

Abstract
Many pathogen life histories include a free-living stage, often with anatomical and physiological adaptations promoting persistence outside of host tissues. More durable particles presumably require that the pathogen metabolize more resources per particle. Therefore, we hypothesize functional dependencies, pleiotropic constraints, between the rate at which free-living particles decay outside of host tissues and other pathogen traits, including virulence, the probability of infecting a host upon contact, and pathogen reproduction within host tissues. Assuming that pathogen strains compete for hosts preemptively, we find patterns in trait dependencies predicting whether or not strain competition favors a highly persistent free-living stage.
Keywords: Host preemption, Infectious-particle longevity, Pathogen evolution, Virulence, Trade-offs
1 Introduction
Pathogen life histories encompass remarkable diversity, but hypothesized functional constraints linking within-host growth rate and between-host infection dynamics suggest organizing principles (Ewald, 1994; Ebert and Mangin, 1997; van Baalen, 2002). Deducing consequences of these constraints leads to predictions that can advance our understanding of host-parasite interactions (Frank, 1996; Gandon et al., 2002; Caraco et al., 2006) and may indicate policies moderating the impact of certain infectious diseases (Dieckmann et al., 2002).
Most analyses of pathogen life histories address direct-contact pathogens, and focus on “transmission-virulence” trade-offs (Massad, 1987; van Baalen and Sabelis, 1995; Ganusov et al., 2002; Alizon and van Baalen, 2005). Direct-contact pathogens are transmitted from an infectious to a susceptible host before the former recovers or dies, since the pathogen rapidly deteriorates outside host tissues. Virulence, the increase in host mortality due to infection (Bull, 1994; Ewald, 1994), may depend functionally on the rate of infection transmission. A pathogen’s within-host replication rate should increase as it accelerates consumption of host resources. For a direct-contact pathogen, the instantaneous rate of transmission between hosts should increase with an increment in the propagule-generation rate. But faster conversion of host resources also can increase the host’s mortality rate, decreasing the expected duration of the infectious period (similar arguments apply to immune-driven recovery). Therefore, excessive virulence might decrease the total number of infections transmitted per infection, so that a trade-off between intensity and duration of disease transmission induces selection for intermediate virulence (Ebert, 1994; Lipsitch and Moxon, 1997; Messenger et al., 1999; van Baalen and Sabelis, 1995; Gandon et al., 2001).
A number of ecologically or economically important pathogens exhibit a more complex life history, where transmission occurs via a distinct free-living stage (Zadoks and Schein, 1979; Evans and Entwistle, 1987; Godfray et al., 1997), rather than by direct contact. A free-living stage may possess morphological and physiological adaptations allowing it to endure lengthy periods without decaying (Anderson and May, 1981; Dwyer, 1992; Setlow, 2007); the pathogen consequently can persist through episodes of low host density (Dwyer, 1994; Bonhoeffer et al., 1996; Gandon 1998). Our paper asks how interactions between the decay rate of free-living infectious particles and other pathogen traits might affect the outcome of competition between different pathogen strains.
1.1 Free-living particles: adaptation for persistence
Pathogens with a free-living stage should be adapted for persistence outside of host tissues (see Dwyer, 1994; Bonhoeffer et al., 1996). Setlow (2007) reviews ensporulation in the bacterial genera Bacillus and Clostridium. Each genus includes human pathogens; B. anthracis is probably the most infamous. In Bacillus species, a complex proteinaceous coat is layered on the spore’s exterior surface. Some species, including the agent of anthrax, then generate an exosporium, composed of proteins and carbohydrates, outside the layered coat. In addition, small, acid-soluble spore proteins are synthesized; they bind to the cell’s DNA and protect it (Driks, 2002). The resulting spore can persist for years, through wet and dry heat, ultraviolet radiation, and exposure to chemicals that would kill the growing form of the same microorganism (Setlow, 2007). However, the ensporulated pathogen’s response to presentation of its host will be slow, and perhaps inefficient, since sporulation processes often must be reversed before growth can occur (Leblanc and Lefebvre, 1984).
The Baculoviruses are probably the largest and most diverse group of DNA viruses; more than 600 have been identified as insect pathogens (Jehle et al., 2006). A number of these viruses have been deployed as biological controls in forestry or agriculture (Godfray et al., 1997). The Baculoviridae comprise two genera, the granuloviruses (GVs) and the nucleopolyhedroviruses (NPVs), distinguished by the structure of their free-living stage (IJkel et al., 2000). Free viral particles must stabilize both their genetic material and receptor-binding sites. Baculoviral virions are embedded in a paracrystalline matrix of proteins (Zhou et al., 1998). The resulting structure is usually termed an occlusion body (Jehle et al., 2006). GV occlusion bodies are small and usually contain only a single virion. NPV occlusion bodies are larger structures containing many virions (IJkel et al., 2000). Occlusion bodies can protect virions for lengthy periods, until consumption by an insect larva leads to virion release and infection (Anderson and May, 1981; Godfray et al., 1997). Presumably, greater investment in spores, occlusion bodies, or similar structures in other microorganisms, will decrease their rate of decay. Our paper addresses plausible costs of a reduced decay rate, with respect to pathogen-strain competitiveness.
1.2 Background
Inclusion of a free-living stage in a pathogen’s life history suggests trait interactions not likely found in directly transmitted pathogens. For example, transmission via free-living particles might relax dependence of the pathogen’s reproductive success on the duration of infected-host survival. As a consequence, selection could favor, or permit, evolution of high virulence (Ewald, 1994). Some recent theoretical results explore questions suggested by this hypothesis; we briefly summarize the models to define a context for our analysis.
Bonhoeffer et al. (1996) assume that an individual host can be infected by at most one pathogen strain. Infected hosts release pathogen particles until they recover or die. Given that infectious-particle production increases with virulence, the authors find that at endemic equilibrium increased persistence of the free-living stage does not imply an increase in the competitively optimal virulence level. However, when host density fluctuates and free-living particles decay quickly, virulence can increase with greater particle persistence; the impact of infectious particle persistence depends on the dynamic context of strain competition (Bonhoeffer et al., 1996).
Gandon’s (1998) model permits coinfection of the same host by two pathogen strains. The addition of within-host competition, modulated by the level of relatedness between pathogens, promotes faster host exploitation and greater virulence. Gandon (1998) analyzes evolutionarily stable virulence and dispersal (which drives between-host transmission). Generally, increased persistence of the pathogen’s free-living stage favors greater virulence, as the general hypothesis suggests.
Day’s (2002) model allows only a single pathogen strain per infected host, and assumes that pathogen-particle production increases with virulence. Disease transmission occurs via direct contact between infectious and susceptible hosts, as well as through free-living particles. The latter may be released continuously for the duration of infection, or as a burst upon death of an infected host (Ebert and Weisser, 1997). Given the death-mediated particle burst, the endemic density of susceptibles depends on particle longevity, and increasing persistence of the free-living stage can favor increased virulence. Kamo and Boots (2004) demonstrate the significance of spatial structure; particles released from an infected host do not disperse beyond a local neighborhood. When pathogen transmission and virulence are linked functionally, low particle persistence (rather than high) can favor greater virulence.
As in Bonhoeffer et al. (1996) and Day (2002), our model assumes that a host can be infected by at most one pathogen strain. Free-living particles may be released during infection, produced as a burst upon death of the host, or both; in this sense, our model resembles Day (2002) and Kamo and Boots (2004). However, our analyses contrast with previous studies in several ways. Most importantly, we consider more general functional dependencies between pathogen traits. In particular, we focus on hypothesized pleiotropic constraints directly involving the rate at which free-living particles decay outside of host tissues. We ask how these constraints might affect pathogen evolution through preemptive strain competition (Bremermann and Thieme, 1989; Rand et al., 1995; Haraguchi and Sasaki, 2000). Between-host competition is preemptive when the first pathogen strain infecting a host prohibits infection of the same individual by any other strain; see Castillo-Chavez and Velasco-Hernandez (1998) or Gandon et al. (2001). Consequently, preemptive competition permits neither coinfection nor superinfection (Nowak and May, 1994; May and Nowak, 1995).
We organize the rest of the paper modularly. First, we present dynamics for a host and pathogen with a free-living stage. We identify equilibrium nodes and the conditions for local stability. Second, we restrict attention to endemic equilibria and focus on pathogen evolution. We hypothesize simple functional dependencies between pathogen traits, emphasizing the rate of decay of free-living pathogen particles, and ask how the shape (concavity or convexity) of those constraints might impact strain competition. Finally, we discuss alternatives and extensions to our model’s assumptions.
2 Host-pathogen dynamics
St represents the density of susceptible hosts at time t. It is the density of infected hosts, and Pt represents the density of free-living pathogen particles. The system’s dynamics reflects the following assumptions; for elaboration, see the Discussion.
Susceptible, but not infected, hosts reproduce.
Susceptible hosts become infected only through contact with free-living pathogen particles (FLP).
No host can be infected by more than one pathogen strain; no infected host recovers.
Depending on parameter values, an infected host may shed FLP during the period of infection, may release a burst of FLP at death, or both may occur.
Given these assumptions, consider the homogeneously mixed dynamics for the host and a single strain:
| (1) |
| (2) |
| (3) |
λ is the maximal birth rate per host, and μ is the host death rate. c (c > 0) imposes crowding, hence host self-regulation. α is the product of the rate at which hosts contact FLP and the probability of infection given contact. υ represents virulence, additional host mortality due to disease. γ is the rate at which FLP are shed from an infected host; β is the FLP burst released upon death of an infected host (Young et al., 2000). ξ is the rate at which free-living particles decay. The decay rate is independent of time (De Paepe and Taddei, 2006); ξ−1 is the expected longevity of each FLP.
We let r = λ − μ > 0, and let θ = μ + υ; θ is the total mortality rate of an infected host. We make a simplification, common in models with free-living particles (e.g., Dwyer, 1992; Day, 2002), and assume that the rate of particle depletion by hosts has negligible impact on particle dynamics. Dwyer (1994), referring to Anderson and May (1981), offers justification for this assumption. The dynamics becomes:
| (4) |
| (5) |
| (6) |
Equation (4) – Equation (6) have two boundary equilibria and one internal (coexistence) equilibrium: (1) extinction of host and pathogen, (2) persistence of the host at self-regulated, disease-free equilibrium, and (3) endemic equilibrium where host and pathogen persist. System extinction, [S*I*P*] = [0, 0, 0], is unstable for any r > 0, which is true by definition. At disease-free equilibrium, host density rests at carrying capacity, [S*I*P*] = [r/c, 0, 0]. At endemic equilibrium we have:
| (7) |
| (8) |
| (9) |
In the absence of functional dependencies between parameters of the dynamics, we can immediately identify direct ecological effects on equilibrium densities. If an infected host does not shed FLP while alive, but an FLP burst is released at the host’s death, then S* = (ξ/αβ). For this case (γ= 0; β > 0), neither susceptible density nor equilibrium infectious-particle density depends directly on virulence. If an infected host sheds FLP, but no burst occurs at the host’s death, S* = (ξθ)/(αγ). In this case (γ > 0; β = 0), the duration of the infectious period has greater significance for the dynamics. The direct effect of increased virulence is reduced density of both infected hosts and infectious particles; susceptible density consequently increases. At internal equilibrium, the direct effect of an increased FLP decay rate ξ would be increased susceptible density. But these direct effects can be misleading; functional dependencies between parameters can alter ecological relationships as selection proceeds. The population dynamics, in turn, defines selection pressures shaping pathogen evolution (van Baalen and Sabelis, 1995; Dieckmann, 2002). So, we proceed by first assuring dynamic stability of the internal equilibrium, and then analyze selection on interdependent pathogen traits.
In passing, we note that if both susceptible and infected hosts reproduce, and compete for the requisite resources, the endemic equilibrium density of susceptibles remains unchanged. That is, S* is still given by equation (7) if all hosts reproduce. Consequently, predictions reached by minimizing S* (see below) may extend beyond the detailed assumptions of our model.
2.1 Ecological stability
The disease-free equilibrium is locally stable if r/c < S*; see the Appendix. This condition implies that sufficiently strong self-regulation in the host population prevents the pathogen from advancing when rare (e.g., Caraco et al. 1998, 2002). The pathogen invades an all-susceptible host population if r/c > S*. Given host density great enough for invasion by the pathogen, the Appendix shows that endemic equilibrium is locally stable if:
| (10) |
Either γ (FLP shed rate) or β (burst size) can go to zero without necessarily destabilizing endemic equilibrium. Substitution in expression (10) yields a simpler condition for local stability of a given endemic equilibrium; expression (10) is true if:
| (11) |
Before turning to pathogen-strain competition, note the ecological effect of r, the host’s growth rate when rare. Figure 1 shows that for r > 0, but small, the host population remans disease-free. At a critical r, which varies inversely with expected particle persistence ξ−1, the pathogen can invade and endemic equilibrium is locally stable. As r continues to increase, the inequality in expression (10) is reversed, and endemic equilibrium becomes unstable. If FLP decay rate varied independently of other pathogen traits, increasing particle persistence could destabilize the disease-free equilibrium and promote pathogen invasion. Continued increase in ξ−1 would eventually destabilize the endemic node and induce density fluctuations.
Fig 1.
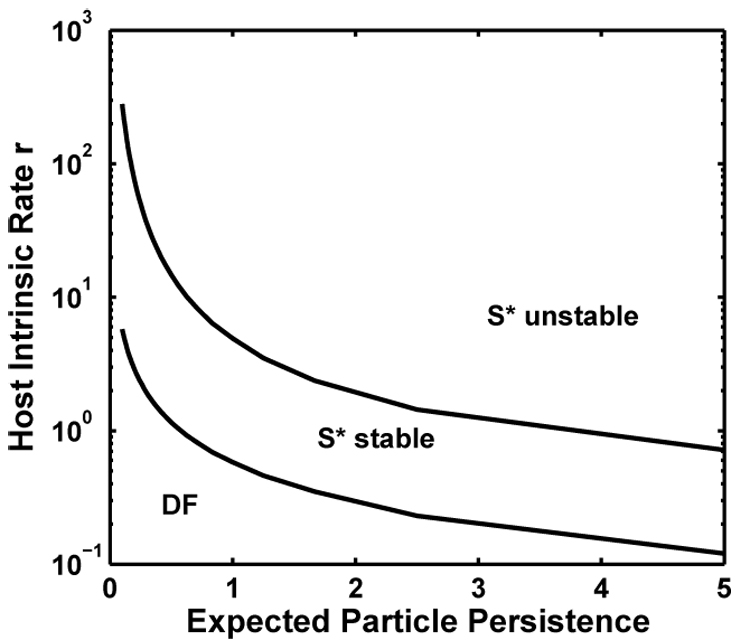
Ecological stability depends on the host’s intrinsic rate of increase, r, and on ξ−1, the expected persistence time of the free-living pathogen particle. Near the origin, the disease-free equilibrium (DF) is stable. In the region identified by S* stable, the endemic equilibrium is locally stable. In the region identified as S* unstable, endemic equilibrium is no longer an attractor. For most values of r where the host is free of disease, sufficient increase in FLP persistence leads to pathogen invasion. Any endemic equilibrium can be destabilized by sufficient increase in r. Parameter values: α = 0.03, β = 15, γ = 15, θ = 0.35, and c = 1.
3 Pathogen strain competition
Hereafter we restrict S* to locally stable, endemic equilibria. We assume that pathogen strains differing in particle-decay rate compete for access to hosts. Our model’s construction implies that different strains compete only between hosts; hence competition is preemptive (Bonhoeffer et al., 1996; van Baalen, 2002). Preemptive competition favors the strain reducing the equilibrium density of susceptibles to a level where other strains infect too few hosts to advance when rare. Consequently, preemptive competition does not admit pathogen-strain coexistence at the epidemiological level (Bremermann and Thieme, 1989; Day, 2002). The strain that can invade, exclude and then repel all other strains exhibits an evolutionarily stable strategy (an ESS; Maynard Smith, 1982). Note, however, that the question of competitive exclusion/coexistence becomes more complex when the dynamics includes either coinfection or superinfection (van Baalen and Sabelis, 1995; Gandon, 1998; Adler and Mosquera, 2000; Pugliese, 2002).
Our analysis concentrates on functional dependencies (pleiotropic constraints) between FLP decay rate ξ and other life-history parameters, since persistence outside of hosts distinguishes free-living pathogens. First, we assume that virulence υ varies as a function of ξ. Second, we let α, a quantification of FLP infectiousness, depend on particle decay rate. Then, we assume that the number of FLP produced per infections varies with decay rate; that is, we take particle-production rate γ and burst size β as functions of ξ. Finally, we vary both infectiousness α and virulence υ simultaneously as functions of ξ. Throughout, we assume ξ ∈ [ξmin, ξmax], recognizing that the expected longevity of FLP can vary from minutes to decades (Leblanc and Lefebvre, 1984; Cano and Borucki, 1995).
3.1 Curse of the pharaoh
Suppose that increasing the expected persistence time of FLP requires that a pathogen convert more host resources per particle produced (see De Paepe and Taddei, 2006). If shed rate and burst size remain constant, the pathogen must then accelerate consumption of the infected host’s resources. The host may be further weakened if toxic by-products of pathogen metabolism accumulate faster (Henderson, 1999; Day, 2002). A likely result would be greater virulence, and we hypothesize a functional dependence where virulence increases with expected persistence time ξ−1. Equivalently, dυ/dξ < 0. We ask if strain competition favors more stable FLP that induce highly virulent infection, or faster-decaying FLP that induce less virulent (perhaps chronic) infection, given the hypothesized trait dependence.
By the monotonicity and sign of the hypothesized dependence, dυ/dξ < 0, we have υ(ξ) ∈ [υ(ξmax), υ(ξmin)] = [υmin, υmax]. For simplicity and specificity, we let υ(ξ) = bυξ−σ; bυ, σ > 0. Virulence declines convexly as FLP decay rate increases; d²υ/dξ² > 0. For any σ > 0, a given reduction in the rate of host-resource consumption decreases virulence more for severe infection than for more benign disease.
The results we list for υ(ξ) should apply qualitatively to similarly shaped functional dependencies between virulence and infectious-particle decay rate. Effectively, for a given reduction in FLP decay rate, we take the increase in expected persistence time (ξ−1), and the consequent increase in virulence, and then translate both to effects on pathogen-strain competitiveness. An increase in FLP decay rate can be favored through strain competition only if trait interactions reduce susceptible density; i.e., when ∂S*/∂ξ < 0. For our choice of υ(ξ), the first step is transparent. If 0 < σ < 1, virulence varies only slowly among pathogen strains, and is relatively large for ξ > 1. But if σ > 1, virulence changes faster among strains and is relatively low for ξ > 1.
1. γ = 0; β > 0. Suppose that no FLP are shed from living hosts, so that pathogen reproduction occurs only as a burst following the death of each infected host (Ebert and Weisser, 1997). Then, S* simplifies to (ξ/αβ). Strain competition minimizes S*, so that any reduction in FLP decay rate is favored, and ξ → ξmin. Given the hypothesized dependence of virulence and FLP persistence, virulence increases; υ(ξ) → υmax.
The notion that high survival of a pathogen’s free-living stage decreases the evolutionary cost of high virulence (Ewald, 1994) is called the “curse of the pharaoh” hypothesis (e.g., Bonhoeffer et al., 1996; Gandon, 1998). When we let γ = 0 in our model, extending the duration of an infected host’s life beyond the minimal time to produce an FLP burst has no value to the pathogen, since no infectious particles are shed before the host dies (Ebert and Weisser, 1997; Day 2002). Increased virulence does not impair strain competitiveness, because S* does not depend directly on υ(ξ). Minimizing the density of susceptibles favors maximal expected persistence of the free-living stage. P*, the equilibrium density of infectious particles, increases with any increase in FLP persistence.
2. γ > 0; β = 0. Suppose now that FLP are shed continuously during the period of infection, and no burst occurs upon death of an infected host. For this case, S* = ξ[μ + υ(ξ)]/αγ. Substituting the functional dependence of virulence on ξ and differentiating yields:
| (12) |
If 0 < σ < 1, then ∂S*/∂ξ > 0. Strain competition always favors a reduced decay rate in the free-living stage, since S* declines. Virulence increases via the functional dependence on ξ. When σ < 1, virulence changes relatively slowly as decay rate varies; see below. Greater FLP persistence increases pathogen-strain competitiveness more than enough to offset reproduction lost through reducing the duration of shedding; ξ → ξmin, and υ(ξ) → υmax.
If σ > 1, virulence increases more rapidly as the FLP decay rate declines. Strains with a given difference in expected free-living persistence time differ more in virulence than is true for σ < 1. Given the hypothesized functional dependence υ(ξ), differentiation indicates that
| (13) |
minimizes S*, and so is an ESS. The corresponding virulence is υ̂ = (μ/bυ)(σ − 1)−1. A greater background host-mortality rate (μ) implies that the evolutionarily stable pathogen has a lower FLP decay rate and a greater virulence. Recall that virulence should increase with the host’s background mortality in directly transmitted pathogens constrained by a infection rate-virulence trait dependence (Sasaki and Iwasa, 1991; Gandon and Michalakis, 2000; Alizon and van Baalen, 2005). When σ > 1 in our model, a reduction in decay rate below increases virulence so rapidly that the loss of pathogen reproduction through shedding imposes a competitive cost exceeding any gain due to longer FLP persistence. Strain competition favors an intermediate virulence, as a consequence of favoring an intermediate persistence of the free-living stage.
When an infected host sheds the pathogen’s free-living stage, but no burst occurs at the host’s death, the outcome of strain competition depends on the value of σ, under our hypothesized υ(ξ). For a clearer biological characterization, consider the ratio of virulence to the FLP’s expected persistence time, ξ−1. Designating the ratio ρ, we have dρ/dξ = (1 − σ)υ. If σ < 1, the ratio of virulence to persistence always increases as ξ increases. Different strains have similar virulences, and selection reduces ρ by favoring any reduction in the FLP decay rate. If σ > 1, the ratio of virulence to persistence always decreases as ξ increases. Virulence differs more among strains, and selection via strain competition favors an intermediate decay rate-virulence combination.
3. γ > 0; β > 0. We now include both shedding and an FLP burst at death of infected hosts. For this case equation (7) gives S*. We set bυ = 1 for notational simplicity; the rescaling has no effect on qualitative predictions. If σ < σcrit, where σcrit > 1, ∂S*/∂ξ > 0. Therefore, ξ → ξmin, and υ(ξ) → υmax under strain competition. For σ < σcrit, differences among strains in virulence are relatively small. Hence, reductions in FLP decay rate are favored; longer persistence of infectious particles increases competitiveness more than is lost by reducing the duration of the shedding period. The value of σcrit depends on β, γ and μ. In particular, σcrit increases as burst size β grows (see below).
Now suppose σ > σcrit, where among-strain variation in virulence is greater. For sufficiently large (ξmax − ξmin), we find two stationary points ξ1 and ξ2. The larger, ξ2, minimizes S* locally, and often globally. S* takes a minimal value at:
| (14) |
where a1 = (γ + βμ) and a2 = (γ + 2βμ). The outcome of strain competition depends on comparison of with boundary conditions. If minimizes S* globally, and so is an ESS; strain competition favors an intermediate decay rate-virulence combination. Otherwise, ξ evolves to either its minimal or maximal value (see Fig. 2); for these cases, larger βμ increases the likelihood that ξ → ξmin.
Fig 2.
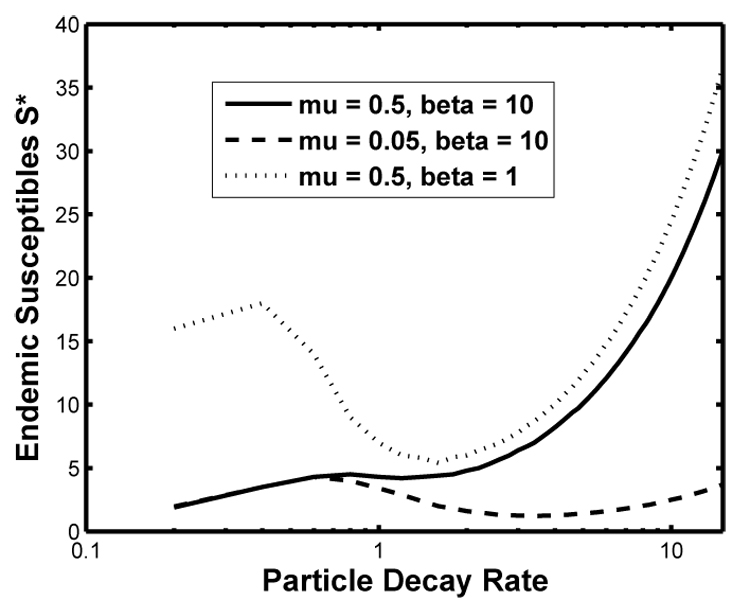
Optimal particle decay rate minimizes endemic susceptible density. For solid line, μ = 0.5 and β = 10. S* reaches a local minimum at , but S* minimized globally at ξmin. Therefore, most competitive strain has minimal FLP decay rate. Dashed line shows effect of reducing host mortality rate μ to 0.05. S* is minimized globally at . Dotted line shows effect of reducing FLP burst size to β = 1. S* is minimized globally at . Other parameter values: α = 0.01, bυ = 1, γ = 20, and σ = 3.
If minimizes S*, virulence at the evolutionarily stable decay rate decreases with σ. When σ is sufficiently large, virulence responds relatively quickly to variation in ξ. Strain competition favors an intermediate decay rate and virulence; at maximal FLP persistence the cost of reproduction lost by reduced shedding diminishes competitiveness. Fixing σ, increases in burst size β, or higher levels of background host mortality (increases in μ) reduce the value to the pathogen of reproducing through shedding; strain competition consequently favors longer FLP persistence and greater virulence.
3.2 Less infectious, more persistent particles
As noted above, complex, rigid structures protect bacterial spores and the infectious stage of many Baculoviruses. Essential for FLP persistence, these structures may reduce the likelihood of responding to host resources/tissues, or may (on average) delay the infectious particle’s response to its host. Therefore, we can imagine that increasing FLP longevity reduces the pathogen’s probability of infecting a susceptible upon contact. In terms of model parameters, dα/dξ > 0. We ask if strain competition favors a highly persistent, but relatively inert free-living stage, or faster decaying, but highly infectious particles.
For any {γ ≥ 0, β > 0} or {γ > 0, β ≥ 0}, susceptible density at endemic equilibrium has the form S* = f(θ, γ, β)ξ/α, where f(θ, γ, β) = ψ is a positive constant. For any continuous functional dependence α(ξ), we have:
| (15) |
We simplify by assuming proportionality: α ∝ ξσ; σ > 0. Then ∂S*/∂ξ ∝ (1 − σ) ψ/α. If σ < 1, α increases as a concave function of ξ. For our assumed functional dependence, concavity implies that ∂S*/∂ξ > 0. Then, ξ → ξmin, and α(ξ) → α(ξmin) = αmin under strain competition. σ < 1 implies that attack rate does not vary strongly among strains with quite different rates of FLP decay. Persistence can be increased with only a relatively small cost due to lower attack rate; strain competition prefers increased longevity, with reduced infectiousness, in the free-living stage.
If σ > 1, α increases as a convex function of ξ; attack rates are now relatively large (for ξ > 1) and vary more among competing pathogen strains. ∂S*/∂ξ < 0 for any such α(ξ), and strain competition reduces S* by increasing FLP decay rate. The outcome of strain competition is reversed; ξ → ξmax, and α(ξ) → αmax. A rapidly decaying, highly infectiousness particle results.
3.3 Less numerous, more persistent particles
Suppose that increasing FLP longevity requires an increase in particle mass; hence, the pathogen must invest more resources per particle. Maintaining the same rate of host-resource consumption (i.e., leaving virulence constant) should then imply that more persistent particles will be less numerous. In our model’s terms, the hypothesized functional dependence becomes dγ/dξ > 0, and/or dβ/dξ > 0. Essentially, the pathogen faces antagonistic pleiotropy between size and number (quality and quantity) of its “offspring” (e.g., Emlen, 1984; Roughgarden, 1998).
1. The cases where (γ = 0, β > 0) or (γ > 0, β = 0) are essentially the same problem. For each, equilibrium susceptible density has the form S* = f(α, θ)ξ/R, where f(α, θ) is a positive constant. R = β if γ = 0, and R = γ if β = 0. S* ∝ ξ/R, and dR/dξ > 0 in both cases. The problem has the same form as (α, ξ)-interaction addressed above. To develop a different approach, we proceed to the general case where both shed rate and burst size are positive.
2. When γ > 0 and β > 0, the expected total number of particles produced per infected host is N = β + γ(μ + υ)−1 = β + (γ/θ). Let m represent the mass of each FLP produced. More massive particles will persist longer, so that ξ−1 ~ m; see De Paepe and Taddei (2006) for an example. We shall approximate N as a function of m, m as a function of ξ, and then write the resulting expression for ∂S*/∂ξ.
De Paepe and Taddei (2006) summarize among-species pattern in bacteriophage life histories (see Bull, 2006). We assume these relationships apply generally. Given that our hypothesized functional dependence leaves α and θ fixed, pathogen generation length is then fixed, and we can take N as surrogate for multiplication rate. De Paepe and Taddei (2006) report that the logarithm of multiplication rate declines linearly in particle mass; the resulting approximation in our model’s terms is N ≈ Nmaxe−ϕm; ϕ > 0. Since ϕ should depend on processes within host tissues, greater suppression of infection by the host should increase ϕ. For a given FLP mass, a larger ϕ results in production of fewer infectious particles.
De Paepe and Taddei (2006) also plot observed estimates of decay rate as a function of particle mass. The data suggest a hyperbolic relationship, so we assume ξm = η; the functional dependence is m(ξ) = η/ξ. The value of η should depend on environmental factors (outside of host tissues) that challenge FLP persistence. η will increase as the environment grows more severe, since a particle of given mass decays faster as η increases.
Given the preceding assumptions, we have:
| (16) |
Then γ + θβ = θNmaxe−ϕη/ξ. Substituting the latter expression into eq. (7) for S* yields:
| (17) |
Differentiating, we find that , an ESS in the sense that it minimizes the approximation for S*. Strain competition should favor particles of intermediate persistence (intermediate mass) produced in intermediate numbers, under the hypothesized functional dependence. As the physical environment becomes more harsh, or as hosts better suppress infection, the most competitive pathogen should increase the number of particles produced per infection, and accept the cost of an increased FLP decay rate.
3.4 A three-trait functional interaction
Here we assume that packaging the free-living stage for extended longevity affects both infectiousness and virulence. An exterior structure protecting each FLP presumably reduces the pathogen’s infectiousness upon contact, as addressed above. The increased investment in FLP exterior protection might deplete host resources faster, and so accelerate infected-host mortality. Therefore, we take ∂α/∂ξ > 0, and ∂υ/∂ξ < 0.
To give our hypothesis some form, we let α(ξ) = ξσα, and let υ(ξ) = ξ−συ; σα, συ > 0. We restrict attention to FLP production via shedding, so that S* = ξ[μ + υ(ξ)]/γα(ξ). Substituting and differentiating, we have:
| (18) |
Suppose that both attack rate and virulence change so slowly relative to change in decay rate that (σα + συ) < 1. Then ∂S* / ∂ξ > 0. Under strain competition, ξ → ξmin, implying that α(ξ) → αmin and υ(ξ) → υmax. Functional relationships where (σα + συ) < 1 favor a highly persistent, slowly reactive free-living stage, that produces highly virulent infections.
Next, we let (σα + συ) > 1, but retain the assumption that (σα < 1). The decay rate exhibts an ESS at:
| (19) |
A few steps show that , assuring that minimizes S*.
Finally, if (σα > 1), then ∂S*/∂ξ < 0. Under strain competition, ξ → ξmax, implying that α(ξ) → αmax and υ(ξ) → υmin. The result is an (almost) avirulent, but highly infectious pathogen that decays very rapidly outside of host tissues. Figure 3 shows an example of each case.
Fig 3.
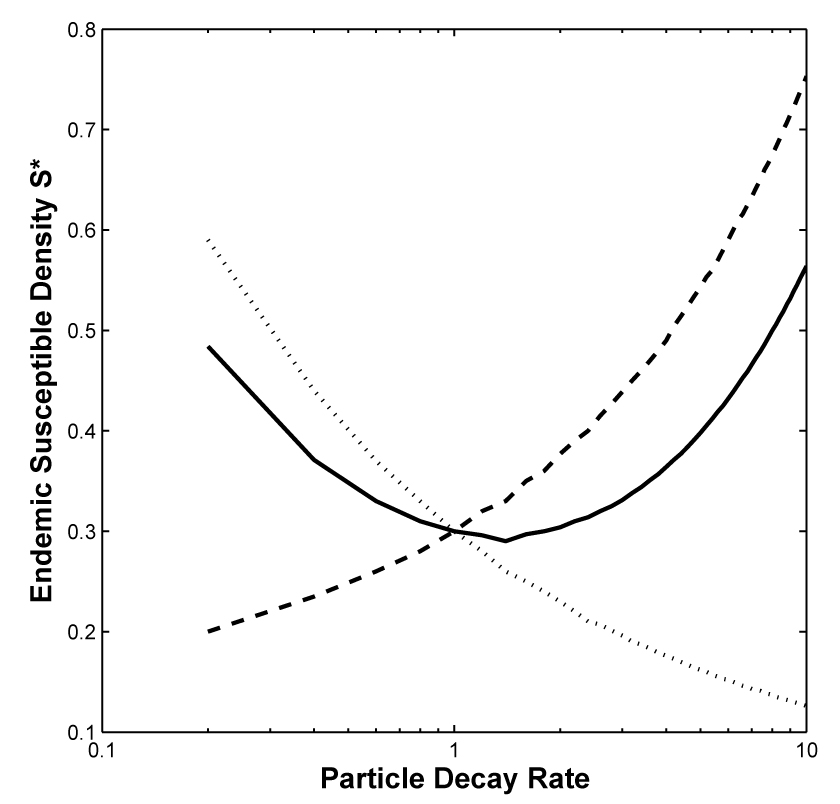
Strain competition minimizes endemic susceptible density. Dashed line: σα = 0.3, and συ = 0.6. Endemic susceptible density S* increases strictly monotonically in decay rate ξ. Solid line: σα = 0.3, and συ = 1.2. Endemic susceptible density S* minimized at , given by equation (16). Dotted line: σα = 1.2, and συ = 0.3. Endemic susceptible density declines strictly monotonically in ξ. Parameters: μ = 0.5, and γ = 5.
Pathogen attack rate and virulence might each depend on decay rate of the free-living form. The assumptions about functional dependence suggest that if both infectiousness and virulence vary relatively slowly with FLP decay rate, strain competition favors a highly resistant free-living stage that produces virulent infection. As infectiousness and virulence together vary more rapidly with a change in FLP decay rate, strain competition favors intermediate persistence of the free-living stage, and intermediate virulence follows. When infectiousness becomes a convex function of decay rate, strain competition favors a highly infectious, rapidly decaying pathogen with low virulence.
4 Discussion
Many viral, bacterial and fungal pathogens have an infectious stage capable of free existence spanning more than one generation of its host (Dwyer, 1994). Ecologically, inclusion of a free-living stage in the pathogen life cycle can separate time scales of within-host reproduction and between-host transmission, complicating the host-pathogen dynamics. Vector-borne pathogens may have to adapt to differing biologies of their host and living vector, and the difference presumably demands evolutionary compromise (Ciota et al., 2007). Pathogens with a free-living stage face parallel challenges, since adaptations advancing the rate of host exploitation might impair the free-living form’s persistence. Given this general assumption, we developed some new, simply stated hypotheses concerning functional interaction of pathogen-life history traits. A few studies assume that increased transmission necessarily increases virulence and then ask how selection might proceed as FLP persistence varies independently (Bonhoeffer et al., 1996). Our approach is different, and more general, in that elements of a set of pathogen life-history traits vary as a function of FLP decay rate itself.
Our analysis yields a number of predictions for pathogen traits evolving in response to preemptive strain competition. Table 1 summarizes the main results. The predicted positive association between FLP persistence and virulence appears in other models (Gandon, 1998; Kamo and Boots, 2004), as a consequence of different assumptions. The intuitive prediction that pathogens reproducing more through a particle burst at host death, and less through shedding during infection, should induce virulent disease has appeared previously (Ebert and Weisser, 1997; Day, 2002; Kamo and Boots, 2004). Half of our predictions follow from hypothesized interaction between FLP decay rate and traits other than virulence; these results apparently are novel.
Table 1.
Summary of predictions.3
| ξ-dependence | Context | Parameter | Prediction |
|---|---|---|---|
| υ ~ ξ−σ | γ = 0, β > 0 | σ > 0 | Persistence ↑ virulence ↑ |
| υ ~ ξ−σ | γ > 0, β = 0 | 0 < σ < 1 | Persistence ↑ virulence ↑ |
| σ > 1 | Persistence ↔ virulence ↔ | ||
| υ ~ ξ−σ | γ,β > 0 | σ small | Persistence ↑ virulence ↑ |
| σ large | ESS varies with parameters | ||
| α ~ ξσ | 0< σ < 1 | Persistence ↑ infectiousness ↓ | |
| σ > 1 | Persistence ↓ infectiousness ↑ | ||
| γ ~ ξσ | β = 0 | 0 < σ < 1 | Persistence ↑ shed rate ↓ |
| σ > 1 | Persistence ↓ shed rate ↑ | ||
| β ~ ξσ | γ = 0 | 0 < σ < 1 | Persistence ↑ burst size ↓ |
| σ > 1 | Persistence ↓ burst size ↑ | ||
| ln[γ + β] ~ ξ−1 | FLP mass ~ ξ−1 | Persistence, FLP mass, FLP number ↔ | |
| Number greater in harsh environment | |||
| 0 < σα + συ < 1 | σα, συ > 0 | Infectiousness ↓ persistence ↑ virulence ↑ | |
| α ~ ξσα; υ ~ ξ−συ | σα + συ > 1 | 0 < σα < 1 | Infectiousness ↔ persistence ↔ virulence ↔ |
| σα + συ > 1 | σα > 1 | Infectiousness ↑ persistence ↓ virulence ↓ | |
First column shows trait’s functional dependence via its scaling with FLP decay rate ξ. υ is virulence, α is infectiousness, γ is shed rate, and β is burst size; σ-values shape trait interactions. ↑ indicates that strain competition favors increase in a trait, ↔ indicates that strain competition favors an intermediate optimal trait value, and ↓ indicates that strain competition favors reduction in a trait.
Several of our hypotheses mirror classic life-history theory in that concavity versus convexity of dependence between fitness-related traits can generate contrasting predictions (Schaffer, 1974; 1978). Taking infectiousness as an example, concavity of functional dependence (d²α/dξ² < 0) implies that strain competition can reduce FLP decay rate (increase persistence) with relatively small reduction in infectiousness. The most competitive strain then minimizes decay rate and infectiousness. A convex functional dependence (d²α/dξ² > 0) implies that infectiousness increases faster than decay rate, and the most competitive pathogen strain maximizes decay rate (minimizes persistence) and maximizes infectiousness.
Our model can be extended to consider pathogen-strain competition under coinfection (May and Nowak, 1995; van Baalen and Sabelis, 1995) where different strains simultaneously exploit the same host individual, and under superinfection (Levin and Pimentel, 1981; Nowak and May, 1994; Castillo-Chavez and Velasco-Hernandez, 1998; Gandon et al., 2002) where one pathogen strain can evict another from individual hosts. Each of these processes adds within-host competition to between-host competition, complicating the evolutionary dynamics (Adler and Mosquera, 2000). Furthermore, since pathogen transmission often occurs only across local neighborhoods (Satō et al., 1994; Duryea et al., 1999; Caraco et al., 2001) a complete theory for evolution of disease requires modeling of spatial processes (Claessen and de Roos, 1995; Caraco et al., 2006; Boots and Mealor, 2007) or contact-network transmission (Keeling, 2005). Not surprisingly, development of hypotheses concerning dependence between traits of pathogens has outpaced empirical tests, but the latter are increasing (e.g., Ebert, 1994; Ebert and Mangin, 1997; Mackinnon and Read, 1999; McKean and Nunney, 2005).
The density of free-living particles resembles, to some extent, a plant population’s seed bank (e.g., Solbrig et al., 1988), and we can draw a parallel between FLP persistence and seed dormancy. Variation in the waiting time until seed gemination may respond adaptively to temporal variation in habitat quality (Venable and Brown, 1988); persistence in the seed bank increases the likelihood that at least some seeds will germinate when conditions for growth are good. FLP persistence may sometimes allow a pathogen to moderate the dynamic risk imposed by host-density fluctuations, so that at least some new infections occur when the density of susceptibles is high.
We close by commenting on some of our assumptions. We let host self-regulation suppress reproduction, while mortality depended only on infection status; see Dwyer (1994) for a different host dynamics. We restricted host reproduction to susceptibles. We defined the virulence parameter υ as an increase in the mortality rate of infected hosts, but loss (or reduction) of fecundity as a consequence of infection constitutes disease virulence. For elaboration, see Haraguchi and Sasaki (2000), or O’Keefe and Antonovics (2002).
Limiting host reproduction to susceptibles articulates our model with host-pathogen systems involving bacteria and bacteriophage (Young et al., 2000; Wang, 2006). For phage that infect E. coli, DePaepe and Taddei (2006) describe a genome-size basis for intraspecific variation in FLP decay rate. Free-living phage virions package their genome within a capsid structure. Charge repulsion between neighboring nucleic-acid segments stresses the capsid’s integrity; a particle decays when internal forces, interacting with the physical environment, destabilize the capsid (Kindt et al., 2001). Smaller genomes (deletion mutants) decrease the internal force, and so increase virion persistence (Rubenstein, 1968; DePaepe and Taddei, 2006). Genome-size differences modulating FLP decay rate might affect other traits, inducing among-strain differences that could be explored experimentally.
We studied pathogen evolution via strain competition, and left the host phenotype static. Genetic variation among hosts sometimes promotes diversity among pathogens (Kirchner and Roy, 2000). In certain fungal infections of plants, different pathogen genotypes specialize on different host genotypes (Crute et al., 1997). More generally, host capacities to resist infection, reduce virulence, and recover form disease may co-evolve with pathogen traits. See, e.g., Frank (1993), Bowers and Hodgkinson (2001), Gandon et al. (2002), Weitz et al. (2005) or Lambrechts et al. (2006).
Finally, we proposed that a pathogen’s elaborated external structure enhances FLP persistence, but can reduce the chance of infecting a susceptible upon contact. However, certain pathogens’ external structures might increase infectiousness through adhesion to host tissues, and may further inhibit the host immune system from attacking the pathogen; see Doering (2000).
5 Acknowledgements
This material is based upon work supported by the National Science Foundation under Grant No. 0342689 (T. Caraco and G. Korniss), and by the National Institutes of Health under Grant No. GM072815 (I.-N. Wang). We thank L. O’Malley, G. Korniss, R. Gallet and G. Dwyer for discussion.
6 Appendix
6.1 Disease-free equilibrium
The Jacobian at the disease-free equilibrium [S*I*P*] = [r/c, 0, 0] is:
The resulting characteristic equation is:
| (20) |
where ρ is an eigenvalue. The first condition for local stability of the disease-free equilibrium requires (r + θ + ξ) > 0, which holds by parameter definitions. The two remaining conditions are fulfilled whenever:
| (21) |
Substituting the definition θ = μ + υ shows that the right-hand side of the last inequality is the density of susceptible hosts at endemic equilibrium; see equation (7) in the text.
6.2 Endemic equilibrium
The Jacobian at endemic equilibrium, equation (7) – equation (9) in the text, is:
where S* = θξ/α(γ + βθ). The associated characteristic equation is:
| (22) |
where ρ is an eigenvalue. The first condition for local stability of endemic equilibrium requires (θ + ξ + cS*) > 0, which holds by definition of model parameters. The second condition reduces to S* < r/c. That is, the density of susceptibles at disease-free equilibrium must be high enough for invasion by the pathogen. The last condition for local stability is:
| (23) |
Substituting the right-hand side of equation (7) for S* yields expression (10) in the text.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
7 References
- Adler FR, Mosquera J. Competition, biodiversity, and analytic functions in spatially structured habitats. Ecology. 2000;81:3226–3232. [Google Scholar]
- Alizon S, van Baalen M. Emergence of a convex trade-off between transmission and virulence. Am. Nat. 2005;165:E155–E167. doi: 10.1086/430053. [DOI] [PubMed] [Google Scholar]
- Anderson RM, May RM. The population dynamics of microparasites and their invertebrate hosts. Phil. Trans. R. Soc. Lond. 1981;B291:451–524. [Google Scholar]
- Bonhoeffer S, Lenski R, Ebert D. The curse of the pharaoh: the evolution of virulence in pathogens with long living propagules. Proc. R. Soc. Lond. 1996;B263:715–721. doi: 10.1098/rspb.1996.0107. [DOI] [PubMed] [Google Scholar]
- Boots M, Mealor M. Local interactions select for lower pathogen infectivity. Science. 2007;315:1284–1286. doi: 10.1126/science.1137126. [DOI] [PubMed] [Google Scholar]
- Bowers RG, Hodgkinson DE. Community dynamics, trade-offs, invasion criteria and the evolution of host resistance to microparasites. J. Theor. Biol. 2001;212:315–331. doi: 10.1006/jtbi.2001.2378. [DOI] [PubMed] [Google Scholar]
- Bremermann H, Thieme H. A competitive exclusion principle for pathogen virulence. J. Math. Biol. 1989;27:179–190. doi: 10.1007/BF00276102. [DOI] [PubMed] [Google Scholar]
- Bull JJ. Virulence. Evolution. 1994;48:1423–1437. doi: 10.1111/j.1558-5646.1994.tb02185.x. [DOI] [PubMed] [Google Scholar]
- Bull JJ. Optimality models of phage life history and parallels in disease evolution. J. Theor. Biol. 2006;241:928–938. doi: 10.1016/j.jtbi.2006.01.027. [DOI] [PubMed] [Google Scholar]
- Cano RJ, Borucki M. Revival and identification of bacterial spores in 25- to 40-million-year-old Dominican amber. Science. 1995;268:1060–1064. doi: 10.1126/science.7538699. [DOI] [PubMed] [Google Scholar]
- Caraco T, Duryea M, Glavankov S, Maniatty W, Szymanski BK. Host spatial heterogeneity and the spread of vector-borne infection. Theor. Pop. Biol. 2001;59:185–206. doi: 10.1006/tpbi.2000.1517. [DOI] [PubMed] [Google Scholar]
- Caraco T, Gardner G, Maniatty W, Deelman E, Szymanski BK. Lyme disease: self-regulation and pathogen invasion. J. Theor. Biol. 1998;193:561–576. doi: 10.1006/jtbi.1998.0722. [DOI] [PubMed] [Google Scholar]
- Caraco T, Glavanakov S, Chen G, Flaherty JE, Ohsumi TK, Szymanski BK. Stage-structured infection transmission and a spatial epidemic: a model for Lyme disease. Am. Nat. 2002;160:348–359. doi: 10.1086/341518. [DOI] [PubMed] [Google Scholar]
- Caraco T, Glavanakov S, Li S, Maniatty W, Szymanski BK. Spatially structured superinfection and the evolution of disease virulence. Theor. Pop. Biol. 2006;69:367–384. doi: 10.1016/j.tpb.2005.12.004. [DOI] [PubMed] [Google Scholar]
- Castillo-Chavez C, Velasco-Hernandez JX. On the relationship between tight coevolution and superinfection. J. Theor. Biol. 1998;192:437–444. doi: 10.1006/jtbi.1998.0661. [DOI] [PubMed] [Google Scholar]
- Ciota AT, Ngo KA, Lovelace AO, Payne AE, Zhou Y, Shi P-Y, Kramer LD. Role of the mutant spectrum in adaptation and replication of West Nile virus. J. Gen. Virol. 2007;88:8656–8874. doi: 10.1099/vir.0.82606-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claessen D, de Roos AM. Evolution of virulence in a host-pathogen system with local pathogen transmission. Oikos. 1995;74:401–413. [Google Scholar]
- Crute IR, Holub EB, Burdon JJ, editors. The Gene-for-Gene Relationship in Plant-Parasite Interactions. Oxon UK: CAB International; 1997. [Google Scholar]
- Day T. Virulence evolution via host exploitation and toxin production in spore-producing pathogens. Ecol. Lett. 2002;5:471–476. [Google Scholar]
- De Paepe M, Taddei F. Viruses’ life history: towards a mechanistic basis of a trade-off between survival and reproduction among phages. PLoS Biol. 2006;4:1248–1256. doi: 10.1371/journal.pbio.0040193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieckmann U. Adaptive dynamics of pathogen-host interactions. In: Dieckmann U, Metz JAJ, Sabelis MW, Sigmund K, editors. Adaptive Dynamics of Infectious Diseases: In Pursuit of Virulence Management. Cambridge, UK: Cambridge University Press; 2002. pp. 39–59. [Google Scholar]
- Dieckmann U, Metz JAJ, Sabelis MW, Sigmund K, editors. Adaptive Dynamics of Infectious Diseases: In Pursuit of Virulence Management. Cambridge, UK: Cambridge University Press; 2002. [Google Scholar]
- Doering TL. How does Cryptococcus get its coat? Trends Microbiol. 2000;8:547–553. doi: 10.1016/s0966-842x(00)01890-4. [DOI] [PubMed] [Google Scholar]
- Driks A. Proteins of the spore core and coat. In: Sonenshen AL, Hoch JA, Losick R, editors. Bacillus subtilis and Its Closest Relatives: From Genes to Cells. New York: ASM Press; 2002. pp. 527–536. [Google Scholar]
- Duryea M, Caraco T, Gardner G, Maniatty W, Szymanski BK. Population dispersion and equilibrium infection frequency in a spatial epidemic. Physica D. 1999;132:511–519. [Google Scholar]
- Dwyer G. On the spatial spread of insect viruses: theory and experiment. Ecology. 1992;73:479–494. [Google Scholar]
- Dwyer G. Density dependence and spatial structure in the dynamics of insect pathogens. Am. Nat. 1994;143:533–562. [Google Scholar]
- Ebert D. Virulence and local adaptation of a horizontally transmitted parasite. Science. 1994;265:1084–1086. doi: 10.1126/science.265.5175.1084. [DOI] [PubMed] [Google Scholar]
- Ebert D, Mangin KL. The influence of host demography on the evolution of virulence in a microsporidian gut parasite. Evolution. 1997;51:1828–1838. doi: 10.1111/j.1558-5646.1997.tb05106.x. [DOI] [PubMed] [Google Scholar]
- Ebert D, Weisser WW. Optimal killing for obligate killers: the evolution of life histories and virulence of semelparous parasites. Proc. R. Soc. Lond. 1997;B264:985–991. doi: 10.1098/rspb.1997.0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emlen JM. Population Biology: The Coevolution of Population Dynamics and Behavior. New York: Macmillan Publishing Company; 1984. [Google Scholar]
- Evans HF, Entwistle PF. Viruses. In: Fuxa JR, Tanada Y, editors. Epizootiology of Insect Diseases. New York: Wiley; 1987. pp. 257–322. [Google Scholar]
- Ewald P. Evolution of Infectious Diseases. Oxford, UK: Oxford University Press; 1994. [Google Scholar]
- Frank SA. Coevolutionary genetics of plants and pathogens. Evol. Ecol. 1993;7:45–75. [Google Scholar]
- Frank SA. Models of parasite virulence. Quart. Rev. Biol. 1996;71:37–78. doi: 10.1086/419267. [DOI] [PubMed] [Google Scholar]
- Gandon S. The curse of the pharaoh hypothesis. Proc. R. Soc. Lond. 1998;B265:1545–1552. doi: 10.1098/rspb.1998.0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandon S, Jansen VAA, van Baalen M. Host life history and the evolution of parasite virulence. Evol. 2001;55:1056–1062. doi: 10.1554/0014-3820(2001)055[1056:hlhate]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Gandon S, Michalakis Y. Evolution of parasite virulence against qualitative or quantitative host resistance. Proc. R. Soc. Lond. 2000;B267:985–990. doi: 10.1098/rspb.2000.1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandon S, van Baalen M, Jansen VAA. The evolution of parasite virulence, superinfection and host resistance. Am. Nat. 2002;159:658–669. doi: 10.1086/339993. [DOI] [PubMed] [Google Scholar]
- Ganusov VV, Bergstrom CT, Antia R. Within-host population dynamics and the evolution of microparasites in a heterogeneous host population. Evol. 2002;56:213–223. doi: 10.1111/j.0014-3820.2002.tb01332.x. [DOI] [PubMed] [Google Scholar]
- Godfray HCJ, O’Reilly DD, Briggs CJ. A model of nucleopolyhedrovirus (NPV) population genetics applied to co-occlusion and the spread of the few polyhedra (FP) phenotype. Proc. R. Soc. Lond. 1997;B264:315–322. [Google Scholar]
- Haraguchi Y, Sasaki A. The evolution of parasite virulence and transmission rate in a spatially structured population. J. Theor. Biol. 2000;203:85–96. doi: 10.1006/jtbi.1999.1065. [DOI] [PubMed] [Google Scholar]
- Henderson B, editor. Cellular Microbology. New York: John Wiley & Sons; 1999. [Google Scholar]
- IJkel WFJ, Westenberg M, Goldbach RW, Blissard GW, Vlak JM, Zuidema D. A novel baculovirus envelope fusion protein with a proprotein convertase cleavage site. Virol. 2000;275:30–41. doi: 10.1006/viro.2000.0483. [DOI] [PubMed] [Google Scholar]
- Jehle JA, Lange M, Wang H, Hu Z, Wang Y, Hauschild R. Molecular identification and phylogenetic analysis of baculoviruses from Lepidoptera. Virol. 2006;346:1890–193. doi: 10.1016/j.virol.2005.10.032. [DOI] [PubMed] [Google Scholar]
- Kamo M, Boots M. The curse of the pharaoh in space: free-living infectious stages and the evolution of virulence in spatially explicit populations. J. Theor. Biol. 2004;231:435–441. doi: 10.1016/j.jtbi.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Keeling M. The implications of network structure for epidemic dynamics. Theor. Pop. Biol. 2005;67:1–8. doi: 10.1016/j.tpb.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Kindt J, Tzlil S, Ben-Shaul A, Gelbart WM. DNA packaging and ejection forces in bacteriophage. Proc. Natl. Acad. Sci. USA. 2001;98:13671–13674. doi: 10.1073/pnas.241486298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchner JW, Roy BA. Evolutionary implications of host-pathogen specificity: the fitness consequences of host life history traits. Evol. Ecol. 2000;14:665–692. [Google Scholar]
- Lambrechts L, Fellous S, Koella JC. Coevolutionary interactions between host and parasite genotypes. Trends Parasit. 2006;22:12–16. doi: 10.1016/j.pt.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Leblanc R, Lefebvre GM. A stochastic model of bacterial spore germination. Bull. Math. Biol. 1984;46:447–460. doi: 10.1007/BF02462018. [DOI] [PubMed] [Google Scholar]
- Levin SA, Pimentel D. Selection for intermediate rates of increase in parasite-host systems. Am. Nat. 1981;117:308–315. [Google Scholar]
- Lipsitch M, Moxon ER. Virulence and transmissibility in microparasites. Trends Microbiol. 1997;5:31–37. doi: 10.1016/S0966-842X(97)81772-6. [DOI] [PubMed] [Google Scholar]
- Mackinnon MJ, Read AF. Genetic relationships between parasite virulence and transmission in the rodent malaria Plasmodium chabaudi. Evolution. 1999;53:689–703. doi: 10.1111/j.1558-5646.1999.tb05364.x. [DOI] [PubMed] [Google Scholar]
- May RM, Nowak M. Coinfection and the evolution of parasite virulence. Proc. R. Soc., Lond. 1995;B261:397–404. doi: 10.1098/rspb.1995.0138. [DOI] [PubMed] [Google Scholar]
- Massad E. Transmission rates and the evolution of pathogenicity. Evol. 1987;41:1127–1130. doi: 10.1111/j.1558-5646.1987.tb05883.x. [DOI] [PubMed] [Google Scholar]
- Maynard Smith J. Evolution and the Theory of Games. Cambridge, UK: Cambridge University Press; 1982. [Google Scholar]
- McKean KA, Nunney L. Bateman’s principle and immunity: phenotypically plastic reproductive strategies predict changes in immunological sex differences. Evolution. 2005;59:1510–1517. [PubMed] [Google Scholar]
- Messenger SL, Molineux IJ, Bull JJ. Virulence evolution in a virus obeys a trade-off. Proc. R. Soc. Lond. 1999;B266:209–215. doi: 10.1098/rspb.1999.0651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak MA, May RM. Superinfection and the evolution of parasite virulence. Proc. R. Soc. Lond. 1994;B255:81–89. doi: 10.1098/rspb.1994.0012. [DOI] [PubMed] [Google Scholar]
- O’Keefe KJ, Antonovics J. Playing by different rules: the evolution of virulence in sterilizing pathogens. Am. Nat. 2002;159:597–605. doi: 10.1086/339990. [DOI] [PubMed] [Google Scholar]
- Pugliese A. On the evolutionary coexistence of parasite strains. Math. Biosci. 2002;177 & 178:355–375. doi: 10.1016/s0025-5564(02)00083-4. [DOI] [PubMed] [Google Scholar]
- Rand DA, Keeling M, Wilson HB. Invasion, stability and evolution to criticality in spatially extended, artificial host-pathogen ecologies. Proc. R. Soc. Lond. 1995;B259:55–63. [Google Scholar]
- Roughgarden J. Primer of Ecological Theory. Upper Saddle River, NJ: Prentice Hall; 1998. [Google Scholar]
- Rubenstein I. Heat-stable mutants of T5 phage. I. The physical properties of the phage and their DNA molecules. Virol. 1968;36:356–376. doi: 10.1016/0042-6822(68)90161-x. [DOI] [PubMed] [Google Scholar]
- Satō K, Matsuda H, Sasaki A. Pathogen invasion and host extinction in lattice structured populations. J. Math. Biol. 1994;32:251–268. doi: 10.1007/BF00163881. [DOI] [PubMed] [Google Scholar]
- Sasaki A, Iwasa Y. Optimal growth schedule of pathogens within a host: switching between lytic and latent cycles. Theor. Pop. Biol. 1991;39:201–239. doi: 10.1016/0040-5809(91)90036-f. [DOI] [PubMed] [Google Scholar]
- Schaffer WM. Optimal reproductive effort in fluctuating environments. Am. Nat. 1974;108:783–790. [Google Scholar]
- Schaffer WM. A note on the theory of reciprocal altruism. Am. Nat. 1978;112:250–253. [Google Scholar]
- Setlow P. I will survive: DNA protection in bacterial spores. Trends Microbiol. 2007;15:172–180. doi: 10.1016/j.tim.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Solbrig OT, Sarandón R, Bossert W. A density-dependent growth model of a perennial herb, Viola frimbiatula. Am. Nat. 1988;131:385–400. [Google Scholar]
- van Baalen M. Contact networks and the evolution of virulence. In: Dieckmann U, Metz JAJ, Sabelis MW, Sigmund K, Law R, Metz H, editors. Adaptive Dynamics of Infectious Disease: In Pursuit of Virulence Management. Cambridge UK: Cambridge University Press; 2002. pp. 85–103. [Google Scholar]
- van Baalen M, Sabelis MW. The dynamics of multiple infection and the evolution of virulence. Am. Nat. 1995;146:881–910. [Google Scholar]
- Venable DL, Brown JS. The selective interactions of dispersal, dormancy, and seed size as adaptations for reducing risk in variable environments. Am. Nat. 1988;131:360–384. [Google Scholar]
- Wang I-N. Lysis timing and bacteriophage fitness. Genetics. 2006;172:17–26. doi: 10.1534/genetics.105.045922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitz JS, Hartman H, Levin SA. Co-evolutionary arms races between bacteria and bacteriophage. Proc. Natl. Acad. Sci., USA. 2005;102:9535–9540. doi: 10.1073/pnas.0504062102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young R, Wang I-N, Roof WD. Phages will out: strategies of host cell lysis. Trends Microbiol. 2000;8:120–128. doi: 10.1016/s0966-842x(00)01705-4. [DOI] [PubMed] [Google Scholar]
- Zadoks JC, Schein RD. Epidemiology and Plant Disease Management. New York: Oxford University Press; 1979. [Google Scholar]
- Zhou CE, Ko R, Maeda S. Polyhedron-like inclusion body formation by a mutant nucleopolyhedrovirus expressing the granulin gene from a granulovirus. Virol. 1998;240:282–294. doi: 10.1006/viro.1997.8927. [DOI] [PubMed] [Google Scholar]