

Abstract
In kidney epithelial cells, an angiotensin II (Ang II) type 2 receptor subtype (AT2) is linked to a membrane-associated phospholipase A2 (PLA2) and the mitogen-activated protein kinase (MAPK) superfamily. However, the intervening steps in this linkage have not been determined. The aim of this study was to determine whether arachidonic acid mediates Ang II’s effect on p21ras and if so, to ascertain the signaling mechanism(s). We observed that Ang II activated p21ras and that mepacrine, a phospholipase A2 inhibitor, blocked this effect. This activation was also inhibited by PD123319, an AT2 receptor antagonist but not by losartan, an AT1 receptor antagonist. Furthermore, Ang II caused rapid tyrosine phosphorylation of Shc and its association with Grb2. Arachidonic acid and linoleic acid mimicked Ang II-induced tyrosine phosphorylation of Shc and activation of p21ras. Moreover, Ang II and arachidonic acid induced an association between p21ras and Shc. We demonstrate that arachidonic acid mediates linkage of a G protein-coupled receptor to p21ras via Shc tyrosine phosphorylation and association with Grb2/Sos. These observations have important implications for other G protein-coupled receptors linked to a variety of phospholipases.
Arachidonic acid release after activation of phospholipase A2 (PLA2) represents one signaling system whereby angiotensin II (Ang II) influences ion transport in renal proximal tubular epithelial cells (1). A unique aspect relates to the fact that a membrane-associated PLA2 appears to be involved rather than cytosolic PLA2 frequently associated with receptor tyrosine kinases (RTKs) and G protein-coupled receptors (GPCR) (2). Recent studies from this laboratory have shown that Ang II-induced arachidonic acid release in kidney epithelial cells mediates extracellular signal-regulated kinase 1 and 2 and c-Jun N-terminal kinase activation (3, 4). Thus, in this paradigm, a GPCR-induced arachidonic acid release is upstream of the mitogen-activated protein kinase (MAPK) superfamily in contrast to the model wherein cPLA2 phosphorylation by MAPK causes activation and arachidonic acid release (5, 6).
Recently, p21ras has been documented to be involved in Gq and phospholipase (PL) C mediated Ang II signaling in cardiac myocytes (7) and vascular smooth muscle cells (8). However, the sequential upstream events linking this GPCR to p21ras were not delineated but seemed to involve the Shc-Grb2-Sos adapter proteins in cardiac myocytes (7). We reasoned that p21ras activation by Ang II in kidney epithelial cells and other cell types may involve arachidonic acid as a critical mediator because arachidonic acid release is common to many signaling pathways (PLA2, PLD, and PLC). Earlier studies indicated that arachidonic acid, its metabolites, and other fatty acids inhibited GTPase-activating proteins (GAPs) thereby allowing p21ras to remain in its active GTP bound form (9, 10).
An alternative mechanism for activation of p21ras involves stimulation of guanine nucleotide exchange activity by tyrosine kinases after exposure to growth factors, cytokines, and/or vasoactive agonists. The process involves the association of SH2/SH3-containing adapter proteins, such as Shc and Grb2, with the guanine nucleotide exchange factor mSos (11–14). The current studies document that both Ang II and arachidonic acid induces GDP-GTP exchange on p21ras in renal proximal tubular cells and provide a mechanism whereby Ang II activated this small GTP binding protein. These studies have important implications for other GPCRs as they establish an important linkage.
METHODS
Materials.
Ang II was obtained from Peninsula Laboratories. Arachidonic acid, linoleic acid, and anti-rat IgG antibody were obtained from Sigma. Losartan (DuP 753) was the generous gift of Ronald D. Smith of DuPont-Merck. PD123319 was purchased from Research Biochemicals International (Natick, MA). Anti-vHras antibody and mepacrine were obtained from Calbiochem, anti-Shc was obtained from Transduction Laboratories (Lexington, KY), and anti-phosphotyrosine was obtained from Santa Cruz Biotechnology and Upstate Biotechnology (Lake Placid, NY).
Cell Culture.
Primary cultures of renal proximal tubular epithelial cells from rabbit were prepared as described previously (15). Briefly, the method involves homogenization of the renal cortex and separation of fully dissociated cells on a discontinuous 30–60% Percoll gradient. Cells with a density of 1.026 g/ml were removed and cultured on Costar tissue culture flasks. The standard medium was a 50:50 mixture of DMEM and Ham’s F-12 media supplemented with 15 mM Hepes buffer, pH 7.35, 600 μg/ml sodium bicarbonate, 100 units/ml penicillin, 100 μg/ml streptomycin, 5 μg/ml insulin, 10 ng/ml epithelial growth factor, 5 μg/ml human transferrin, 0.5 μM hydrocortisone, and 5% fetal bovine serum. These cells have been shown to be derived mainly from the proximal tubule (15). Subconfluent cells were passaged after 2 weeks and replated on 100-mm cell culture Petri dishes. They were used in the experiment as subconfluent monolayers.
Metabolic Labeling, Immunoprecipitation, and Determination of Guanine Nucleotides Bound to p21ras.
The monolayer of renal proximal tubular cells was labeled with [32P]orthophosphate (NEN) at 0.1 mCi/ml (1 Ci = 37 GBq) in phosphate-free DMEM (GIBCO) without serum for 14 hr. After stimulation with Ang II or fatty acids, cells were lysed (50 mM Tris, pH 7.4/150 mM NaCl/20 mM MgCl2/1% Triton X-100/1 mM phenylmethanesulfonyl fluoride/1 mM Na orthovanadate/10 μg/ml leupeptin/10 μg/ml aprotinin) plus anti-vHras antibody (10 μg per sample, Calbiochem) and they were collected in microcentrifuge tubes. The cell lysate was clarified by centrifugation at 14,000 rpm for 10 min. Immunoprecipitation was for 90 min in 4°C using anti-rat IgG precoupled to protein A-Sepharose CL-4B (Pharmacia). Immunoprecipitates were washed three times with 1 ml of the lysis buffer followed by 1 ml of PBS. Nucleotide was eluted with 0.75 M KH2PO4 (pH 3.4), 0.2 mM GTP, and 0.2 mM GDP at 90°C in heating block for 10 min. Separation of eluted nucleotide was on polyethyleneimine (PEI)-cellulose flexible plates (Fisher Scientific) run in 0.75 M KH2PO4 (pH 3.4). The positions of GTP and GDP standards were determined under UV light. Radiolabeled nucleotides were quantitated by collecting the GTP and GDP from the plate and counting by a liquid scintillation system (Beckman LS 5801). p21ras activity is expressed by percentage of GTP bound ras in 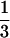 GTP and ½ GDP bound ras [(
GTP and ½ GDP bound ras [(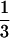 GTP/
GTP/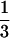 GTP + ½ GDP) × 100%].
GTP + ½ GDP) × 100%].
Immunoprecipitation and Immunoblot Analysis.
The monolayer of renal proximal tubular cells was deprived of serum for 14 hr before initiating experiments. After stimulation with Ang II or fatty acids, cells were lysed as previously described. Antibodies were added to cell lysates and incubated at 4°C for 60 min with gentle agitation. Immune complexes were collected with protein A Sepharose 4B (Pharmacia) at 4°C for 30 min, washed gently in cold lysis buffer, and analyzed by SDS/PAGE. For Western blot analysis, protein samples separated on SDS/PAGE gels were blotted onto nitrocellulose membrane (Schleicher & Schuell). The membranes were then blocked with 5% milk in washing buffer (10 mM Tris, pH 7.4/150 mM NaCl/0.1% Tween 20) for 2 hr, incubated with primary antibodies for 2 hr, washed six times for 1 hr, incubated with secondary antibodies for 1 hr, and washed another six times for 1 hr. Specific antibody signals were detected with an enhanced chemiluminescence kit (Amersham).
RESULTS
Arachidonic Acid and Ang II Activate p21ras in Renal Proximal Tubular Cells.
To examine the roles of p21ras in signaling, epithelial cells were stimulated with either arachidonic acid (20 μM) or Ang II (1 μM) for 5 and 15 min, and cell lysates were subjected to immunoprecipitation with p21ras antibody. Guanine nucleotides bound to the p21ras immunoprecipitates were separated by TLC (Fig. 1A). The basal level of GTP-bound p21ras was the 8.6% that increased to 17.7% and 16.5% after exposure to Ang II (1 μM) and arachidonic acid (20 μM) for 5 min, respectively. At 15 min, values for Ang II and arachidonic acid decreased to 12.3% and 13.6%, respectively (Fig. 1B) and were not significantly different from control.
Figure 1.

Activation of p21ras by arachidonic acid and Ang II in rabbit renal proximal tubular epithelial cells. (A) The cells were labeled with [32P]orthophosphate and stimulated with arachidonic acid (20 μM) and Ang II (1 μM) for 0 (control), 5, and 15 min. p21ras was immunoprecipitated and bound guanine nucleotides were separated by TLC. A very small amount of GTP bound ras was detected in the control. (B) Quantitation of GTP and GDP bound ras by liquid scintillation. The GTP and GDP of each sample on TLC plate were cut out and placed in scintillation vials. Five milliliters of counting mixture was added in each vial and counted in Beckman LS 5801 scintillation system. p21ras activation was expressed as (1/3ras-GTP/1/3ras-GTP + 1/2ras GDP) × 100%. The results are expressed as mean ± SE from five experiments for control and 5-min stimulation with arachidonic acid and Ang II and three experiments for 15-min stimulation with arachidonic acid and Ang II. AA, arachidonic acid. ∗∗, P < 0.01 compared with control.
In subsequent experiments, cells were evaluated after 5-min exposure to agonists. Linoleic acid at 30 μM had an effect similar to 20 μM arachidonic acid (data not shown).
Ang II Stimulation of p21ras Was Blocked by the AT2-Selective Antagonist PD123319 and the PLA2 Inhibitor Mepacrine.
To discriminate which ANG II receptor subtype was involved in p21ras stimulation, both the AT1 and AT2 receptor blockers were used. Losartan (an AT1 blocker) and PD123319 (an AT2 blocker) were added at a concentration of 100 μM for 20 min before the addition of 1 μM Ang II. The AT2-selective antagonist PD123319, but not the AT1-selective antagonist losartan inhibited Ang II-induced stimulation of p21ras (Fig. 2). These results support a role for the AT2 receptor as a mediator of p21ras activation. To confirm a role of PLA2 in this paradigm, we used mepacrine, a PLA2 inhibitor, and observed that Ang II activation of p21ras was abrogated by the inhibitor (Fig. 3A).
Figure 2.

The effect of Ang II type 1 and 2 receptor antagonists on p21ras activation in rabbit renal proximal epithelial cells. The proximal tubular epithelial cells labeled with [32P]orthophosphate were pretreated with losartan (LSTN) or PD123319 (PD) at 100 μM for 20 min before stimulation with Ang II (1 μM) for 5 min. p21ras was immunoprecipitated and bound guanine nucleotides were separated by TLC and quantitated by liquid scintillation. p21ras activation was expressed as Fig. 1. The results are expressed as mean ± SE illustrating four experiments. ∗, P < 0.05; ∗∗, P < 0.01 compared with control.
Figure 3.

Blockage of p21ras activation and tyrosine phosphorylation of Shc by mepacrine in rabbit renal proximal tubular epithelial cells. (A) The proximal epithelial cells labeled with [32P]orthophosphate were pretreated for 5 min with mepacrine (100 μM) before stimulation with Ang II (1 μM) for 5 additional min. p21ras was immunoprecipitated and bound guanine nucleotides were separated by TLC and quantitated by liquid scintillation. p21ras activation was expressed as mean ± SE (n = 3). ∗∗, P < 0.01 compared with control. (B) Cells were stimulated with 1 μM Ang II for 5 min after pretreatment with mepacrine (20 and 100 μM) or diluent for 5 min. The cell lysates were immunoprecipitated with an anti-Shc polyclonal antibody. The immunoprecipitates were analyzed by SDS/PAGE and immunoblotting using antibodies to phosphotyrosine (a) and Shc (b). I.P., immunoprecipitation; ptyr, phosphotyrosine.
Arachidonic Acid and Ang II Stimulated Shc Tyrosine Phosphorylation and Its Association with Grb2.
Adapter proteins containing the SH2 domain, such as Shc, link RTKs and GPCRs to the guanine nucleotide exchange factor Sos for activation of p21ras (11-14). Therefore, we examined whether a similar mechanism is associated with arachidonic acid and Ang II activation of p21ras. Cell lysates were immunoprecipitated with Shc antibody, separated by SDS/PAGE, and transferred to nitrocellulose membrane. Blots were subsequently probed with phosphotyrosine and Shc antibodies, respectively. Fig. 4 illustrates that both arachidonic acid and Ang II caused an increase in tyrosine phosphorylation of Shc. Moreover, arachidonic acid-induced tyrosine phosphorylation of p52 Shc persisted longer than that by Ang II. Using photodensitometer to quantitate Shc tyrosine phosphorylation in three individual experiments, Ang II’s effect peaked at 3 min 112% above the basal and returned to control in 30 min (34% above basal). Arachidonic acid stimulation peaked at 10 min 187% above basal and persisted at 30 min (115% above basal) (data not shown). Ang II-induced-tyrosine phosphorylation of Shc was abrogated by the PLA2 inhibitor mepacrine at a concentration as low as 20 μM (Fig. 3B).
Figure 4.

Arachidonic acid and Ang II induced tyrosine phosphorylation of Shc in renal proximal epithelial cells. The cells were stimulated with either 20 μM arachidonic acid or 1 μM Ang II for the indicated times. Cell lysates were immunoprecipitated with an anti-Shc polyclonal antibody. The immunoprecipitates were analyzed by SDS/PAGE and immunoblotting by using antibodies to phosphotyrosine (A) and Shc (B) as indicated. I.P., immunoprecipitation; ptyr, phosphotyrosine.
To determine whether arachidonic acid and Ang II-induced Shc tyrosine phosphorylation leads to its association with Grb2, cell lysates were immunoprecipitated with Grb2 antibody, separated by SDS/PAGE and transferred to nitrocellulose membrane. Blots were probed with anti-Shc antibody and demonstrated that not only was more Shc protein coimmunoprecipitated with Grb2 protein, but a band shift was also observed after stimulating the cells with arachidonic acid and Ang II (Fig. 5). As observed previously, 20 μM arachidonic acid exhibited a greater magnitude of effect than 1 μM Ang II and persisted longer.
Figure 5.

Arachidonic acid and Ang II induced the association of Shc with Grb2 in renal proximal tubular epithelial cells. The cells were stimulated with either 20 μM arachidonic acid or 1 μM Ang II for indicated times. The cell lysates were immunoprecipitated with an anti-Grb2 polyclonal antibody. The immunoprecipitates were analyzed by SDS/PAGE and immunoblotting using antibodies to Shc (A) and Grb2 (B). I.P., immunoprecipitation; PTCL, positive control of total cell lysate.
Arachidonic Acid and Ang II Induce Association of Shc with p21ras.
To examine whether Shc forms a complex with p21ras after exposure to arachidonic acid and Ang II, lysates immunoprecipitated with p21ras were subjected to SDS/PAGE and Western blot analysis followed by immunoblotting with the anti-Shc antibody. Shc was immunoprecipitated with p21ras after stimulation with both arachidonic acid and Ang II (Fig. 6). Arachidonic acid induced a phosphorylation shift of Shc that lasted for 30 min, whereas Ang II-induced shift diminished after 10 min. This pattern is the same as Shc tyrosine phosphorylation stimulated by the two agents (Fig. 4).
Figure 6.

Arachidonic acid and Ang II induced association of Shc with p21ras in renal proximal tubular epithelial cells. The cells were stimulated with either 20 μM arachidonic acid or 1 μM Ang II for the indicated times. The cell lysates were immunoprecipitated with an anti-ras antibody. The immunoprecipitates were analyzed by SDS/PAGE and immunoblotting used antibodies to Shc (A), and ras (B). The positions of Shc and ras are indicated on the left. I.P., immunoprecipitation; ptyr, phosphotyrosine.
DISCUSSION
Activation of p21ras was observed to be linked to RTKs (16, 17). A physical association occurs between the tyrosine kinase insert of RTK and the adapter protein Shc that in turn associates with Grb2 and Sos to initiate the exchange of GDP for GTP on p21ras (13). It is now clear that a number of GPCRs, such as the Ang II receptor, activate p21ras through the Shc/Grb2/Sos complex (7). Our data demonstrate that Ang II activates this complex and that leads to the exchange of GDP for GTP on p21ras. The activation of PLA2 and release of arachidonic acid are critical because downstream signaling is blocked by a PLA2 inhibitor (Fig. 3 A and B) and arachidonic acid and linoleic acid mimic Ang II’s effect. This represents an unusual mechanism of cross-talk between a GPCR and RTK through the release of arachidonic acid, an important lipid mediator in many signaling systems (PLA, PLC, and PLD, etc.). Moreover, our observations demonstrate the association of p21ras with Shc after the epithelial cell challenge with either Ang II or arachidonic acid. The time-course of this association shows a striking parallel to the activation of p21ras. These observations support the conclusion that formation of the active complex of proteins that initiates the exchange of GDP for GTP on p21ras is caused by a lipid second messenger. However, it is not clear how arachidonic acid induces the activation of the Shc-Grb2-Sos complex and its association with p21ras. One possibility is that arachidonic acid may activate a tyrosine kinase thereby inducing phosphorylation of Shc. Alternatively, a RTK may be involved in this fatty acid- mediated signaling. These possibilities need to be addressed further in future studies.
This study also demonstrates Ang II-mediated activation of p21ras via an AT2 receptor subtype because the effect was blocked by the AT2 antagonist, PD123319 but not by losartan, the AT1 antagonist (Fig. 2). In general, AT2 receptors represent about 14% of the Ang II receptor population in adult kidney with the remainder being AT1 subtype (18). The distribution of renal AT2 receptor subtypes has not been precisely determined with the exception of the apical receptor on proximal tubular epithelial cells and glomerular endothelial cells (1, 19). Signaling linked to AT2 receptor includes activation of MAPK phosphatase, NO, guanylate cyclase, and PLA2 (1, 20, 21, 22). The current studies broaden our understanding of AT2-mediated signaling and document modulation of p21ras in proximal tubular epithelium.
It has been shown that the activity of p21ras is regulated through binding of guanine nucleotides to p21ras. Active p21ras is bound to GTP, whereas inactive p21ras is bound to GDP (16). The ratio of GTP/GDP bound to p21ras is regulated by both guanine nucleotide exchange factors, which catalyze GDP release and GTP binding (11–13), and the GTPase-activating protein, which stimulates the intrinsic p21ras GTPase activity (9, 10). It has been reported that arachidonic acid and other fatty acids can activate ras by inhibition of GTPase-activating proteins; however, this paradigm has not been show to be a functional signaling model in intact cells (9). Furthermore, the concentrations of arachidonic acid and other fatty acids used were significantly higher than those used in the present study (9, 10).
Recent advances have shown that receptors coupled to heterotrimeric guanine triphosphate-binding protein (G protein) stimulate p21ras through G βγ subunits (23), but the subsequent intervening molecules are still poorly defined. Although there is a linkage of GPCRs to the MAPK signaling pathway through phosphatidylinositol 3-kinase γ, signaling appears to require a tyrosine kinase, Shc, Grb2, Sos, Ras, and Raf (24). Several Src-related tyrosine kinases have been proposed to link GPCRs to MAPK, including Lyn and Syk (25). Our studies provide an alternative model with arachidonic acid as an essential element.
Studies from this laboratory have demonstrated that the apical AT2 receptor in renal proximal tubular epithelial cells is linked to a membrane-associated PLA2 that initiates arachidonic acid release (1, 2). Arachidonic acid, in turn, has been documented to activate extracellular signal-regulated kinase 1 an 2 and c-Jun N-terminal kinase (3, 4). Thus, this represents a different model from earlier studies that have documented that MAPK phosphorylates and activates cPLA2 and mimics agonist-mediated effects in a variety of cell types in vitro (5, 6). We explored the alternative hypothesis that the Shc-Grb2-Sos pathway links arachidonic acid to activation of p21ras. Our results demonstrate that arachidonic acid activates p21ras through tyrosine phosphorylation of Shc and its interaction with Grb2 and p21ras (Fig. 4–6). Inhibition of PLA2 by mepacrine blocks arachidonic acid release in cells stimulated with Ang II (1) and abrogates Shc tyrosine phosphorylation and p21ras activation (Fig. 3 A and B). This is important as we have established an alternative linkage in signal transduction between GPCRs and activation of p21ras by arachidonic acid and other fatty acids.
In conclusion, our finding demonstrates that Ang II’s activation of p21ras occurs via AT2-mediated signaling in renal proximal tubular epithelial cells. Moreover, arachidonic acid mediates Ang II-induced tyrosine phosphorylation of the Shc and formation of Shc/Grb2/Sos complex and subsequent association with, and activation of, p21ras. These observations have significant implications for GPCR-medicated signaling in that arachidonic acid is an important lipid mediator linked to a variety of signaling pathway.
Acknowledgments
We thank Pearl M. Whitley and Nnennaya Nkemere for the expertise assistance with cell isolation and tissue culture. This work was supported by National Institutes of Health Grant HL41618.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: MAPK, mitogen-activated protein kinase; Ang II, angiotensin II; PL, phospholipase; GPCRs, G protein-coupled receptors; RTK, receptor tyrosine kinase; AT2 receptor, Ang II subtype 2 receptor.
References
- 1.Jacobs L S, Douglas J G. Hypertension. 1996;28:663–668. doi: 10.1161/01.hyp.28.4.663. [DOI] [PubMed] [Google Scholar]
- 2.Harwalkar S, Chang C H, Dulin N O, Douglas J G. Hypertension. 1997;31:809–814. doi: 10.1161/01.hyp.31.3.809. [DOI] [PubMed] [Google Scholar]
- 3.Dulin N O, Sorokin A, Harwalkar S, Douglas J G. J Am Soc Nephrol. 1995;6:789. (abstr.). [Google Scholar]
- 4.Cui X L, Douglas J G. Proc Natl Acad Sci USA. 1997;94:3771–3776. doi: 10.1073/pnas.94.8.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin L-L, Wartmann M, Lin A Y, Knopf J L, Seth A, Davis R J. Cell. 1993;72:269–278. doi: 10.1016/0092-8674(93)90666-e. [DOI] [PubMed] [Google Scholar]
- 6.Nemenoff R A, Winitz S, Qian N-X, Putten V V, Johnson G L, Heasley L E. J Biol Chem. 1993;268:1960–1964. [PubMed] [Google Scholar]
- 7.Sadoshima J, Izumo S. EMBO J. 1996;15:775–787. [PMC free article] [PubMed] [Google Scholar]
- 8.Eguchi S, Matsumoto T, Motley E D, Utsunomiya G, Inagami T. J Biol Chem. 1996;271:14169–14175. doi: 10.1074/jbc.271.24.14169. [DOI] [PubMed] [Google Scholar]
- 9.Tsai M H, Yu C L, Wei F S, Stacey D W. Science. 1989;243:522–526. doi: 10.1126/science.2536192. [DOI] [PubMed] [Google Scholar]
- 10.Bollag G, McCormick F. Nature (London) 1991;351:576–579. doi: 10.1038/351576a0. [DOI] [PubMed] [Google Scholar]
- 11.Buday L, Downward J. Cell. 1993;73:611–620. doi: 10.1016/0092-8674(93)90146-h. [DOI] [PubMed] [Google Scholar]
- 12.Buday L, Downward J. Mol Cell Biol. 1993;13:1903–1910. doi: 10.1128/mcb.13.3.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Egan S E, Giddings B W, Brooks M W, Buday L, Sizeland A M, Weinberg R A. Nature (London) 1993;363:45–51. doi: 10.1038/363045a0. [DOI] [PubMed] [Google Scholar]
- 14.Medema R H, de Vries-Smits A M, van der Zon G C, Maassen J A, Bos J L. Mol Cell Biol. 1993;13:155–162. doi: 10.1128/mcb.13.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Welsh C, Dubyak G, Douglas J G. J Clin Invest. 1988;81:710–719. doi: 10.1172/JCI113376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boguski M S, McCormick F. Nature (London) 1993;366:643–654. doi: 10.1038/366643a0. [DOI] [PubMed] [Google Scholar]
- 17.Smith M R, DeGudicibus S J, Stacey D W. Nature (London) 1986;320:540–543. doi: 10.1038/320540a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ernsberger P, Zhou J, Damon J G, Douglas J G. Am J Physiol. 1992;263:F411–F416. doi: 10.1152/ajprenal.1992.263.3.F411. [DOI] [PubMed] [Google Scholar]
- 19.Wolf G, Ziyadeh F N, Thaiss F, Tomaszewski J, Caron R J, Wenzel U, Zahner G, Helmchen U, Stah R A K. J Clin Invest. 1997;100:1047–1058. doi: 10.1172/JCI119615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siragy H M, Carey R M. J Clin Invest. 1997;100:264–269. doi: 10.1172/JCI119531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakajima M, Hutchinson H G, Fujinaga M, Hayashida W, Zhang L, Pratt R E, Dzau V J. Proc Natl Acad Sci USA. 1995;92:10663–10667. doi: 10.1073/pnas.92.23.10663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siragy H M, Carey R M. J Clin Invest. 1996;97:1978–1982. doi: 10.1172/JCI118630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crespo P, Xu N, Simonds W F, Gutkind J S. Nature (London) 1994;369:418–420. doi: 10.1038/369418a0. [DOI] [PubMed] [Google Scholar]
- 24.Lopez-Ilasaca M, Crespo P, Pellici P G, Gutkind J S, Wetzker R. Science. 1997;275:394. doi: 10.1126/science.275.5298.394. [DOI] [PubMed] [Google Scholar]
- 25.Wan Y, Kurosaki T, Huang X-Y. Nature (London) 1996;380:541–544. doi: 10.1038/380541a0. [DOI] [PubMed] [Google Scholar]