mice results in a substantial increase in the mobility and a significant decrease in the inter-bouton density of axonal mitochondria. Hence, it is likely that different effects on synaptic vesicle depletion may reflect different amount of energy consumption at neuromuscular junctions and hippocampal synapses and/or different mitochondrial phenotypes in axons and at terminals.
Given that one-third of axonal mitochondria are normally motile in hippocampal neurons, immobilization of nearly all of axonal mitochondria by overexpressing SNPH might affect axonal and/or synaptic physiology. This may provide a cellular explanation for the previous observation that overexpression of SNPH in autaptic hippocampal neurons partially reduced neurotransmission (Lao et al., 2000). Alternatively, the acute synaptic effect after injection of the coiled-coil domain of SNPH into presynaptic superior cervical ganglion neurons in culture may be attributed to a competitively interference with synaptic vesicle release given that the sequence of this domain is approximately 80% identical to the syntaxin-binding domain of Ocsyn (Safieddine et al., 2002) and syntabulin (Su et al., 2004). In the current study, we generated a SNPH-specific antibody against the sequence between residues 225-428. The specificity of this antibody was further confirmed in the snph (−/−) mice (Figure S7C-E). Using the antibody, we consistently found that SNPH is predominantly targeted to axonal mitochondria.
Presynaptic structure and function are highly plastic and undergo spontaneous and activity-dependent remodeling, thereby changing the demand for mitochondria in axons and at nerve terminals. Mitochondrial balance between motile and stationary phase is a possible target of regulation by intracellular signals and synaptic activity. How are motile mitochondria recruited to the SNPH-dependent stationary pool in response to neuronal activity and synaptic modification? Identification of SNPH as a docking protein provides a molecular target for such regulation. Future studies using the snph knockout mice model will provide molecular and cellular details on how SNPH regulates mitochondrial motility and presynaptic function.
EXPERIMENTAL PROCEDURES
A more detailed version of the Experimental Procedures can be found in the Supplemental Information.
Hippocampal Neuron Culture and Transfection
Hippocampi were dissected from postnatal day 1-3 Sprague Dawley rat pups. For immunocytochemistry, low-density cultures of rat hippocampal neurons were cultured. Transfection was performed at 6-8 days in vitro using the calcium phosphate method. Following transfection, cells were cultured for an additional 4-7 days before imaging or immunocytochemistry.
Mitochondrial Motility Study
All imaging was performed using a Zeiss LSM 510 META confocal microscope with a C-Apochromat 40x/1.2W Corr objective. Time-lapse imaging was performed in the perfusion system (0.4 ml per minute) with Tyrode's solution at 37°C. A motile mitochondrion was counted only if the displacement was at least 5 μm. The total number of mitochondria was defined as the number of mitochondria in each frame. Measurements not noted are presented as mean±SEM. Statistical analyses for unpaired t tests were performed using software OriginPro (OriginLab) and nonparametric statistical tests were performed using GNU Octave, such as the chi-square (χ2) test or the Mann-Whitney test (U test).
A Simple Model for Mitochondrial Motility Studies
SNPH-tagged mitochondria have a binominal B (n, ρ) distribution, where n is the total number of examined mitochondria and ρ is the proportion of the SNPH-tagged mitochondria. The mean of the proportions of the SNPH-tagged mitochondria is an unbiased estimator of the ρ in statistical analysis (Moore and McCabe, 1993), and experimentally estimated as 65%±3% (1126 out of 1727 mitochondria associated with SNPH) under the 99% confidence interval (Figure 3F). If nρ and n(1-ρ) are both larger than or equal to 10, the proportion of the SNPH-tagged mitochondria has approximately normal N(ρ,ρ(1-ρ)/n) distribution, and the equation (critical value z equal to 1.645 when confidence level C is 90%) can be used to calculate confidence levels. In those mitochondrial motility studies, 90% confidence level yields a value of 62%±15% by the equation using the edge value 27 (Figure 3G).
Electrophysiology
Hippocampal neurons were grown to DIV14-15 in culture and dual patch-clamp recordings were performed with whole-cell configuration at room temperature. Both neurons were voltage clamped at −70 mV and membrane currents were digitized by Digidata 1322A and acquired using MultiClamp 700B amplifier with pCLAMP 9.2 (Axon instruments). Master 8 (AMPI) was used to generate trains of high frequency stimulation. Extracellular recording solution contained (in mM): 145 NaCl, 3 KCl, 10 Hepes, 2 CaCl2, 8 glucose, and 2 MgCl2 2 ATP-Na2 (pH 7.3, 300 mOsm). Patch pipettes (Sutter Instrument) with resistance of 4-8 mega-ohms were tip-filled and then backfilled with internal solution containing (in mM): 146.5 K-gluconate, 7.5 KCl, 9 NaCl, 1 MgCl2, 10 Hepes, and 0.2 EGTA (pH 7.2, 300 mOsm). 1 μM TTX and 50 μM picrotoxin were added to the extracellular solution to record miniature AMPA currents at holding potential. Data were analyzed by MiniAnalysis (Synaptosoft).
Calcium Imaging and Data Analysis
Calcium imaging was performed using Fluo-4 NW dye (Invitrogen) in modified Tyrode's solution (in mM: 10 Hepes, 10 glucose, 3 KCl, 145 NaCl, 1.2 CaCl2, 1.2 MgCl2, 0.5 kynurenic acid, and 2.5 Probenecid, pH 7.4) at 37°C. Kynurenic acid (Sigma-Aldrich) was added to reduce glia calcium wave, to block postsynaptic response, and to prevent recurrent excitation during stimulation. Probenecid (Invitrogen) was applied for inhibition of Fluo-4 NW extrusion. For imaging presynaptic boutons, neurons were transfected with DsRed-monomer-synaptophysin. Time-lapse images for single bouton were collected around 70×30 pixels resolution (12 bit) and taken at 20 Hz to meet Nyquist sampling rate for 10 Hz stimulation. Calcium images were analyzed using “Plot Z-axis Profile” function of ImageJ (NIH). The changes in fluorescent intensity of Fluo-4 NW over baseline (ΔF/F0) were averaged and plotted. Peak values of intracellular [Ca2+] levels (ΔF/F0) were calculated by averaging 20 frames of calcium images during last 10 stimuli. OriginPro was used for sigmoid fitting for initial rising phases, linear fitting for steady state plateaus, and single exponential decay fitting for decay phases.
Supplementary Material
Acknowledgements
We thank the following people for their help: S. Das and C. Smith for the initial identification of mitochondrial targeting of SNPH; R. Youle, L-G. Wu, J. Diamond, G. Chen, E. Jonas, Q. Cai for helpful discussions; C. Gerwin for critical reading of the manuscript; R. S. Petralia and Y. X. Wang for mice perfusion; Electron Microscopy Facility (NINDS) and DNA Sequencing Facility (NINDS). We thank three anonymous reviewers for constructive suggestions to strengthen the study. Behavioral study was conducted by NeuroDetective Inc. under service contract. P-Y. Pan is a graduate student of the NIH-Shanghai JiaoTong University Joint Ph.D. Program in Neuroscience. This work was supported by intramural research program of NINDS, NIH (Z-H. S).
All animal experiments have been conducted in accordance with the NIH Animal Use Guidelines.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aiello GL, Bach-y-Rita P. The cost of an action potential. J. Neurosci. Methods. 2000;103:145–149. doi: 10.1016/s0165-0270(00)00308-3. [DOI] [PubMed] [Google Scholar]
- Belles B, Hescheler J, Trube G. Changes of membrane currents in cardiac cells induced by long whole-cell recordings and tolbutamide. Pflugers Arch. 1987;409:582–588. doi: 10.1007/BF00584657. [DOI] [PubMed] [Google Scholar]
- Benshalom G, Reese TS. Ultrastructural observations on the cytoarchitecture of axons processed by rapid-freezing and freeze-substitution. J. Neurocytol. 1985;14:943–960. doi: 10.1007/BF01224806. [DOI] [PubMed] [Google Scholar]
- Berthold CH, Fabricius C, Rydmark M, Andersén B. Axoplasmic organelles at nodes of Ranvier. I. Occurrence and distribution in large myelinated spinal root axons of the adult cat. J. Neurocytol. 1993;22:925–940. doi: 10.1007/BF01218351. [DOI] [PubMed] [Google Scholar]
- Billups B, Forsythe ID. Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J. Neurosci. 2002;22:5840–5847. doi: 10.1523/JNEUROSCI.22-14-05840.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Gerwin C, Sheng Z-H. Syntabulin-mediated anterograde transport of mitochondria along neuronal processes. J. Cell Biol. 2005;170:959–969. doi: 10.1083/jcb.200506042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chada SR, Hollenbeck PJ. Nerve growth factor signaling regulates motility and docking of axonal mitochondria. Curr. Biol. 2004;14:1272–1276. doi: 10.1016/j.cub.2004.07.027. [DOI] [PubMed] [Google Scholar]
- Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Das S, Boczan J, Gerwin C, Zald PB, Sheng Z-H. Regional and developmental regulation of syntaphilin expression in the brain: a candidate molecular element of synaptic functional differentiation. Brain Res. Mol. Brain Res. 2003;116:38–49. doi: 10.1016/s0169-328x(03)00212-2. [DOI] [PubMed] [Google Scholar]
- Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911–921. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- Glater EE, Megeath LJ, Stowers RS, Schwarz TL. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J. Cell Biol. 2006;173:545–557. doi: 10.1083/jcb.200601067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M, Marin L, Charlton MP, Atwood HL, Zinsmaier KE. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron. 2005;47:379–393. doi: 10.1016/j.neuron.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Górska-Andrzejak J, Stowers RS, Borycz J, Kostyleva R, Schwarz TL, Meinertzhagen IA. Mitochondria are redistributed in Drosophila photoreceptors lacking milton, a kinesin-associated protein. J. Comp. Neurol. 2003;463:372–388. doi: 10.1002/cne.10750. [DOI] [PubMed] [Google Scholar]
- Hirokawa N. Cross-linker system between neurofilaments, microtubules, and membranous organelles in frog axons revealed by the quick-freeze, deep-etching method. J. Cell Biol. 1982;94:129–142. doi: 10.1083/jcb.94.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Takemura R. Kinesin superfamily proteins and their various functions and dynamics. Exp. Cell Res. 2004;301:50–59. doi: 10.1016/j.yexcr.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ. The pattern and mechanism of mitochondrial transport in axons. Front Biosci. 1996;1:d91–102. doi: 10.2741/a118. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ, Saxton WM. The axonal transport of mitochondria. J. Cell Sci. 2005;118:5411–5419. doi: 10.1242/jcs.02745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubley MJ, Locke BR, Moerland TS. The effects of temperature, pH, and magnesium on the diffusion coefficient of ATP in solutions of physiological ionic strength. Biochim. Biophys. Acta. 1996;1291:115–121. doi: 10.1016/0304-4165(96)00053-0. [DOI] [PubMed] [Google Scholar]
- Jonas E. BCL-xL regulates synaptic plasticity. Mol. Interv. 2006;6:208–222. doi: 10.1124/mi.6.4.7. [DOI] [PubMed] [Google Scholar]
- Jung D, Filliol D, Miehe M, Rendon A. Interaction of brain mitochondria with microtubules reconstituted from brain tubulin and MAP2 or TAU. Cell Motil Cytoskeleton. 1993;24:245–255. doi: 10.1002/cm.970240405. [DOI] [PubMed] [Google Scholar]
- Lao G, Scheuss V, Gerwin CM, Su Q, Mochida S, Rettig J, Sheng Z-H. Syntaphilin: a syntaxin-1 clamp that controls SNARE assembly. Neuron. 2000;25:191–201. doi: 10.1016/s0896-6273(00)80882-x. [DOI] [PubMed] [Google Scholar]
- Leterrier JF, Rusakov DA, Nelson BD, Linden M. Interactions between brain mitochondria and cytoskeleton: evidence for specialized outer membrane domains involved in the association of cytoskeleton-associated proteins to mitochondria in situ and in vitro. Microsc. Res. Tech. 1994;27:233–261. doi: 10.1002/jemt.1070270305. [DOI] [PubMed] [Google Scholar]
- Levy M, Faas GC, Saggau P, Craigen WJ, Sweatt JD. Mitochondrial regulation of synaptic plasticity in the hippocampus. J Biol Chem. 2003;278:17727–17734. doi: 10.1074/jbc.M212878200. [DOI] [PubMed] [Google Scholar]
- Ligon LA, Steward O. Movement of mitochondria in the axons and dendrites of cultured hippocampal neurons. J. Comp. Neurol. 2000;427:340–350. doi: 10.1002/1096-9861(20001120)427:3<340::aid-cne2>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Lindén M, Nelson BD, Loncar D, Leterrier JF. Studies on the interaction between mitochondria and the cytoskeleton. J. Bioenerg. Biomembr. 1989;21:507–518. doi: 10.1007/BF00762522. [DOI] [PubMed] [Google Scholar]
- Mattson M. Mitochondrial Regulation of Neuronal Plasticity. Neurochem. Res. 2007;32:707–715. doi: 10.1007/s11064-006-9170-3. [DOI] [PubMed] [Google Scholar]
- Miller KE, Sheetz MP. Axonal mitochondrial transport and potential are correlated. J. Cell Sci. 2004;117:2791–2804. doi: 10.1242/jcs.01130. [DOI] [PubMed] [Google Scholar]
- Moore DS, McCabe GP. Introduction to the Practice of Statistics. W.H. Freeman and company; New York: 1993. [Google Scholar]
- Morris RL, Hollenbeck PJ. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J. Cell Sci. 1993;104:917–927. doi: 10.1242/jcs.104.3.917. [DOI] [PubMed] [Google Scholar]
- Pannese E, Procacci P, Ledda M, Arcidiacono G, Frattola D, Rigamonti L. Association between microtubules and mitochondria in myelinated axons of Lacerta muralis. A quantitative analysis. Cell Tissue Res. 1986;245:1–8. doi: 10.1007/BF00218080. [DOI] [PubMed] [Google Scholar]
- Price RL, Lasek RJ, Katz MJ. Microtubules have special physical associations with smooth endoplasmic reticula and mitochondria in axons. Brain Res. 1991;540:209–216. doi: 10.1016/0006-8993(91)90509-t. [DOI] [PubMed] [Google Scholar]
- Rapaport D. Finding the right organelle. Targeting signals in mitochondrial outer-membrane proteins. EMBO Rep. 2003;4:948–952. doi: 10.1038/sj.embor.embor937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds IJ, Rintoul GL. Mitochondrial stop and go: signals that regulate organelle movement. Sci. STKE. 2004;251:PE46. doi: 10.1126/stke.2512004pe46. [DOI] [PubMed] [Google Scholar]
- Rowland KC, Irby NK, Spirou GA. Specialized synapse-associated structures within the calyx of Held. J. Neurosci. 2000;20:9135–9144. doi: 10.1523/JNEUROSCI.20-24-09135.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safieddine S, Ly CD, Wang Y, Wang CY, Kachar B, Petralia RS, Wenthold RJ. Ocsyn, a novel syntaxin-interacting protein enriched in the subapical region of inner hair cells. Mol. Cell Neurosci. 2002;20:343–353. doi: 10.1006/mcne.2002.1120. [DOI] [PubMed] [Google Scholar]
- Shepherd GM, Harris KM. Three-dimensional structure and composition of CA3->CA1 axons in rat hippocampal slices: implications for presynaptic connectivity and compartmentalization. J. Neurosci. 1998;18:8300–8310. doi: 10.1523/JNEUROSCI.18-20-08300.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DS, Järlfors U, Cayer ML. Structural cross-bridges between microtubules and mitochondria in central axons of an insect (Periplaneta americana) J. Cell Sci. 1977;27:255–272. doi: 10.1242/jcs.27.1.255. [DOI] [PubMed] [Google Scholar]
- Stokin GB, Goldstein LS. Axonal transport and Alzheimer's disease. Annu. Rev. Biochem. 2006;75:607–627. doi: 10.1146/annurev.biochem.75.103004.142637. [DOI] [PubMed] [Google Scholar]
- Stowers RS, Megeath LJ, Górska-Andrzejak J, Meinertzhagen IA, Schwarz TL. Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron. 2002;36:1063–1077. doi: 10.1016/s0896-6273(02)01094-2. [DOI] [PubMed] [Google Scholar]
- Su Q, Cai Q, Gerwin C, Smith CL, Sheng Z-H. Syntabulin is a microtubule-associated protein implicated in syntaxin transport in neurons. Nat. Cell Biol. 2004;6:941–953. doi: 10.1038/ncb1169. [DOI] [PubMed] [Google Scholar]
- Tang Y, Zucker RS. Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron. 1997;18:483–491. doi: 10.1016/s0896-6273(00)81248-9. [DOI] [PubMed] [Google Scholar]
- Verstreken P, Ly CV, Venken KJT, Koh T, Zhou Y, Bellen HJ. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005;47:365–378. doi: 10.1016/j.neuron.2005.06.018. [DOI] [PubMed] [Google Scholar]
- Werth JL, Thayer SA. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J. Neurosci. 1994;14:348–356. doi: 10.1523/JNEUROSCI.14-01-00348.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, He XP, Russell J, Lu B. Ca2+ influx-independent synaptic potentiation mediated by mitochondrial Na(+)-Ca2+ exchanger and protein kinase C. J. Cell Biol. 2003;163:511–523. doi: 10.1083/jcb.200307027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.