

Abstract
In response to Toll-like receptor ligands, dendritic cells (DCs) dramatically enhance their antigen presentation capacity by stabilizing at the cell-surface MHC II molecules. We demonstrate here that, in human monocyte-derived DCs, the RING-CH ubiquitin E3 ligase, membrane-associated RING-CH I (MARCH I), promotes the ubiquitination of the HLA-DR β-chain. Thus, in nonactivated DCs, MARCH I induces the surface internalization of mature HLA-DR complexes, therefore reducing their stability and levels. We further demonstrate that the maturation-dependent down-regulation of MARCH I is a key event in MHC class II up-regulation at the surface of LPS-activated DCs. MARCH I is, therefore, a major regulator of HLA-DR traffic, and its loss contributes to the acquisition of the potent immunostimulatory properties of mature human DCs.
Keywords: antigen presentation, ubiquitin ligase, endocytosis, LPS, TLR
Activation of dendritic cells (DCs) by invading pathogens involves a process of maturation that converts an immature DC (iDC) to a mature DC (mDC) and a subsequent Toll-like receptor (TLR)-mediated immune response. This process is accompanied by a marked cellular reorganization (1, 2). Redistribution of MHC class II molecules (MHC class II) from late endosomal compartments to the plasma membrane (3) represents one of the major functional events observed during the acquisition of the immunostimulatory function by mDCs. Recently in mouse DCs, ubiquitination of lysine-225 in the cytoplasmic tail of MHC II β-chain has been shown to be determinant for the targeting and accumulation of the mature form of MHC II in the lysosomes of iDCs (4, 5). Interestingly, ubiquitination of MHC II in murine DCs ceases on maturation, thus contributing to the increased level of MHC II at the cell surface (4, 5). Membrane-associated RING-CH (MARCH) proteins represent a family of E3 ubiquitin ligases, which all contain a variant catalytical RING-finger domain (RING-CH domain, C4HC3) located at the N terminus (6–10). It has been recently reported that transgenic overexpression of MARCH VIII/c-MIR in mouse splenic antigen-presenting cells (APCs) leads to internalization of surface CD86/B7-2 and MHC II, and subsequent lysosomal degradation (10). Furthermore, deletion of MARCH I, another E3 ubiquitin ligase, promoted an increase of surface MHC II levels in mouse B cells, although a direct implication of this ligase in the process of MHC II internalization was excluded (11). We identify here human MARCH I as a potent regulator of HLA-DR surface expression and explore its role during human monocyte-derived DC (MoDC) activation. We further show that MARCH I expression is lost at late stages of DC maturation and thereby participates in the stabilization of peptide-loaded HLA-DR at the cell surface. Our observations suggest that MARCH I is essential to coordinate MHC II-restricted antigen presentation during human DC activation by TLR ligands.
Results
MHC Class II Ubiquitination Is Regulated in MoDCs.
The subcellular localization of MHC class II (HLA-DR) in CD14+ MoDCs changes dramatically when cells are exposed to LPS (12, 13). As determined by confocal microscopy, iDCs accumulate HLA-DR molecules in LAMP1+ late endosomes and lysosomes. On activation by LPS, rapid relocalization of MHC II to the cell surface is observed (4–8 h) [supporting information (SI) Fig. 7A]. Recently, this process has been shown to be associated with a loss of ubiquitination on the class II β-chain in mouse bone marrow-derived DCs (bmDCs) (4, 5). We investigated whether the control of HLA-DR traffic was also mediated by ubiquitination in human MoDCs. HLA-DR molecules were immunoprecipitated at different times of maturation by using peptide-loaded HLA-DR-specific L243 antibodies (Fig. 1A) (see SI Materials and Methods). On immunoblotting with the P4D1 anti-ubiquitin antibody, HLA-DR ubiquitination was detected in immature MoDCs. In addition to the characteristic polyubiquitinated proteins ladder, monoubiquitinated MHC II (≈37 kDa) appeared to be the prevalent form found in human iDCs. Interestingly, HLA-DR ubiquitination rapidly decreased with maturation and was fully abrogated after 16 h. Fig. 1B shows immunoprecipitated HLA-DR αβ dimers (left lanes), which were boiled and further immunoprecipitated by using the anti HLA-DR β-chain-specific antibody XD5 (right lanes). Immunoblot detection with P4D1 of the two different immunoisolates confirmed that reversible MHC II ubiquitination occurs specifically and probably uniquely on HLA-DR β-chain (37-kDa band). The ratio between ubiquitinated and unmodified HLA-DR forms indicated that only a relatively small proportion of all of the β-chain molecules were modified at steady state. Immunoprecipitation of total lysates with an anti-invariant chain antibody failed to reveal any ubiquitination of invariant chain–MHC class II complexes, indicating that in humans only mature peptide-loaded HLA-DR complexes are regulated by β-chain ubiquitination (SI Fig. 7B).
Fig. 1.

HLA-DR ubiquitination is down-regulated during MoDC maturation. (A) MoDCs were stimulated with LPS, and HLA-DR (L243) was immunoprecipitated and immunoblotted by using either anti-ubiquitin or anti-MHC II (XD5) antibodies. The asterisks represent nonspecific bands. (B) HLA-DR molecules from immature MoDCs were first immunoprecipitated with L243 antibody (left lanes). L243 immunoprecipitated material was then boiled and reimmunoprecipitated with XD5 (β-chain-specific) antibody (right lanes) before SDS/PAGE. Immunoblots of the immunoprecipitated material with anti-ubiquitin, anti-HLA-DR β-chain (XD5), or anti-HLA-DR α-chain specific antibodies are shown (Top, Middle, and Bottom, respectively). The asterisks indicate nonspecific bands.
Overexpression of MARCH I Down-Regulates Surface MHC Class II.
MARCH proteins are E3 ubiquitin ligases that have been reported to down-regulate several surface molecules including transferrin receptor (TfR), MHC class I and II, and CD86 (7, 10). We evaluated the capacity of different human MARCH ligases to control HLA-DR surface expression through transfection of class II transactivator (CIITA)-expressing HeLa cells (14). Different mRNAs, coding for MARCH I through MARCH IX fused with an eGFP moiety were transfected in HeLa-CIITA cells, before FACS analysis (Fig. 2A Upper). Surface HLA-DR levels were affected by the overexpression of MARCH I-eGFP and also, as expected, by MARCH VIII-eGFP (10). We thereby tested whether a single lysine residue in the cytoplasmic tail of the MHC-II β-chain was required by MARCH I to induce HLA-DR surface down-regulation. We transiently expressed in HEK293T cells the HLA-DR α-chain together with two different HLA-DR β cDNA constructs, containing either a wild-type (β) or a mutated cytoplasmic tail, with an alanine replacement at position 225 (βK225A). On MARCH I expression, down-regulation was heavily impaired for βK225A-bearing MHC II dimers (Fig. 2A Lower), thus identifying lysine-225 as a target residue for MARCH I-driven ubiquitination on HLA-DR.
Fig. 2.

MARCH I down-regulates surface MHC class II through ubiquitination. (A) FACS analysis of HLA-DR surface level in MARCH-eGFP (MARCH I–IX) transfected HeLa-CIITA cells (Upper) and in MARCH I-eYFP transfected HEK293T cells expressing HLA-DRα and either WT HLA-DRβ or mutant HLA-DRβ K225A (Lower). (B) FACS analysis of surface HLA-DR, CD86, and CD1a in MoDCs electroporated with either eGFP or MARCH-eGFP mRNAs. MHC II surface level is clearly decreased in MARCH I-transfected MoDCs (gray-filled curve) compared with eGFP alone (black-filled curve). Thin lines, isotype controls. (C) Immunoprecipitation of mature MHC II from MARCH I-transfected DCs followed by immunoblotting with anti-ubiquitin and anti-MHC II antibodies. The asterisk represents nonspecific bands. (D) Confocal microscopy analysis of MARCH I-eGFP transfected MoDCs. Mature MHC II molecules (L243, red) are found in HLA-DM-positive compartments (blue) in MARCH I-eGFP-transfected MoDCs, whereas they accumulate at the plasma membrane in eGFP or MARCH II-eGFP transfected cells. eGFP labeling is not shown, because its expression level is too low to allow confocal microscopy detection in MoDCs.
To confirm the relevance of MARCH I in mediating MHC II down-regulation in human DCs, surface expression of peptide-loaded MHC II was analyzed by FACS after MARCH I-eGFP mRNAs transfection. Despite a modest MARCH I expression (SI Fig. 8), surface levels of mature MHC II were strongly down-regulated in transfected MoDCs (Fig. 2B). Surface CD86 up-regulation, normally occurring on DC maturation, was also prevented. Yet, MARCH I targets a restricted number of surface proteins because the lipid–antigen presenting molecule CD1a was not affected by its overexpression. Consistently with the down-regulation of surface MHC II, MARCH I overexpression in MoDCs also induced an important increase in DR β-chain ubiquitination (Fig. 2C). Although the transfection procedure was found to induce DC maturation and the subsequent reduction of MHC II ubiquitination (control eGFP), MARCH I expression restored ubiquitination levels, which were quantitatively and qualitatively very similar to the ladder observed in nontransfected iDCs.
Immunofluorescence confocal microscopy of MARCH I-overexpressing MoDCs revealed an enhanced accumulation of mature HLA-DR in late endosomal/lysosomal compartments (Fig. 2D). L243 mAb, being specific for Ii chain-free αβ HLA-DR dimers, labels predominantly the plasma membrane of DCs at all maturation stages. On MARCH I overexpression, we could observe a loss of L243-positive MHC II plasma membrane staining and an increased colocalization with HLA-DM compared with eGFP- and MARCH II-transfected cells. MARCH I is therefore also likely to promote late endosomal sorting of mature MHC II dimers.
We followed the fate of surface MHC II molecules on cold binding of L243 antibody at the cell surface, followed by incubation at 37°C for different times, and subsequent cell lysis and immunoprecipitation (Fig. 3A). Quantification of precipitated material by immunoblot revealed that MARCH I rapidly promotes a 40% reduction of L243-bound MHC II in transfected DCs. This loss likely represents a rapid degradation of internalized surface MHC II, consistent with a prolonged exposure to the acidic environment of late endocytic compartments. However, the dissociation of the immune complex could lead to an overestimation of the efficiency of this process.
Fig. 3.

MARCH I overexpression promotes MHC class II late-endosomal targeting. (A) L243 antibodies were cold-bound to the surface of MARCH I-eGFP- or eGFP-transfected cells, before internalization at 37°C for different times. Cells were lysed and L243 immunoprecipitates were monitored for MHC class II β-chain by Western blot followed by digital quantification. Data are representative of three independent experiments. (B) Confocal immunofluorescence microscopy showed L243 detection (red) after 1 h of uptake in eGFP- and MARCH I-eGFP transfected MoDCs. Mature HLA-DR reached HLA-DM+ lysosomes (blue) more efficiently in MARCH I-eGFP transfected cells.
Indeed, confocal microscopy analysis indicated that L243-bound DR molecules reached endosomal compartments more efficiently in MARCH I-transfected than control DCs (Fig. 3B). Heterologous expression of MARCH I therefore induces a dominant “immature-like” DC phenotype, likely via increased MHC II ubiquitination and subsequent targeting to the endocytic pathway.
MARCH I Expression Is Regulated During DC Maturation.
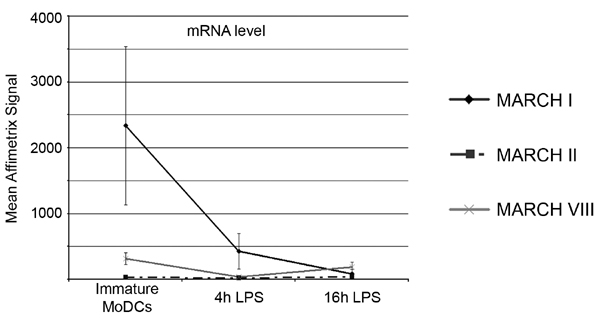
Based on our observations on MARCH I activity and its role in MHC II traffic, we reasoned that this ligase may be submitted to transcriptional regulation specifically on LPS-driven maturation. mRNA extraction and RT-PCR of all MARCH transcripts were performed in different human cell lines, and CD14+ monocytes and LPS-treated MoDCs (Fig. 4A). MARCH mRNAs were expressed in all cell types tested, with the exception of MARCH I and IV. Strikingly, MARCH I expression, although restricted to professional APCs, was not detected in mature DCs (Fig. 4A, lane 2). Real-time quantitative PCR confirmed that MARCH I transcription was rapidly (4 h) and strongly down-regulated (20-fold after 24 h) on LPS-induced maturation (Fig. 4B). MARCH II and VIII mRNA levels, however, showed no significant variations during maturation. Similar results were obtained testing cells from several different blood donors and were in full agreement with data generated by an independent Affymetrix pangenome chip analysis of maturing MoDCs mRNA (SI Fig. 9).
Fig. 4.

MARCH I is down-regulated during MoDCs maturation. (A) Expression profile of different MARCH mRNAs was analyzed by RT-PCR in the indicated cells. From left to right, immature MoDCs, LPS-stimulated MoDCs, HeLa, HeLa CIITA, Daudi B cells, and CD14+ monocytes are shown. (B) Expression levels of MARCH I, II, and VIII mRNA were compared among the indicated times of MoDC LPS maturation by using real-time quantitative PCR. (C) Protein levels of MARCH I were analyzed by immunoprecipitation followed by immunoblot during LPS stimulation of MoDCs. Actin from unbound fraction is shown as control.
Because MARCH protein expression is poorly detectable in MoDCs, MARCH I was enriched by immunoprecipitation before immunoblot detection with antibodies validated in cells transfected with MARCH-eGFP chimeras (SI Fig. 10). In agreement with its decreased transcription, the level of MARCH I protein was found to be reduced after 4 h of maturation and nondetectable after 24 h (Fig. 4C). MARCH I down-regulation is therefore a previously undescribed feature of LPS-driven MoDC maturation and is tightly correlated with the loss of MHC II ubiquitination and its increased surface stabilization.
MARCH I Regulates Surface MHC Class II Levels During DC Activation.
Our observations, mainly based on overexpression experiments, implicate MARCH I in the ubiquitination and the control of surface MHC II molecule internalization in iDCs. To study further the effects of endogenous MARCH I activity on MHC II and CD86 dynamic in DCs, we artificially down-regulated MARCH I expression in iDCs by using nucleoporation of specific siRNA. The efficiency and selectivity of the RNAi procedure in transfected cells was established by following MARCH I-eGFP chimera expression by FACS and immunoblot (SI Fig. 10).
Artificial MARCH I mRNA depletion in iDCs (equivalent to the down-modulation levels reached on 4 h of LPS treatment) resulted in a clear increase of HLA-DR and CD86 surface levels, whereas CD1a expression remained constant (Fig. 5 A and B). We tested next the effect of MARCH I depletion on HLA-DR ubiquitination in iDCs. MHC II complexes were immunoprecipitated with L243 from cells submitted to MARCH I RNAi, and HLA-DR ubiquitination was monitored by immunoblot (Fig. 5C). MARCH I depletion resulted in an important reduction (50%) of ubiquitinated HLA-DR, consistent with the concomitant increase in MHC II and CD86 surface levels observed by FACS (Fig. 5A). MARCH I transcriptional and translational down-regulation during MoDCs activation appears therefore to be a major event leading to the inhibition of MHC II ubiquitination and surface stabilization in mature human DCs.
Fig. 5.

RNA interference of MARCH I expression leads to a decrease of MHC II ubiquitination. (A) MoDCs were transfected with synthetic 21-mer-siRNAs specific for MARCH I or nonrelevant (NR) siRNA and analyzed by FACS for mature MHC II, CD86, and CD1a surface levels. MARCH I (black lines) and nonrelevant siRNA (dotted lines) transfected cells, and isotype control antibodies (gray lines) are shown. Data are representative of 10 experiments performed on different blood donors. (B) RNAi efficiency was measured by quantitative PCR comparing natural mRNA levels for MARCH I in maturing (4- and 24-h LPS) and siRNA-treated immature DCs (NR and MARCH I), relative to the expression level in iDCs. (C) Mature MHC II from siRNA-treated MoDCs was immunoprecipitated and immunoblotted by using either anti-ubiquitin or anti-MHC II antibodies. Mono- and polyubiquitinated MHC II forms are clearly decreased in MARCH I siRNA-treated cells. Western blot of one of three independent experiments used for digital quantification is shown. Quantification was performed normalizing ubiquitination level to MHC class II signal.
MARCH I Controls MHC Class II Internalization.
To determine at which level MHC class II transport might be affected by MARCH I depletion, we established the subcellular localization of HLA-DR molecules by confocal microscopy in MARCH I RNAi-treated iDCs (Fig. 6A). In addition to the expected late endosomal localization (HLA-DM+) observed normally in iDCs, MARCH I depletion induced the accumulation of HLA-DR molecules in EEA1+ sorting endosomes and at the plasma membrane. This result was confirmed by quantification of colocalization of MHC II either with HLA-DM- or with EEA1-positive compartments by using the Pearson coefficient method (Fig. 6B). The early endosomal accumulation of HLA-DR is in agreement with MARCH I subcellular distribution (SI Fig. 11), which is mostly restricted to early endosomes and to the plasma membrane of transfected cells. In addition, endocytosis inhibitors such as bafilomycin and dynasore [recently characterized as a cell-permeable inhibitor of dynamin (15)] counteract MARCH I activity by preventing MHC II surface down-regulation in transfected HeLa-CIITA cells (SI Fig. 11), thus confirming that this ligase plays a major role in surface MHC II internalization and/or early endosomal sorting.
Fig. 6.

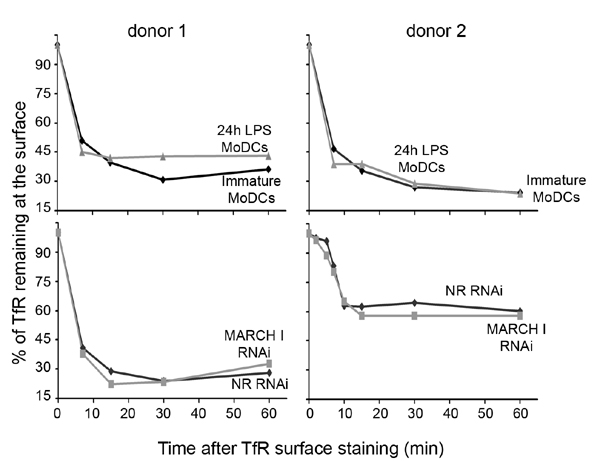
MARCH I depletion promotes MHC class II early-endosomal retention in MoDCs. (A) Confocal microscopy analysis of MARCH I-depleted cells. MHC II (ISCR3 mAb, green) fails to reach lysosomal compartments (blue), being retained in early endosomes (red) and at cell surface, whereas it accumulates in lysosomes in nonrelevant (NR) siRNA-treated cells. (B) Quantification of molecules colocalization using the ImageJ image analysis software and Pearson coefficient determination. A low Pearson coefficient is indicative of the absence of staining overlap. Histograms represent means ± SEM of Pearson coefficient between MHC II and HLA-DM (black) or MHC II and EEA1 (gray) on three independent experiments. (C) FACS quantification of L243 antibody uptake by immature and mature (24-h LPS-activated) MoDCs or by nonrelevant and MARCH I siRNA-transfected MoDCs (lower line). MoDCs were stained with L243 antibody for 30 min at 4°C, warmed at 37°C for indicated times, and stained with a secondary anti-mouse antibody at 4°C before FACS analysis. The kinetics of surface MHC II internalization in MARCH I siRNA-transfected cells is strikingly similar to the one observed in LPS stimulated-MoDCs. Results for two representative blood donors are shown (C and D).
We finally evaluated the impact of MARCH I depletion on the internalization rate of mature HLA-DR, monitoring the percentage of L243 antibody remaining at the surface of maturing MoDCs over time (Fig. 6C Upper). As anticipated, MHC II–antibody complex internalization was significantly reduced in cells treated with LPS for 24 h, whereas nonactivated DCs displayed an efficient endocytic activity. When MARCH I was artificially down-regulated in iDCs (Fig. 6C Lower), the reduction of MHC II internalization was strikingly similar to the situation observed in mature DCs. Indeed, the uptake over 1 h of the L243 antibody was decreased by 50% (shown here for two representative blood donors), thus confirming the importance of MARCH I in mediating surface internalization of mature MHC II molecules. The same experiment performed with an anti-TfR antibody failed to reveal any difference in its endocytosis rate in absence of MARCH I, thus confirming the specificity of this E3 ligase for MHC II (SI Fig. 12).
Discussion
We analyzed here the molecular mechanism leading to the maturation-dependent HLA-DR and CD86 surface up-regulation required by human DCs to acquire their immunostimulating properties. We have identified the down-regulation of the E3 ubiquitin ligase MARCH I as a major biochemical event leading to MHC II surface stabilization during DC activation. We showed that HLA-DR β-chain ubiquitination is promoted by MARCH I in human immature MoDCs. MARCH I favors the direct internalization of peptide-loaded MHC II from the cell surface, likely weakening the antigen presentation capacity of immature DCs. The maturation-dependent loss of MARCH I prevents the ubiquitination-mediated internalization of HLA-DR resulting in full DC activation. Consistent with this hypothesis, the depletion of MARCH I in iDCs reduces dramatically MHC II ubiquitination levels and enhances its plasma membrane stabilization, thus recapitulating DC functional maturation.
We demonstrated the existence of human ubiquitinated HLA-DR molecules, indicating that mono- and polyubiquitination are evolutionally conserved endosomal sorting signals required for the regulation of MHC II traffic. Although down-regulation of cell-surface HLA-DR is almost complete in MARCH I-transfected DCs and large amounts of MHC-II molecules are detected in lysosomes, only a very small fraction of total DR was found to be ubiquitinated. This suggests that MHC II molecules may be efficiently deubiquitinated, most probably in late endosomes as shown for other cargo molecules (16).
Recent work on the effect of MARCH I deletion in mouse B cells excluded a direct implication of MARCH I in the process of MHC II internalization (11). Our divergent finding on MARCH I role in the earliest steps of MHC II endocytosis may be due to the different experimental settings, in particular the utilization of RNA interference to deplete MARCH I in human DCs instead of a full genetic inactivation as in KO mice, which could lead to compensatory mechanisms. In addition, we could show that pharmacological inhibition of dynamin-mediated surface internalization with dynasore (SI Fig. 11) impaired MARCH I-induced MHC II down-regulation, further supporting an implication of this ligase in HLA-DR surface uptake. Moreover, consistent with the mouse KO data, we find that human MARCH I is also involved in MHC II early endosomal sorting, as suggested by HLA-DR accumulation in EEA+ compartments in MARCH I-depleted cells.
Several ubiquitin ligases are likely to be involved in MHC II internalization and endosomal targeting. In particular, the role of MARCH VIII, which is ubiquitously expressed (7), remains to be investigated. Its ubiquitous tissue expression pattern, taken together with the fact that MARCH VIII does not undergo any major maturation-dependent transcriptional regulation in DCs, suggests that MARCH VIII may have other functions in this cell type. Like most E3 ubiquitin-ligases, MARCH proteins have multiple substrates and both MARCH I and VIII have been shown to down-regulate also Fas (CD95) and TfR (7). The main physiological substrates of MARCH VIII may therefore be others than MHC-II molecules. It would be rather intriguing to test whether, in DCs, MARCH I and MARCH VIII could cooperate to internalize MHC II and CD86. At this stage, we cannot formally exclude the existence of a target molecule serving as an intermediate between MARCH I and MHC II. The complete loss of MARCH I mRNA on LPS-stimulation suggests nevertheless that this enzyme occupies a central and dominant position in the control of MHC II traffic in human DCs, and other professional APCs.
The existence of different MARCH ubiquitin ligases in DCs, each of them specific for distinct targets and endocytic steps, might ensure the correct sorting of neosynthesized and peptide-loaded MHC II complexes, and immunomodulatory molecules (e.g., CD86). The tolerogenic properties of DCs harboring a relatively weak stimulatory activity (17, 18) may be linked to the rapid internalization of surface MHC II complexes and CD86. Indeed, previous studies report that tolerogenic cytokines, such as IL-10, can interfere with DC maturation by impairing surface accumulation of MHC II and costimulatory molecules (19). The fate of MARCH ligases on DC exposure to tolerogenic cytokines will have therefore to be carefully investigated.
Materials and Methods
Cell Culture.
To promote differentiation into iDC, purified CD14+ cells (0.5 × 106 cells per ml) were plated in six-well plates (2 × 106 cells per well) and cultured in RPMI medium 1640 supplemented with 10% FCS, nonessential amino acids, penicillin/streptomycin at 100 ng/ml (>1,000 units/ml), recombinant human GM-CSF, and 20 ng/ml (>100 units/ml) IL-4 for 5 days (both from PeproTech).
Immunocytochemistry.
DCs were harvested and let adhere on 1% Alcian blue-treated coverslips for 10 min at 37°C, fixed with 3% paraformaldehyde in PBS for 10 min at room temperature, permeabilized with 0.1% saponin in PBS/5% FCS/100 mM glycine for 10 min at room temperature, and stained 1 h with indicated primary antibody. All Alexa secondary antibodies were from Molecular Probes. Immunofluorescence and confocal microscopy (using microscope model LSM 510; Carl Zeiss MicroImaging) were performed as described in ref. 20.
Cell Transfection.
mRNA HeLa-CIITA transfections were performed by using Lipofectamine2000 Reagents (Invitrogen) following the manufacturer's instructions. For mRNA transfections, immature DCs were harvested at day 6 of culture and resuspended in the specified Amaxa electroporation buffer (Human Dendritic Cell Nucleofector kit; Amaxa) to a final concentration of 3 × 107 cells per ml. A total of 15 μg of in vitro transcribed mRNA was mixed with 0.1 ml of cell suspension, transferred to a 2.0-mm electroporation cuvette, and nucleofected with an Amaxa Nucleofector apparatus as indicated by the manufacturer.
mRNA Quantification by Real-Time RT-PCR.
PCR was carried out in complete SYBR Green PCR buffer (PE Biosystems) by using 200 nM of each specific primer. A total of 20 μl of PCR mix was added to 5 μl of cDNA template, and the amplification was tracked via SYBR Green incorporation by using a Stratagene sequence detection system. cDNA concentration in each sample were normalized by using GAPDH. A nontemplate control was also routinely performed.
Antibodies.
Anti-mature HLA-DR (L243 clone; BD Pharmingen) (21); rabbit polyclonal anti-HLA-DR (DRAB; P. Cresswell, New Haven, CT); anti-HLA-DR (clone ISCR3); anti-HLA-DR β-chain specific (clone XD5); rabbit polyclonal anti-HLA-DRα; anti-CD74 (clone PIN-1 and clone BU45); anti-MARCH I and anti-MARCH VIII, both a generous gift from Laurent Coscoy (University of California, Berkeley, CA) and Klaus Früh (Oregon Health and Science University, Portland, OR); anti-ubiquitin (clone P4D1; Covance); anti-GFP (clone JL-8; Clontech); rabbit polyclonal anti-HLA-DM (Lars Karlson, San Diego, CA); anti-EEA1 (clone 14), anti-human LAMP-1 (clone H4A3) from BD Transduction Laboratory; anti-TfR (clone OKT9; M. Vidal, Montpellier, France); anti-CD1a (clone BL6) from Beckman Coulter; allophycocyanin-conjugated L243 and biotin-conjugated anti-CD86 (clone IT2.2) from BD Pharmingen.
Supplementary Material
ACKNOWLEDGMENTS.
We are particularly indebted to Laurent Coscoy, Peter Cresswell, and Klaus Früh for antibody and reagents gift and to Marie-Claude Bourgeois-Daigneault for sharing unpublished results. We also thank the PICsL Imaging Core Facility for expert technical assistance. This work was supported by grants from La Ligue Nationale Contre le Cancer. A.D.G. is supported by a fellowship from the Centre National de la Recherche Scientifique. J.T. is supported by a Canadian Institutes for Health Research–Institut National de la Santé et de la Recherche Médicale International Scientific Exchanges fellowship. M.C. is supported by the Swiss National Research Fund. P.P. is part of the European Molecular Biology Organization Young Investigator Program and of the DC-THERA FP6 NoE.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0708874105/DC1.
References
- 1.Mellman I, Steinman RM. Cell. 2001;106:255–258. doi: 10.1016/s0092-8674(01)00449-4. [DOI] [PubMed] [Google Scholar]
- 2.Reis e Sousa C. Nat Rev Immunol. 2006;6:476–483. doi: 10.1038/nri1845. [DOI] [PubMed] [Google Scholar]
- 3.Pierre P, Turley SJ, Gatti E, Hull M, Meltzer J, Misra A, Inaba K, Steinman RM, Mellman I. Nature. 1997;388:787–792. doi: 10.1038/42039. [DOI] [PubMed] [Google Scholar]
- 4.van Niel G, Wubbolts R, Ten Broeke T, Buschow SI, Ossendorp FA, Melief CJ, Raposo G, van Balkom BW, Stoorvogel W. Immunity. 2006;25:885–894. doi: 10.1016/j.immuni.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 5.Shin JS, Ebersold M, Pypaert M, Delamarre L, Hartley A, Mellman I. Nature. 2006;444:115–118. doi: 10.1038/nature05261. [DOI] [PubMed] [Google Scholar]
- 6.Lehner PJ, Hoer S, Dodd R, Duncan LM. Immunol Rev. 2005;207:112–125. doi: 10.1111/j.0105-2896.2005.00314.x. [DOI] [PubMed] [Google Scholar]
- 7.Bartee E, Mansouri M, Hovey Nerenberg BT, Gouveia K, Fruh K. J Virol. 2004;78:1109–1120. doi: 10.1128/JVI.78.3.1109-1120.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coscoy L. Nat Rev Immunol. 2007;7:391–401. doi: 10.1038/nri2076. [DOI] [PubMed] [Google Scholar]
- 9.Ishido S, Wang C, Lee BS, Cohen GB, Jung JU. J Virol. 2000;74:5300–5309. doi: 10.1128/jvi.74.11.5300-5309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohmura-Hoshino M, Matsuki Y, Aoki M, Goto E, Mito M, Uematsu M, Kakiuchi T, Hotta H, Ishido S. J Immunol. 2006;177:341–354. doi: 10.4049/jimmunol.177.1.341. [DOI] [PubMed] [Google Scholar]
- 11.Matsuki Y, Ohmura-Hoshino M, Goto E, Aoki M, Mito-Yoshida M, Uematsu M, Hasegawa T, Koseki H, Ohara O, Nakayama M, et al. EMBO J. 2007;26:846–854. doi: 10.1038/sj.emboj.7601556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cella M, Engering A, Pinet V, Pieters J, Lanzavecchia A. Nature. 1997;388:782–787. doi: 10.1038/42030. [DOI] [PubMed] [Google Scholar]
- 13.Pierre P, Mellman I. Cell. 1998;93:1135–1145. doi: 10.1016/s0092-8674(00)81458-0. [DOI] [PubMed] [Google Scholar]
- 14.Bania J, Gatti E, Lelouard H, David A, Cappello F, Weber E, Camosseto V, Pierre P. Proc Natl Acad Sci USA. 2003;100:6664–6669. doi: 10.1073/pnas.1131604100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dev Cell. 2006;10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 16.Katzmann DJ, Odorizzi G, Emr SD. Nat Rev Mol Cell Biol. 2002;3:893–905. doi: 10.1038/nrm973. [DOI] [PubMed] [Google Scholar]
- 17.Steinman RM, Hawiger D, Nussenzweig MC. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 18.Banchereau J, Steinman RM. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 19.Koppelman B, Neefjes JJ, de Vries JE, de Waal Malefyt R. Immunity. 1997;7:861–871. doi: 10.1016/s1074-7613(00)80404-5. [DOI] [PubMed] [Google Scholar]
- 20.Lelouard H, Gatti E, Cappello F, Gresser O, Camosseto V, Pierre P. Nature. 2002;417:177–182. doi: 10.1038/417177a. [DOI] [PubMed] [Google Scholar]
- 21.Roche PA, Cresswell P. Nature. 1990;345:615–618. doi: 10.1038/345615a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}