

Abstract
Cell–cell fusion is critical to the normal development of certain tissues, yet the nature and degree of conservation of the underlying molecular components remains largely unknown. Here we show that the two guanine-nucleotide exchange factors Brag2 and Dock180 have evolutionarily conserved functions in the fusion of mammalian myoblasts. Their effects on muscle cell formation are distinct and are a result of the activation of the GTPases ARF6 and Rac, respectively. Inhibition of ARF6 activity results in a lack of physical association between paxillin and β1-integrin, and disruption of paxillin transport to sites of focal adhesion. We show that fusion machinery is conserved among distinct cell types because Dock180 deficiency prevented fusion of macrophages and the formation of multinucleated giant cells. Our results are the first to demonstrate a role for a single protein in the fusion of two different cell types, and provide novel mechanistic insight into the function of GEFs in the morphological maturation of multinucleated cells.
Introduction
Fusion of cells to form multinucleated syncytia is fundamental to the development and maintenance of diverse tissues in eukaryotic organisms. The incorporation of additional nuclei in mammalian skeletal muscle cells, multinucleated giant cells, osteoclasts, and trophoblast tissue serves to augment tissue size and function (Potgens et al., 2004; Chen and Olson, 2005; Quinn and Gillespie, 2005). In skeletal muscle, fusion results in the generation of elongated fibers, with nuclei oriented longitudinally, providing instruction for contractile protein production within specific domains along the fiber (Pavlath et al., 1989; Horsley and Pavlath, 2004). In contrast, multinucleated cells derived from macrophages have globular forms, with centrally located nuclear clusters, a morphology thought to serve important roles in foreign body removal (Vignery, 2005). How syncytial cells incorporate nuclei in a controlled fashion and attain particular morphologies enabling unique functions remains poorly understood.
Studies in Drosophila and Caenorhabditis elegans have identified three phases of cell fusion: recognition and adhesion, cytoskeletal rearrangement, and membrane merger (Chen et al., 2007). Impeding the first phase of fusion by eliminating or blocking protein function has led to the identification of an array of surface proteins in myoblasts and in macrophages responsible for cell-specific adhesion and recognition of appropriate fusion partners (Greve and Gottlieb, 1982; Rosen et al., 1992; Charlton et al., 2000; Gorza and Vitadello, 2000; Schwander et al., 2003; Vignery, 2005; Jansen and Pavlath, 2006).
In the later stages of fusion, cytoskeletal rearrangement is responsible for architectural reorganization and coordinates the formation of functional syncytia. Screening of Drosophila mutants has implicated guanine-nucleotide exchange factors (GEFs) in the development of skeletal muscle by demonstrating failure of fusion of muscle cells in embryos that carry mutations in the GEFs Myoblast city (Erickson et al., 1997) and Loner (Chen et al., 2003). These GEFs are components of two separate signal transduction pathways, both predicted to converge on the GTPase Rac (Chen et al., 2007). Likewise in zebrafish, components of the Dock1-Crk-Rac pathway (Moore et al., 2007), and the Kirre-Rst pathway (Srinivas et al., 2007), the role of which in unclear in mammals, have been shown to function in fast-twitch muscle fiber formation. The roles of both Brag2 and Dock180 have yet to be studied in mammalian cell–cell fusion.
The mammalian homologue of Myoblast city, Dock180 is a Src-homology 3 (SH3) protein that interacts with the adaptor protein Crk, and activates Rac1 by direct binding (Hasegawa et al., 1996; Kiyokawa et al., 1998). Biochemical characterization of Dock180 placed it in the CDM family of GEF proteins (Cote and Vuori, 2002), which have varying, context-dependent roles ranging from membrane ruffling and cell migration to phagocytosis (Cheresh et al., 1999; Albert et al., 2000; Gumienny et al., 2001). The mammalian homologue of Loner is Brag2/GEP100, a GEF of ADP ribosylation factor 6 (ARF6). In addition to the Sec7 domain common to other ARF family GEFs, Brag2 contains a nuclear localization signal and an IQ motif (Someya et al., 2001). Concordant with the known functions of ARF6 (D'Souza-Schorey and Chavrier, 2006), the roles of Brag2 in mammalian nonmuscle cells involve the regulation of cell adhesion by controlling β1-integrin endocytosis and E-cadherin redistribution (Dunphy et al., 2006; Hiroi et al., 2006).
Evidence suggests that Loner activation of ARF6 is responsible for transport of Rac to sites of cell fusion in Drosophila (Chen et al., 2003). Transport of Rac in vesicles to the plasma membrane has also been observed in mammalian cells, and found to be partly dependent on chemically activated ARF6 (Boshans et al., 2000). Regulation of Rac activity is involved in differentiation of myoblasts (Heller et al., 2001; Samson et al., 2007), cell shape determination, and tissue invasion by macrophages and neutrophils (Pestonjamasp et al., 2006; Wheeler et al., 2006). Studies that have attempted to characterize the role of Rac in myoblast differentiation and cell cycle withdrawal have arrived at contradicting conclusions (Meriane et al., 2000, 2002) partly because of the wide array of cellular processes influenced by Rho/Rac GTPases (Hall, 2005).
We have used RNA interference in cultured mammalian cells to study how Brag2 and Dock180 control the formation of multinucleated cells. We investigated two different cell types, myoblasts and macrophages, which fuse under different conditions, and have disparate morphologies (Helming and Gordon, 2007). Our results suggest that these two GEFs are required for the formation of large multinucleated structures characteristic of each cell type. However, we provide evidence for divergent downstream mechanisms each responsible for distinct components of cellular morphology as well as the initiation of differentiation irrespective of fusion. This paper is the first to document a shared component of fusion machinery among different fusogenic cell types, implying that cell fusion has common evolutionary ancestry.
Results
Brag2 and Dock180 expression and silencing in myoblasts
We analyzed the mRNA levels of Brag2 and Dock180 in C2C12 myoblasts during proliferation (growth media [GM]) and after d 4 in differentiation media (DM). Semi-quantitative (Fig. 1 A) and real-time RT-PCR (Fig. 1 C) indicate a fourfold induction of Brag2 levels during differentiation, while there is no significant change in Dock180 mRNA levels.
Figure 1.

Expression and knockdown of Brag2 and Dock180 in C2C12 myoblasts. (A) Brag2 (1,100 bp) and Dock180 (2,485 bp) expression during growth (GM) and differentiation (DM) as shown by RT-PCR. GAPDH (250 bp). (B) Total RNA from cell lines Brag2Si, Dock180Si, and DKD, was harvested after d 4 in DM, and tested for Brag2 and Dock180 expression by RT-PCR. (C) Quantitative PCR analysis of Brag2 (dark gray) and Dock180 (light gray) expression in GM and d 4 DM. Values are normalized to the expression in CntSi cells (white). (D) Quantitative PCR analysis of Brag2 and Dock180 expression from Brag2Si, Dock180Si, and DKD cells. Values are normalized to the expression of CntSi and Cnt2Si cells. Error bars indicate the mean ± SE of three independent determinations.
Stable cell lines deficient in Brag2 and Dock180 were created using restriction enzyme–generated siRNAs (REGS) (Sen et al., 2004). Several short hairpin RNA (shRNA) candidates were retrovirally delivered to myoblasts, and mRNA expression levels were verified by PCR after puromycin selection. Fig. 1 B shows knockdown of Brag2 and Dock180 using the shRNAs that were chosen for this study. Both Brag2Si and Dock180Si cell lines exhibited >70% reduction in expression when compared with cells treated with control shRNA (CntSi) (Fig. 1 D). A double knockdown (DKD) cell line was generated by infecting Brag2Si cells with Dock180Si retrovirus (Fig. 1, B and D). The Cnt2Si cell line underwent equal rounds of infection with control shRNAs as the DKD line. Protein levels of Brag2 and Dock180 at DM d 4 were verified by Western analysis (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200707191/DC1). In total, five myoblast cell lines were generated, two control lines expressing control shRNAs, and three cell lines deficient in the expression levels of Brag2, Dock180, or both genes.
Brag2- and Dock180-deficient muscle cells share a similar decrease in fusion index but have distinct myotube morphologies
Mutation of Brag2 and Dock180 homologues leads to aberrant embryonic muscle development in Drosophila (Erickson et al., 1997; Chen et al., 2003). This has been attributed to defects in cell fusion; however, the impact of Brag2 and Dock180 deficiency in established myoblasts has not been investigated. We compared the five cell lines actively proliferating to each other and to uninfected myoblasts for morphological defects, and found comparable myoblast morphology among all (Fig. S2 A, available at http://www.jcb.org/cgi/content/full/jcb.200707191/DC1). Next, we analyzed cell cycle progression using BrdU incorporation and found no difference among the cell lines (Fig. S2 B). Thus, Brag2 and Dock180 are dispensable for myoblast morphology and proliferation. In addition, retroviral infection and selection did not overtly affect the growth properties of these cells.
To determine if Brag2 and Dock180 play roles in mammalian myoblast fusion, 5.0 × 105 cells were seeded in DM on collagen-coated plates. Media was changed daily, and on d 3, ArabinoseC (Cozzarelli, 1977) was added in order to remove proliferating cells and improve the visibility of myotube morphology. In the CntSi and Cnt2Si cell lines the majority of myoblasts had fused by d 3 in DM, and these myotubes increased in size over the next days. After d 6 in DM, cells were analyzed by immunofluorescence for expression of myosin heavy chain (MHC). Uncharacteristic myotube morphology was apparent when comparing myotubes from Brag2Si, Dock180Si, and DKD cells to the controls (Fig. 2 A). Control cells formed robust myotubes that elongated past 800 μm in length with a diameter of over 30 μm, evenly distributed nuclei, striations, and contractility. In contrast, the majority Brag2Si cells that differentiated failed to form myotubes containing three or more nuclei, as indicated by the presence of a majority of single or double nucleated cells. When larger myotubes did form by the Brag2Si cells (Fig. 2, A and D) they produced “stubby” syncytia, termed bragballs, in which elongation failed to occur and nuclei remained centrally clustered. A majority of Dock180Si cells also failed to fuse. However, in direct contrast to Brag2Si, Dock180Si myotubes elongated appropriately (Fig. 2, A and D), but formed structures comparatively destitute of nuclei that resemble atrophic myotubes (Stevenson et al., 2005). The most drastic fusion defect was observed in the DKD cell line. These cells only fused to form simple structures (Fig. 2 A) and exhibited a discernible decrease in the proportion of MHC-positive cells (Fig. 2 D). The rare multinucleated cells strongly resembled bragballs, but had an even more pronounced ball-like morphology lacking even the most rudimentary elongated structures expected of myotubes.
Figure 2.

Brag2- and Dock180-deficient cells have similar fusion index but disparate myotube morphologies. (A) Immunofluorescence images from each cell line, after d 6 in DM. Cells were labeled with primary antibody to MHC and secondary Alexa 488 (green) and Hoechst 33258 (blue). Bar, 50 μm. (B) Total fusion index analysis representing the number of nuclei in multinucleated myotubes divided by total number of nuclei in a field, with a myotube defined by at least three nuclei. A minimum of 4,000 nuclei were counted from random fields of each line at d 6 in DM. P value was determined with a t test, in which CntSi served as control for Brag2Si and Dock180Si cells and Cnt2Si served as control for DKD (*, P < 0.00001). (C) Fusion index analysis indicating the formation of di-, tri-, tetra-, or penta+ multinucleated myotubes after d 6 in DM. All error bars indicate the mean ± SE of at least four independent determinations (*, P < 0.00001). (D) Representative immunofluorescence images of d 6 DM fields used in the fusion index analysis. Bar, 200 μm. (E) β-gal complementation assay of fusion of Brag2Si, Dock180Si, and C2C12 control cells containing equal populations of α- and ω-fragments over the course of 5 d in DM. Background luminescence determined by C2C12 control cells containing α- and ω-fragments of β-gal seeded under proliferation conditions. (n = 3).
To quantify the fusion defects, 3.7 × 106 cells were seeded in triplicate for fusion index analysis. After d 6 in DM, the fusion index was determined by counting the numbers of nuclei within MHC+ cells as a percentage of total nuclei present in each of the randomly captured fields (Fig. 2 D). Despite striking morphological differences, Brag2Si and Dock180Si cells share a similar fusion index of just over 20%, whereas the DKD fusion index is slightly lower at 15% (Fig. 2 B). This represented a decrease in fusion index of 30 and 35%, respectively, compared with control cells. The aberrant morphology observed in the knockdown cell lines suggested that the common fusion index analysis, which assigns the label of “myotube” to any cell containing more than three nuclei, understates the fusion defect occurring in the Brag2- and Dock180-deficient cells. Although a three-nucleated cell indicates the occurrence of fusion, it is not representative of a functional mammalian myotube, which is capable of incorporating hundreds of nuclei per myofiber in culture and in vivo. Thus, we quantified the occurrence of a range of myotube sizes from the di-nucleated to five or more (penta+) nucleated structures. (Fig. 2 C). These data clearly show that there is no disparity among the formation of di-, tri-, or even tetra-nucleated structures, but that the difference observed in the total fusion index can be accredited to the formation of large, penta+ nucleated myotubes, which is strongly impeded in each of the knockdowns. These results indicate that Brag2 and Dock180 are essential for the formation of mature myotubes, but that each has a distinct effect on cell morphology.
To analyze whether fusion defects observed in culture are retained in vivo, we injected GFP+ cells from each of the cell lines into the tibialis anterior (TA) of CB17/SCID mice. 1 wk after injection we analyzed cross sections of the TA from each of the injected animals. Control myoblasts fuse regularly at the site of injection, as can be seen by the presence of GFP+-laminin–bound myofibers (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200707191/DC1). However, both Brag2- and Dock180-deficient cells fail to form GFP+ myofibers and lead to the disruption of the existing musculature as indicated by the dearth of laminin-bound structures at the injection site. Thus, GEFs are necessary for the fusion of myoblasts to existing myotubes, a function distinct from their known role in the formation of developing muscle in Drosophila and zebrafish (Chen et al., 2003; Moore et al., 2007).
β-Galactosidase complementation reveals early and late fusion defects
The fusion index as determined by blind counting trials of random image fields has disadvantages such as limited sample size. It is also tedious, potentially contributing to human error. The fusion index analysis at d 6 DM attributed the fusion defect we observed in Brag2Si and Dock180Si cells primarily to myotube growth. To verify this result by a quantitative biochemical approach, we took advantage of β-galactosidase (β-gal) enzymatic complementation, which has been previously used in mammalian cells to examine cell fusion (Mohler and Blau, 1996) and protein–protein interactions (Rossi et al., 2000; Wehrman et al., 2002). Brag2Si, Dock180Si, and C2C12 cells were split in two sub-populations, each of which was infected with retroviral vectors encoding one of the two complementing β-gal fragments (Wehrman et al., 2005). The infected sub-populations, one carrying the α-peptide-GFP and the other carrying the ω-deletion-GFP were expanded separately and FACS sorted to equilibrate the levels of expression of each fragment. The two sub-populations were mixed in equal proportions before seeding in 96-well plates (n = 3). Fusion, assayed as a bioluminescence reading every 24 h for 5 d, demonstrated a decrease in enzymatic activity in the Brag2Si and Dock180Si cells, confirming the fusion index results. Similar to the fusion index analysis, the two knockdown cell lines exhibit very similar levels of enzyme activity over the course of the experiment. However, β-gal analysis revealed that the kinetics of fusion differed at two separate periods: early stages between d 1 and 3 in DM and later stages between d 4 and 5 in DM (Fig. 2 E). During these time points Brag2- and Dock180-deficient cells show smaller factors of luminescence increase than controls (C2C12 = 1.4248, Brag2Si = 1.3585, Dock180Si = 1.3581), which indicates that Brag2Si and Dock180Si myotube maturation does not progress normally after early myotube formation. However, the β-gal fusion analysis also indicates an additional fusion defect early in the course of differentiation for the two knockdown cell lines.
Delayed cell cycle withdrawal and impaired differentiation contribute to the early fusion defect in Brag2Si and Dock180Si cells
Fusing cells were visualized by time-lapse microscopy over the course of 2–4 d in DM. When compared with the fusion of the CntSi cells (Video 1), the formation of the Brag2Si bragballs (Video 2) and thin Dock180Si myotubes (Video 3) is evident. However, during the first 24–36 h of image capture, large numbers of Dock180Si, Brag2Si, and especially DKD cells (Video 4) die and detach, indicative of aberrant proliferation in the presence of ArabinoseC (Videos are available at http://www.jcb.org/cgi/content/full/jcb.200707191/DC1). Furthermore, Brag2Si and Dock180Si cells injected into the TA showed tremendous proliferation, as can be seen by the large numbers of GFP+ cells at the site of injection when compared with the control cells (Fig. S4).
BrdU labeling experiments were designed to determine the extent of differentiation deficiency in the knockdowns. Myoblasts were seeded sparsely, 1.0 × 104 cells per 6-cm plate (n = 9). After 24 h in DM, cells were labeled with BrdU for 6 h (Zhang et al., 2007). It is clearly evident, as shown in Fig. 3 A, that the number of cells per field was higher in the DKD cells, indicating proliferation despite DM conditions. BrdU analysis confirms this observation because 67 ± 3.9% of DKD cells are BrdU positive when compared with the 35 ± 4.6% from Cnt2Si, 24 h after serum withdrawal. This phenotype was shared by Brag2Si cells (63 ± 6.4%), and to a lesser extent by the Dock180Si cells (52 ± 3.5%) (Fig. 3 B).
Figure 3.

Delayed cell cycle withdrawal and differentiation in Brag2- and Dock180-deficient myoblasts. (A) Merged laser-scanning confocal image of representative fields of Cnt2Si and DKD myoblasts after 24 h in DM, labeled for BrdU (green) and with To-Pro (blue). BrdU+ nuclei are colored in teal in the merged image. Bar, 100 μm. (B) BrdU incorporation for each stable cell line after 24 h in DM and a 6-h BrdU incubation. (*P < 0.001). (C) Immunofluorescence of representative image fields of Cnt2Si and DKD myoblasts after 72 h in DM, labeled for myogenin (red), MHC (green), and with Hoechst 33258 (blue). Bar, 50 μm. (D) Differentiation as measured by myogenin (*, P < 0.0006) and MHC (**, P < 0.003; ***, P < 0.03) expression for each stable cell line after 72 h in DM. In all cases, error bars indicate the mean ± SE of three independent determinations, in which a minimum of 1,000 nuclei were counted per trial.
Cell cycle withdrawal, an early step in differentiation, is followed by the induction of myogenin, signifying terminal differentiation (Sabourin and Rudnicki, 2000). Further evidence of late skeletal muscle differentiation is the expression of structural proteins such as MHC. We analyzed all cell lines for the expression of these myogenic factors. Cells were seeded sparsely in order to avoid cell–cell contact, and maintained in DM for 72 h, after which they were double stained for myogenin and MHC. Control cells showed the highest levels of myogenin expression at 54 ± 5.3%, whereas in Brag2Si (18 ± 2.9%) and Dock180 (23 ± 3.2%) fewer differentiation events occurred (Fig. 3 D). DKD cells further confirmed the BrdU trend by exhibiting the lowest levels of myogenin expression in only 15 ± 3.5% total nuclei. Representative images are displayed in Fig. 3 C. MHC expression followed a similar pattern to myogenin, with control cells expressing the highest levels of MHC. The percentage of differentiated cells likely represents an underestimate of the proportion of cells originally capable of differentiation because cells that failed to differentiate continue to cycle. These data indicate that Brag2 and Dock180 are required in myoblasts for efficient cell cycle exit and induction of the differentiation program, and segregates these functions from fusion because these experiments were performed at low density. It is possible that the early fusion defect observed in the β-gal complementation assay reflects a failure to induce the fusion-related portion of the differentiation program in Brag2- and Dock180-deficient cells, but these experiments suggest additional roles for these molecules in muscle differentiation.
Brag2 and Dock180 lead to deficient ARF6 and Rac1 activation, respectively, during myoblast fusion
Brag2 and Dock180 are responsible for the activation of GTPases ARF6 and Rac1, respectively (Kiyokawa et al., 1998; Someya et al., 2001). Furthermore, in Drosophila, Loner is believed to modulate the Dock180/Rac1 pathway, leading to membrane ruffling and myoblast fusion (Chen et al., 2003). We assessed total Rac1 and ARF6 protein levels by Western analysis in all cell lines, and found no difference (Fig. 4, A and D). To determine if Rac1 and ARF6 activation is affected in Brag2Si, Dock180Si, and DKD cells, we performed a Rac-GTP pull-down assay, using PAK-GST and an ARF6-GTP assay using GGA3-GST fusion proteins with lysates from cells at d 3 in DM. As shown in Fig. 4 B, a dramatic decrease in Rac1-GTP levels was observed in the Dock180Si and the DKD cells, but not in the Brag2Si cells. The levels of Rac-GTP were quantified with CntSi serving as the normalized control. Dock180Si displayed the lowest levels of Rac-GTP (43 ± 14.4 a.u.), followed by DKD (61 ± 14.9 a.u.), both of which were significantly lower than the controls CntSi and Cnt2Si, respectively (Fig. 4 C). Brag2 deficiency caused a marked decrease in the ARF6 activity levels, which was also observed in the DKD cells (Fig. 4 E). Unexpectedly, there was a decrease in ARF6-GTP levels in the Dock180-deficient cells (73 ± 4.7 a.u.), intermediate when compared with the control and the Brag2Si (42 ± 13.2 a.u.) cells (Fig. 4 F). Thus, as in Drosophila, Dock180 modulates Rac activation during mammalian myoblast fusion, whereas Brag2 regulates ARF6 activity. Our results indicate that potential crosstalk between these two GEFs occurs at the level of ARF6 activation, which was moderately impeded in Dock180Si cells.
Figure 4.

Brag2 and Dock180 deficiency decreases GTPase activity. (A) Western blot analysis of total expression of Rac in each cell line after d 3 in DM. Rac migrates as a 21-kD protein and GAPDH migrates as 35-kD protein. (B) Western blot indicating the level of activated Rac, as detected in lysates at d 3 in DM using a pull-down assay with GST-fused PAK protein-binding domain. Lysates from CntSi cell line were treated with GTPγS and GDP for positive and negative controls, respectively. (C) The histogram represents the levels of GTP-Rac as determined by three independent experiments. All samples were normalized to total protein and Brag2Si, Dock180Si, Cnt2Si, and DKD values are also normalized to the Rac-GTP levels observed in CntSi. (D) Western blot analysis of total expression of ARF6 after d 3 in DM. ARF6 migrates as a 20-kD protein. (E) Western blot indicating the level of activated ARF6, as detected in lysates at d 3 in DM using a pull-down assay with GST-fused GGA3 protein-binding domain, which specifically binds ARF6-GTP. (F) The histogram represents the levels of ARF6-GTP as determined by three independent experiments. All samples were normalized to total protein and Brag2Si, Dock180Si, Cnt2Si, and DKD values are also normalized to the ARF6-GTP levels observed in CntSi at d 3 in DM.
Brag2Si myoblasts exhibit defects in paxillin localization during differentiation
Morphological analysis revealed that although Dock180 was dispensable for myotube elongation, loss of Brag2 resulted in a failure to elongate. This prompted an analysis of cell adhesion molecule expression. Although cellular levels of E-cadherin and M-cadherin were unaffected in either knockdown, Dock180 deficiency resulted in a twofold increase in β1-integrin expression (Fig. 5). We also analyzed the localization and protein levels of paxillin during differentiation and fusion. Paxillin is a multi-domain scaffold protein that recruits structural and signaling molecules to focal adhesions where it performs critical roles in transducing adhesion and growth factor signals to elicit changes in the cytoskeleton and gene expression (Brown and Turner, 2004). It has been suggested that ARF6 activity may be required to establish paxillin localization at focal adhesions (Kondo et al., 2000).
Figure 5.
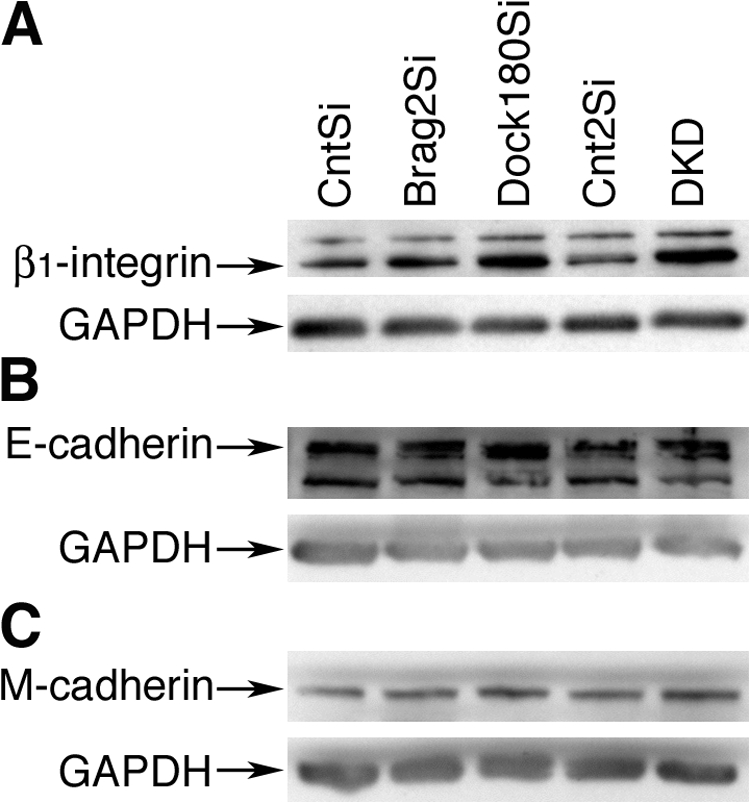
Dock180 deficiency in myotubes increases cellular levels of β1-integrin. (A) Western blot analysis of total protein levels of β1-integrin at d 6 in DM. β1-integrin migrates as a 130-kD protein and loading control GAPDH migrates as 35-kD protein. (B) Western blot analysis of total protein levels of E-cadherin. E-cadherin migrates as a 120-kD protein; the 90-kD band likely reflects digestion of the extracellular domain during lysate preparation. (C) Western blot analysis of M-cadherin total protein levels. M-cadherin migrates as a 130-kD protein.
All cell lines were sparsely seeded for 72 h in DM, and then stained for myogenin and paxillin. All myogenin-positive cells were analyzed for redistribution of paxillin. The images in Fig. 6 A show the expression of myogenin and redistribution of paxillin in CntSi cells. In contrast, in the Brag2Si line, the cells that express myogenin do not acquire elongated myocyte morphology and paxillin in these cells fails to redistribute, remaining diffusely localized. We quantified the distribution of paxillin in myogenin-positive cells. In CntSi and Cnt2Si cells, 76 ± 9.9% and 75 ± 1.2% of the myogenin-positive cells, respectively, redistributed paxillin, whereas only 22 ± 2.4% of the Brag2Si cells and 23 ± 10.6% of DKD cells behaved similarly. Concordant with its effect on ARF6 activation, Dock180Si cells exhibited an intermediate phenotype with 45 ± 7.7% expressing myogenin and redistributing paxillin (Fig. 6 B). To determine if aberrant paxillin expression occurs, we tested expression of paxillin by Western blot in lysates from cells after d 5 in DM (Fig. 6 C). The results in lanes 2, 3, and 5 indicate that in the case of Brag2Si, Dock180Si, and DKD cells there is only a slight decrease in the protein levels of paxillin when compared with the two controls.
Figure 6.

Brag2-deficient myoblasts exhibit aberrant paxillin localization during differentiation. (A) Representative images of CntSi and Brag2Si myoblasts after d 3 in DM depicted in phase contrast and immunofluorescence, labeled with Hoechst 33258 (blue), myogenin (red), paxillin (yellow), and merged. Bar, 50 μm. (B) Paxillin redistribution in myogenin-positive cells, d 3 in DM. At least 1,000 myogenin-positive nuclei were counted for each experimental trial, and the error bars indicate the mean ± SE of three independent determinations (*, P < 0.01). (C) Western blot analysis of paxillin expression after d 5 in DM under fusion conditions. Paxillin migrates as a 65-kD protein.
Paxillin fails to colocalize with ARF6 in early fusion of Brag2-deficient myoblasts
We tested whether the defects in paxillin localization were retained during early fusion of Brag2Si myoblasts and whether this observation was due to a disruption in ARF6 or Rac1 localization. Analysis of Rac1 localization during early fusion failed to distinguish any differences between the cytoplasmic distribution of Rac1 in control myotubes, bragballs, or Dock180-deficient thin myotubes (Fig. S5, available at http://www.jcb.org/cgi/content/full/jcb.200707191/DC1). We examined the localization of endogenous ARF6 and paxillin after d 3 in DM. In the CntSi cells, ARF6 and paxillin were diffusely dispersed in the cytoplasm of nascent myotubes, but colocalized regularly at the leading edges of myocytes, depicted by the arrows in Fig. 7 A. In contrast, in Brag2Si cells, ARF6 was primarily perinuclear and concentrated clusters of paxillin (Fig. 7 B, arrowheads) formed in several areas of the cytoplasm. These alterations in ARF6 and paxillin localization are primarily evident in Brag2Si myocytes that have attained the bragball morphology. Similar to our observations during differentiation, Dock180-deficient early myotubes exhibited an intermediate morphology, where clusters of paxillin (Fig. 7 C, arrowheads) were evident, but less frequent and generally smaller. Dock180Si myocytes elongated to comparable levels with the CntSi cells and normal ARF6 localization was observed throughout the entire length of these elongated cells, in several of which colocalization of paxillin and ARF6 was observed (Fig. 7 C, arrows). Thus, our data suggest that appropriate localization of paxillin is dependent on the localization of ARF6.
Figure 7.

ARF6 and paxillin fail to colocalize during early fusion in Brag2-deficient cells. (A) Representative images of CntSi, (B) Brag2Si, and (C) Dock180Si myoblasts after d 3 in DM depicted in phase contrast and immunofluorescence, labeled for ARF6 (green), paxillin (red), and merged with Hoechst 33258 (blue). Arrowheads point to clusters of paxillin observed in Brag2 and to a lesser extent in Dock180 knockdowns. Arrows point to colocalization sites of ARF6 and paxillin in the merged fields. Bar, 50 μm.
Brag2-deficient cells exhibit limited association between ARF6 and paxillin
Our immunofluorescence data suggest that ARF6 colocalizes with paxillin; however, in order to verify whether physical association between the two occurs, we performed paxillin immunoprecipitation (IP) during d 3 in DM (Fig. 8 A). In CntSi lysates, anti-paxillin immunoprecipitates contained abundant ARF6 protein, as well as β1-integrin as compared with immunoprecipitates with IgG. The interaction between paxillin and β1-integrin, and its importance for the establishment of focal adhesions has been previously described (Brown and Turner, 2004). Next, we tested whether these paxillin interactions are altered in the GEF knockdowns (Fig. 8 B). Our data indicate that the interaction of paxillin with ARF6 and β1-integrin during myoblast fusion is severely disrupted in Brag2Si and to a slightly lesser extent in the Dock180Si cells when compared with controls (Fig. 8 B, lanes 2 and 3). Considering that the majority of ARF6 in Brag2Si myotubes is in the GDP-bound state, the lack of interaction between paxillin and ARF6 correlates with the levels of ARF6-GTP as observed in the ARF6 activity assay (Fig. 4 E). Collectively, these results support a role for activated ARF6 in the transport of paxillin to cellular sites crucial for the maintenance of structural integrity during myotube formation.
Figure 8.

Paxillin physical interaction with ARF6 and β1-integrin is disrupted in Brag2 and Dock180-deficient cells. (A) Cell lysates of CntSi myoblasts in d 3 DM under fusion conditions were immunoprecipitated using anti-Paxillin antibody and mouse IgG serum as negative control. Immunoblotting was performed for ARF6 (20-kD) and β1-integrin (130- and 100-kD). 5% of lysate extract was immunoblotted for paxillin (65-kD) and GAPDH (35-kD) in order to verify equal amounts of cell lysates. (B) Cell lysates of each cell line were immunoprecipitated using anti-Paxillin antibody and immunoblotted for presence of ARF6 and β1-integrin. 1% of lysate extract was immunoblotted for paxillin and GAPDH in order to verify equal amounts of cell lysates.
Dock180 knockdown impairs macrophage fusion and multinucleated giant cell formation
A fundamental question is whether fusion machinery is conserved among different cell types. We investigated fusion in the hematopoietic lineage, where fusion of macrophages results in the formation of osteoclasts and multinucleated giant cells (MNGCs). In contrast to muscle, no genetically tractable, in vitro model has been described to study macrophage fusion. Thioglycollate-induced activation of intraperitoneal macrophages is a standard source of fusogenic macrophages (Helming and Gordon, 2007). While occasional fusion events producing only di- or tri-nucleated cells occur in the absence of interleukin-4 (IL-4) (Fig. 9, Bi) upon culturing with IL-4 (McInnes and Rennick, 1988), macrophages form MNGCs within 48 h. We analyzed the mRNA expression of Brag2 and Dock180 in IP-harvested macrophages cultured with or without IL-4. Similar levels of Brag2 and Dock180 mRNA were expressed before and after addition of IL-4 (Fig. 9, Ai).
Figure 9.

Macrophages deficient in Dock180 fail to form MNGCs. (A) (i) Brag2 (1,100 bp) and Dock180 (745 bp) expression in thioglycollate-induced IP-harvested macrophages before and after treatment with IL-4 as indicated by RT-PCR analysis. β-actin (300 bp). (ii) Semi-quantitative RT-PCR showing expression of Dock180 in CD11b+Gr1+ monocytes obtained from GFP+ peripheral blood of PVR-Cnt-BM and PVR-Dock180Si-BM mice 4 wk after transplantation. (iii) Semi-quantitative RT-PCR showing expression of Dock180 in CD11b+ IP-macrophages of PVR-Cnt-BM and PVR-Dock180Si-BM mice 6 wk after transplantation under fusion conditions. (B) Phase-contrast and immunofluorescence images of IP-harvested macrophages from: (i) PVR-Cnt-BM macrophages serving as negative fusion control cultured without IL-4; (ii) GFP− cells from PVR-Dock180Si-BM representing the endogenous macrophage population cultured with IL-4; (iii) GFP+ cells from PVR-Cnt-BM representing the transplanted control population cultured with IL-4; and (iv) GFP+ cells from PVR-Dock180Si-BM representing the transplanted Dock180-deficient population cultured with IL-4. MNGC formation can be distinguished by the cluster of nuclei with a large surrounding area of cytoplasm, the extent of which is indicated by dashed lines. (C) Fusion index analysis in macrophages from each of the four populations shown in B. Two multinucleated categories are analyzed and a minimum of 1,000 nuclei were counted for each fusion assay. Error bars indicate the mean ± SE of four independent fusion assays. P values were determined by t test between the two positive fusion controls and the PVR-Dock180Si-BM macrophages (*, P < 0.03; **, P < 0.0005). (D) Comparison of fusion indexes, of three or more nuclei, in myoblasts and macrophages deficient in Dock180. Error bars indicate the mean ± SE of four independent fusion assays (*, P < 0.00001; **, P < 0.0005).
Intraperitoneal-harvested macrophages are terminally differentiated, precluding the establishment of stable cell lines, and limiting the amount of material available for experiments. To overcome this problem, we modified the vREGS backbone by adding the MSCVpgkEGFP expression cassette in place of the puromycin resistance gene. Into this modified construct, named PVR, we subcloned the same shRNAs used for knockdown of Brag2 and Dock180 in myoblasts. After large-scale viral production, we infected bone marrow (BM) with virus containing the Brag2Si (PVR-Brag2Si-BM) and Dock180Si (PVR-Dock180Si-BM) as well as PVR control virus (PVR-Cnt-BM), following the procedure described in Kalberer et al. (2000). Peripheral blood analysis for GFP+ hematopoietic reconstitution was performed by flow cytometry 4 wk after transplantation of donor BM into lethally irradiated mice. Myeloid GFP+CD11b+Gr1+, cells were sorted from PVR-Brag2Si-BM and PVR-Dock180Si-BM mice as well as PVR-Cnt-BM control mice. Knockdown of Dock180 was confirmed in the sorted PVR-Dock180Si-BM myeloid cells (Fig. 9, Aii), and knockdown of Brag2 was also verified in the PVR-Brag2Si-BM myeloid cells (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200707191/DC1). The recipient mice were allowed to recuperate from the peripheral bleeding for 2 wk, after which time they were given IP injections of thioglycollate. Only one out of eight PVR-Brag2Si-BM mice survived longer than 6 wk after BM transplantation, and the sole survivor demonstrated low bone marrow reconstitution, not yielding enough GFP+ macrophages for a fusion assay. In contrast, 12 of 12 PVR-Cnt-BM mice, from three independent experimental trials, and 6 of 7 PVR-Dock180Si-BM mice, from two independent trials, survived the reconstitution period and were treated with thioglycollate.
Despite high levels of reconstitution, some endogenous macrophages remained in the intraperitoneal cavity at the time of thioglycollate treatment because a combination of GFP+ and GFP− CD11b+ cells were sorted from the intraperitoneal lavage of both PVR-Dock180Si-BM and PVR-Cnt-BM mice. Both GFP+ and GFP− macrophage populations from PVR-Dock180Si-BM mice were seeded for fusion analysis. Knockdown of expression of Dock180 was confirmed in GFP+ PVR-Dock180Si-BM macrophages seeded for fusion (Fig. 9, Aiii).
MNGCs formed readily in both control populations of the uninfected, endogenous GFP− PVR-Dock180Si-BM and in the transduced, transplanted GFP+ PVR-Cnt-BM cells, but only in the presence of IL-4 (Fig. 9, Bii and Biii). In the PVR-Dock180Si-BM macrophages most of the cells did not fuse. A few tri-nucleated cells were observed, and formation of MNGCs was rare (Fig. 9, Biv). After the first intraperitoneal lavage, survival surgery was performed and 4 wk later the procedure was repeated. In both instances, fusion index was analyzed 4 d after the addition of IL-4 and the results are shown in Fig. 9 C. As in the analysis of myoblast fusion, both tri+ and penta+ nucleated cells were counted. PVR-Dock180Si-BM showed a marked decrease in fusion, 15 ± 1.6% for tri+ nucleated cells and only 5.6 ± 1.7% for penta+ nucleated cells, when compared with both control populations, the fusion index of which exceeded 42% under both fusion criteria.
Despite contrasting morphological appearances and distinct cellular functions, there is a similar defect in the fusion of myoblasts and macrophages deficient in Dock180 (Fig. 9 D). Thus, our results indicate that Dock180 contributes to the development of fully mature multinucleated cells in different models and cell lineages.
Discussion
Whether the formation of multinucleated cells by fusion evolved independently in different cell types or if there exist common underlying mechanisms is unknown. In this study we have investigated the mammalian homologues of Loner and Myoblast city, which have previously been found to be essential in the development of the embryonic musculature in Drosophila (Erickson et al., 1997; Chen et al., 2003). Our data show that the functions of Brag2 and Dock180 are evolutionarily conserved and involve control of differentiation and cell shape in addition to fusion, via different mechanisms directed by their respective GTPases. Moreover, this study identifies Dock180 as a common molecular component that controls cell fusion in different cell types, providing the first evidence of shared fusion machinery.
Two assays to quantitate myoblast fusion, one by observation and the other biochemical, complemented each other by permitting the detection of morphological differences and by removing observer bias and error, respectively. In light of the failure of Brag2- and Dock180-deficient myoblasts to fuse to existing muscle fibers in vivo, the low level of fusion observed in vitro leads us to conclude that whereas Brag2 and Dock180 are required for myotube maturation, early fusion in mammalian myotubes is not dependent on Brag2 or Dock180. However, it cannot be ruled out that incomplete knockdown, a ubiquitous caveat of RNAi experiments, may have contributed to the low level of fusion observed.
Whereas Dock180 and Brag2 have not previously been linked to muscle cell differentiation aside from fusion, our analysis of individual cells at early stages of differentiation indicated that myoblasts deficient in either Brag2 or Dock180 failed to efficiently exit the cell cycle. These cells were likewise impaired in their ability to induce the transcription factor myogenin or the contractile protein MHC. Suppression of myogenin promoter activity has been previously observed with dominant-negative mutants of Rac (Takano et al., 1998). However, the role of Rac in myoblast differentiation is complex and remains poorly understood, as other recent studies suggest that a drop in Rac activity is essential for cell cycle withdrawal (Heller et al., 2001; Charrasse et al., 2007). This implies that inappropriate cycling of cells deficient in Dock180 may occur though Rac-independent mechanisms, possibly involving interactions of Dock180 with Crk and other signaling components involved in cell cycle regulation.
In further support of Rac independent control of differentiation, Brag2 deficiency did not affect Rac activation, yet its effects on cell cycle withdrawal and differentiation were more severe when compared with Dock180. Cells deficient in both Dock180 and Brag2 more closely mimicked the differentiation and morphological defects observed in Brag2-deficient cells. Although ARF6 has not previously been linked to cell cycle, it has been recently shown that Brag2 can localize to the nucleus (Dunphy et al., 2007), where its functions in cell cycle regulation have not been characterized to date. Their similar effects on cell cycle withdrawal and initiation of differentiation suggest that Brag2 and Dock180 function in a common pathway possibly through ARF6.
The similar defects early in differentiation notwithstanding, disparity between Brag2 and Dock180 are highlighted by the striking morphological differences between the two during fusion, demonstrating unique roles of each in myotube maturation. Defects in skeletal muscle maturation have been previously reported in myoferlin null mice, which formed myotubes with significantly reduced myofiber cross-sectional area and muscle mass (Doherty et al., 2005). Our data indicate that Brag2 and Dock180 are dispensable for myoblasts proliferation but that they play critical roles in the orchestration of skeletal muscle formation, especially in the later stages of myotube growth. It is possible that the different morphological observations relate to the distinct roles that each GEF carries out in the regulation of its respective GTPase. We have shown that Brag2 deficiency, and to a lesser extent Dock180 deficiency, leads to ARF6 inactivity whereas Dock180 deficiency exclusively leads to a drop in Rac activity. Furthermore, with respect to Dock180, these results are in agreement with recent results in which Trio, another GEF with dual specificity for Rac and Rho, severely impairs myoblast fusion when its levels are decreased by RNAi (Charrasse et al., 2007). Unlike Trio, Dock180 has no cross-specificity for Rho, thus it may be the combination of both Rac and Rho inactivation, which results in the severe fusion defect of Trio deficiency. Time-lapse analysis showed that in contrast to the Dock180-deficient cells, absence of Brag2 rendered nascent myotubes unable to extend and adhere to the surface. This unique Brag2 phenotype combined with previous results, which indicate that overexpression of an ARF6 GAP PAG3 decreases the recruitment of paxillin to focal adhesions (Kondo et al., 2000), led us to analyze the localization of paxillin. During differentiation and fusion, distribution of paxillin was severely disrupted in myogenin-positive Brag2Si and DKD cells. A failure to redistribute paxillin upon differentiation accounts for the adhesion defect observed in bragballs, where a newly formed multinucleated cell is incapable of creating or maintaining functional focal adhesions, causing collapse into the bragball morphology.
Our immunoprecipitation and immunofluorescence data identify an association between paxillin and ARF6. Furthermore, there is a strong correlation between a deficiency in ARF6 activity and improper localization of paxillin during fusion. The intermediate drop in ARF6 activity observed in Dock180Si cells, according to our model (Fig. 10), accounts for the moderate paxillin distribution defect observed during differentiation and the minor paxillin clustering observed during fusion. It is possible that the observed increase in total β1-integrin protein levels in Dock180-deficient cells combined with normal cell motility during differentiation (Video 3) could account for the ability of Dock180Si myotubes to elongate. Further analysis will determine if other structural components of focal adhesions such as vinculin and focal adhesion kinase are specifically transported by ARF6 activation after differentiation, and how integrin levels affect morphology in Dock180-deficient cells.
Figure 10.

Model for the roles of Brag2 and Dock180 in cell–cell fusion. Upon receipt of signals from fusion receptors on the cell surface Brag2 activates ARF6, which in turn transports paxillin to sites of focal adhesion, where it complexes with integrins thereby maintaining the structural integrity required during myotube maturation. Dock180 regulates Rac-GTP activity thus primarily contributing to lamellipodia formation and cytoskeletal rearrangement. Dock180 can activate ARF6, thus in part functioning in paxillin transport and possibly integrin recycling.
In contrast to skeletal muscle, there is little known about the intracellular mechanisms of cell fusion in nonmuscle cell types (Vignery, 2005). Knockdown in primary macrophages, which are differentiated cells not amenable to passage in culture, required introduction of shRNA constructs in dividing hematopoietic stem cells (HSCs). In the case of Brag2, an unexpected role for this molecule in HSC biology was revealed by the repeated inability to achieve bone marrow reconstitution using HSCs infected with the Brag2 shRNA. Further experiments will determine the function of Brag2 in HSC homing, but defects in adhesion and niche recognition are two potential explanations. Generation of shRNA-expressing intraperitoneal macrophages was possible in the case of Dock180. Deficiency in Dock180 prevented the formation of MNGCs and the rare fusion events that did occur resulted in cells that had low nuclear number, and aberrant morphology that lacked the characteristic centrally located nuclear cluster common in the control MNGCs. From our data, it is apparent that the loss of Dock180 leads to a significant fusion defect in both myoblasts and macrophages. These findings are the first to identify a role for a single molecule in fusion in different lineages, and the results suggest that muscle and macrophages use redundant intracellular machinery during the fusion process.
It is becoming apparent that the dynamic morphological state of a cell reflects the particular balance of GEFs present at a given time. For example, a recent study (Yeh et al., 2007) found that the introduction of single, engineered GEFs and their domains could induce morphological changes including the formation of filopodia or lamellipodia. This study and others (Lee et al., 2001; Dunphy et al., 2006; Francis et al., 2006; Otani et al., 2006; Charrasse et al., 2007) suggest that cell morphology is subject to manipulation by the repression or expression of particular GEF activities. We show that GEFs control the differentiation-specific morphology of skeletal muscle cells and MNGCs. The effects of individual GEFs are unique, an example being the markedly different impact of Dock180 and Brag2 on muscle morphology. In addition, their functions are cell type specific as indicated by analyses of a variety of nonfusion-related functions of Dock180 and Brag2 in mammalian cells (Hasegawa et al., 1996; Dunphy et al., 2006; Hiroi et al., 2006; Handa et al., 2007). Conversely, our experiments demonstrate that there are fundamental GEF-controlled fusion processes that are conserved across lineages, underscored by the similar role of Dock180 in fusion of different cell types. This paper furthers our understanding of the generation of multinucleated cells and the GEF-mediated intracellular functions responsible for their formation and maintenance.
Materials and methods
Cell culture
C2C12 mouse myoblasts were cultured at 10% CO2 37°C in DMEM supplemented with 20% FBS (GM). Myoblasts were differentiated for fusion under low mitogenic conditions in DMEM supplemented with 2% ES, (DM) on collagen (Sigma-Aldrich) coated plates, under the following seeding densities: 6-cm plate, 5 × 105 cells; 10-cm plate, 1.5 × 106 cells; 15-cm plate, 3.7 × 106 cells. DM was replaced every 24 h for a total of 7 d. Where indicated, ArabinoseC (10−5 M) was added to DM at d 2 and/or d 3 during differentiation.
shRNA stable cell line generation
shRNAs to knockdown Dock180 and Brag2 expression were generated using the REGS method and inserted into vREGS expression vector as described (Sen et al., 2004). Ecotropic phoenix cells were transfected using FuGENE 6 (Roche) with vREGS constructs. C2C12 cells were infected with viral supernatants containing polybrene (5 μg/ml−1) and centrifuged for 30 min at 2,000 g. Cells were infected two rounds and selected on puromycin (1 μg/ml−1) 48 h after last infection. After verification of knockdown by RT-PCR and DM morphology analysis, the following shRNA-REGS constructs were selected for each gene. Listed are the shRNA nucleotides that correspond to the Brag2: GGAGTTGCCGCCTGACCGTGGGT and Dock180: GTGAATGAGGTACAGCGATTCG mouse cDNA sequences. As infection and shRNA control (CntSi), REGS-GFP-489 described in Sen et al. (2004) was used. Three separate stable cell lines were generated on C2C12 cells for each of the selected shRNAs. For Brag2+Dock180 DKD generation, cells verified in knockdown of Brag2 expression were infected two extra rounds with virus expressing the Dock180 shRNA. As DKD control, the CntSi stable cell line was infected two extra rounds with the control REGS-GFP-489 shRNA.
Semi-quantitative and quantitative RT-PCR
RNA was harvested from stable cell lines in GM or DM d 4 by RNeasy mini kit (QIAGEN) and 500 ng of total RNA was used in semi-quantitative RT-PCR analysis with Superscript III One-Step RT-PCR (Invitrogen) for Brag2 (5′: GTCACATGCAGCACAGTCCCCCTTCCTT; 3′: AGCTTCCGGCAGTCCTTCTCCCTCTACG) and Dock180 (5′:TCATCATGGAGACGCTGCTT; 3′: TCGTCTCCTCTGCCTCCTTC). The RT-PCR conditions for each analysis were as described in manufacturer's instructions, with annealing temperatures of 63°C for 30 s. For RT-PCR controls, 50 ng of total RNA were used, with primers for GAPDH (5′: CACTGAGCATCTCCCTCACA; 3′: TGGGTGCAGCGAACTTTATT) and β-actin (5′: TTTGAGACCTTCAACACCCCAGCC; 3′: AATGTCACGCACGATTTCCCGC) with annealing temperatures of 55°C for 30 s. For Brag2 and Dock180 26, 28, and 30 cycles were tested, for GAPDH and β-actin 19, 21, and 23 cycles were tested. Semi-quantitative RT-PCR of CD11b+ and Gr1+ cells FACS sorted from peripheral blood was performed as described above except that 50 ng of total RNA used, while in the RT-PCR of harvested IP macrophages 200 ng of total RNA was used. Numbers of cycles analyzed for each experiment was 37, 39, and 41. For qRT-PCR 1 μg of total RNA from GM or DM d 4 stable cell lines was used to synthesize single-stranded cDNA using first Strand cDNA Synthesis kit (Roche). 1 μl of the cDNA synthesis reaction mix was used with Platinum SYBR Green kit from Invitrogen in the real-time thermocycler from Corbett Research Rotor-Gene RG3000 for quantitative analysis of mRNA gene expression. New primers were designed for Brag2 (5′: CTCCAGCCTCAAAAAGGAGTC; 3′: CTAGGCTTAGGAGCACAGCACT) and Dock180 (5′: TTAAGAACCTCATCGGGAAGAA; 3′: TCCAGAAATCTCTTGTTCAGCA) specifically for real-time analysis.
Immunofluorescence
Stable cell lines were seeded for fusion or differentiation analysis in triplicate. Cells seeded for fusion as described above were fixed every day for 6 d with 1.5% paraformaldehyde 15 min at room temperature (RT), permeabilized with 0.3% Triton 10 min at RT, and blocked with 20% FBS for 30 min at RT. For differentiation analysis, cells were seeded 2.0 × 104 in 6-cm collagen-coated plates and treated as described above after 72 h in DM. Primary antibodies for MHC (Chemicon) diluted 1:50 in blocking buffer, incubated at RT for 30 min; secondary Alexa-488 GtαMs 1:1,000 (Invitrogen). Primary antibodies for myogenin (Santa Cruz Biotechnology, Inc.) diluted 1:100, incubated at RT for 1 h; secondary Alexa-546 GtαRb 1:1,000. Primary antibodies for paxillin (BD or Santa Cruz Biotechnology, Inc.) diluted 1:100, incubated at RT for 1 h; secondary Alexa-488 GtαMs 1:1,000 or Alexa-647 GtαRb 1:1,000. Primary antibodies for Rac (Upstate Biotechnology) 1:50 and ARF6 (AbCam) 1:50 incubated at RT for 1 h secondary Alexa-488 GtαMs 1:1,000. Nuclear staining of cells with Hoechst 33258 (Sigma-Aldrich) diluted 1:10,000 and incubated at RT for 15 min. Cells were imaged with Carl Zeiss Axioplan2 using 40x water immersion objective, or Carl Zeiss Axiovert 200M using NeoFluar 10 or 20x objectives and an ORCA-ER C4742-95 (Hamamatsu Photonics) digital camera. Openlab 4.0.2 and Volocity 3.6.1 (Improvision) were the software used for image acquisition. Images were composed and edited in Openlab 4.0.2 or Photoshop 7.0 (Adobe). Background was reduced using contrast adjustments and color balance was performed to enhance colors. All modifications were applied to the whole image.
β-Gal complementation fusion analysis
C2C12 control cells and stable cell lines Brag2Si and Dock180Si were split and each fraction was retrovirally infected with virus containing constructs expressing either the β-gal fragment α-GFP on the MFG vector backbone or ω-GFP on the pwz1 vector backbone modified to include GFP as a fusion to the C terminus of the ω-fragment. All infected cells were cell sorted by flow cytometry (DIVA-Van, Becton Dickinson) for GFP expression. Before seeding for fusion analysis, the α-GFP and ω-GFP Brag2Si cells were trypsinized and mixed in equal proportions and seeded in 96-well plates, 10,000 per well, 24 wells total. Five plates were seeded for fusion in this manner and analysis of β-gal activity was measured as described (Wehrman et al., 2005) daily for a total of 5 d using GAL-screen substrate reagents (Applied Biosystems). Luminescence was measured in a Tropix TR717 luminometer. The exact process was performed for the C2C12 control and Dock180Si cells coinciding with the Brag2Si analysis described above. Background luminescence reading was measured each day with C2C12 control cells seeded 10,000 per well 6 h before sample analysis.
Bone marrow MSCV transduction and transplantation
Retroviral vector design for BM transductions was based on the vREGS backbone, in which the puromycin resistance gene was replaced by MSCVpgkEGFP via digestion by AgeI and ClaI (pVR-GFP). The siRNAs verified for successful knockdown of Brag2 and Dock180 in myoblasts were inserted into cloning site of pVR-GFP. Large-scale retrovirus production was performed 24 h after double-transfection of each construct + pCL-eco into 293T cells. Bone marrow transduction was performed as described (Kalberer et al., 2000). In brief, 6- to 8-wk-old mice (C57BL/6) mice were used as donor (male) and recipients (female). Donor mice were injected with 15 mg/ml of 5-fluorouracil (Sicor) per 10 g of mouse weight. Harvested BM cells were prestimulated for 24 h in IL6, IL3, and SCF cytokine cocktail (Peprotech), then infected for two rounds with MSCV retrovirus on RetroNectin (10 μg/ml) (Takara) coated plates in the presence of 5 μg protamine sulfate (Sigma-Aldrich). Transduced BM cells were isolated by sorting for GFP expression (DIVA-Van, Becton Dickinson) 24 h after last infection, and 50,000–200,000 cells were IV injected into lethally irradiated recipient mice, a total of 18 Gy of whole-body irradiation administered in split dosage. 4 wk after transplantations, recipient mice were inspected for hematopoietic reconstitution by flow cytometry of GFP+ peripheral blood (PB) followed by analysis of the presence of myeloid (Gr1, CD11b) and lymphoid (B220, CD3) lineage markers (BD PharMingen). Cells in the myeloid compartment of GFP+ PB were sorted and RNA was harvested for RT-PCR analysis.
IP macrophage harvest and fusion assay
Recipient mice confirmed for GFP+ PB reconstitution were harvested for resident macrophages as described (Helming and Gordon, 2007). In brief, recipient mice were IP injected with 1 mL of sterilized Thioglycollate medium brewer modified (BD). IP lavage was performed 4 d after injections with ice-cold PBS. Harvested cells were sorted for GFP expression, and 5.0 × 105 cells were seeded for fusion analysis on Permanox-coated SonicSeal slide wells (Nalge Nunc). Seeded cells were cultured in Opti-MEM (GIBCO BRL) supplemented with 10% FBS, and to induce MNGC formation 5 ng/ml of IL-4 (Peprotech) was added to the media. Cells were analyzed for fusion after culturing for 96 h. After the lavage, survival surgery was performed with a success rate of 100%, and 3-4 wk after initial procedure, recipient mice were reinjected with thioglycollate and a second IP lavage and fusion analysis was performed.
BrdU analysis
BrdU labeling and detection kit (Roche) was used on controls and Brag2Si, Dock180Si and DKD C2C12 stable cell lines, which were seeded 10,000 cells/6 cm on collagen coated plates for differentiation analysis. After 24 h in DM, BrdU labeling reagent was added to the cells with fresh DM media for an additional 6 h, after which the cells were fixed and detected for immunofluorescence as per manufacturer's instructions. Following BrdU detection procedure, cell nuclei were labeled with To-Pro (invitrogen) in PBS. Cells were imaged using laser-scanning confocal microscope (LSM510 Axiovert 200M; Carl Zeiss) using NeoFluar 10x objective lenses and maximum optical section with the LSM software. After acquisition, images were composed and edited in Photoshop 7.0 as described above.
GTP activity assays and immunoblotting
Rac-activity assay was essentially performed as per manufacturer's instructions of Rac Activation Assay (Cytoskeleton, Inc.). In brief, controls and Brag2Si, Dock180Si, and DKD C2C12 stable cell lines were seeded for fusion analysis in 15-cm collagen-coated plates. After d 3 in DM, cells were lysed at room temperature in 50 mM Tris, pH 7.5, 10 mM MgCl2, 0.3 M NaCl, and 2% IGEPAL. Lysates were cleared by centrifugation for 5 min at 6,000 rpm at 4°C, and protein quantification was determined by Bradford assay (Bio-Rad). 5 mg of each protein lysate was incubated with GST-PAK PBD protein beads (25 μg) for 1 h at 4°C with agitation. PAK PBD beads were pelleted by centrifugation and resuspended in 15 μl of Laemmli buffer (Sigma-Aldrich). ARF6 activity assay was performed similarly but using GST-GGA3 PBD beads for incubation with the d 3 DM lysates of each of the five stable cell lines. Western blot analysis for Rac and ARF6 activity and other protein samples was conducted using NuPage system from invitrogen. Samples were loaded in 10% Bis-Tris precast gel, resolved by running with MES or MOPS SDS buffer, and transferred onto Immobilon-P membranes (Amersham). Membranes were then incubated with antibodies against Rac1 (1:250; Cytoskeleton, Inc.), AFR6 (1:500; Abcam), Paxillin (1:5,000; BD Transduction Laboratories), β1-integrin (1:80; R&D Systems), E-cadherin (1:200; Santa Cruz Biotechnology, Inc.), M-cadherin (1:1,000; BD Transduction Laboratories), and GAPDH (1:10,000; Santa Cruz Biotechnology, Inc.). Membranes were then incubated with HRP-conjugated secondary antibodies (1:5,000; Zymed Laboratories) and detected by ECL or ECL+ detection reagents (Amersham).
Immunoprecipitation
Cell lysates from d 3 DM of each of the five stable cell lines were prepared as described above, in addition they were pre-cleared with 30 μl of protein-G agarose (Calbiochem). 2 mg of pre-cleared lysate was incubated with 2 μg of anti-paxillin antibody or mouse IGg serum overnight at 4°C with agitation, followed by incubation at 4°C with agitation with 30 μl of protein-G agarose for 2 h. Precipitates were washed for four rounds in lysis buffer supplemented with increasing concentrations of NaCl (150–300 mM). Immunoprecipitates were pelleted by centrifugation and resuspended in 15 μl of Laemmli buffer (Sigma-Aldrich); immunoblotting was performed as described above.
Time lapse
Controls and Brag2Si, Dock180Si, and DKD C2C12 stable cell lines were seeded for fusion 5.0 × 104 cells/well in 24-well collagen-coated plates. ArabinoseC (Sigma-Aldrich) was added to the cells d 2 and d 3 after the transition to DM media. Cells were imaged using Zeiss Axiovert 200M equipped with time-lapse apparatus CTI-Controller 3700 Digital; Tempcontrol 37–2 digital; scanning stage Incubator XL 100/135 (PECON). Frames were captured every 15 min for a total of 60 h, encompassing d 2, 3, and 4 during myoblast differentiation and fusion. Images were acquired and analyzed using Volocity 3.6.1 (Improvision).
Immunocytochemistry
1 wk after injection, TA muscles were dissected and immersed in PBS/0.5% EM-grade PFA (Polysciences) for 2 h at RT followed by overnight immersion in PBS with 20% sucrose at 4°C. Fixed tissue was embedded in optimal cutting temperature compound (Tissue-Tek; Sakura Finetek), snap-frozen in isobutene and liquid nitrogen, and sectioned at 10 μm thickness. Tissue sections were blocked in PBS/20% normal goat serum/0.3% Triton X-100 (blocking buffer); incubated with primary antibodies (diluted in blocking buffer; 2 μg/ml rabbit anti-GFP (Invitrogen), 3 μg/ml rat anti-laminin-2 (Upstate Biotechnology), and incubated with 10 μg/ml of the secondary-conjugated antibodies anti–rabbit-Alexa488 and anti–rat-Alexa546 (Invitrogen). Sections were then washed overnight and mounted with Fluoromount G. Images of muscle transverse sections were acquired using an epifluorescent microscope (Axioplan2; Carl Zeiss), Fluar 20x/0.75 objective lenses, and a digital camera (ORCA-ER C4742-95; Hamamatsu Photonics). The software used for acquisition was OpenLab 4.0.2 (Improvision). All images were composed and edited in Photoshop 7.0 (Adobe). Background was reduced using brightness and contrast adjustments, and color balance was performed to enhance colors. All the modifications were applied to the whole image using Photoshop 7.0 (Adobe).
Online supplemental material
Several figures and videos throughout this article are marked as supplementary. This material is available online and includes figures depicting the depletion of Brag2 and Dock180 protein levels, analysis of the morphology and BrdU incorporation for each stable cell line during active growth in myoblast conditions, as well as analysis of the fusion potential of Brag2- and Dock180-deficient myoblasts in vivo, when injected in the TA of immunodeficient mice. Furthermore, time-lapse visualization of each stable cell line during d 2, 3, and 4 while in DM conditions is also available online. All online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200707191/DC1.
Supplementary Material
Acknowledgments
We would like to thank Eric Olson and Elizabeth Chen for providing us with the mRNA sequence of hLoner/Brag2; Margaret Vaughan for her gift of Brag2/GEP100 antibodies; Siamon Gordon for help with the IP macrophage harvest protocol; James Casanova and Valerie Weaver for her gift of GST-GGA3; and Penny Gilbert for her help with the ARF6-GTP activity assay. We also thank Alessandra Sacco and Yan Ling Liao for comments and useful discourse on this manuscript, Adam Palermo for discussion and statistical analysis, Tom Wehrman for the β-gal constructs, George Sen for lending his expertise with REGS, and Peggy Kraft for help with sectioning.
K.V. Pajcini was supported by National Institutes of Health (NIH) Training Grants 5T32 HD007249 and 5T32 AI07328. J.H. Pomerantz was supported by NIH National Research Service Award AF051678. H.M. Blau's research is supported by NIH grants AG009521, EB005011, HD018179, AG020961, and AG024987, and by the Baxter Foundation.
K.V. Pajcini and J.H. Pomerantz contributed equally to this paper.
Abbreviations used in this paper: ARF6, ADP ribosylation factor 6; β-gal, β-galactosidase; BM, bone marrow; DKD, double knockdown; DM, differentiation media; GEF, guanine-nucleotide exchange factor; GM, growth media; IL-4, interleukin-4; MHC, myosin heavy chain; MNGC, multinucleated giant cell; shRNA, short hairpin RNA.
References
- Albert, M.L., J.I. Kim, and R.B. Birge. 2000. alphavbeta5 integrin recruits the CrkII-Dock180-rac1 complex for phagocytosis of apoptotic cells. Nat. Cell Biol. 2:899–905. [DOI] [PubMed] [Google Scholar]
- Boshans, R.L., S. Szanto, L. van Aelst, and C. D'Souza-Schorey. 2000. ADP-ribosylation factor 6 regulates actin cytoskeleton remodeling in coordination with Rac1 and RhoA. Mol. Cell. Biol. 20:3685–3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, M.C., and C.E. Turner. 2004. Paxillin: adapting to change. Physiol. Rev. 84:1315–1339. [DOI] [PubMed] [Google Scholar]
- Charlton, C.A., W.A. Mohler, and H.M. Blau. 2000. Neural cell adhesion molecule (NCAM) and myoblast fusion. Dev. Biol. 221:112–119. [DOI] [PubMed] [Google Scholar]
- Charrasse, S., F. Comunale, M. Fortier, E. Portales-Casamar, A. Debant, and C. Gauthier-Rouviere. 2007. M-Cadherin activates Rac1 GTPase through the Rho-GEF trio during myoblast fusion. Mol. Biol. Cell. 18:1734–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, E.H., and E.N. Olson. 2005. Unveiling the mechanisms of cell-cell fusion. Science. 308:369–373. [DOI] [PubMed] [Google Scholar]
- Chen, E.H., B.A. Pryce, J.A. Tzeng, G.A. Gonzalez, and E.N. Olson. 2003. Control of myoblast fusion by a guanine nucleotide exchange factor, loner, and its effector ARF6. Cell. 114:751–762. [DOI] [PubMed] [Google Scholar]
- Chen, E.H., E. Grote, W. Mohler, and A. Vignery. 2007. Cell-cell fusion. FEBS Lett. 581:2181–2193. [DOI] [PubMed] [Google Scholar]
- Cheresh, D.A., J. Leng, and R.L. Klemke. 1999. Regulation of cell contraction and membrane ruffling by distinct signals in migratory cells. J. Cell Biol. 146:1107–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cote, J.F., and K. Vuori. 2002. Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. J. Cell Sci. 115:4901–4913. [DOI] [PubMed] [Google Scholar]
- Cozzarelli, N.R. 1977. The mechanism of action of inhibitors of DNA synthesis. Annu. Rev. Biochem. 46:641–668. [DOI] [PubMed] [Google Scholar]
- Doherty, K.R., A. Cave, D.B. Davis, A.J. Delmonte, A. Posey, J.U. Earley, M. Hadhazy, and E.M. McNally. 2005. Normal myoblast fusion requires myoferlin. Development. 132:5565–5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Souza-Schorey, C., and P. Chavrier. 2006. ARF proteins: roles in membrane traffic and beyond. Nat. Rev. Mol. Cell Biol. 7:347–358. [DOI] [PubMed] [Google Scholar]
- Dunphy, J.L., R. Moravec, K. Ly, T.K. Lasell, P. Melancon, and J.E. Casanova. 2006. The Arf6 GEF GEP100/BRAG2 regulates cell adhesion by controlling endocytosis of beta1 integrins. Curr. Biol. 16:315–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunphy, J.L., K. Ye, and J.E. Casanova. 2007. Nuclear functions of the Arf guanine nucleotide exchange factor BRAG2. Traffic. 8:661–672. [DOI] [PubMed] [Google Scholar]
- Erickson, M.R., B.J. Galletta, and S.M. Abmayr. 1997. Drosophila myoblast city encodes a conserved protein that is essential for myoblast fusion, dorsal closure, and cytoskeletal organization. J. Cell Biol. 138:589–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis, S.A., X. Shen, J.B. Young, P. Kaul, and D.J. Lerner. 2006. Rho GEF Lsc is required for normal polarization, migration, and adhesion of formyl-peptide-stimulated neutrophils. Blood. 107:1627–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorza, L., and M. Vitadello. 2000. Reduced amount of the glucose-regulated protein GRP94 in skeletal myoblasts results in loss of fusion competence. FASEB J. 14:461–475. [DOI] [PubMed] [Google Scholar]
- Greve, J.M., and D.I. Gottlieb. 1982. Monoclonal antibodies which alter the morphology of cultured chick myogenic cells. J. Cell. Biochem. 18:221–229. [DOI] [PubMed] [Google Scholar]
- Gumienny, T.L., E. Brugnera, A.C. Tosello-Trampont, J.M. Kinchen, L.B. Haney, K. Nishiwaki, S.F. Walk, M.E. Nemergut, I.G. Macara, R. Francis, et al. 2001. CED-12/ELMO, a novel member of the CrkII/Dock180/Rac pathway, is required for phagocytosis and cell migration. Cell. 107:27–41. [DOI] [PubMed] [Google Scholar]
- Hall, A. 2005. Rho GTPases and the control of cell behaviour. Biochem. Soc. Trans. 33:891–895. [DOI] [PubMed] [Google Scholar]
- Handa, Y., M. Suzuki, K. Ohya, H. Iwai, N. Ishijima, A.J. Koleske, Y. Fukui, and C. Sasakawa. 2007. Shigella IpgB1 promotes bacterial entry through the ELMO–Dock180 machinery. Nat. Cell Biol. 9:121–128. [DOI] [PubMed] [Google Scholar]
- Hasegawa, H., E. Kiyokawa, S. Tanaka, K. Nagashima, N. Gotoh, M. Shibuya, T. Kurata, and M. Matsuda. 1996. DOCK180, a major CRK-binding protein, alters cell morphology upon translocation to the cell membrane. Mol. Cell. Biol. 16:1770–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller, H., E. Gredinger, and E. Bengal. 2001. Rac1 inhibits myogenic differentiation by preventing the complete withdrawal of myoblasts from the cell cycle. J. Biol. Chem. 276:37307–37316. [DOI] [PubMed] [Google Scholar]
- Helming, L., and S. Gordon. 2007. Macrophage fusion induced by IL-4 alternative activation is a multistage process involving multiple target molecules. Eur. J. Immunol. 37:33–42. [DOI] [PubMed] [Google Scholar]
- Hiroi, T., A. Someya, W. Thompson, J. Moss, and M. Vaughan. 2006. GEP100/BRAG2: activator of ADP-ribosylation factor 6 for regulation of cell adhesion and actin cytoskeleton via E-cadherin and alpha-catenin. Proc. Natl. Acad. Sci. USA. 103:10672–10677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsley, V., and G.K. Pavlath. 2004. Forming a multinucleated cell: molecules that regulate myoblast fusion. Cells Tissues Organs. 176:67–78. [DOI] [PubMed] [Google Scholar]
- Jansen, K.M., and G.K. Pavlath. 2006. Mannose receptor regulates myoblast motility and muscle growth. J. Cell Biol. 174:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalberer, C.P., R. Pawliuk, S. Imren, T. Bachelot, K.J. Takekoshi, M. Fabry, C.J. Eaves, I.M. London, R.K. Humphries, and P. Leboulch. 2000. Preselection of retrovirally transduced bone marrow avoids subsequent stem cell gene silencing and age-dependent extinction of expression of human beta-globin in engrafted mice. Proc. Natl. Acad. Sci. USA. 97:5411–5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyokawa, E., Y. Hashimoto, S. Kobayashi, H. Sugimura, T. Kurata, and M. Matsuda. 1998. Activation of Rac1 by a Crk SH3-binding protein, DOCK180. Genes Dev. 12:3331–3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo, A., S. Hashimoto, H. Yano, K. Nagayama, Y. Mazaki, and H. Sabe. 2000. A new paxillin-binding protein, PAG3/Papalpha/KIAA0400, bearing an ADP-ribosylation factor GTPase-activating protein activity, is involved in paxillin recruitment to focal adhesions and cell migration. Mol. Biol. Cell. 11:1315–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S.H., M. Eom, S.J. Lee, S. Kim, H.J. Park, and D. Park. 2001. BetaPix-enhanced p38 activation by Cdc42/Rac/PAK/MKK3/6-mediated pathway. Implication in the regulation of membrane ruffling. J. Biol. Chem. 276:25066–25072. [DOI] [PubMed] [Google Scholar]
- McInnes, A., and D.M. Rennick. 1988. Interleukin 4 induces cultured monocytes/macrophages to form giant multinucleated cells. J. Exp. Med. 167:598–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meriane, M., P. Roux, M. Primig, P. Fort, and C. Gauthier-Rouviere. 2000. Critical activities of Rac1 and Cdc42Hs in skeletal myogenesis: antagonistic effects of JNK and p38 pathways. Mol. Biol. Cell. 11:2513–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meriane, M., S. Charrasse, F. Comunale, A. Mery, P. Fort, P. Roux, and C. Gauthier-Rouviere. 2002. Participation of small GTPases Rac1 and Cdc42Hs in myoblast transformation. Oncogene. 21:2901–2907. [DOI] [PubMed] [Google Scholar]
- Mohler, W.A., and H.M. Blau. 1996. Gene expression and cell fusion analyzed by lacZ complementation in mammalian cells. Proc. Natl. Acad. Sci. USA. 93:12423–12427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, C.A., C.A. Parkin, Y. Bidet, and P.W. Ingham. 2007. A role for the Myoblast city homologues Dock1 and Dock5 and the adaptor proteins Crk and Crk-like in zebrafish myoblast fusion. Development. 134:3145–3153. [DOI] [PubMed] [Google Scholar]
- Otani, T., T. Ichii, S. Aono, and M. Takeichi. 2006. Cdc42 GEF Tuba regulates the junctional configuration of simple epithelial cells. J. Cell Biol. 175:135–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlath, G.K., K. Rich, S.G. Webster, and H.M. Blau. 1989. Localization of muscle gene products in nuclear domains. Nature. 337:570–573. [DOI] [PubMed] [Google Scholar]
- Pestonjamasp, K.N., C. Forster, C. Sun, E.M. Gardiner, B. Bohl, O. Weiner, G.M. Bokoch, and M. Glogauer. 2006. Rac1 links leading edge and uropod events through Rho and myosin activation during chemotaxis. Blood. 108:2814–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potgens, A.J., S. Drewlo, M. Kokozidou, and P. Kaufmann. 2004. Syncytin: the major regulator of trophoblast fusion? Recent developments and hypotheses on its action. Hum. Reprod. Update. 10:487–496. [DOI] [PubMed] [Google Scholar]
- Quinn, J.M., and M.T. Gillespie. 2005. Modulation of osteoclast formation. Biochem. Biophys. Res. Commun. 328:739–745. [DOI] [PubMed] [Google Scholar]
- Rosen, G.D., J.R. Sanes, R. LaChance, J.M. Cunningham, J. Roman, and D.C. Dean. 1992. Roles for the integrin VLA-4 and its counter receptor VCAM-1 in myogenesis. Cell. 69:1107–1119. [DOI] [PubMed] [Google Scholar]
- Rossi, F.M., B.T. Blakely, C.A. Charlton, and H.M. Blau. 2000. Monitoring protein-protein interactions in live mammalian cells by beta-galactosidase complementation. Methods Enzymol. 328:231–251. [DOI] [PubMed] [Google Scholar]
- Sabourin, L.A., and M.A. Rudnicki. 2000. The molecular regulation of myogenesis. Clin. Genet. 57:16–25. [DOI] [PubMed] [Google Scholar]
- Samson, T., C. Will, A. Knoblauch, L. Sharek, K. von der Mark, K. Burridge, and V. Wixler. 2007. Def-6, a guanine-nucleotide exchange factor for Rac1, interacts with the skeletal muscle integrin chain alpha 7A and influences myoblast differentiation. J. Biol. Chem. 282:15730–15742. [DOI] [PubMed] [Google Scholar]
- Schwander, M., M. Leu, M. Stumm, O.M. Dorchies, U.T. Ruegg, J. Schittny, and U. Muller. 2003. Beta1 integrins regulate myoblast fusion and sarcomere assembly. Dev. Cell. 4:673–685. [DOI] [PubMed] [Google Scholar]
- Sen, G., T.S. Wehrman, J.W. Myers, and H.M. Blau. 2004. Restriction enzyme-generated siRNA (REGS) vectors and libraries. Nat. Genet. 36:183–189. [DOI] [PubMed] [Google Scholar]
- Someya, A., M. Sata, K. Takeda, G. Pacheco-Rodriguez, V.J. Ferrans, J. Moss, and M. Vaughan. 2001. ARF-GEP(100), a guanine nucleotide-exchange protein for ADP-ribosylation factor 6. Proc. Natl. Acad. Sci. USA. 98:2413–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas, B.P., J. Woo, W.Y. Leong, and S. Roy. 2007. A conserved molecular pathway mediates myoblast fusion in insects and vertebrates. Nat. Genet. 39:781–786. [DOI] [PubMed] [Google Scholar]
- Stevenson, E.J., A. Koncarevic, P.G. Giresi, R.W. Jackman, and S.C. Kandarian. 2005. Transcriptional profile of a myotube starvation model of atrophy. J. Appl. Physiol. 98:1396–1406. [DOI] [PubMed] [Google Scholar]
- Takano, H., I. Komuro, T. Oka, I. Shiojima, Y. Hiroi, T. Mizuno, and Y. Yazaki. 1998. The Rho family G proteins play a critical role in muscle differentiation. Mol. Cell. Biol. 18:1580–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignery, A. 2005. Macrophage fusion: the making of osteoclasts and giant cells. J. Exp. Med. 202:337–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrman, T., B. Kleaveland, J.H. Her, R.F. Balint, and H.M. Blau. 2002. Protein-protein interactions monitored in mammalian cells via complementation of beta-lactamase enzyme fragments. Proc. Natl. Acad. Sci. USA. 99:3469–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrman, T.S., C.L. Casipit, N.M. Gewertz, and H.M. Blau. 2005. Enzymatic detection of protein translocation. Nat. Methods. 2:521–527. [DOI] [PubMed] [Google Scholar]
- Wheeler, A.P., C.M. Wells, S.D. Smith, F.M. Vega, R.B. Henderson, V.L. Tybulewicz, and A.J. Ridley. 2006. Rac1 and Rac2 regulate macrophage morphology but are not essential for migration. J. Cell Sci. 119:2749–2757. [DOI] [PubMed] [Google Scholar]
- Yeh, B.J., R.J. Rutigliano, A. Deb, D. Bar-Sagi, and W.A. Lim. 2007. Rewiring cellular morphology pathways with synthetic guanine nucleotide exchange factors. Nature. 447:596–600. [DOI] [PubMed] [Google Scholar]
- Zhang, F., J.H. Pomerantz, G. Sen, A.T. Palermo, and H.M. Blau. 2007. Active tissue-specific DNA demethylation conferred by somatic cell nuclei in stable heterokaryons. Proc. Natl. Acad. Sci. USA. 104:4395–4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}