

Abstract
Novel 5-aza-ellipticine derivatives were synthesized and tested as antitumor agents. The new compounds were prepared more readily than the analogous ellipticine derivatives, which are known to be potent anti-tumor agents Although the novel 5-aza-ellipticine derivatives are not as biologically active as their corresponding ellipticine analogues, the new compounds represent a new, readily accessible class of heteroaromatic catalytic inhibitors of topoisomerase II and possible anti-tumor agents.
Keywords: 5-Aza-ellipticine, Anti-tumor agents, Topoisomerase II, Ellipticine
Ellipticine (1) was first identified in 1959 as a compound in the leaves of an Australian evergreen, Ochrosia elliptica labil (Apocynaceae family).1 Ellipticine is one of many carbazole alkaloids that exhibit biological, especially anti-tumor, activities.2 Many synthetic variants of the natural product were made and tested for anti-tumor activity. The most likely target of ellipticine is topoisomerase II, whose inhibition results in the observed cytotoxicity. However, the low water solubility of ellipticine at physiological pH, as well as its systemic toxicity, prevents its use as a therapeutic agent. Several simple structural modifications to ellipticine derivatives gave compounds with increased toxicity (such as a methoxy substituent at the C9 position and a methyl substituent at the C11 position).3,4
Previously, the novel ellipticine derivative (2) was coupled to heptagastrin, which binds with high affinity to the gastrin/cholecystokinin type B receptor.5 The hypothesis was that since CCK-B receptors are over-expressed in some gastrointestinal cancers,6 the heptagastrin conjugate could be delivered very selectively to CCK-B positive cancer cells by receptor-mediated endocytosis. We demonstrated that heptagastrin conjugate was delivered to lysozomes in CCK-B receptor positive cells where it was processed to release the cytotoxic ellipticine derivative. Compound 2 is cytotoxic to tumor cells at low nanomolar concentrations but is relatively nonselective, while the heptagastrin conjugate, while slightly less toxic to sensitive cells, was completely selective to only those cells that expressed the CCK-B receptor. Compound 2 was synthesized in thirteen steps with an overall 7% yield. Recently, Zhang et al.7 reported the synthesis of 5-aza analogues of ellipticines (3) via an interesting radical cyclization reaction. We wished to examine the anti-tumor properties of these novel compounds because their synthesis was more readily accomplished than the original ellipticines and because they offered the possibility of additional elaboration of the original structures.
In this study, novel 5-aza-ellipticine derivatives were synthesized, which contain a C9 methoxy substituent and a C11 methyl substituent. Consistent with known structure–activity relationships of ellipticine, many of these novel 5-aza-ellipticine derivatives inhibited the growth of HCT116 cells, with approximately the same potency as ellipticine.
The synthesis of the 1-chloro-5-aza-ellipticine (7) was accomplished in six steps with a 6.7% overall yield (Scheme 1) using a modification of the procedure developed by Zhang et al.7,14,15 The precursor 416 was converted to the isocyanate 5 using triphosgene.17 The radical cycloaddition of 5 with phosphorane 6 occurs at elevated temperature and is accompanied by precipitation of triphenylphosphine oxide.18 The yield of this reaction is only 17%, but since the precursors are readily available, the final product can be prepared in quantity, in spite of the low overall yield.
Scheme 1.
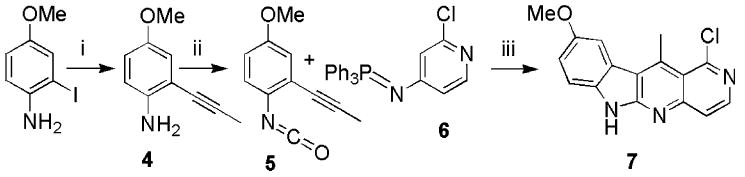
Reagents and conditions: (i) Pd(PPh3)2Cl2, i-Pr2NEt, CuI, propyne, DMF, 86%; (ii) triphosgene, toluene, 96%; (iii) benzene, p-xylene, 17%.19
Several modifications of 7 were carried out. The chlorine in position 1 was readily replaced by three amine side chains that could be utilized in conjugation reactions with peptide ligands (Scheme 2). Data from our laboratory and others suggest that the basic side chains frequently enhance the activity of intercalating drugs, probably by binding to the minor groove of DNA.8-11
Scheme 2.
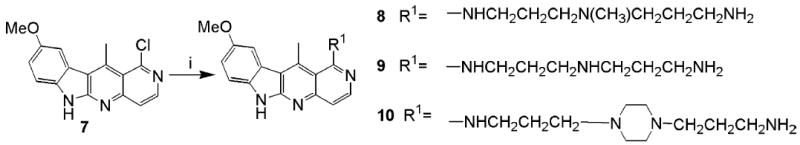
Reagents and conditions: (i) for 8, 3,3′-diamino-N-methyldipropylamine; for 9, N-(3-aminopropyl)-1,3-propanediamine; for 10, 1,4-bis(3-aminopropyl)piperazine.20
We also prepared derivatives where the endocyclic nitrogen at position 5 was quaternized with a methyl group (Scheme 3). The introduction of a positive charge in the molecule improved water solubility but we also expected profound difference in the biological activity. The synthesis was accomplished by quaternization of the 1-chloro derivative. The side chains were attached directly without purification because the quaternary salt was very unstable. However, during the purification of both 11 and 12, some of the chloro compound was hydrolyzed to 13, which was also isolated.21 It is interesting to note that hydrolysis of the chloro substituent was especially facile in compounds where the 5-position was quaternized, perhaps by enhancement of Meisenheimer complexes (Scheme 4).
Scheme 3.
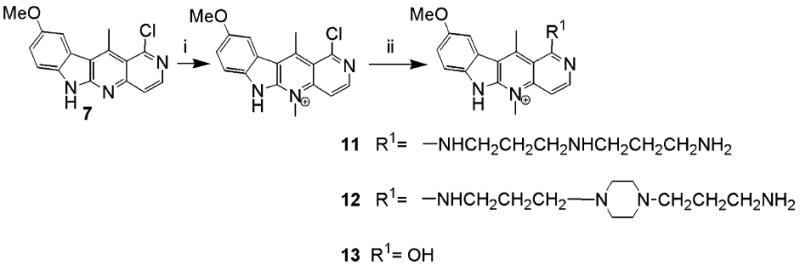
Reagents: (i) methyl trifluoromethane sulfonate, DCM; (ii) for 11 and 13, N-(3-aminopropyl)-1,3-propanediamine; for 12, 1,4-bis(3-aminopropyl)piperazine.
Scheme 4.
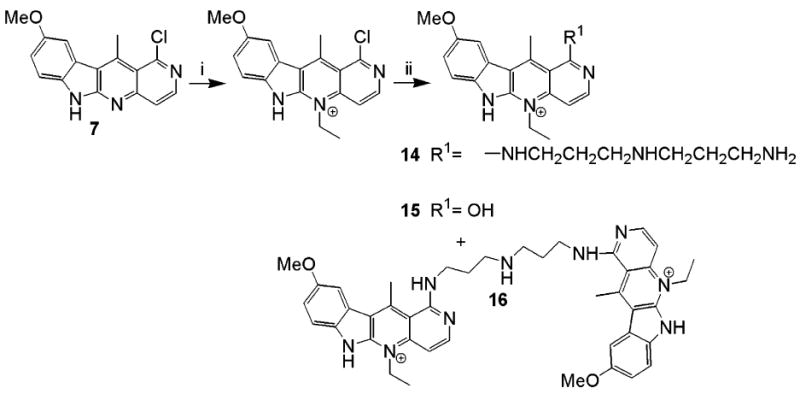
Reagents: (i) ethyl trifluoromethane sulfonate, DCM; (ii) N-(3-aminopropyl)-1,3-propanediamine.
Ethylation of 7 also resulted in very unstable products, but replacement of the chloro group by a polyamine side produced a stable product 14, along with the hydrolysis product 15 and the dimer 16.22
In vitro toxicity of the novel 5-aza-ellipticine derivatives was determined by the MTT assay12 using human colon adenocarcinoma cells HCT116 and murine NIH 3T3 cells. The latter was of interest because we had previously transfected the CCK-B receptor into this cell line and showed that this line was sensitive to ellipticine derivatives.5 The results (Table 1) indicate that some of the novel compounds were moderately active cytotoxic agents (IC50 values in the sub-micromolar range). The 1-chloro substituted 5-aza-ellipticine derivative, 7, did not inhibit cell growth. Compounds 8 and 9 while more potent than 7, were an order of magnitude less active than the corresponding ellipticine derivative 2. Interestingly, compound 12, which has a piperizine-containing side chain, was inactive, in contrast to other agents where that structural feature enhanced activity. The 5-aza ellipticine derivative with the 5-ethyl substituent, 14, had an order of magnitude greater potency than the derivative with the 5-methyl substituent 11. In contrast, the 5-methyl derivative, 13, was an order of magnitude more toxic than the 5-ethyl derivative, 15. The one dimer tested, 16, has approximately the same potency as the monomer 8. These data suggest that appropriately substituted 5-aza-ellipticine derivatives may produce molecules with interesting anti-tumor properties (Fig. 1).
Table 1.
Cell growth inhibition by 5-aza-ellipticine derivatives
| Compound | IC50 (μM) on HCT116 cells | IC50 (μM) on 3T3 cells |
|---|---|---|
| Ellipticine (1) | 0.3 | 0.003 |
| (2) | 0.07 | 0.02 |
| 7 | >1 | >1 |
| 8 | 0.3 | 0.6 |
| 9 | N/A | 1 |
| 11 | 2 | N/A |
| 12 | >10 | N/A |
| 13 | 0.2 | N/A |
| 14 | 0.4 | N/A |
| 15 | 3 | N/A |
| 16 | 0.3 | N/A |
N/A, not tested.
Figure 1.
Ellipticines.
Ellipticine 1 is known to be an inhibitor of topoisomerase II (topoII).13 We therefore tested the ability of 8, an active derivative of the 5-aza-ellipticine series, to inhibit the de-catenation of catenated kinetoplast DNA (kDNA) by topoisomerase II. Topoisomerase II-mediated kDNA de-catenation assays were performed using the the manufacturer’s recommended procedure (TopoGen, Inc., Port Orange, FL). The de-catenation reaction requires a double strand break and re-ligation, a hallmark of topoisomerase II. Ellipticine 1 was chosen as a positive control. The data in Figure 2 indicate that ellipticine 1 as well as the 5-aza-ellipticine derivative 8 are potent catalytic inhibitors of the enzyme.
Figure 2.
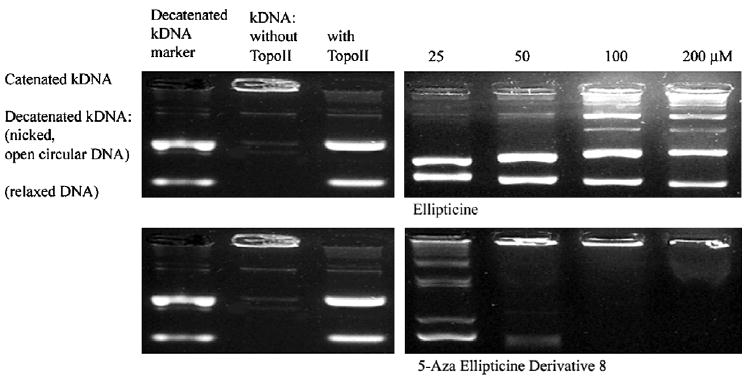
Effect of ellipticines on catalytic activity of topoisomerase II in vitro. Under these conditions, the catenated kinetoplast DNA does not move from the gel origin. Two bands appear in the gel when the topoisomerase catalyzes the double strand break and re-ligation of the DNA. Concentration-dependent delay in the appearance of the two bands correlates with inhibition of topoisomerase II.
It is interesting to note that while 1 and 8 were about equally potent against HCT116 colon cancer cells in vitro, compound 8 is actually a more potent catalytic inhibitor of topoisomerase II.
In conclusion, 5-aza-ellipticine derivatives are readily available compounds that have some interesting anti-tumor properties. Although the currently available derivatives are not as active as the corresponding ellipticines, this new heteroaromatic class of compounds should continue to be examined because of the strong indication of anti-tumor activity and some clear advantages (ease of synthesis and possibly better PK/PD properties) over other polycyclic heteroaromatic anti-tumor agents. It is likely that different substitution pattern on the basic 5-aza-ellipticine scaffold may yield compounds with better anti-cancer activity.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. We also thank the Biophysics Resource of the Structural Biophysics Laboratory, NCI-Frederick, for assistance with LC–MS studies.
References and notes
- 1.Garbett NC, Graves DE. Curr Med Chem Anticancer Agents. 2004;4:149. doi: 10.2174/1568011043482070. [DOI] [PubMed] [Google Scholar]
- 2.Knolker HJ, Reddy KR. Chem Rev. 2002;102:4303. doi: 10.1021/cr020059j. [DOI] [PubMed] [Google Scholar]
- 3.Peczynska-Czoch W, Pognan F, Kaczmarek L, Boratynski J. J Med Chem. 1994;37:3503. doi: 10.1021/jm00047a008. [DOI] [PubMed] [Google Scholar]
- 4.Ducrocq C, Wendling F, Tourbez-Perrin M, Rivalle C, Tambourin P, Pochon F, Bisagni E, Chermann JC. J Med Chem. 1980;23:1212. doi: 10.1021/jm00185a012. [DOI] [PubMed] [Google Scholar]
- 5.Czerwinski G, Tarasova NI, Michejda CJ. Proc Natl Acad Sci USA. 1998;95:11520. doi: 10.1073/pnas.95.20.11520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kusyk CJ, McNiel NO, Johnson LR. Am J Physiol. 1986;251:G597. doi: 10.1152/ajpgi.1986.251.5.G597. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Q, Shi C, Zhang HR, Wang KK. J Org Chem. 2000;65:7977. doi: 10.1021/jo000978e. [DOI] [PubMed] [Google Scholar]
- 8.Moron J, Rautureau M, Huel C, Pierre A, Berthier LK, Atassi G, Bisagni E. Anticancer Drug Des. 1993;8:399. [PubMed] [Google Scholar]
- 9.Escude C, Nguyen CH, Mergny JL, Sun JS, Bisagni E, Garestier T, Helene C. J Am Chem Soc. 1995;117:10212. [Google Scholar]
- 10.Cholody WM, Hernandez L, Hassner L, Scudiero DA, Djurickovic DB, Michejda CJ. J Med Chem. 1995;38:3043. doi: 10.1021/jm00016a007. [DOI] [PubMed] [Google Scholar]
- 11.Cholody WM, Kosakowska-Cholody T, Michejda CJ. Cancer Chemother Pharmacol. 2001;47:241. doi: 10.1007/s002800000234. [DOI] [PubMed] [Google Scholar]
- 12.MTT assay procedure was preformed as previously described Cholody WM, Kosakowska-Cholody T, Hollingshead MG, Hariprakasha HK, Michejda CJ. J Med Chem. 2005;48:4474. doi: 10.1021/jm048946x.
- 13.Monnot M, Mauffret O, Simon V, Lescot E, Psaume B, Saucier JM, Charra M, Belehradek J, Jr, Fermandjian S. J Biol Chem. 1991;266:1820. [PubMed] [Google Scholar]
- 14.Unless indicated, reagents were commercially available and used without further purification. Flash chromatography was performed on silica under argon pressure or on an ISCO CombiFlash Companion instrument and Silicycle 230–400 mesh cartridges. HPLC purification was performed using preparative XTerra® Prep RP18 column from Waters (19 mm id × 300 mm, 10 μm pore size). Acetonitrile/water gradient with 0.1% TFA was used as the eluent for all purifications. Mass spectroscopic analysis was preformed using a Agilent Series 1100 MSD HPLC–MS with atmospheric pressure ionization electrospray (API-ES). The colums used were the Agilent Poroshell 300 SB-C18 (2.1 mm id × 75 mm, 5 μm pore size) and the Zorbax 300 SB-C18 (2.1 mm id × 150 mm, 5 μm pore size). NMR’s were recorded on Varian 200 or 400 MHz instruments. The de-catenation assay was preformed using a kit from TopoGen, Inc. (Port Orange, FL)
- 15.Compounds 8–10 were the products of nucleophilic aromatic substitution on 7 under standard conditions. Because the C1 position on 7 was the only site with an activated leaving group for nucleophilic substitution, the structures were verified using HPLC–ESI-MS. The compounds identified were either the starting material 7 or the product 8, 9 or 10. Compounds 11–16 were the products of nucleophilic aromatic substitution on either the 5-methylated or 5-ethylated derivative of 7. Byproducts that were formed were identified with the products by using HPLC–ESI-MS
- 16.Synthetic procedure for 4. m-Iodophenol, 1, (5.5 g, 25 mmol) was dissolved in water containing KOH (1.4 g, 25 mmol). A cold solution of diazotized sulfanilic acid (5.09, 27.5 mmol) was added to the mixture. The mixture stood for a 30 min and then sodium hydrosulfite (12 g, 69 mmol) was added. The solution changed color immediately upon addition of the sodium hydrosulfite and the solution was stirred at 45 °C for 15 min. Diethyl ether was added to the mixture, the mixture was filtered and the filtrate was concentrated. The solid was recrystallized from hot water to yield needles. Methyl iodide was added (0.98 ml, 15.7 mmol) to 4-amino-3-iodophenol (3.905 g,16.6 mmol) in 109 ml DMF in the presence of CsCO3 (13.45 g, 41.3 mmol). The reaction was left at room temperature for 48 h. The mixture was then diluted with water, extracted with diethyl ether, washed with water, then dried over MgSO4 and filtered. The product was isolated after flash column chromatography on silica gel (diethyl ether/h exane 3:7) to yield a light brown oil. NMR (CDCl3) 1H δ7.21 (1H, d, J = 2.8 Hz), 6.77 (1H, dd, J = 8.7, 2.8 Hz), 6.70 (1H, d, J = 8.7 Hz), 3.72 (3H, s). An outgassed (argon) solution of Pd(PPh3)2Cl2 (0.017 g, 0.024 mmol) and CuI (0.009 g, 0.047 mmol), N,N-diisopropylethylamine (0.35 ml, 2.0 mmol) and 4-amino-3-iodo-1-methoxybenzene (0.200 g, 0.80 mmol) in 5 ml DMF was prepared. Gaseous propyne (290 ml, 12.9 mmol) was introduced with a syringe and the reaction was stirred at room temperature for 12 h. The reaction mixture was diluted with 20 ml dichloromethane and washed with 20 ml saturated NH4Cl. The organic layer was washed with water and dried over Na2SO4. The organic layer was concentrated and subjected to flash chromatography on silica gel with diethylether/hexanes 3:7. NMR (CDCl3) 1H δ 6.81 (1H, d, J = 2.9 Hz), 6.72 (1H, dd, J = 8.7, 2.9 Hz), 6.63 (1H, D, J = 8.7 Hz), 3.72 (3H, s), 2.11 (3H, s)
- 17.Synthesis of 5. Triphosgene (0.3182 g, 1.1 mmol) was dissolved in 6 ml freshly dried anhydrous toluene. Compound 4 (0.471 g, 1.6 mmol) was added to anhydrous TEA (0.81 ml, 5.8 mmol) in 9 ml anhydrous toluene and this solution was added dropwise to the triphosgene solution. The mixture was stirred at room temperature for 30 min and concentrated. The residual white solid was dissolved in 10% diethyl ether in hexanes and filtered through a short silica gel column. NMR (CDCl3) 1H δ 6.91 (1H, d, J = 8.9 Hz), 6.89 (1H, d, J = 2.9 Hz), 6.75 (1H, dd, J = 8.9, 3.0 Hz), 3.76 (3H, s), 2.12 (3H, s)
- 18.Synthesis of 6. Fresh, anhydrous TEA (1.72 ml, 12.3 mmol) was added dropwise to a solution of Ph3PBr2 (2.6 g, 6.2 mmol), 4-amino-2-chloropyridine (0.7192 g, 5.6 mmol), in 42 ml freshly dried anhydrous p-xylene. The solution was heated at 105 °C for 5 h and concentrated. The reaction mixture was purified by chromatography on a silica gel column using 20% diethyl ether in chloroform as the eluant. NMR (CDCl3) 1H δ 7.79 (1H, dd, J = 5.7, 1.0 Hz), 7.47–7.74 (15H, m), 6.59 (1H, d, J = 2.0 Hz), 6.46, (1H, dd, J = 5.7, 1.5 Hz). 13C 161.2, 151.5, 148.6, 151.5, 151.4, 132.6, 132.5, 132.4, 129.6, 129.1, 128.9, 128.6, 117.8, 117.7, 117.6, 117.5
- 19.Synthesis of 7. A solution of 6 (0.339 g, 0.87 mmol) and 5 (0.187 g, 1.0 mmol) in 10 ml fresh anhydrous benzene was heated at 60 °C for 1 h. The solution was filtered through a short silica gel column using argon pressure and the column was additionally washed with fresh anhydrous p-xylene. The benzene was evaporated and the mixture in residual xylene was heated at 180 °C for 12 h. The xylene was removed by evaporation and the residue was fractionated by low pressure chromatography on silica gel with ethyl acetate in hexane using a gradient from 0% to 100% in 30 min. The crude product was purified by second low pressure chromatography on silica gel using diethyl ether in chloroform with a gradient from 0% to 100% in 60 min as the eluant. NMR (CDCl3) 1H δ 8.19 (1H, d, J = 6.0 Hz), 8.01 (1H, s), 7.85 (2H, dd,) 7.44–7.70 (60H, m), 7.31 (1H, dd, J = 9.1, 2.8 Hz), 7.01 (1H, d, J = 2.7 Hz), 3.92 (3H, s), 2.46 (3H, s)
- 20.Synthesis of 8. Compound 7 (0.0093 g, 0.03 mmol) was stirred with 3,3′-diamino-N-methyldipropylamine (1 ml, 6.2 mmol) under argon at 150 °C for 46 h. The mixture was concentrated and purified by flash chromatography on basic alumina with methanol in chloroform with a 0–5% gradient in 80 min. The mixture was dissolved in 1:1 acetonitrile/water and purified by preparative HPLC on a C18 column using a water/acetonitrile solvent system. The purity of the compound was established by HPLC–ESI-MS. Compounds 9 and 10 were prepared in an analogous manner
- 21.Synthesis of 11 and 13. A solution of 7 (0.0062 g, 0.02 mmol) in anhydrous dichloromethane was treated with methyl trifluoromethane sulfonate (2.8 μl, 0.025 mmol) and stirred at room temperature for 24 h, concentrated, and dissolved directly in an excess of N-(3-aminopropyl)-1,3-propanediamine and stirred for 1 h at room temperature. The product was purified by reverse phase preparative HPLC using a C18 column and water/ acetonitrile solvent system. The side product, 13, was isolated from the HPLC separation. The purity of the compounds were established by HPLC–ESI-MS. A similar procedure was used for the preparation of compound 12
- 22.Synthesis of 14, 15, and 16. A solution of 7 (0.0055 g, 0.019 mmol) in anhydrous dichloromethane was treated with ethyl trifluoromethanesulfonate (5 μl, 0.039 mmol) and stirred for 160 h at 30 °C. Additional 5 μl portions of ethyl trifluoromethanesulfonate and 1 ml portions anhydrous dichloromethane were added every 48 h. The product was concentrated and used directly in a reaction with N-(3-aminopropyl)-1,3-propanediamine (3.2 μl, 0.022 mmol). The mixture dissolved in 1 ml THF was stirred for 1 h at room temperature. The products were separated and purified by reverse phase preparative HPLC using a C18 column and water/acetonitrile solvent system. Compounds 16 and 15 were side products of the reaction. The purity of the compounds was determined by HPLC–ESI-MS