

Abstract
Invasive cancer cells form dynamic adhesive structures associated with matrix degradation called invadopodia. Calpain 2 is a calcium-dependent intracellular protease that regulates adhesion turnover and disassembly through the targeting of specific substrates such as talin. Here, we describe a novel function for calpain 2 in the formation of invadopodia and in the invasive abilities of breast cancer cells through the modulation of endogenous c-Src activity. Calpain-deficient breast cancer cells show impaired invadopodia formation that is rescued by expression of a truncated fragment of protein tyrosine phosphatase 1B (PTP1B) corresponding to the calpain proteolytic fragment, which indicates that calpain modulates invadopodia through PTP1B. Moreover, PTP1B activity is required for efficient invadopodia formation and breast cancer invasion, which suggests that PTP1B may modulate breast cancer progression through its effects on invadopodia. Collectively, our experiments implicate a novel signaling pathway involving calpain 2, PTP1B, and Src in the regulation of invadopodia and breast cancer invasion.
Introduction
Integrin-mediated interactions between the extracellular matrix and the actin cytoskeleton play a central role in regulating cell migration in both normal and pathological processes, such as tumor metastasis. Many adherent cell types, such as fibroblasts, form integrin-mediated adhesions, known as focal adhesions, that undergo cycles of assembly and disassembly during cell migration. Focal adhesions are generally found in less motile cells and function to tether actin-containing stress fibers. In contrast, motile and invasive cells, including myeloid cells and invasive cancer cells, frequently form specialized integrin-mediated adhesive structures known as podosomes or matrix-degrading invadopodia (Buccione et al., 2004; Linder, 2007). Dynamic regulation of invadopodia in carcinoma cells is likely critical for the invasive capacity of metastatic cancer cells. However, an understanding of the mechanisms that regulate invadopodia dynamics in cancer cells remains limited.
First identified in fibroblasts transformed with the Rous-sarcoma virus v-Src oncogene (Tarone et al., 1985), podosomes and invadopodia contain many of the cytoskeletal and signaling proteins found in focal adhesions, including talin, vinculin, and paxillin (Linder and Aepfelbacher, 2003). In contrast to focal adhesions, podosomes and invadopodia are not associated with stress fibers but instead contain several actin regulatory proteins, including cortactin, gelsolin, Wiskott-Aldrich syndrome protein, and the actin-nucleating Arp 2/3 complex (Buccione et al., 2004). Furthermore, a hallmark of the invasive podosomes or invadopodia formed in metastatic cancer cells is the capacity for matrix degradation through the localized secretion of ECM-degrading matrix metalloproteases (Linder and Aepfelbacher, 2003).
There has been substantial recent progress in defining the molecular mechanisms that regulate podosome dynamics (Calle et al., 2004; Yamaguchi and Condeelis, 2007). For example, Src tyrosine kinases and the actin-binding protein cortactin are critical regulators of podosome formation and turnover (Luxenburg et al., 2006). The intracellular, calcium-dependent, thiol proteases calpains have also recently been implicated in regulating podosome disassembly in dendritic cells (Calle et al., 2006) and osteoclasts (Marzia et al., 2006). Calpains cleave many adhesion-associated and actin regulatory proteins including talin, FAK, paxillin, cortactin, and protein tyrosine phosphatase 1B (PTP1B; Frangioni et al., 1993; Franco and Huttenlocher, 2005). PTP1B is a key positive regulator of Src kinase activity (Bjorge et al., 2000) and is expressed at elevated levels in several human cancers (Bentires-Alj and Neel, 2007).
Our previous studies have demonstrated roles for calpains in the disassembly of focal adhesions in fibroblasts through the limited proteolysis of the cytoskeletal protein talin (Franco et al., 2004b). In addition to its role in adhesion disassembly, recent evidence suggests a critical role for calpain in cell migration through the regulation of membrane protrusion (Franco et al., 2004a; Nuzzi et al., 2007). Furthermore, the ubiquitous calpain isoform calpain 2 is up-regulated in breast cancer and its expression correlates with increased invasive properties of tumors (Libertini et al., 2005). However, the mechanism of calpain effects on cancer cell invasion or its role in regulating invadopodia dynamics in cancer cells has not been previously explored.
In this study, we examined the role of calpains in invadopodia formation and dynamics in the invasive breast adenocarcinoma MTLn3 cell line. Our findings implicate a novel signaling pathway involving calpain 2, PTP1B, and Src in the regulation of invadopodia dynamics and breast cancer cell invasion.
Results
Calpain 2 is necessary for invadopodia formation and invasion
Previous studies have demonstrated that the highly metastatic rat mammary adenocarcinoma (MTLn3) cells form invadopodia when plated on fibronectin (FN)- or gelatin-coated dishes and that the formation of these structures correlates with their metastatic potential (Yamaguchi et al., 2005). Calpains have been implicated in cancer cell migration and invasion (Mamoune et al., 2003; Libertini et al., 2005; Carragher et al., 2006). To determine if invadopodia formation is affected by calpain 2 expression, calpain 2–deficient MTLn3 cell lines were generated using RNA interference (Franco et al., 2004a). Using two different targeting vectors, calpain 2 expression levels were reduced to ∼30% of control MTLn3 cells, and invadopodia formation was assayed in control and calpain 2–deficient MTLn3 cells plated on FN or FN-coated gelatin substrates (Fig. 1 A). Interestingly, we observed a significant reduction in the number of invadopodia formed in calpain 2–deficient MTLn3 cells on both FN and FN-coated gelatin (Fig. 1 A). To confirm that the actin and cortactin-containing structures represent membrane projections on the ventral cell surface, confocal imaging was performed. Accordingly, the formation of invadopodia on the ventral cell surface was impaired in the calpain 2–deficient cell lines as compared with control cells by confocal imaging (Fig. 1 B). Calpain inhibition with cell-permeable calpain inhibitors also impaired invadopodia formation (unpublished data), which suggests that calpain protease activity is necessary for invadopodia formation.
Figure 1.

Calpain is necessary for invadopodia formation. (A) Cell lysates from MTLn3 cells stably expressing control or calpain 2 siRNA were analyzed by Western blotting and probed for calpain 2 and actin as a loading control. MTLn3 cells expressing control or calpain 2si (targets A and B) were cultured on FN-coated glass coverslips and stained with anti-cortactin antibody (green) and rhodamine phalloidin (red). Quantification of cortactin and actin containing invadopodia is expressed as the mean number of invadopodia per cell for FN-coated and gelatin/FN-coated coverslips. Data are mean ± SEM of three independent experiments. *, P < 0.05 compared with control cells. Bar, 10 μm. (B) Confocal images demonstrate actin (red) and cortactin (green) staining at protrusive structures on the ventral cell surface of control or calpain 2 si cells.
To determine if calpain is required for matrix degradation, calpain-deficient cells were plated on Oregon green 488–labeled gelatin-coated coverslips. The cortactin-containing structures generally associated with sites of matrix degradation in control cells, which suggests that the majority of cortactin-containing dotlike structures in MTLn3 cells represent invadopodia (Fig. 2 A). The formation of invadopodia was significantly impaired in the calpain 2–deficient cells, although the few structures that formed in calpain-deficient cells retained their matrix degrading function (Fig. 2 A and not depicted). To further assess the invasive potential of calpain 2–deficient breast cancer cells, both calpain 2–deficient cell lines were plated on Matrigel-coated membranes and assayed for their ability to invade through the membrane. Both calpain 2–deficient cell lines demonstrated a significant reduction (approximately twofold) in invasiveness as compared with control cells (Fig. 2 B). Together, these findings suggest that calpain 2 is important for both the formation of invadopodia and invasive capacity of breast cancer cells.
Figure 2.

Calpain 2 is required for ECM degradation and invasion. (A) MTLn3 cells expressing control or calpain 2 siRNA were cultured on Oregon green 488 gelatin–coated coverslips and stained with anti-cortactin antibody. Arrows indicate areas of degradation that colocalize with cortactin containing invadopodia. Quantification of the percentage of cells containing any ECM-degrading invadopodia is shown. Data are mean ± SEM of three independent experiments. *, P < 0.02 compared with control cells. (B) MTLn3 cells expressing control or calpain 2si were plated on Matrigel-coated membranes and assayed for their ability to invade through the membrane. Invasion is shown relative to control cells. Data are mean ± SEM of three independent experiments. *, P < 0.04 compared with control cells. Bars: (A) 10 μm; (B) 1mm.
Time-lapse analysis of invadopodia dynamics in calpain 2–deficient MTLn3 cells
Previous studies have implicated calpain 2 in the turnover of focal adhesions in fibroblast-like cells (Bhatt and Huttenlocher, 2003; Franco et al., 2004b) and podosomes in osteoclasts (Marzia et al., 2006) and dendritic cells (Calle et al., 2006). To determine the effect of calpain 2 on invadopodia dynamics in MTLn3 cells, time-lapse microscopy was performed using control and calpain 2–deficient MTLn3 cells that express the invadopodia markers GFP-cortactin or GFP-actin. Calpain 2–deficient cells formed less GFP-cortactin or GFP-actin containing invadopodia than control cells despite similar efficiency of transgene expression (Figs. 3 and S1, available at http://www.jcb.org/cgi/content/full/jcb.200708048/DC1). Although the number of invadopodia was reduced, there were sufficient numbers of invadopodia in calpain 2–deficient cell lines to assay their dynamics. Live fluorescence imaging of GFP-actin or GFP-cortactin in these cells revealed the persistence of actin and cortactin in invadopodia for extended durations in calpain 2–deficient cells as compared with control cells with cells plated on FN (Fig. 3, A and B; and Videos 1–4) or FN-coated gelatin (not depicted). Plots of GFP-actin or GFP-cortactin fluorescence intensity as a function of time were used to calculate rate constants for invadopodia assembly and disassembly (Fig. 3, A and B). Control cells formed invadopodia with dynamics that were similar to those in previously published studies (Buccione et al., 2004; Yamaguchi et al., 2005). In contrast, we found that the mean rates of assembly and disassembly of invadopodia were decreased in calpain 2–deficient cells (Fig. 3, A and B). For the purposes of our study, we quantified the dynamics of short-lived precursor invadopodia, although similar calpain effects were observed with long-lived invadopodia (unpublished data). Interestingly, our findings suggest a novel role for calpain 2 in regulating assembly of invadopodia with a greater than twofold reduction in assembly rates of invadopodia in calpain 2–deficient cells (Fig. 3). Together, these findings indicate that calpain 2 is required for both the efficient assembly and disassembly of invadopodia.
Figure 3.

Invadopodia assembly and disassembly rates are reduced in calpain 2–deficient MTLn3 cells. (A) GFP-actin or (B) GFP-cortactin were transiently transfected into MTLn3 control and calpain 2 siRNA lines. Cells were plated on FN-coated glass-bottomed dishes and analyzed by time-lapse fluorescence microscopy. The time-lapse montage of calpain-deficient cells expressing GFP-actin (A) or GFP-cortactin (B) demonstrates that GFP-actin or GFP-cortactin persist at invadopodia for longer durations in calpain-deficient MTLn3 cells. Arrows indicate representative invadopodia. Duration measurements and assembly and disassembly rates are shown. Data show means ± SEM from four independent experiments. Asterisks indicate statistical significance compared with control based on a t test (*, P < 0.03; **, P < 0.05). See Videos 1–4 (available at http://www.jcb.org/cgi/content/full/jcb.200708048/DC1). Bars, 10 μm.
Expression of a calpain 2–resistant cortactin impairs invadopodia disassembly
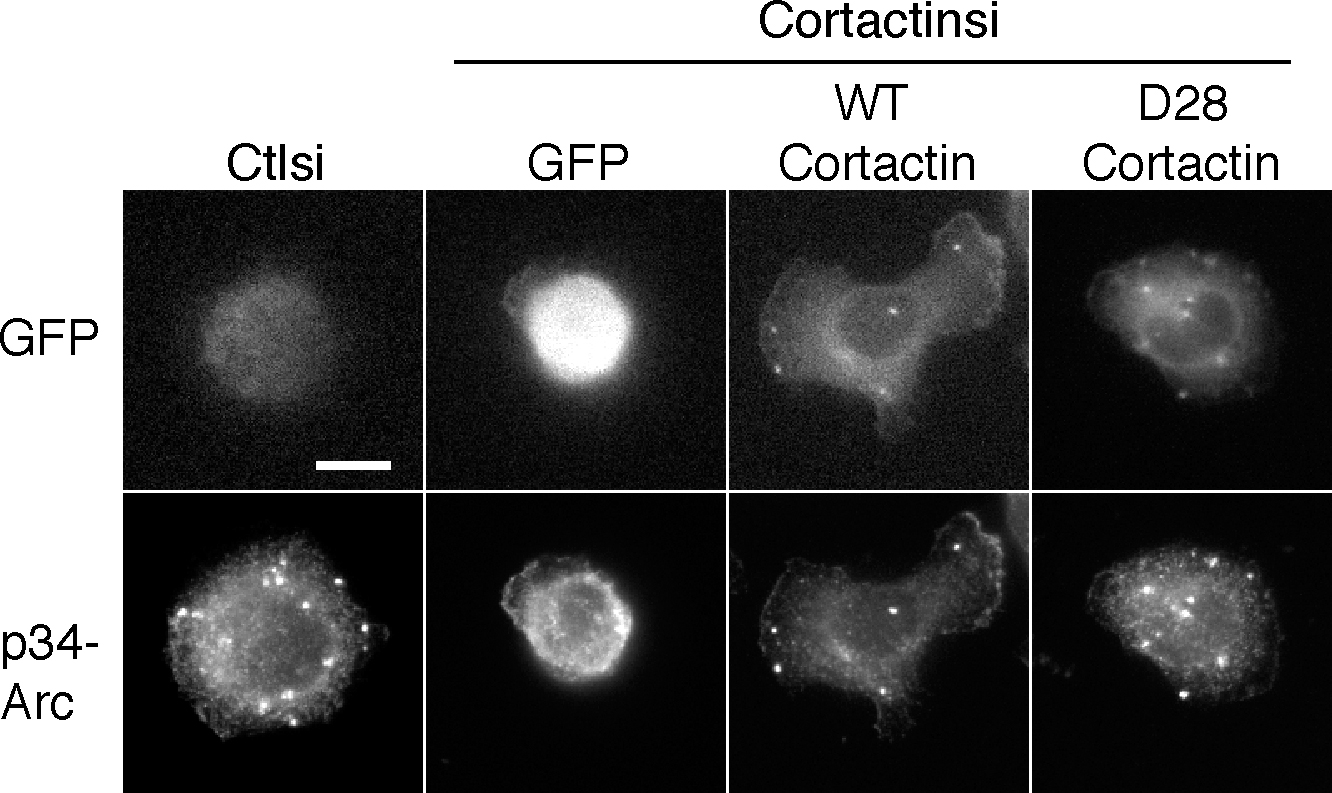
Previous studies have implicated direct proteolysis of cortactin by calpain 2 in the regulation of membrane protrusion dynamics (Perrin et al., 2006). To determine if the direct proteolysis of cortactin modulates the formation of invadopodia, we used a mutant form of cortactin, GFP–cortactin D28, which is resistant to calpain-mediated proteolysis (Perrin et al., 2006). Cortactin-deficient MTLn3 cells were generated using RNA interference with two different targeting vectors that reduced cortactin expression to ∼20% of control cells (Fig. 4). In accordance with previous studies (Clark et al., 2007; Webb et al., 2007), invadopodia formation was impaired in cortactin-deficient MTLn3 cells that express cortactin-specific siRNA targets as compared with control siRNA (Fig. 4 A). Both wild-type and calpain-resistant GFP–cortactin D28 rescued invadopodia formation in cortactin-deficient MTLn3 cells, indicating that calpain-mediated proteolysis of cortactin is not necessary for the formation of invadopodia (Fig. 4 B). This rescue was confirmed by staining for both actin and Arp 2/3 invadopodia markers (Figs. 4 B and S2, available at http://www.jcb.org/cgi/content/full/jcb.200708048/DC1). These findings suggest that calpain 2 regulates invadopodia formation independent of its effects on cortactin proteolysis.
Figure 4.

Calpain 2–mediated proteolysis of cortactin is not necessary for invadopodia formation but mediates disassembly of cortactin from invadopodia. (A) Cell lysates from MTLn3 cells stably expressing control or cortactin siRNA were analyzed by Western blotting and probed for cortactin and ERK as a loading control. MTLn3 cells expressing control or cortactin siRNA were cultured on gelatin/FN-coated coverslips and stained with anti–p34-Arc antibody and rhodamine phalloidin. Quantification of invadopodia is expressed as the mean number of invadopodia per cell. *, P < 0.05 compared with control cells. (B) Wild-type (WT) or calpain-resistant (D28) GFP-cortactin was transiently transfected into MTLn3 control and cortactin siRNA lines, cultured on gelatin/FN-coated coverslips, and stained with rhodamine phalloidin. Quantification of invadopodia is expressed as the mean number of invadopodia per cell. *, P < 0.05 compared with control cells. (C) GFP-cortactin WT or GFP-cortactin D28 expressing cells were plated on FN-coated glass-bottomed dishes and analyzed by time-lapse microscopy. Time-lapse montage of cortactin-deficient MTLn3 cells expressing GFP-cortactin WT or GFP-cortactin D28 demonstrates that GFP-cortactin D28 persists at invadopodia for longer durations than GFP-cortactin WT. Duration measurements and assembly and disassembly rates are shown. Arrows indicate representative invadopodia. Asterisk indicates statistical significance based on t test (P < 0.01). See Videos 5 and 6 (available at http://www.jcb.org/cgi/content/full/jcb.200708048/DC1). Data are means ± SEM from three independent experiments. Bars, 10 μm.
To determine if cortactin proteolysis regulates its dynamics at invadopodia, live fluorescence imaging was performed using cortactin-deficient MTLn3 cells that express either GFP-cortactin or GFP–cortactin D28. Live imaging revealed that calpain-resistant cortactin, GFP–cortactin D28, persisted at invadopodia approximately twice as long as wild-type GFP-cortactin (Fig. 4 C and Videos 5 and 6, available at http://www.jcb.org/cgi/content/full/jcb.200708048/DC1). We measured assembly and disassembly rates of wild-type GFP-cortactin and GFP–cortactin D28 at invadopodia and found that although assembly rates were slightly decreased, the rate of invadopodia disassembly was reduced approximately twofold with GFP–cortactin D28 as compared with wild-type GFP-cortactin (Fig. 4 C). These findings suggest that cortactin proteolysis is necessary for its efficient disassembly from invadopodia. Furthermore, these results suggest, in a manner analogous to the role of calpain proteolysis of talin in focal adhesion disassembly (Franco et al., 2004b), that calpain proteolysis of cortactin may be an important mechanism by which invadopodia are disassembled.
Calpain 2 functions both upstream and downstream of Src to regulate invadopodia
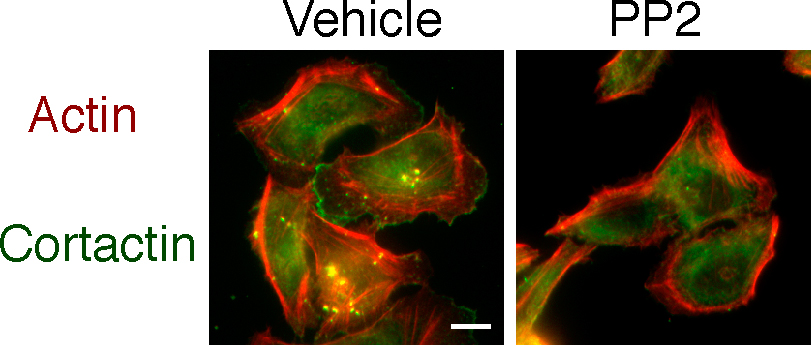
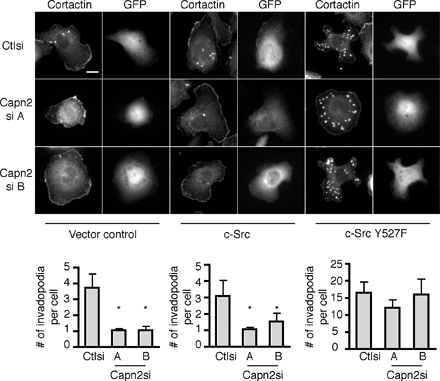
Previous studies indicate that Src tyrosine kinases function at an early step in the formation of podosomes and invadopodia. Accordingly, we found that Src activity was necessary for invadopodia formation in MTLn3 cells because the Src inhibitor PP2 impaired invadopodia formation (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200708048/DC1). To address whether calpain functions upstream or downstream of Src tyrosine kinases, we determined if calpain 2 was required for invadopodia formation in cells that express constitutively active v-Src. In accordance with previous publications, there was a substantial increase in the number of invadopodia formed in control MTLn3 cells transformed with v-Src (Fig. 5 A). Surprisingly, there was no significant difference in invadopodia numbers in v-Src transformed calpain 2–deficient MTLn3 cells compared with v-Src transformed control cells, indicating that calpain 2 is not required for invadopodia formation downstream of v-Src (Fig. 5 A). Expression of a constitutively active c-Src Y527F but not wild-type c-Src also rescued invadopodia formation (Fig. S4). Together, these findings suggest that calpain 2 functions upstream of c-Src kinase to regulate invadopodia formation in MTLn3 breast cancer cells but is not necessary for invadopodia formation in the context of v-Src transformed cells.
Figure 5.

Calpain 2 regulates invadopodia dynamics downstream of transforming v-Src kinase in MTLn3 breast cancer cells. (A) Control and calpain 2 knockdown MTLn3 cells expressing v-Src kinase were cultured on FN-coated glass coverslips and stained with anti-cortactin antibody and rhodamine phalloidin. Quantification of cortactin and actin containing invadopodia is expressed as the mean number of invadopodia per cell. Data shown are means ± SEM of three independent experiments. (B) GFP-cortactin was transfected into v-Src transformed MTLn3 control or calpain 2 siRNA lines. Cells were plated on FN-coated glass-bottomed dishes and analyzed by time-lapse fluorescence microscopy. Fluorescent images of v-Src transformed control or calpain 2–deficient cells demonstrate that GFP-cortactin persists at invadopodia for longer durations in the absence of calpain 2. Arrows indicate representative GFP-cortactin containing invadopodia. Duration measurements and assembly and disassembly rates ± SEM are shown. Asterisks indicate statistical significance compared with control cells based on t test (P < 0.05). See Videos 7 and 8 (available at http://www.jcb.org/cgi/content/full/jcb.200708048/DC1). Bars, 10 μm.
To further elucidate how calpain 2 affects invadopodia dynamics in v-Src transformed MTLn3 cells, time-lapse microscopy was performed using control and calpain 2–deficient v-Src transformed MTLn3 cells. Live fluorescent imaging of GFP-cortactin in these cells demonstrated enhanced invadopodia assembly in v-Src transformed cells in agreement with previous studies (Tarone et al., 1985; Tatin et al., 2006). However, the duration of invadopodia was enhanced approximately twofold in calpain 2–deficient cells transformed with v-Src, which suggests that, although calpain 2 is not necessary for invadopodia formation, it modulates invadopodia turnover in v-Src transformed cells. Accordingly, invadopodia assembly rates in v-Src transformed control and calpain 2–deficient cells were found to be approximately equal (Fig. 5 B). However, disassembly rates of invadopodia were reduced approximately twofold in calpain 2–deficient as compared with control v-Src transformed cells. Furthermore, our findings implicate cortactin proteolysis downstream of v-Src in invadopodia disassembly (Fig. S5 and not depicted). Collectively, these data suggest that calpain 2 functions downstream of Src to regulate invadopodia dynamics and disassembly at least in part through cortactin proteolysis. Furthermore, our findings also implicate a novel role for calpain 2 in invadopodia formation upstream of c-Src.
Calpain and PTP1B modulate c-Src activity
To determine if calpain 2 regulates endogenous c-Src kinase activity in MTLn3 cells, c-Src expression and activity were analyzed in calpain 2–deficient cells. There was no difference in expression of endogenous c-Src in control and calpain 2–deficient MTLn3 cells (Fig. 6 A). However, adhesion-induced c-Src activity was decreased by approximately twofold in calpain 2–deficient cell lines compared with control cells (Fig. 6 A). These findings support the hypothesis that calpain 2 functions upstream of c-Src to regulate Src activity and thereby invadopodia formation in MTLn3 cells.
Figure 6.

Calpain 2 and PTP1B regulate Src kinase activity and phosphorylation on tyrosine 529 in MTLn3 breast cancer cells. (A) Lysates from MTLn3 cells expressing control or calpain 2 siRNA were probed for total Src expression. Immunoprecipitations were performed on lysates from MTLn3 cells stably expressing control or calpain 2 siRNA using Src-specific antibodies. Kinase activity assays were performed and the autoradiograph is shown. Quantification of Src activity relative to total Src in either control or calpain 2si lysates from three separate experiments ± SEM is shown. Asterisks indicate statistical significance compared with control based on t test (P < 0.01). (B) MTLn3 cells stably expressing control or calpain 2 siRNA were analyzed by immunoblotting and probed for total Src and phospho-Y529. As a positive control, wild-type MTLn3 cells were treated with 200 nM CII, a cell-permeable PTP1B inhibitor, or vehicle before lysis. Quantification of phospho-Y529 levels normalized to total Src ± SEM of three independent experiments is shown. Asterisks indicate statistical significance compared with control based on one-way ANOVA (P < 0.05). (C) Cell lysates from MTLn3 cells stably expressing control or PTP1B siRNA were analyzed by Western blotting and probed for calpain 2 and vinculin as a loading control. PTP1Bsi cell lysates were analyzed by immunoblot and probed for total Src and phospho-Y529. Quantification of phospho-Y529 levels normalized to total Src ± SEM of three independent experiments is shown. Asterisks indicate statistical significance compared with control based on one-way ANOVA (P < 0.05). (D) Src expression was analyzed by Western blotting in control and PTP1Bsi cells. Immunoprecipitations were performed as described in A on PTP1Bsi cells. Kinase activity assays were performed and the autoradiograph is shown. Quantification of Src activity relative to total Src in either control or PTP1Bsi lysate from three separate experiments is shown as the means ± SEM. The asterisk indicates statistical significance compared with control based on a t test (P < 0.05). Numbers to the left of gel blots indicate molecular mass standards in kD.
An attractive candidate to mediate calpain effects on Src activity is PTP1B, a known calpain substrate. Previous studies have reported that PTP1B activates endogenous c-Src in breast cancer cells by the dephosphorylation of its inhibitory tyrosine 529 (Bjorge et al., 2000). Accordingly, it has been reported that the cell-permeable, PTP1B inhibitor compound II (CII) enhances phosphorylation of tyrosine 529 and reduces c-Src activity (Liang et al., 2005). We also found that the PTP1B inhibitor CII increased phosphorylation at tyrosine 529 using a phospho-specific antibody that detects phosphorylation at tyrosine 529. The increase in phosphorylation at tyrosine 529 was only modest with an ∼1.4-fold increase compared with vehicle control-treated cells (Fig. 6 B). However, these modest increases in phosphorylation are consistent with what has previously been found in other cell types with PTP1B inhibition (Liang et al., 2005). To determine if calpain modulates phosphorylation at tyrosine 529, the calpain 2–deficient cell lines were tested. We found similar increases in phosphorylation at this inhibitory tyrosine with a 1.4–1.5-fold increase in phosphorylation, which suggests that calpain 2 may modulate Src activity through the regulation of PTP1B activity (Fig. 6 B).
Previous studies have demonstrated that PTP1B regulates c-Src activity in breast cancer cells (Bjorge et al., 2000). To determine if PTP1B also regulates the activity of endogenous c-Src in MTLn3 cells, PTP1B-deficient MTLn3 cell lines were generated using RNA interference (Franco et al., 2004a). We had difficulty generating stable cell lines that were deficient in PTP1B but generated partial knockdown lines that had PTP1B levels reduced by 40–50% (Fig. 6 C). PTP1B-deficient MTLn3 cells displayed enhanced phosphorylation at tyrosine 529 (Fig. 6 C) and reduced activity of c-Src as compared with control cells (Fig. 6 D). Collectively, our findings indicate that both calpain 2 and PTP1B modulate Src phosphorylation at tyrosine 529 and regulate the activity of endogenous c-Src.
Calpain 2–mediated proteolysis of PTP1B results in enhanced phosphatase activity
Previous studies have reported that calpains cleave PTP1B in vivo (Frangioni et al., 1993; Ezumi et al., 1995) and enhance its phosphatase activity (Frangioni et al., 1993). The putative calpain proteolytic site involves truncation of ∼8 kD from the C terminus of PTP1B between residues 360 and 381 (Fig. 7 A). To examine the effects of calpain 2–mediated proteolysis on PTP1B activity, we generated full-length His-tagged PTP1B and His-tagged C-terminal truncations at amino acids 360 and 381 (Fig. 7 A). Purified pools were assayed for phosphatase activity using the widely used phosphatase substrate paranitrophenyl phosphate (pNPP) as described previously (Peters et al., 2003). In agreement with previously published reports, the His-tagged PTP1B truncations demonstrated enhanced phosphatase activity compared with full-length PTP1B in vitro (Fig. 7 A; Frangioni et al., 1993). To further characterize the effects of calpain proteolysis on PTP1B activity in vitro, both the full-length PTP1B and the truncation mutants were treated with purified calpain 2 and assayed for phosphatase activity. Calpain 2 enhanced the activity of full-length PTP1B by ∼1.7-fold but had no effect on the activity of the truncation mutants, which further supports a role for calpain proteolysis in the regulation of PTP1B activity (Fig. 7 B). This enhancement in PTP1B activity required the presence of calcium and was blocked by the calpain inhibitor ALLM (Fig. 7 B).
Figure 7.

Calpain 2 proteolysis of PTP1B results in enhanced PTP1B activity. (A) Schematic of full-length and truncated His-tagged PTP1B constructs. The arrow indicates the approximate site of calpain 2 cleavage. Equal amounts of purified PTP1B constructs were assayed for phosphatase activity against pNPP as described in Materials and methods. Activity of truncated PTP1B constructs is expressed relative to the full-length protein. Activity data are shown as the means ± SEM from three separate experiments. Asterisks indicate statistical significance compared with full-length PTP1B activity based on a t test (P < 0.0001). (B) Purified full-length (FL) or PTP1B truncated at amino acid 381 were incubated in the presence or absence of purified calpain 2 and assayed for phosphatase activity. The asterisk indicates statistical significance compared with untreated PTP1B activity based on a t test (P < 0.001). Error bars represent the SEM of at least three different determinations.
Truncated PTP1B rescues invadopodia formation in calpain 2–deficient cells
To determine if PTP1B is cleaved by calpain 2 in vivo, we exogenously expressed a tagged form of PTP1B in control or calpain 2–deficient HEK-293 cells. Human embryonic kidney (HEK) cells were used to obtain high levels of exogenously expressed PTP1B to facilitate observation of calpain 2–mediated cleavage. We observed substantial proteolysis of exogenously expressed GFP-PTP1B-HA in control but not calpain 2–deficient HEK cells (Fig. 8 A). Cell lysates isolated from control HEK cells treated with ionomycin generated a proteolytic fragment ∼8 kD smaller than the full-length, tagged PTP1B (Fig. 8 A). Similarly, we observed modest proteolysis of exogenously expressed FLAG-PTP1B in control MTLn3 cells and decreased PTP1B proteolysis in both calpain 2–deficient cell lines (Fig. 8 B). To determine if calpain 2 regulates PTP1B activity in MTLn3 cells, FLAG-PTP1B was immunoprecipitated from control or calpain-deficient MTLn3 cells treated with ionomycin and assayed for phosphatase activity. Despite similar expression levels, FLAG-PTP1B activity was significantly decreased in both calpain 2 knockdown cell lines compared with the control cells (Fig. 8 B). This is the first study that indicates that calpain 2 is the likely isoform required for this proteolytic modification in vivo. These findings indicate that PTP1B is cleaved by calpain 2 in vivo, and this proteolytic modification results in the generation of an activated form of PTP1B. This is especially intriguing in light of the role of PTP1B in activating c-Src in breast cancer cells by dephosphorylating the inhibitory tyrosine 529.
Figure 8.

Calpain 2 regulates PTP1B activity in vivo. (A) HEK cells transfected with GFP-PTP1B-HA were analyzed by Western blot after treatment with vehicle or ionomycin to stimulate calpain activity and blotted for GFP, calpain 2, talin, and actin as a loading control. Blots shown are representative of three independent experiments. The arrow indicates a calpain 2–dependent PTP1B cleavage product. (B) FLAG-PTP1B was transiently transfected into MTLn3 control and calpain 2 siRNA lines. Immunoprecipitations were performed on cells expressing FLAG-PTP1B after treatment with ionomycin. Shown are the relative amounts of FLAG-PTP1B that were pulled down. The arrow with an asterisk indicates a calpain 2–dependent PTP1B cleavage product. Blots shown are representative of three independent experiments. PTP1B cleavage was quantified for control and calpain 2 siRNA lines as a percentage of total FLAG-PTP1B expression and is shown relative to control cells. (left) Asterisks indicate statistical significance compared with control cells based on one-way ANOVA (P < 0.05). Immunoprecipitations were assayed for phosphatase activity against pNPP as described in Materials and methods. Activity of immunoprecipitated PTP1B constructs in MTLn3 calpain 2 siRNA lines is expressed relative to the control cell line. Activity data are shown as the means ± SEM from three separate experiments. (right) Asterisks indicate statistical significance based on a t test (P < 0.002). (C) MTLn3 cells were transfected with GFP-PTP1B truncated at amino acid 360 or GFP alone. Transfected cells were plated on FN-coated glass coverslips and stained with anti-cortactin antibody. Invadopodia formation was quantified by determining the mean number of invadopodia per cell. Data represent means ± SEM from three independent experiments. *, P < 0.05 compared with GFP-expressing control cells. Bar, 10 μm.
Our findings indicate that calpain 2–mediated proteolysis of PTP1B results in an approximate 8-kD C-terminal truncation and enhanced phosphatase activity. To determine if this truncated form of PTP1B can rescue invadopodia formation in calpain 2–deficient MTLn3 cells, GFP control or the GFP-tagged PTP1B 360 truncation mutant were expressed in control or calpain 2–deficient cells and assayed for invadopodia formation (Fig. 8 C). In accordance with our reported findings (Fig. 1), calpain 2–deficient cells that expressed control GFP displayed impaired invadopodia formation as compared with cells that express control siRNA. In contrast, calpain 2–deficient cells that expressed GFP-PTP1B 360 showed a substantial increase in the number of invadopodia as compared with control cells (Fig. 8 C). Together, these findings indicate that a truncated PTP1B, based on the predicted calpain proteolytic fragment, rescues invadopodia formation in calpain 2–deficient MTLn3 cells.
PTP1B regulates invadopodia formation and breast cancer invasion
Recent studies implicate a critical role for PTP1B in breast cancer progression in mouse models (Bentires-Alj and Neel, 2007; Julien et al., 2007). To determine if PTP1B regulates breast cancer invasion, we tested the effects of PTP1B inhibition using a PTP1B inhibitor or PTP1B-deficient cell lines on the formation of invadopodia and invasion across Matrigel-coated membranes. We found that invadopodia formation was impaired in MTLn3 cells treated with the cell-permeable PTP1B inhibitor CII (Fig. 9 A). In accordance with our findings using CII, we found that invadopodia formation was also reduced approximately twofold in the PTP1B-deficient cell lines (Fig. 9 B). To determine if PTP1B is required for invasion, PTP1B-deficient cell lines were plated on Matrigel-coated membranes and assayed for their ability to migrate through the membrane. Despite only a 50% knockdown, the cell line most deficient in PTP1B exhibited a twofold inhibition of cell invasion compared with control cells (Fig. 9 C). Furthermore, treatment with the PTP1B inhibitor CII also impaired invasion of MTLn3 breast cancer cells by approximately twofold (Fig. 9 C). Together, the findings implicate PTP1B as a novel component of a signaling pathway that regulates invadopodia formation and invasion in breast cancer cells.
Figure 9.

PTP1B is necessary for efficient invadopodia formation and invasion. (A) MTLn3 cells cultured on FN-coated glass coverslips were stained with anti-cortactin antibody and rhodamine phalloidin. Cells were cultured in the presence of vehicle or the cell-permeable PTP1B inhibitor CII. Quantification of invadopodia is expressed as the mean number of invadopodia per cell. Data are mean ± SEM of three independent experiments. *, P < 0.01 compared with control cells. (B) MTLn3 cells expressing control or PTP1Bsi were cultured on FN-coated glass coverslips and stained with anti-cortactin antibody and rhodamine phalloidin. Quantification of invadopodia is expressed as the mean number of invadopodia per cell. Data are mean ± SEM of three independent experiments. *, P < 0.01 compared with control cells. (C, left) MTLn3 cells were cultured in the presence of vehicle or CII and assayed for their ability to invade through the membrane. Invasion is shown relative to control cells. Data are mean ± SEM of three independent experiments. *, P < 0.05. (right) MTLn3 cells expressing control or PTP1B siRNA were plated on Matrigel-coated membranes and assayed for their ability to invade through the membrane. Invasion is shown relative to control cells. Data are mean ± SEM of three independent experiments. *, P < 0.02 compared with control cells. Bars: (A and B) 10 μm; (C) 1 mm.
Discussion
Invasive cancer cells form dynamic protrusive structures known as invadopodia that are associated with ECM degradation and increased tumor cell invasiveness. Here, we describe a novel function for the intracellular, calcium-dependent protease calpain 2 in the dynamic regulation of invadopodia both upstream and downstream of Src tyrosine kinases in metastatic breast cancer cells. Although calpains have been implicated in podosome turnover (Calle et al., 2006; Marzia et al., 2006), this is, to our knowledge, the first finding that calpain 2 is necessary for the formation of invadopodia. We also provide evidence to suggest that calpain 2–mediated proteolysis and activation of PTP1B is an important mechanism by which calpain 2 regulates c-Src activity and invadopodia formation in breast cancer cells. Finally, our findings indicate that PTP1B is a novel component of the regulatory pathway that modulates invadopodia formation through the regulation of Src activity. Collectively, our findings identify a novel signaling pathway involving calpain 2, PTP1B, and Src that regulates invadopodia formation and invasion of metastatic breast cancer cells.
Both invadopodia and podosomes are dynamic, actin-containing structures that share similar components but have been reported to have distinct kinetics and dynamics. Time-lapse analysis of invadopodia formation has shown that invadopodia, in contrast to podosomes, are formed de novo at the periphery of the cell with lifetimes that are generally longer than podosomes, ranging from minutes to hours (Buccione et al., 2004; Yamaguchi et al., 2005; Gimona and Buccione, 2006). We performed time-lapse microscopy of MTLn3 cells to characterize the dynamics of “short-lived” invadopodia or podosome-like structures in MTLn3 breast cancer cells; however, calpain effects on assembly and disassembly of both “short-lived” and “long-lived” structures were found to be similar (unpublished data). These structures displayed dynamic assembly and disassembly as well as lateral mobility with lifetimes that averaged ∼10 min. Calpain inhibition resulted in extended durations of invadopodia (mean ∼17 min) and impaired both the dynamic assembly and disassembly of these structures. It has been suggested that precursor invadopodia are more motile on the ventral cell membrane and that stabilization of invadopodia or decreased mobility is associated with enhanced matrix degradation (Gimona and Buccione, 2006); however, no significant differences in invadopodia mobility were observed between control and calpain 2–deficient cells (unpublished data). This suggests that although calpain 2–deficient cells were significantly impaired in their ability to degrade matrix and invade through Matrigel, this defect is likely a result of the inability of calpain 2–deficient cells to efficiently form invadopodia and not a defect in stabilization of precursor structures.
Previous studies have established a role for calpain proteases in the turnover of integrin-mediated adhesions in migrating cells (Huttenlocher et al., 1997; Bhatt and Huttenlocher, 2003) with the specific involvement of talin proteolysis in focal adhesion disassembly (Franco et al., 2004b). Recent progress has also implicated calpain protease activity in the regulation of podosome disassembly in dendritic cells (Calle et al., 2006) and osteoclasts (Marzia et al., 2006). However, in these studies, the involvement of specific substrates was not demonstrated, although several calpain substrates, including cortactin, Wiskott-Aldrich syndrome protein, and Pyk2, have been shown to localize to podosomes (Calle et al., 2006; Linder, 2007). In this study, we show that calpain 2 is involved in the disassembly of invadopodia at least in part through the targeted proteolysis of cortactin. Although expression of cortactin is required for invadopodia assembly, calpain-resistant cortactin was able to rescue invadopodia formation in cortactin-deficient MTLn3 cells, which suggests that calpain proteolysis of cortactin is not essential for the formation of invadopodia. However, expression of the calpain-resistant cortactin impaired the disassembly of cortactin from invadopodia, indicating that cortactin proteolysis is likely a mechanism by which invadopodia disassemble. Collectively, our findings suggest that calpain 2 functions downstream of Src tyrosine kinases to regulate invadopodia disassembly through the targeted proteolysis of specific effector substrates such as cortactin.
Substantial evidence has implicated calpain 2 in the regulation of adhesion disassembly downstream of the transforming oncogene v-Src. Studies by Carragher et al. (2002) demonstrated that v-Src transformation induces the calpain-dependent proteolysis of specific substrates. In further studies, calpain 2 was demonstrated to be a critical effector downstream of v-Src in regulating adhesion disassembly, cell morphology, and cell cycle progression (Carragher et al., 2004). In agreement with these findings, invadopodia in v-Src transformed calpain 2–deficient MTLn3 cells had substantially reduced disassembly rates but not assembly rates compared with control cells. The importance of cortactin proteolysis downstream of transforming v-Src is further supported by the impaired disassembly of calpain-resistant cortactin in v-Src transformed cells (unpublished data). It will be interesting to determine if cortactin proteolysis is also critical for calpain effects on the disassembly of podosomes in other cell types.
In this work, we show that calpain 2 functions upstream of endogenous c-Src to positively regulate Src activity and invadopodia formation. Previous studies have shown that Src activity is necessary for invadopodia formation (Chen, 1989; Spinardi and Marchisio, 2006) and that its activity is modulated downstream of integrin-mediated adhesion and by the activity of growth factor signaling pathways such as EGF receptor signaling. EGF receptor activation stimulates invadopodia formation and invasion of MTLn3 cells (Yamaguchi et al., 2005) and has also been reported to activate calpain 2 through an extracellular signal-regulated kinase (ERK)-dependent pathway (Glading et al., 2002). We now provide direct evidence that calpain 2 may be a critical link between integrin and/or EGF receptor signaling and the activation of Src kinase activity in the formation of invadopodia. Calpain 2 depletion by siRNA inhibited the activation of endogenous c-Src activity by integrin-mediated adhesion and impaired invadopodia formation in breast cancer cells. Furthermore, expression of activated v-Src or c-Src rescued invadopodia formation in calpain 2–deficient cells, which suggests that calpain 2 functions upstream of Src to regulate invadopodia formation.
We have identified a potential mechanism for calpain 2 regulation of c-Src activity through the proteolysis of the ubiquitously expressed tyrosine phosphatase PTP1B. PTP1B has been shown to interact with integrin complexes (Arregui et al., 1998) and localizes to early cell matrix adhesion sites (Hernandez et al., 2006). PTP1B can promote c-Src activation by the direct dephosphorylation of the inhibitory phosphate tyrosine 529 (Bjorge et al., 2000; Liang et al., 2005). In contrast to c-Src, for which calpain 2–dependent proteolysis was not detected in vivo, PTP1B was proteolyzed in a calpain 2–dependent manner, resulting in the generation of a stable PTP1B fragment that has enhanced phosphatase activity (Frangioni et al., 1993). Accordingly, we found that calpain 2–deficient cells had increased phosphorylation at the c-Src inhibitory tyrosine 529 that is regulated by PTP1B, and that a PTP1B fragment corresponding to the calpain proteolytic fragment rescued invadopodia formation in calpain 2–deficient cells. It is interesting that we detected substantially enhanced phosphatase activity with the PTP1B truncations, which are similar in size to the calpain 2 cleavage fragment, and that this activity was not further modified by the activity of calpain 2. These findings suggest that calpain 2 functions as a positive regulator of PTP1B activity. This is in contrast to a recent study that indicates that calpain 1 is a negative regulator of PTP1B function in platelets (Kuchay et al., 2007). Our findings also indicate that PTP1B is a positive regulator of c-Src in breast cancer cells. We show that reduction of PTP1B expression in MTLn3 breast cancer cells enhanced phosphorylation at the inhibitory tyrosine 529 and reduced endogenous c-Src activity. Accordingly, PTP1B inhibition impaired invadopodia formation in breast cancer cells, identifying PTP1B as novel component of the signaling machinery involved in regulating invadopodia formation. Collectively, these findings implicate a novel pathway through calpain 2, PTP1B, and c-Src that regulates invadopodia formation in breast cancer cells.
Both PTP1B and calpain 2 are overexpressed in breast tumors, with studies suggesting that >70% of human breast cancers overexpress PTP1B (Wiener et al., 1994). Recent evidence has also implicated PTP1B as a key factor involved in the acceleration of breast cancer progression in mouse models (Bentires-Alj and Neel, 2007; Julien et al., 2007; Tonks and Muthuswamy, 2007). For example, PTP1B depletion or inhibition impairs ErbB2-induced mammary tumorigenesis and the development of lung metastasis in mouse models (Julien et al., 2007). However, the mechanism by which PTP1B facilitates breast cancer progression has not been elucidated. Our findings implicate a role for PTP1B in the modulation of endogenous c-Src activity, providing an attractive mode of action because elevated c-Src activity has been detected in many breast cancer cell lines (Bjorge et al., 2000). Moreover, we provide evidence that PTP1B is a novel component of the signaling machinery involved in regulating invadopodia formation and breast cancer invasion. Together, these findings suggest that modulation of calpain 2/PTP1B function may provide a novel therapeutic target for breast cancer. The availability of cell-permeable calpain 2 and PTP1B inhibitors supports the utility of this approach.
In summary, our findings provide novel mechanistic insight into the functional roles of calpain 2/PTP1B/c-Src signaling in the formation of invadopodia in metastatic breast cancer cells. These findings support a critical role for regulated intracellular proteolysis by calpains functioning in concert with targeted phosphorylation/dephosphorylation events in the modulation of intracellular signaling pathways through Src. Our findings indicate that calpains play critical roles both upstream and downstream of Src tyrosine kinases by targeting distinct substrates that modulate the formation and turnover of invadopodia (Fig. 10). It is likely that calpain effects are context dependent because calpain 2 is not necessary for invadopodia formation in breast cancer cells transformed with v-Src. Furthermore, calpains target other substrates at invadopodia, including talin, that may also contribute to calpain effects on invadopodia dynamics. Future studies should shed additional light on the role of this pathway in regulating the temporal and spatial dynamics of invadopodia formation and maturation into matrix-degrading structures that mediate breast cancer cell invasion and metastasis.
Figure 10.

Model for calpain 2 involvement in invadopodia dynamics. Integrin engagement and/or EGF receptor activation enhances calpain activity. Calpain cleaves PTP1B, resulting in enhanced phosphatase activity. PTP1B removes the inhibitory phosphate from Y529 of Src, thereby activating Src kinase activity and initiating invadopodia formation. Calpain 2 also functions downstream of Src kinase activity by regulating the disassembly of cortactin from invadopodia.
Materials and methods
Reagents
FN was purified from human plasma by affinity chromatography as described previously (Ruoslahti et al., 1982). α-MEM, Ham's F12, Oregon green 488 gelatin conjugate, and rhodamine phalloidin were obtained from Invitrogen. ALLM and ALLN were obtained from EMD and used at a concentration of 50 μg/ml. Enolase and anti-FLAG M2 agarose beads were obtained from Sigma-Aldrich. The cell-permeable, potent, and selective PTP1B inhibitor CII was synthesized as described previously (Xie et al., 2003). Specific pharmacological properties for the cell-permeable inhibitor have been reported previously (Xie et al., 2003; Liang et al., 2005). Ni-NTA Superflow was obtained from QIAGEN. pNPP was obtained from Santa Cruz Biotechnology, Inc..
Cell culture and transfection
MTLn3 cells were a gift from J. Condeelis and J. Segall (Albert Einstein College of Medicine, New York, NY). Cells were cultured in α-MEM supplemented with 5% FBS and antibiotics as described previously (Segall et al., 1996). HEK cells were obtained from the American Type Culture Collection and cultured as recommended. Transfections of MTLn3 cells were performed with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Cells, plated at 105 per well in 6-well plates, were transfected with 2 μg DNA and 4 μl Lipofectamine 2000 for 45 min to 1 h. Cells were cultured for 48 h before being analyzed. Retroviral transfection was performed as described previously (Franco et al., 2004a). Transfections of HEK cells were performed using calcium phosphate precipitation. In brief, 10-cm plates containing HEK cells at ∼80% confluency were transfected with 12 μg of DNA for 6 h. Cells were cultured for 48 h before being analyzed.
Antibodies and constructs
Anti–calpain 1 and 2 antibodies were obtained from Triple Point Biologics, Inc.; anti–p34-Arc (subunit of Arp 2/3), anti-Src antibody (clone GD11), and anti–phospho-tyrosine antibody (4G10) were obtained from Millipore; anti–phospho-Src (Y529), anti-ERK, and anti-p38 antibodies were obtained from Invitrogen; anti-PTP1B was obtained from R&D Systems; anti-cortactin (clone 4F11) was a gift from A. Weaver (Vanderbilt University, Nashville, TN); and anti-Src (clone 327), anti-vinculin (clone h-VIN1), anti-talin (clone 8d4), anti-FLAG (clone M2), and anti-actin (clone AC-15) antibodies were obtained from Sigma-Aldrich. Alexa Fluor 680 goat anti–mouse IgG secondary antibody and anti-GFP antibody was obtained from Invitrogen. IRDye 800CW goat anti–rabbit IgG secondary antibody was obtained from Rockland Immunochemicals, Inc. Anti–mouse IgG (ChromaPure, whole molecule) was obtained from Jackson ImmunoResearch Laboratories. Rhodamine red-X goat anti–mouse IgG was obtained from Invitrogen.
Constructs and siRNA
The pSUPER.retro (Oligoengine) RNA interference system was used to achieve stable expression of siRNAs. Oligonucleotides targeted to calpain 2 or PTP1B mRNA as well as a nonsilencing control were synthesized by Integrated DNA Technologies, annealed, and cloned into the pSUPER.retro.puro vector according to manufacturer's instructions. Retroviral transfection was performed as described previously (Franco et al., 2004a). Wild-type MTLn3 cells were infected at 32°C for 6 h and allowed to recover in growth medium for 24 h before selection with 1 μg/ml puromycin for 4–5 d. Target sequences for calpain 2 in MTLn3 cells: control, 5′-TTCTCCGAACGTGTCACGT-3′; Capn2 si-A, 5′-AGGCCTATGCCAAGATCAA-3′; and Capn2 si-B, 5′-GAATGGCGATTTCTGCATC-3′. Target sequences for PTP1B in MTLn3 cells: PTP1B si-A, 5′-GCTGACACTGATCTCTGAA-3′; and PTP1Bsi-B, 5′-CAGGAGGAGCCTTGGTGTC-3′. Target sequences for human calpain 2 have been described previously (Su et al., 2006). Target sequences for cortactin: control, 5′-TTCTCCGAACGTGTCACGT-3′; cortactin si-A, 5′-CAGATGTGGAAAGCTTCTG-3′; and cortactin si-B, 5′-CGCTTCGAGAGAATGTCTT-3′. The FPGV v-Src construct was a gift from M. Frame (University of Glasgow, Glasgow, Scotland). The enhanced GFP (EGFP)-PTP1B-HA construct was a gift from C. Arregui (University of San Martin, Buenos Aires, Argentina). LCNX–c-Src was a gift from P. Keely (University of Wisconsin, Madison, WI) and a GFP tag was added to the N terminus, which was amplified by PCR with the following primers: 5′-ACTCAGATCTATGGGGAGCAGCAAGAGC-3′ and 5′-CTCAGAATTCCTATAGGTTCTCTCCAGG-3′. EGFP-actin was obtained from Clontech Laboratories, Inc. EGFP-cortactin and calpain-resistant EGFP-cortactin D28 have been described previously (Perrin et al., 2006). RcRSV-cSrcY527F was a gift from A. Reynolds (Vanderbilt University, Nashville, TN) and was excised with ApaI and EcoRI and subsequently religated into pCDNA 3.1(+) (Invitrogen). A Kozak methionine initiator sequence and a Flag tag were added to the N terminus of rat PTP1B, which was amplified by PCR with the following primers: 5′-CTCGGATCCGCCACCATGGACTACAAGGACGACGATGACAAGAAGCTTGGGATGGAAATGGAGAAGGAATTCGAG-3′ and 5′-CTCTAGACTCGAGCGGCCGCTCAGTGAAAACATACCCGGTAACAG-3′. The PCR product was digested with BamHI and NotI and subsequently ligated into the corresponding sites of pCDNA3.1(+) (Invitrogen).
Immunoblot analysis
Cells were plated in α-MEM on dishes coated with 10 μg/ml FN and incubated at 37°C under 5% CO2 for 3 h. Cells were lysed in modified RIPA buffer (20 mM Hepes, pH 7.2, 150 mM NaCl, 2 mM EDTA, 0.5% deoxycholate, 1% NP-40, 0.2 mM PMSF, 1 μg/ml pepstatin, 2 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 mM sodium orthovanadate) on ice and clarified by centrifugation. For phospho-Src analysis, cells were lysed in 50 mM Tris, pH 7.5, 1% Triton X-100, 5 mM EGTA, 150 mM NaCl, 10 mM sodium phosphate, 10 mM sodium fluoride, 5 mM iodoacetic acid, 1 mM benzamidine, 10 μg/ml aprotinin, and 10 μg/ml leupeptin on ice for 30 min and clarified by centrifugation; then, 10 mM DTT was added to the lysate after protein concentration determination (Liang et al., 2005). Protein concentrations were determined using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific) according to the manufacturer's instructions. Equal amounts of total protein were denatured in SDS sample buffer, run on 4–20% gradient SDS-polyacrylamide gels, and transferred to nitrocellulose. Western blots were imaged and quantified with an infrared imaging system (Odyssey; LI-COR Biosciences). For analysis of cortactin cleavage, v-Src transformed cells were lysed and analyzed as described previously (Perrin et al., 2006).
Immunofluorescence
Glass coverslips were acid-washed and coated with 10 μg/ml FN or a 0.2% gelatin/FN (10 μg/ml) mix as described previously (Artym et al., 2006). Cells were plated on coverslips in α-MEM culture medium and allowed to adhere for 3 h. The cell permeable PTP1B inhibitor CII was added to culture medium at a final concentration of 500 nM. The Src kinase inhibitor PP2 was added to culture medium at a final concentration of 2 μM. Cells were fixed in 3.7% formaldehyde for 10 min, permeabilized with 0.2% Triton X-100 for 10 min, and blocked in 5% goat serum. The cells were incubated with primary antibodies and rhodamine phalloidin for 30 min and then incubated with fluorophore-conjugated secondary antibodies for 30 min. Coverslips were imaged using a 60× 1.40 oil objective on an inverted microscope (1X-70; both from Olympus) Images were acquired with a cooled charge-coupled device (CCD) camera (CoolSNAP fx; Photometrics) and captured into MetaVue imaging software v6.2 (MDS Analytical Technologies). Error bars in all figures represent the SEM of at least three different determinations. 50–100 cells were counted in each determination. Cells were scored as positive for invadopodia formation if dotlike structures contained both cortactin and actin or actin and p34-Arc, and were quantified by counting the number of structures per cell.
Protein expression and purification
PCR primers (5′-AAACTCGAGATGGAGATCGAGAAG-3′ and 5′-GGGGGTACCTCAATGAAAACATAC-3′) containing XhoI and KpnI sites were used to generate the PTP1B sequence for insertion into the pTrcHis vector (Invitrogen). To generate PTP1B C-terminal truncations, stop codons were introduced at amino acid 360 and 381 using site-directed mutagenesis (Stratagene). Native purification of the fusion proteins was performed over Ni-NTA metal chelating sepharose using previously described methods (Cortesio and Jiang, 2006). Purity of PTP1B pools was analyzed by silver stain (Bio-Rad Laboratories) according to the manufacturer's instructions.
Phosphatase activity assay
Purified PTP1B activity was measured against pNPP using slow kinetics at an absorbance of 405 nm and a Multilabel HTS counter (Victor V; PerkinElmer). PTP1B (2 μg of purified pools) was assayed in 50 mM Tris, pH 7.5, and 10 mM DTT containing 10 mM pNPP. To determine the effects of calpain 2–mediated cleavage of PTP1B, 2 μg of purified PTP1B was incubated with or without 200 ng of purified calpain 2 (EMD) for 30 min at 37°C before activity assay. To verify the importance of calpain 2 activity for PTP1B cleavage, calcium was omitted from the cleavage reaction or 200 μg/ml ALLM was added before the 30-min incubation. As a control, 2 mM sodium orthovanadate was added to activity assay to verify measured activity was caused by a phosphatase. To determine the effects of in vivo PTP1B proteolysis on PTP1B activity, FLAG-PTP1B was immunoprecipitated from equal amounts of protein lysate and assayed in 50 mM Tris, pH 7.5, and 10 mM DTT containing 10 mM pNPP using slow kinetics at an absorbance of 405 nm. PTP1B activity was then normalized to total levels of FLAG-PTP1B as assessed by immunoblot.
Fluorescent gelatin degradation assay
Oregon green 488–conjugated gelatin-coated coverslips were prepared as described previously (Artym et al., 2006). In brief, cells were cultured on coated coverslips for 6–12 h and immunostained with anti-cortactin antibody. Coverslips were imaged as described in the Immunofluorescence section. Error bars in all figures represent the SEM of at least three separate experiments. At least 50 cells were counted in each experiment. Cells were scored as positive for invadopodia formation if at least one cortactin-containing dotlike structure colocalized with black spots of degradation.
Invasion assay
Cell invasion experiments were performed with 8-μm porous chambers coated with Matrigel (BD Bioscences) according to the manufacturer's instructions. 5% FBS was used as chemoattractants in the lower compartment. Cells were serum starved in culture media containing 1% FBS for 3–4 h before the assay allowed to invade through the Matrigel membrane for 24 h. The invasive cells underneath were fixed and stained. To determine the effect of PTP1B inhibition, cells were cultured in the presence of 500 nM CII for 3 h before and during the invasion assay.
Time-lapse fluorescent microscopy
Fluorescence imaging of live cells was performed using a 60× 1.40 oil objective on an inverted microscope (1X-70; Olympus) housed in a closed system to maintain temperature at 37°C. Glass-bottom dishes (35 mm) were coated with 10 μg/ml FN for 1 h at 37°C. For microscopy experiments, cells were plated in Ham's F12 with 5% fetal bovine serum and 20 mM Hepes, pH 7.2, and allowed to adhere for 2–3 h. Fluorescent images were collected using a CoolSNAP fx cooled CCD camera and captured into MetaVue v6.2 every minute for 1 h as described previously (Franco et al., 2004b).
Confocal microscopy
Confocal imaging was performed using a 60× 1.40 oil objective on a laser scanning confocal microscope (C1; Nikon). Images were acquired using a digital monochrome CCD camera system (CoolSNAP-ES; Photometrics) controlled by Metamorph software (MDS Analytical Technologies) and the resulting z stacks were deconvolved using Autodeblur/autovisualize software (MediaCybernetics).
Quantification of invadopodia dynamics
Time-lapse sequences from live fluorescent imaging were first subjected to high-pass filtration based on the “water” algorithm (Zamir et al., 1999) to remove diffuse background fluorescence. Next, the dynamics of fluorescently tagged cortactin or actin constructs were quantified as described previously (Franco et al., 2004b; Webb et al., 2004). Time-lapse microscopy was performed on MTLn3 calpain 2si (target B) and cortactin si (target A). For each rate constant, measurements were made on a total of 40–60 invadopodia in four to six separate cells from at least three separate experiments. Duration measurements were made for these same invadopodia by counting the amount of time lapsed between the first and last frames in which an individual invadopodium was observed. All measurements shown are the mean ± SEM.
v-Src transformation of MTLn3 control and calpain 2 knockdown cell lines
The FPGV vector containing v-Src kinase was transfected into retroviral packaging cells. Retroviral infection of MTLn3 cells was performed as described previously (Franco et al., 2004a). MTLn3 cells were infected at 32°C for 6 h and allowed to recover in growth medium for 24 h before selection with 100–250 μg/ml G418 (Invitrogen) for 7–10 d. Immunoblot analysis of Src protein levels and total phospho-tyrosine content was used to confirm v-Src transformation.
Immunoprecipitation and Src activity assay
For immunoprecipitations, MTLn3 cells were plated onto 10-cm dishes coated with 10 μg/ml FN. Cells were incubated for 3 h at 37°C in 5% CO2, washed one time with PBS, and lysed with ice-cold RIPA buffer. Lysates were cleared of insoluble material by centrifugation and 400 μg of cleared lysate was used for the immunoprecipitation step. Next, 1 μg of anti-Src (Millipore) and 1 μg of anti-Src (Sigma-Aldrich) antibodies or 2 μg of mouse IgG were added to the cleared lysates and incubated at 4°C with tilting for 2 h. 20 μl of protein G–Sepharose (GE Healthcare) was then added to immunoprecipitations and incubated at 4°C with tilting for 1 h. Samples were then washed three times with RIPA buffer and split into equal volumes for Western blot analysis or a kinase assay. For immunoblot analysis, immunoprecipitated Src was extracted from beads with Laemmli SDS sample buffer and boiling for 10 min. Samples were run on an 8% SDS-polyacrylamide gel and transferred to nitrocellulose. Western blots were imaged and quantified with an Odyssey infrared imaging system. Kinase assays were performed as described previously (Chang et al., 1998) and were terminated with Laemmli SDS sample buffer and boiling for 10 min. Reactions were then run on an 8% SDS-polyacrylamide gel, dried, and exposed to a phosphoimage screen for 15–20 h.
In vivo calpain 2 proteolysis of PTP1B
HEK or MTLn3 cells were transfected with EGFP-PTP1B-HA and FLAG-PTP1B, respectively. HEK cells were lysed 48 h later with 50 mM Tris, pH 7.6, 0.5 M NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100, and 0.5 mM MgCl2. MTLn3 cells were lysed 48 h later with 50 mM Hepes, pH 7.2, 0.25 M sucrose, 5 mM EDTA, and 1% Triton X-100. Calpain activity was stimulated before lysis using 1 μM ionomycin (Sigma-Aldrich) and 10 mM CaCl2. Calpain 2–dependent talin cleavage has been described previously (Franco et al., 2004b) and was used as a positive control.
Statistics
For statistical comparison, the two-tailed, paired t test or one-way analysis of variance (ANOVA) test were used, with p-values of <0.05 considered significant.
Online supplemental material
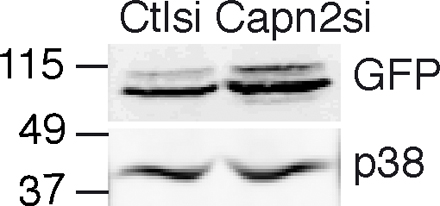
Fig. S1 corresponds to Fig. 3 B and demonstrates levels of transiently expressed GFP-cortactin in control and calpain 2 siRNA-expressing MTLn3 cells. Fig. S2 corresponds to Fig. 4 B and demonstrates the rescue of invadopodia formation in MTLn3 cells stably expressing cortactin siRNA using p34-Arc as an invadopodia marker. Fig. S3 demonstrates that inhibition of Src kinase activity with PP2 abolishes invadopodia formation in MTLn3 cells. Fig. S4 corresponds to Fig. 5 and demonstrates that the constitutively active c-Src mutant Y527F can rescue invadopodia formation in calpain 2–deficient cells, whereas wild-type c-Src cannot. Fig. S5 corresponds to Fig. 5 and demonstrates decreased cortactin proteolysis in v-Src transformed MTLn3 cells expressing calpain 2 siRNA compared with control cells. Video 1 shows dynamics of GFP-actin containing invadopodia in MTLn3 control cells. Video 2 shows dynamics of GFP-actin containing invadopodia in MTLn3 calpain 2 knockdown cells. Video 3 shows dynamics of GFP-cortactin containing invadopodia in MTLn3 control cells. Video 4 shows dynamics of GFP-cortactin containing invadopodia in MTLn3 calpain 2 knockdown cells. Video 5 shows dynamics of invadopodia in MTLn3 cortactin knockdown cells expressing wild-type GFP-cortactin. Video 6 shows dynamics of invadopodia in MTLn3 cortactin knockdown cells expressing calpain-resistant GFP-cortactin D28. Video 7 shows dynamics of GFP-cortactin containing invadopodia in v-Src transformed MTLn3 control cells. Video 8 shows dynamics of GFP-cortactin containing invadopodia in v-Src transformed MTLn3 calpain 2 knockdown cells. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200708048/DC1.
Supplemental Material
Acknowledgments
We would like to acknowledge funding from American Cancer Society (grant RSG-03-245-01-CSM) and the National Cancer Institute (grant R01CA85862-06 to A. Huttenlocher and grant R01CA69202 to Z.Y. Zhang).
Abbrevations used in this paper: ANOVA, analysis of variance; CCD, charge-coupled device; CII, compound II; ERK, extracellular signal-regulated kinase; FN, fibronectin; HEK, human embryonic kidney; pNPP, paranitrophenyl phosphate; PTP1B, protein tyrosine phosphatase 1B.
References
- Arregui, C.O., J. Balsamo, and J. Lilien. 1998. Impaired integrin-mediated adhesion and signaling in fibroblasts expressing a dominant-negative mutant PTP1B. J. Cell Biol. 143:861–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artym, V.V., Y. Zhang, F. Seillier-Moiseiwitsch, K.M. Yamada, and S.C. Mueller. 2006. Dynamic interactions of cortactin and membrane type 1 matrix metalloproteinase at invadopodia: defining the stages of invadopodia formation and function. Cancer Res. 66:3034–3043. [DOI] [PubMed] [Google Scholar]
- Bentires-Alj, M., and B.G. Neel. 2007. Protein-tyrosine phosphatase 1B is required for HER2/Neu-induced breast cancer. Cancer Res. 67:2420–2424. [DOI] [PubMed] [Google Scholar]
- Bhatt, A.K., and A. Huttenlocher. 2003. Dynamic imaging of cell-substrate contacts. Methods Enzymol. 361:337–352. [DOI] [PubMed] [Google Scholar]
- Bjorge, J.D., A. Pang, and D.J. Fujita. 2000. Identification of protein-tyrosine phosphatase 1B as the major tyrosine phosphatase activity capable of dephosphorylating and activating c-Src in several human breast cancer cell lines. J. Biol. Chem. 275:41439–41446. [DOI] [PubMed] [Google Scholar]
- Buccione, R., J.D. Orth, and M.A. McNiven. 2004. Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat. Rev. Mol. Cell Biol. 5:647–657. [DOI] [PubMed] [Google Scholar]
- Calle, Y., H.C. Chou, A.J. Thrasher, and G.E. Jones. 2004. Wiskott-Aldrich syndrome protein and the cytoskeletal dynamics of dendritic cells. J. Pathol. 204:460–469. [DOI] [PubMed] [Google Scholar]
- Calle, Y., N.O. Carragher, A.J. Thrasher, and G.E. Jones. 2006. Inhibition of calpain stabilises podosomes and impairs dendritic cell motility. J. Cell Sci. 119:2375–2385. [DOI] [PubMed] [Google Scholar]
- Carragher, N.O., M.A. Westhoff, D. Riley, D.A. Potter, P. Dutt, J.S. Elce, P.A. Greer, and M.C. Frame. 2002. v-Src-induced modulation of the calpain-calpastatin proteolytic system regulates transformation. Mol. Cell. Biol. 22:257–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carragher, N.O., B.D. Fonseca, and M.C. Frame. 2004. Calpain activity is generally elevated during transformation but has oncogene-specific biological functions. Neoplasia. 6:53–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carragher, N.O., S.M. Walker, L.A. Scott Carragher, F. Harris, T.K. Sawyer, V.G. Brunton, B.W. Ozanne, and M.C. Frame. 2006. Calpain 2 and Src dependence distinguishes mesenchymal and amoeboid modes of tumour cell invasion: a link to integrin function. Oncogene. 25:5726–5740. [DOI] [PubMed] [Google Scholar]
- Chang, B.Y., K.B. Conroy, E.M. Machleder, and C.A. Cartwright. 1998. RACK1, a receptor for activated C kinase and a homolog of the beta subunit of G proteins, inhibits activity of src tyrosine kinases and growth of NIH 3T3 cells. Mol. Cell. Biol. 18:3245–3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, W.T. 1989. Proteolytic activity of specialized surface protrusions formed at rosette contact sites of transformed cells. J. Exp. Zool. 251:167–185. [DOI] [PubMed] [Google Scholar]
- Clark, E.S., A.S. Whigham, W.G. Yarbrough, and A.M. Weaver. 2007. Cortactin is an essential regulator of matrix metalloproteinase secretion and extracellular matrix degradation in invadopodia. Cancer Res. 67:4227–4235. [DOI] [PubMed] [Google Scholar]
- Cortesio, C.L., and W. Jiang. 2006. Mannan-binding lectin-associated serine protease 3 cleaves synthetic peptides and insulin-like growth factor-binding protein 5. Arch. Biochem. Biophys. 449:164–170. [DOI] [PubMed] [Google Scholar]
- Ezumi, Y., H. Takayama, and M. Okuma. 1995. Differential regulation of protein-tyrosine phosphatases by integrin alpha IIb beta 3 through cytoskeletal reorganization and tyrosine phosphorylation in human platelets. J. Biol. Chem. 270:11927–11934. [DOI] [PubMed] [Google Scholar]
- Franco, S.J., and A. Huttenlocher. 2005. Regulating cell migration: calpains make the cut. J. Cell Sci. 118:3829–3838. [DOI] [PubMed] [Google Scholar]
- Franco, S., B. Perrin, and A. Huttenlocher. 2004. a. Isoform specific function of calpain 2 in regulating membrane protrusion. Exp. Cell Res. 299:179–187. [DOI] [PubMed] [Google Scholar]
- Franco, S.J., M.A. Rodgers, B.J. Perrin, J. Han, D.A. Bennin, D.R. Critchley, and A. Huttenlocher. 2004. b. Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nat. Cell Biol. 6:977–983. [DOI] [PubMed] [Google Scholar]
- Frangioni, J.V., A. Oda, M. Smith, E.W. Salzman, and B.G. Neel. 1993. Calpain-catalyzed cleavage and subcellular relocation of protein phosphotyrosine phosphatase 1B (PTP-1B) in human platelets. EMBO J. 12:4843–4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimona, M., and R. Buccione. 2006. Adhesions that mediate invasion. Int. J. Biochem. Cell Biol. 38:1875–1892. [DOI] [PubMed] [Google Scholar]
- Glading, A., D.A. Lauffenburger, and A. Wells. 2002. Cutting to the chase: calpain proteases in cell motility. Trends Cell Biol. 12:46–54. [DOI] [PubMed] [Google Scholar]
- Hernandez, M.V., M.G. Sala, J. Balsamo, J. Lilien, and C.O. Arregui. 2006. ER-bound PTP1B is targeted to newly forming cell-matrix adhesions. J. Cell Sci. 119:1233–1243. [DOI] [PubMed] [Google Scholar]
- Huttenlocher, A., S.P. Palecek, Q. Lu, W. Zhang, R.L. Mellgren, D.A. Lauffenburger, M.H. Ginsberg, and A.F. Horwitz. 1997. Regulation of cell migration by the calcium-dependent protease calpain. J. Biol. Chem. 272:32719–32722. [DOI] [PubMed] [Google Scholar]
- Julien, S.G., N. Dube, M. Read, J. Penney, M. Paquet, Y. Han, B.P. Kennedy, W.J. Muller, and M.L. Tremblay. 2007. Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nat. Genet. 39:338–346. [DOI] [PubMed] [Google Scholar]
- Kuchay, S.M., N. Kim, E.A. Grunz, W.P. Fay, and A.H. Chishti. 2007. Double knockouts reveal that protein tyrosine phosphatase 1B is a physiological target of calpain-1 in platelets. Mol. Cell. Biol. 27:6038–6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, F., S.Y. Lee, J. Liang, D.S. Lawrence, and Z.Y. Zhang. 2005. The role of protein-tyrosine phosphatase 1B in integrin signaling. J. Biol. Chem. 280:24857–24863. [DOI] [PubMed] [Google Scholar]
- Libertini, S.J., B.S. Robinson, N.K. Dhillon, D. Glick, M. George, S. Dandekar, J.P. Gregg, E. Sawai, and M. Mudryj. 2005. Cyclin E both regulates and is regulated by calpain 2, a protease associated with metastatic breast cancer phenotype. Cancer Res. 65:10700–10708. [DOI] [PubMed] [Google Scholar]
- Linder, S. 2007. The matrix corroded: podosomes and invadopodia in extracellular matrix degradation. Trends Cell Biol. 17:107–117. [DOI] [PubMed] [Google Scholar]
- Linder, S., and M. Aepfelbacher. 2003. Podosomes: adhesion hot-spots of invasive cells. Trends Cell Biol. 13:376–385. [DOI] [PubMed] [Google Scholar]
- Luxenburg, C., L. Addadi, and B. Geiger. 2006. The molecular dynamics of osteoclast adhesions. Eur. J. Cell Biol. 85:203–211. [DOI] [PubMed] [Google Scholar]
- Marzia, M., R. Chiusaroli, L. Neff, N.Y. Kim, A.H. Chishti, R. Baron, and W.C. Horne. 2006. Calpain is required for normal osteoclast function and is down-regulated by calcitonin. J. Biol. Chem. 281:9745–9754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamoune, A., J.H. Luo, D.A. Lauffenburger, and A. Wells. 2003. Calpain-2 as a target for limiting prostate cancer invasion. Cancer Res. 63:4632–4640. [PubMed] [Google Scholar]
- Nuzzi, P.A., M.A. Senetar, and A. Huttenlocher. 2007. Asymmetric localization of calpain 2 during neutrophil chemotaxis. Mol. Biol. Cell. 18:795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin, B.J., K.J. Amann, and A. Huttenlocher. 2006. Proteolysis of cortactin by calpain regulates membrane protrusion during cell migration. Mol. Biol. Cell. 17:239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, G.H., S. Branner, K.B. Moller, J.N. Andersen, and N.P. Moller. 2003. Enzyme kinetic characterization of protein tyrosine phosphatases. Biochimie. 85:527–534. [DOI] [PubMed] [Google Scholar]
- Ruoslahti, E., E.G. Hayman, M. Pierschbacher, and E. Engvall. 1982. Fibronectin: purification, immunochemical properties, and biological activities. Methods Enzymol. 82 Pt A:803–831. [DOI] [PubMed] [Google Scholar]
- Segall, J.E., S. Tyerech, L. Boselli, S. Masseling, J. Helft, A. Chan, J. Jones, and J. Condeelis. 1996. EGF stimulates lamellipod extension in metastatic mammary adenocarcinoma cells by an actin-dependent mechanism. Clin. Exp. Metastasis. 14:61–72. [DOI] [PubMed] [Google Scholar]
- Spinardi, L., and P.C. Marchisio. 2006. Podosomes as smart regulators of cellular adhesion. Eur. J. Cell Biol. 85:191–194. [DOI] [PubMed] [Google Scholar]
- Su, Y., Z. Cui, Z. Li, and E.R. Block. 2006. Calpain-2 regulation of VEGF-mediated angiogenesis. FASEB J. 20:1443–1451. [DOI] [PubMed] [Google Scholar]
- Tarone, G., D. Cirillo, F.G. Giancotti, P.M. Comoglio, and P.C. Marchisio. 1985. Rous sarcoma virus-transformed fibroblasts adhere primarily at discrete protrusions of the ventral membrane called podosomes. Exp. Cell Res. 159:141–157. [DOI] [PubMed] [Google Scholar]
- Tatin, F., C. Varon, E. Genot, and V. Moreau. 2006. A signalling cascade involving PKC, Src and Cdc42 regulates podosome assembly in cultured endothelial cells in response to phorbol ester. J. Cell Sci. 119:769–781. [DOI] [PubMed] [Google Scholar]
- Tonks, N.K., and S.K. Muthuswamy. 2007. A brake becomes an accelerator: PTP1B–a new therapeutic target for breast cancer. Cancer Cell. 11:214–216. [DOI] [PubMed] [Google Scholar]
- Webb, D.J., K. Donais, L.A. Whitmore, S.M. Thomas, C.E. Turner, J.T. Parsons, and A.F. Horwitz. 2004. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 6:154–161. [DOI] [PubMed] [Google Scholar]
- Webb, B.A., L. Jia, R. Eves, and A.S. Mak. 2007. Dissecting the functional domain requirements of cortactin in invadopodia formation. Eur. J. Cell Biol. 86:189–206. [DOI] [PubMed] [Google Scholar]
- Wiener, J.R., B.J. Kerns, E.L. Harvey, M.R. Conaway, J.D. Iglehart, A. Berchuck, and R.C. Bast Jr. 1994. Overexpression of the protein tyrosine phosphatase PTP1B in human breast cancer: association with p185c-erbB-2 protein expression. J. Natl. Cancer Inst. 86:372–378. [DOI] [PubMed] [Google Scholar]
- Xie, L., S.Y. Lee, J.N. Andersen, S. Waters, K. Shen, X.L. Guo, N.P. Moller, J.M. Olefsky, D.S. Lawrence, and Z.Y. Zhang. 2003. Cellular effects of small molecule PTP1B inhibitors on insulin signaling. Biochemistry. 42:12792–12804. [DOI] [PubMed] [Google Scholar]
- Yamaguchi, H., and J. Condeelis. 2007. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim. Biophys. Acta. 1773:642–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi, H., M. Lorenz, S. Kempiak, C. Sarmiento, S. Coniglio, M. Symons, J. Segall, R. Eddy, H. Miki, T. Takenawa, and J. Condeelis. 2005. Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J. Cell Biol. 168:441–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamir, E., B.Z. Katz, S. Aota, K.M. Yamada, B. Geiger, and Z. Kam. 1999. Molecular diversity of cell-matrix adhesions. J. Cell Sci. 112:1655–1669. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}