

Abstract
MicroRNAs (miRNAs) are small RNAs of 19 to 25 nucleotides that are negative regulators of gene expression. To determine whether miRNAs are associated with cytogenetic abnormalities and clinical features in acute myeloid leukemia (AML), we evaluated the miRNA expression of CD34+ cells and 122 untreated adult AML cases using a microarray platform. After background subtraction and normalization using a set of housekeeping genes, data were analyzed using Significance Analysis of Microarrays. An independent set of 60 untreated AML patients was used to validate the outcome signatures using real-time polymerase chain reaction. We identified several miRNAs differentially expressed between CD34+ normal cells and the AML samples. miRNA expression was also closely associated with selected cytogenetic and molecular abnormalities, such as t(11q23), isolated trisomy 8, and FLT3-ITD mutations. Furthermore, patients with high expression of miR-191 and miR-199a had significantly worse overall and event-free survival than AML patients with low expression (overall survival: miR-191, P = .03; and miR-199a, P = .001, Cox regression). In conclusion, miRNA expression in AML is closely associated with cytogenetics and FLT3-ITD mutations. A small subset of miRNAs is correlated with survival.
Introduction
Acute myeloid leukemia (AML) is a cytogenetically and molecularly heterogeneous disorder characterized by differentiation arrest and malignant proliferation of clonal myeloid precursors.1 Patients with intermediate- and poor-risk cytogenetics represent the majority of AML; chemotherapy-based regimens fail to cure most of these patients, and stem-cell transplantation is frequently the treatment of choice.2,3 Because allogeneic stem-cell transplantation is not an option for many patients with high risk leukemia for a variety of reasons, there is a critical need to improve our understanding of the biology of these leukemias to develop novel therapies
MicroRNAs (miRNAs) are noncoding RNAs of 19 to 25 nucleotides in length that regulate gene expression by inducing translational inhibition and cleavage of their target mRNAs through base pairing to partially or fully complementary sites.4 miRNAs are involved in critical biologic processes, including development, cell differentiation, stress response, apoptosis, and proliferation.4 Recently, miRNA expression has been linked to hematopoiesis and cancer.5–11 In mice, the ectopic expression of miR-181 in hematopoietic progenitor cells led to proliferation in the B-cell compartment.5 Likewise, important roles for miRNAs have been found during human granulocytic, erythrocytic, and megakaryocytic differentiation.6–8 The first report linking miRNAs and cancer involved chronic lymphocytic leukemia (CLL).9 A cluster of 2 miRNAs, miR-15a and miR-16-1, was found to be located in the minimal region of deletion (∼30 kb) at 13q14 and to be deleted or down-regulated in approximately 60% of CLL samples.9 Further studies confirmed the widespread involvement of miRNAs in cancer.10,11 Little is known, however, about miRNA expression in AML. Here, we analyzed a large set of AML patients with predominantly intermediate and poor prognosis using miRNA microarrays to investigate whether miRNA expression is associated with clinical features, cytogenetic abnormalities, and outcome.
Methods
This study was performed under institutional review board approvals from the M. D. Anderson Cancer Center, Thomas Jefferson University, and The Ohio State University.
Patients and cell samples
Pretreatment bone marrow and blood samples from 182 patients with newly diagnosed AML were obtained from the Cell and Tissue Bank at M. D. Anderson Cancer Center (MDACC; (n = 172) and Thomas Jefferson University (n = 10). A total of 122 AML samples were used to analyze the miRNA expression using a microarrays platform, whereas 60 untreated AML samples were used to validate the outcome signatures using quantitative real-time polymerase chain reaction (RT-PCR; Table 1). A second cohort of 54 AML patients with relapsed (n = 34) or refractory (n = 20) disease obtained from the MDACC was used to sought differences in the miRNA expression between newly diagnosed and relapsed/primary refractory AML patients (Table S7, available on the Blood website; see the Supplemental Materials link at the top of the online article). Informed consent was obtained from the patients in accordance with the Declaration of Helsinki to procure and bank the cells for future research according to institutional guidelines. Patient's samples were prepared by Ficoll-Hypaque (Sigma-Aldrich, St Louis, MO) gradient centrifugation, enriched for leukemic cells by CD3/CD19 depletion (MACS; Miltenyi Biotec, Auburn, CA) and cryopreserved.12 Cytogenetic analyses of the samples were performed at diagnosis or at relapse, using unstimulated short-term (24-, 48-, and 72-hour) cultures with or without a direct method and G-banding. The criteria used to describe a cytogenetic clone and description of karyotype followed the recommendations of the International System for Human Cytogenetic Nomenclature.13 At least 20 bone marrow metaphase cells were analyzed in patients designated as having a normal karyotype. FLT3 in tandem duplication (ITD) and activation loop D835 mutations were performed on most of the samples as previously described.14 The first cohort of 122 AMLs was treated within a variety of institutional review board-approved protocols open at the MDACC during the collection period, including idarubicin with 2 different cytarabine combinations (n = 53; protocol 91004 and 10193), high dose ARA-C (n = 20) containing regimens (protocols 330139 and 202074), DCTER (n = 5; protocol 202089), and investigational drugs, such as PKC 412 and interleukin-11 (n = 24; protocols 201591 and 20202). All 4 patients with acute promyelocytic leukemia received regiments containing all-trans-retinoic acid. The majority of the 60 patients in the validation cohort (78%) were treated with the same idarubicin and cytarabine regimen (n = 47; protocol 91003), high dose ARA-C containing regimens (n = 5; protocols 330139 and 202074) and other investigational agents, such as PKC 412 and interleukin-11 (n = 6; protocols 201591 and 20202). Blood mature granulocytes and monocytes and bone marrow CD71+ selected erythrocyte precursors from 4 healthy donors were purchased from Allcells (Emeryville, CA). Bone marrow CD34+ progenitors from 10 healthy donors were purchased from Allcells. In vitro differentiated megakaryocytes were obtained as previously described.8
Table 1.
Clinical and cytogenetic characteristics of newly diagnosed AML patients
| Characteristic | Microarray cohort (n = 122) | Quantitative RT-PCR (outcome validation) cohort (n = 60) |
|---|---|---|
| Age, y | ||
| Median | 60.3 | 59 |
| Range | 18-86 | 24-80 |
| Sex, no. (%) | ||
| Female | 47 (38) | 29 (48) |
| Male | 75 (62) | 31 (52) |
| White cell count, ×103/L | ||
| Median | 45.27 | 53.7 |
| Range | 0.7-278 | 1.3-273 |
| Bone marrow blasts, % | ||
| Median | 62 | 70.2 |
| Range | 20-99 | 25-93 |
| WHO classification, no. (%) | ||
| AML with recurrent genetic abnormalities | ||
| t(15;17)(q22;q12) | 4 (3.3) | 0 |
| inv(16)(p13q22)/ t(16;16)(p13;q22) | 4 (3.3) | 0 |
| t(6;11) or t(9;11) | 9 (7.5) | 3 (5) |
| AML with multilineage dysplasia | 29 (24) | 7 (12) |
| AML and MDS, therapy-related | 12 (10) | 8 (13) |
| AML not otherwise categorized | ||
| AML minimally differentiated | 5(4) | 5 (8.3) |
| AML without maturation | 9 (7.5) | 9 (15) |
| AML with maturation | 10 (8) | 12 (20) |
| AML myelomonocytic leukemia | 21 (17) | 4 (6.6) |
| Acute monoblastic and monocytic leukemia | 8 (6.5) | 9 (15) |
| Acute erythroid leukemia | 6 (5) | 0 |
| Acute megakaryoblastic leukemia | 3 (2.5) | 0 |
| Not categorized* | 2 (1.5) | 3 (5) |
| Cytogenetics,† no. (%) | ||
| Normal karyotype | 45 (37) | 29 (48) |
| +8 | 5 (4) | 1 (1.6) |
| Complex karyotype | 23 (20) | 12 (20) |
| Other karyotypes | 26 (19) | 14 (24) |
| Not done | 6 (5) | 1 (1.6) |
| FLT3 status,‡ no. (%) | ||
| FLT3-ITD+ | 17 (18) | 14 (27) |
| FLT3-wt | 73 (82) | 38 (73) |
| FLT3-D835 | 2 (2) | 2 (4) |
| Not done | 32 | 8 |
| Status at last follow-up, no. (%)§ | ||
| Dead | 81 (66) | 42 (70) |
| Alive | 41 (33) | 18 (30) |
No statistically significant differences were observed between the two set of patients (microarrays vs quantitative RT-PCR) by t test and χ2, except for the category of AML without maturation (χ2, P = .03). All the values represent frequencies (%).
Those AML cases do not fulfill criteria for inclusion in one of the previously described subgroups.
Other cytogenetics groups not otherwise categorized in the WHO classification. A total of 116 of 122 patients from the microarray cohort and 59 of 60 patients from the quantitative RT-PCR cohort had at least 20 or more metaphases analyzed by conventional karyotype. Complex karyotype is defined as more than or equal to 3 chromosomal abnormalities.
Not all the patients had FLT3 analyzed. The percentages shown are in relationship to the total number of patients with FLT3 mutation studies.
The median follow-up for alive patients in the 122 AML patients is 100 weeks (range, 1-586 weeks) and in the 60 AML cohorts is 124 weeks (range, 7-278 weeks).
RNA extraction and miRNA microarray experiments
RNA extraction and miRNA microchip experiments were performed as previously described.15 The miRNA microarray is based on a one-channel system.15 The chips contain gene-specific oligonucleotide probes, spotted by contacting technologies and covalently attached to a polymeric matrix (Document S1; ArrayExpress database at EBI for the miRNA oligonucleotide probe sequences).
Real-time quantification of miRNAs
The single-tube TaqMan miRNA assays were used to detect and quantify mature miRNAs as previously described16 using PCR 9700 Thermocycler ABI Prism 7900HT and the sequence detection system (Applied Biosystems, Foster City, CA). Normalization was performed with let-7i. let-7-i was chosen because it had the lowest expression variability in the microarray patient dataset. Comparative real-time PCR was performed in triplicate, including no-template controls. Relative expression was calculated using the comparative Ct method.17
Data analysis
Microarray images were analyzed using GENEPIX PRO. Average values of the replicate spots of each miRNA were background subtracted; log2 transformed and normalized using a set of housekeeping genes (Table S1) and the BRB Array tools (http://linus.nci.nih.gov/BRB-ArrayTools.html). Absent calls were threshold to 22 (4.5 in log2 scale) before statistical analysis. This level is the average minimum intensity level detected above background in miRNA chip experiments. In 2 class comparisons (eg, CD34 vs AML), differentially expressed miRNAs were identified using the adjusted t test procedure within the Significance Analysis of Microarrays (SAM).18 The SAM Excel plug-in used here calculated a score for each gene on the basis of the change in expression relative to the standard deviation of all measurements. Because this was a multiple test, permutations were performed to calculate the false discovery rate (FDR) or q value. miRNAs with FDRs lower than 5% and fold changes larger than 2 were considered for further analysis. To investigate miRNAs that correlated with quantitative variables (eg, white cell counts), we used quantitative regression analysis within SAM. The microarray dataset was deposited at Array-Express (http://www.ebi.ac.uk/arrayexpress), array accession E-TABM-405.
Survival analysis and definitions
Overall survival (OS) was calculated from the time of diagnosis until the date of death (censoring for alive patients at the time of the last follow-up) and event-free survival (EFS) from the time of diagnosis until relapse or death (censoring for patients who were alive at the time of the last follow-up). In the first cohort of 122 AML patients, we used the SAM method, which involved a modified Cox proportional-hazards maximum-likelihood score, to identify a set of miRNAs whose expression significantly correlated with the duration of survival. Next we validated these miRNAs in an independent cohort of 60 newly diagnosed AML patients (Table 1) using quantitative RT-PCR. Univariate Cox proportional hazard method was used in this validation set of 60 patients to identify miRNAs associated with OS and EFS. Multivariate proportional-hazards analysis was then used to asses whether miRNAs could predict outcome independently from other factors (eg, cytogenetics and FLT-ITD+) using the R 2.4.0 software. To select best among all the multivariate models, we used the Akaike Information Criteria. Kaplan-Meier plots were used to display the association of miRNA with outcome. To generate the Kaplan-Meier plots, miRNA levels, measured by quantitative RT-PCR, were converted into discrete variables by splitting the samples into 2 classes (high and low expression, according to the median expression in the full set of samples). Survival curves were obtained for each group and compared using the log-rank test.
Statistical analysis
Fisher exact test, t test, and χ2 were used to compare baseline characteristics and average miRNA expression between groups of patients. All reported P values were 2-sided and obtained using the SPSS software package (SPSS 15.0 for Windows).
Results
AML patients reveal a distinct spectrum of miRNA expression compared with normal CD34+ progenitor cells
We compared 122 newly diagnosed AML samples (Table 1) with CD34+ cells from 10 normal donors for differential miRNA expression using a previously described and validated miRNA microarray platform.15 We identified 26 down-regulated miRNAs and none up-regulated in AML samples compared with CD34+ normal cells (Table 2). To validate these results, we performed quantitative RT-PCR for 7 of the down-regulated miRNAs (miR-126, miR-130a, miR-135, miR-93, miR-146, miR-106b, and miR-125a) using a subset of randomly chosen AML samples and 4 CD34+ samples obtained from different donors. As shown in (Figure 1A,B), we confirmed the down-regulation of the above miRNAs in AML samples with respect to the bone marrow CD34+ progenitors, except for miR-135.
Table 2.
MiRNAs down-regulated in 122 newly diagnosed AML patients with respect to CD34+ cells obtained from 10 healthy donors
| MicroRNA | SAM score* | Fold change | FDR, %† |
|---|---|---|---|
| hsa-miR-126 | −3.28 | 0.21 | 0 |
| hsa-miR-130a | −2.91 | 0.29 | 0 |
| hsa-miR-135 | −2.55 | 0.38 | 0 |
| hsa-miR-93 | −2.52 | 0.08 | 0 |
| hsa-miR-146 | −2.47 | 0.41 | 0 |
| hsa-miR-106b | −2.43 | 0.36 | 0 |
| hsa-miR-224 | −2.39 | 0.32 | 0 |
| hsa-miR-125a | −2.18 | 0.52 | 0 |
| hsa-miR-92 | −2.13 | 0.46 | 0 |
| hsa-miR-106a | −2.12 | 0.46 | 0 |
| hsa-miR-95 | −2.07 | 0.04 | 0 |
| hsa-miR-155 | −2.03 | 0.49 | 0 |
| hsa-miR-25 | −2.01 | 0.51 | 0 |
| hsa-miR-96 | −1.94 | 0.25 | 0 |
| hsa-miR-124a | −1.92 | 0.37 | 0 |
| hsa-miR-18 | −1.89 | 0.38 | 0 |
| hsa-miR-20 | −1.87 | 0.51 | 0 |
| hsa-let-7d | −1.81 | 0.48 | 0 |
| hsa-miR-26a | −1.76 | 0.48 | 0 |
| hsa-miR-222 | −1.71 | 0.51 | 0 |
| hsa-miR-101 | −1.67 | 0.51 | 0 |
| hsa-miR-338 | −1.54 | 0.31 | 0 |
| hsa-miR-371 | −1.51 | 0.38 | 0 |
| hsa-miR-199b | −1.44 | 0.03 | 0 |
| hsa-miR-29b | −1.41 | 0.12 | 0 |
| hsa-miR-301 | −1.37 | 0.47 | 0 |
SAM identifies genes with statistically significant changes in expression by assimilating a set of gene-specific scores (ie, paired t tests). Each gene is assigned a score on the basis of its change in gene expression relative to the standard deviation of repeated measurements for that gene. Genes with scores greater than a threshold are deemed potentially significant.
The percentage of such genes identified by chance is the q value or false discovery rate.
Figure 1.

miRNAs down-regulated in AML samples with respect to CD34+cells and mature and hematopoietic precursors. (A,B) We selected the most differentiated miRNAs according to SAM score and fold change and measured them in a random group of 6 AML patients and 4 CD34 samples obtained from healthy donors by quantitative RT-PCR. Results are presented as fold change of the miRNA expression in AML samples with respect to the CD34+ expression from one healthy donor after normalization with let-7i and 2Δ Ct conversion18 (thin bars represent standard deviations). The difference in miRNA expression between the 4 CD34s and all the 6 AML patients was statistically significant by the t test: miR-106a (P = .001), miR125a (P = .001), miR-126 (P = .001), miR-93(P = .001), miR-130a (P = .006), miR-146 (P = .001), except for miR-135 (P = .38). (C) Average miRNA expression (from 4 different healthy donors) of peripheral blood mature granulocytes and monocytes and bone marrow committed (erythrocytic and megakaryocytic) precursors and 6 AML patients compared with that of CD34+ cells after normalization and 2Δ Ct conversion. The results are presented as fold change, with respect to the CD34+ cells, average miRNA expression. The down-regulation of miRNA expression in mature peripheral blood cells and committed precursors with respect to CD34 cells was statistically significant by t test (P < .05).
In addition, to validate the results of the microarray platform, we performed quantitative RT-PCR for 42 miRNAs whose expression was high, intermediate, and low on the chip in 12 randomly chosen AML samples. As shown in Figure S1, the miRNA levels measured by the microarray and those measured by quantitative RT-PCR highly correlated (r = 0.92, P < .001), thereby validating the microarray platform as an analytical tool to measure miRNA expression.
A subset of miRNAs is associated with specific hematopoietic lineages
miRNA expression has been shown to be informative of the hematopoietic developmental lineage and differentiation stage of tumors.11 To determine how levels of the miRNAs most differentially expressed between AML samples and CD34+ cells related to the different hematopoietic lineages, we assessed the expression levels of 5 of 26 miRNAs (chosen according to the SAM scores) in a panel of human hematopoietic cells, which included mature granulocytes, monocytes, and erythrocyte and megakaryocyte precursors by quantitative RT-PCR. Among the miRNAs down-regulated in AML compared with normal CD34+ cells, miR-126, miR-130a, miR-93, miR-125a, and miR-146 were also significantly down-regulated in mature and precursor hematopoietic cells (Figure 1C).
miRNA-181a is down-regulated in AML with multilineage dysplasia
AML with multilineage dysplasia (MLD) occurs most frequently in older patients and is often associated with unfavorable cytogenetic profile and response to therapy.19 To investigate whether this group has a characteristic miRNA profile, we compared untreated AML patients with “de novo” or primary AML (n = 79) with respect to AML patients with MLD (n = 29) as defined by the WHO classification of AML.19 Using SAM, we identified only the down-regulation of miR-181a in AML with MLD (FDR 0%, FC > 2, SAM score of −1.68). Then we compared untreated “de novo” samples (n = 79) to untreated patients with therapy related AML (n = 12) and identified 3 up-regulated miRNAs in therapy-related AML patients (miR-190, miR-9, and miR-188, all with FDR of 0%, FC > 1.8, SAM score of > 1.8). We did not detect any significant difference of miRNA expression between AML with MLD and therapy-related AML.
miRNAs correlate positively to white blood cell and blast counts
We investigated whether miRNAs are associated with pretreatment patient characteristics, such as age, sex, white blood cell (WBC) count, bone marrow, or peripheral blood blast percentage using SAM quantitative analysis as described in “Methods.” We detected a positive correlation of several miRNAs (all with FDR of 0% and high SAM quantitative scores > 2), including miR-155 and miR-181b for WBC, peripheral and bone marrow blasts percentage, miR-30b and miR-30c for WBC, and bone marrow blast percentage and miR-25 for circulating blast percentages (Table S2).
miRNA signatures associated with defined cytogenetic subgroups
To identify miRNAs associated with known cytogenetic abnormalities in AML, we studied 116 pretreatment AML samples with known karyotype. SAM was used to detect miRNAs differentially expressed between defined cytogenetic groups versus other karyotypes, including normal karyotype. Because some cytogenetics subgroups were predominantly hybridized in one batch (eg, t(11q23) and normal karyotype), we validated the signatures using quantitative RT-PCR.
11q23 balanced translocations
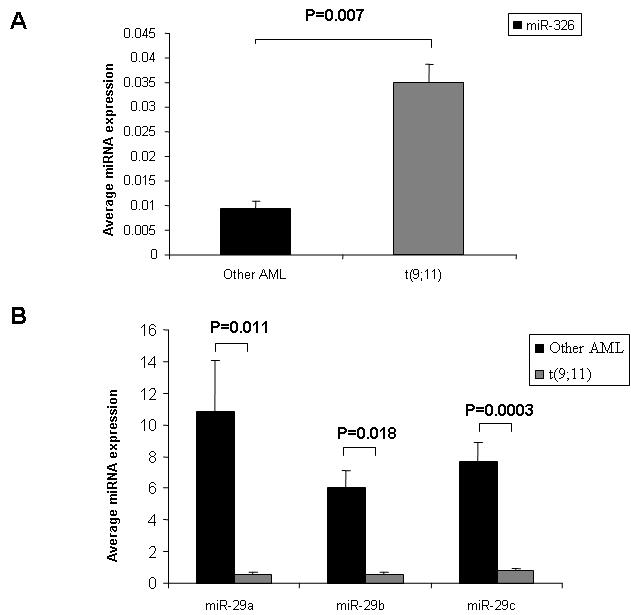
We identified 8 miRNAs up-regulated (miR-326, miR-219, miR-194, miR-301, miR-324, miR-339, miR-99b, miR-328) and 14 down-regulated (miR-34b, miR-15a, miR-29a, miR-29c, miR-372, miR-30a, miR-29b, miR-30e, miR-196a, let-7f, miR-102, miR-331, miR-299, miR-193) in patients with t(11q23) (n = 9) versus all other AML patients (Table S3). We validated the microarray results for selected miRNAs (chosen by higher SAM scores) using patient samples from the outcome validation signature cohort (non t(11q23), n = 10; and t(9;11), n = 3) by quantitative RT-PCR (Figure S2). Among the miRNAs down-regulated in balanced 11q23 translocation patients, many are tumor suppressor miRNAs that target critical oncogenes, that is, miR-34b (CDK4 and CCNE2),20 miR-15a (BCL-2),21 the let-7 family (RAS),22 the miR-29 family (MCL-1 and TCL-1),23,24 miR-372 (LATS2),25 and miR-196 (HOX-A7, HOX-A8, HOX-D8, HOX-B8).26 Next we asked whether miRNA expression differed between patients with t(6;11) (n = 4) and t(9;11) (n = 5). Sixteen miRNAs were up-regulated in patients with t(6;11) (Table S4), including the antiapoptotic miR-21, which targets the tumor suppressor PTEN27 and miR-26a and b, which target the TGFb1 regulator SMAD1.28 Down-regulation of SMAD1 has been suggested to be involved in the deregulation of TGFb1 associated with oncogenesis.29
Trisomy 8
The signature obtained using SAM was comprised of 42 up-regulated and no down-regulated miRNAs in patient samples with isolated trisomy 8 (n = 5) compared with all other AML patients with other karyotype after removing patients with secondary trisomy 8 (n = 5; Table S5). Among the up-regulated miRNAs, miR-124a and miR-30d are located at 8p21 and 8q23, respectively, suggesting that a gene dosage effect may play a role in their up-regulation. Interestingly, miR-124a targets the myeloid transcription factor CEBPA.30 Further work would be needed to investigate the role of miR-124a in the modulation of CEBPA in AML patients.
AML with normal karyotype
We first compared normal karyotype AML (NK-AML) patients to AML patients with abnormal karyotypes. We identified a signature in NK-AML composed of 10 up-regulated miRNAs (miR-10a, miR-10b, miR-26a, miR-30c, let-7a-2, miR-16–2, miR-21, miR-181b, miR-368, and miR-192) and 13 down-regulated miRNAs (miR-126, miR-203, miR-200c, miR-182, miR-204, miR-196b, miR-193, miR-191, miR-199a, miR-194, miR-183, miR-299, and miR-145) (Table S6). This signature was not predictive of NK-AML, probably because of the molecular heterogeneity of this subgroup (data not shown). We validated the microarray results for selected miRNAs using patient samples from the outcome validation signature cohort (NK-AML, n = 12; and abnormal karyotype AML, n = 22 by quantitative RT-PCR; Figure S3).
MiR-155 is overexpressed in FLT3-ITD mutations in AML patients
To identify miRNAs associated with the presence of FLT3-ITD mutations (FLT3-ITD+) in AML we first compared untreated AML patients with FLT3-ITD+ (n = 17) versus FLT3-wt (n = 73), excluding for FLT3-D835 mutations (n = 2) using SAM. We found 3 miRNAs up-regulated in FLT3-ITD+, miR-155 (3.1-fold), miR-10a (2.5-fold), and miR-10b (2.27-fold), all with FDR of 0 and SAM score above 2. There were not enough patients with FLT3-D 835 mutations (n = 2) to perform a statistical evaluation. We validated these results in an independent set of AML patients (16 patients from the outcome signature validation group) using quantitative RT-PCR. AML patients with FLT3-ITD+ (n = 4) had again higher miR-155 expression than FLT3-wt patients (n = 12, P = .007, t test; Figure 2).
Figure 2.

MiR-155 expression in AML with FLT3-ITD mutations. Average miR-155 expression in AML patients with FLT3-WT (n = 12) and FLT3-ITD positive mutations (n = 4) measured by quantitative RT-PCR. The miRNA expression between the different groups was compared using t test (SPSS).
miRNA expression in relapsed and primary refractory AML patients
By using our miRNA platform, we further investigated miRNA profiles of 54 patients with relapsed (n = 34) or primary refractory AML (n = 20; Table S7). This independent cohort of treated patient samples was obtained from patients different from the initial 122 cohort. No major differences between untreated (n = 122) and treated patients (n = 54) were detectable (data not shown). Using this set of 54 treated patients, we analyzed miRNA expression among the different cytogenetics and molecular subgroups (eg, AML with t(11q23) vs other karyotypes, FLT3-ITD+ vs FLT3-wt, etc) using SAM. Similar miRNAs signatures to those of the untreated patients were obtained (Tables S8–S10), thereby supporting the hypothesis that miRNA expression is largely driven by cytogenetics.
miRNAs associated with the outcome
We investigated survival and miRNA expression in 122 newly diagnosed AML patients. Here, we identified a small number of miRNAs with a FDR lower than 1% and a SAM survival score (Cox regression) higher than 2. All the identified genes, miR-199a, miR-199b, miR-191, miR-25, and miR-20a, when overexpressed, adversely affected OS. To validate this prognostic miRNA signature, we measured miR-199a, miR-191, miR-25, and miR-20a with a different technique (quantitative RT-PCR) in an independent group of 60 newly diagnosed AML patients (Table 1). Univariate Cox proportional hazard analysis was performed to determine the association of each miRNAs to OS and EFS. We confirmed the significant associations for miR-199a and miR-191 to OS (miR-199a, P = .001; miR-191, P = .03) and EFS (miR-199a, P = .002; miR-191, P = .02). We could not validate the association of miR-20 and miR-25 to OS (miR-20 P = .92; miR-25, P = .07) and EFS (miR-20, P = .8; miR-25, P = .07). To further confirm and display graphically the association of these miRNAs with outcome, miRNA expression levels measured by quantitative RT-PCR were converted into discrete variables by splitting the samples into 2 classes (high and low expression, according to the median expression in the full set of 60 samples), and Kaplan-Meier survival plots were generated. Patients with high expression of miR-199a and miR-191 were found to have significant shorter OS (Figure 3) and EFS (miR-199a, P = .002; and miR-191, P = .02, log-rank test).
Figure 3.

miRNAs associated with overall survival in newly diagnosed patients with AML. Kaplan-Meier estimates of overall survival for 60 AML patients with high or low expression of miR-191(A) and miR-199a (B) detected by quantitative RT-PCR. The log-rank test was used to compare differences between survival curves.
As expected, adverse cytogenetics at diagnosis, defined by the Cancer and Leukemia Group B criteria,31 was associated with OS and EFS by univariate Cox analysis (both with P < .001). Other characteristics, such as age (P = .48), white blood cells (P = .92), and FLT3-ITD+ (P = .2), were not significantly associated with OS nor EFS in this independent set of 60 AML patients (data not shown). The reasons behind the lack of survival association between FLT3-ITD mutations and our cohort of newly diagnosed patients may be the result of the number of patients with missing data (ie, no FLT3 test, n = 8) and the rather advanced age of the population studied (median = 59 years). Contrary to young AML patients, FLT3-ITD mutations have not been found associated with poor outcome in elderly patients with AML.32
To assess whether miRNAs could predict outcome independent from other factors (eg, cytogenetics), first we build a purely clinical model to predict OS and EFS using a Cox proportional hazard model, allowing any possible clinical covariates (WBC, FLT3-status, cytogenetics, and age). After applying the Akaike Information Criteria to eliminate redundant terms from the model, cytogenetics provided the best predictor for OS (hazard ratio = 3.87; 95% confidence interval, 1.83-8.18, P < .001) and EFS (hazard ratio = 3; 95% confidence interval, 1.47-6.10, P = .002). Then, we added the 4 miRNAs (miR-20a, miR-25, miR-191, and miR-199) as dichotomous miRNA variables (high or low miRNA expression, according to the median expression in the full set of samples) to the best clinical model. The best model keeps miR-191, miR-199, and cytogenetics for both OS and EFS (Table 3).
Table 3.
Influence of miRNAs on the clinical multivariate model for outcome prediction
| Hazard ratio | 95% CI | P | |
|---|---|---|---|
| Overall survival* | |||
| miR-191 high expression | 2.35 | 1.18-4.75 | .01 |
| miR-199 high expression | 2.81 | 0.96-8.23 | .05 |
| Adverse cytogenetics | 2.31 | 0.85-6.29 | .09 |
| Event-free survival† | |||
| miR-199 high expression | 2.57 | 0.89-2.57 | .08 |
| miR-191 high expression | 2.29 | 1.15-4.55 | .01 |
| Adverse cytogenetics | 1.78 | 0.66-1.49 | .24 |
Likehood ratio: 26.1 on 3 df (P < .001).
Likelihood ratio: 20.76 on 3 df (P < .001).
Discussion
In this study, we used a microarray platform to perform genome wide miRNome analysis of AML samples and normal progenitor CD34+ cells. Most miRNAs were down-regulated in AML patients with respect to CD34+ cells. Two recent studies have suggested widespread miRNA down-regulation during in vitro differentiation of CD34+ cells to several lineages.8,33 Our data confirmed that the most down-regulated miRNAs in AML with respect to CD34+ cells were also down-regulated in healthy precursors and mature peripheral blood myeloid cells, suggesting that a subset of miRNAs in leukemia follow closely the differentiation patterns of miRNA expression in normal hematopoiesis.
Here, we identified molecular signatures associated with several cytogenetic groups. The 2 strongest signatures were those associated with balanced 11q23 translocations and isolated trisomy 8. The down-regulation of miR-196, known to regulate HOX genes26 in patients harboring 11q23 translocations, suggests a novel mechanism to explain the up-regulation of several HOX genes in these patients. Using the microarray platform, we were also able to distinguish between t(6;11) and t(9;11). Among the up-regulated miRNAs in t(6;11), miR-21 has been found overexpressed in many solid tumors.10 Another study indicated that miR-21 targets PTEN,27 an important tumor suppressor, and antisense inhibition of miR-21 induces apoptosis of tumor cells “in vitro” and suppresses tumor growth in a xenograft mouse model.34 Aberrant expression of oncomiRs, such as miR-21 and miR-26 in t(6;11), may explain the worse prognosis of this subgroup of patients.31
In contrast, miR-29 family members, down-modulated in balanced 11q23 translocations target the oncogene TCL124 and MCL1,23 a critical apoptosis regulator found up-regulated in cells that are resistant to a variety of chemotherapeutic agents.35 Moreover, other miR-29 family members are down-regulated in high risk CLL36 and lung cancer.37
Interestingly, miR-155 was found to be up-regulated in AML patients with high white count and FLT3-ITD mutations. This miRNA has been recently described to block “in vitro” human myeloid colony formation,38 halt megakaryopoiesis,38 and induce B-cell lymphoma and leukemia in mice.39
In this study, there were few patients with favorable cytogenetics, such as inv(16) [4] and t(15;17) [4]. We were not able to identify any characteristic miRNA signature in these 2 groups of AML patients. The lack of correlation may be the result of heterogeneity within the groups and/or to the small sample size.
Although this study was not designed to answer whether miRNAs could predict outcome, here we describe a miRNA signature significantly associated with OS and EFS. Several observations strengthen our results. First, we identified miRNAs associated with survival despite the overall poor prognosis and short survival of the patients studied here, where outcome differences could be difficult to demonstrate. Second, high expression of miR-199a and miR-191 was also identified in patients with isolated trisomy 8, a subgroup of AML, which is associated with poor outcome.31 Third, the outcome signature is constituted of up-regulated miRNAs in common with the shared signatures of 6 solid tumors (eg, miR-20, miR-25, miR-199a, and miR-191).10 Further studies will be required to address the accuracy and independent prognostic significance of these miRNAs in predicting outcome.
In conclusion, our study demonstrates that a subset of miRNAs is clearly deregulated in AML and associated with cytogenetic groups and outcome.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health (PO1CA76259 and PO1CA81534, C.M.C.; P01 CA055164, M.A., S.M.K.; CA16058, C.D.B.), the Paul and Mary Haas Chair in Genetics (M.A.), Lauri Strauss Discovery grant (R.G.), Kimmel Cancer Research Foundation grant and CLL Global Research Foundation Grant (G.A.C.), AIRC and PRRIITT Regione Emilia Romagna GebbaLab grants (S.V.), and by the Leukemia Clinical Research Foundation (C.D.B.). Microarray analysis was in part performed using BRB Array Tools developed by Dr Richard Simon and Amy Peng Lam.
Footnotes
The online version of this article contains a data supplement.
Presented in part in abstract form at the 48th annual meeting of the American Society of Hematology, Orlando, FL, December 11, 2006: abstract 152.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: R.G. and S.V. performed experiments, analyzed the data, and wrote the manuscript; C.-G.L. performed the microarray experiments; C.F.C. and K.C. analyzed the microarray data; T.P., F.P., and M.F. performed experiments; H.A. performed and analyzed quantitative RT-PCR data; T.N., N.F., S.M.K., H.K., and M.A. provided patient samples and clinical data and designed research; and G.M., C.D.B., G.A.C., and C.M.C. designed research and edited the manuscript. All authors critically reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Carlo Croce, Ohio State University, CCC, Wiseman Hall, Room 385, 400 12th Ave, Columbus, OH 43210; e-mail: Carlo.croce@osumc.edu.
References
- 1.Lowenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med. 1999;341:1051–1062. doi: 10.1056/NEJM199909303411407. [DOI] [PubMed] [Google Scholar]
- 2.Burnett AK. Current controversies: which patients with acute myeloid leukemia should receive bone marrow transplantation? An adult theater's view. Br. J Haematol. 2002;118:357–364. doi: 10.1046/j.1365-2141.2002.03698.x. [DOI] [PubMed] [Google Scholar]
- 3.Drobyski WR. The role of allogeneic transplantation in high-risk acute myeloid leukemia. Leukemia. 2004;10:1565–1568. doi: 10.1038/sj.leu.2403482. [DOI] [PubMed] [Google Scholar]
- 4.Bartel D. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 5.Chen CZ, Li L, Lodish H, Bartel D. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–86. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- 6.Fazi F, Rosa A, Fatica A, et al. A mini-circuitry comprising microRNA-223 and transcription factors NFI-A and C/EBPa regulates human granulopoiesis. Cell. 2005;123:819–831. doi: 10.1016/j.cell.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 7.Felli N, Fontana L, Pelosi L, et al. MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via kit receptor down-modulation. Proc Natl Acad Sci U S A. 2005;102:18081–18086. doi: 10.1073/pnas.0506216102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garzon R, Pichiorri F, Palumbo T, et al. MicroRNAs fingerprints during human megakaryocytopoiesis. Proc Natl Acad Sci U S A. 2006;103:5078–5083. doi: 10.1073/pnas.0600587103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calin GA, Dumitru CD, Shimizu M, et al. Frequent deletions and down-regulation of micro-RNA genes miR-15 and miR-16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99:15524–15529. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Volinia S, Calin G, Liu CG, et al. A microRNA expression signature in human solid tumors defines cancer targets. Proc Natl Acad Sci U S A. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 12.Kornblau SM, Womble M, Cade JS, et al. Comparative analysis of the effects of sample source and test methodology on the assessment of protein expression in acute myelogenous leukemia. Leukemia. 2005;19:1550–1557. doi: 10.1038/sj.leu.2403845. [DOI] [PubMed] [Google Scholar]
- 13.Shaffer LG, Tommerup N, editors. Basel, Switzerland: S. Karger; 2005. ISCN 2005: An International System for Human Cytogenetic Nomenclature. [Google Scholar]
- 14.Kiyoi H, Naoe T, Nakano Y, et al. Prognostic implication of FLT3 and N-RAS gene mutations in acute myeloid leukemia. Blood. 1999;93:3074–3080. [PubMed] [Google Scholar]
- 15.Liu CG, Calin GA, Meloon B, et al. An oligonucleotide microchip for genomic-wide microRNA profiling in human and mouse tissues. Proc Natl Acad Sci U S A. 2004:11755–11760. doi: 10.1073/pnas.0403293101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen C, Ridzon DA, Broomer AJ, et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33:e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C (T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 18.Tusher VG, Tibshirani R, Chu G. Significant analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jaffe ES, Harris NL, Stein H, Vardinam JW, editors. Lyon, France: IARC; 2001. World Health Organization Classifications of Tumors. Pathology and Genetics of Tumors of Haematopoietic and Lymph Tissues. [Google Scholar]
- 20.He L, He X, Lim LP, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cimmino A, Calin GA, Fabbri M, et al. MiR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson SM, Grosshans H, Shingara J, et al. RAS is regulated by the let-7 microRNA family. Cell. 120:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 23.Mott JL, Kobayashi S, Bronk SF, Gores GJ. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene. 2007;26:6133–6140. doi: 10.1038/sj.onc.1210436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pekarsky Y, Santaman U, Cimmino A, et al. Tcl1 expression in chronic lymphocytic leukemia is regulated by miR-29 and miR-181. Cancer Res. 2006;66:11590–11593. doi: 10.1158/0008-5472.CAN-06-3613. [DOI] [PubMed] [Google Scholar]
- 25.Voorhoeve PM, Le Sage C, Schrier M, et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell. 2006;124:1169–1181. doi: 10.1016/j.cell.2006.02.037. [DOI] [PubMed] [Google Scholar]
- 26.Yekta S, Shih IH, Bartel DP. MicroRNA-directed cleavage of HOXB8 mRNA. Science. 2004;304:594–596. doi: 10.1126/science.1097434. [DOI] [PubMed] [Google Scholar]
- 27.Meng F, Henson R, Lang M, et al. Involvement of human micro-RNA in growth and response to chemotherapy in human cholangiocarcinoma cell lines. Gastroenterology. 2006;130:2113–2129. doi: 10.1053/j.gastro.2006.02.057. [DOI] [PubMed] [Google Scholar]
- 28.Lewis B, Shih I, Jones-Rhoades M, Bartel D, Burge C. Prediction of mammalian microRNA targets. Cell. 2003:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 29.Ryu B, Kern SE. The essential similarity of TGFbeta and activin receptor transcriptional responses in cancer cells. Cancer Biol Ther. 2003;2:164–170. doi: 10.4161/cbt.2.2.276. [DOI] [PubMed] [Google Scholar]
- 30.Lim LP, Lau LC, Garrett-Engele P, et al. Microarray analysis shows that some microRNAs down-regulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- 31.Zhou P, Qian L, Kozopas KM, et al. Mcl-1, a Bcl-2 family member, delays the death of hematopoietic cells under a variety of apoptosis-inducing conditions. Blood. 1997;89:630. [PubMed] [Google Scholar]
- 32.Stirewalt DL, Kopecky KJ, Meshinchi S, et al. FLT3, RAS, and TP53 mutations in elderly patients with acute myeloid leukemia. Blood. 2001;97:3589–3595. doi: 10.1182/blood.v97.11.3589. [DOI] [PubMed] [Google Scholar]
- 33.Felli N, Pelosi E, Beta R, et al. Lineage-specific expression and functional relevance of microRNA genes in normal hematopoiesis [abstract]. Blood. 2006;106:11a. Abstract 2263. [Google Scholar]
- 34.Si ML, Zhu S, Wu H, Lu Z, Wu F, Mo YY. MiR-21-mediated tumor growth Oncogene. 2007;26:2799–2803. doi: 10.1038/sj.onc.1210083. [DOI] [PubMed] [Google Scholar]
- 35.Byrd JC, Mrózek K, Dodge RK. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461). Blood. 2002;100:4325–4336. doi: 10.1182/blood-2002-03-0772. [DOI] [PubMed] [Google Scholar]
- 36.Calin GA, Ferracin M, Cimmino A, et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005;353:1793–1801. doi: 10.1056/NEJMoa050995. [DOI] [PubMed] [Google Scholar]
- 37.Yanaihara N, Caplen N, Bowman E, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006;9:189–198. doi: 10.1016/j.ccr.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 38.Georgantas G, Hildreth R, Morisot S, et al. CD34+ hematopoietic stem-cell progenitor cell microRNA expression and function: a circuit diagram of differentiation control. Proc Natl Acad Sci U S A. 2007;104:2750–2755. doi: 10.1073/pnas.0610983104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Costinean S, Zanesi N, Pekarsky Y, et al. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in Eμ-miR155 transgenic mice. Proc Natl Acad Sci U S A. 2006;103:7024–7029. doi: 10.1073/pnas.0602266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}