

Abstract
When a growing cell expands, lipids and proteins must be delivered to its periphery. Although this phenomenon has been observed for decades, it remains unknown how the secretory pathway responds to growth signaling. We demonstrate that control of Golgi phosphatidylinositol-4-phosphate (PI(4)P) is required for growth-dependent secretion. The phosphoinositide phosphatase SAC1 accumulates at the Golgi in quiescent cells and down-regulates anterograde trafficking by depleting Golgi PI(4)P. Golgi localization requires oligomerization of SAC1 and recruitment of the coat protein (COP) II complex. When quiescent cells are stimulated by mitogens, SAC1 rapidly shuttles back to the endoplasmic reticulum (ER), thus releasing the brake on Golgi secretion. The p38 mitogen-activated kinase (MAPK) pathway induces dissociation of SAC1 oligomers after mitogen stimulation, which triggers COP-I–mediated retrieval of SAC1 to the ER. Inhibition of p38 MAPK abolishes growth factor–induced Golgi-to-ER shuttling of SAC1 and slows secretion. These results suggest direct roles for p38 MAPK and SAC1 in transmitting growth signals to the secretory machinery.
Introduction
Cell growth relies on proportional delivery of proteins and lipids to the cell periphery. How the secretory pathway is regulated in response to growth signaling is unknown. Recent studies have shown that phosphoinositide (PI) lipids are essential regulators of intracellular membrane traffic (Di Paolo and De Camilli, 2006; for review see De Matteis et al., 2005). At the Golgi, a resident pool of phosphatidylinositol-4-phosphate (PI(4)P) promotes anterograde transport of secretory proteins (Walch-Solimena and Novick, 1999; Wang et al., 2003; Godi et al., 2004). Site-specific function of specific lipid kinases is essential for localized PI(4)P production at the Golgi (Godi et al., 1999; Wang et al., 2003; Weixel et al., 2005). Conversely, organelle-specific turnover by lipid phosphatases prevents the random equilibration of PI(4)P, thus maintaining spatially restricted Golgi pools of this PI (Hedin et al., 2004; Tahirovic et al., 2005).
Studies in yeast and mammals have shown that the transmembrane lipid phosphatase SAC1 is responsible for PI(4)P turnover at the ER and Golgi (Foti et al., 2001; Schorr et al., 2001; Rohde et al., 2003). Yeast Sac1p associates with the ER-specific enzyme dolicholphosphate-mannose synthase Dpm1p (Faulhammer et al., 2005). In contrast, ER localization of mammalian SAC1 is mediated by the canonical coatomer complex coat protein (COP) I binding motif missing in the yeast homologue (Rohde et al., 2003). It is unknown, however, how the distribution of SAC1 between the ER and Golgi is achieved. In addition, the cellular function of mammalian SAC1 at the Golgi has not been defined. Lipids and proteins must cooperate to regulate the transport and sorting functions performed by the Golgi. We hypothesized that PI levels at the Golgi may be regulated in a cell growth–dependent manner, thus coupling lipid signaling and anterograde traffic to cell proliferation. In this study, we analyzed the role of the lipid phosphatase SAC1 in mitogen-stimulated control of secretion.
Results
Growth-dependent shuttling of SAC1 between the ER and Golgi
We first analyzed the intracellular localization of human SAC1 in primary human fibroblasts by confocal fluorescence microscopy. During standard growth conditions in the presence of high serum, a large proportion of SAC1 colocalized with the ER marker protein disulfide isomerase (PDI; Fig. 1 A). Some SAC1 was also present at the Golgi, as shown by colocalization with the Golgi protein TGN46 (Fig. 1 A). When the cells were starved in the presence of low serum, a large proportion of SAC1 accumulated at the Golgi (Fig. 1 B). SAC1 shuttling from the ER to the Golgi occurred within 24–48 h during starvation (Fig. 1 C), likely coinciding with the transition to quiescence (Larsson et al., 1985). In contrast, serum addition to quiescent cells caused rapid translocation of SAC1 to the ER with a t 1/2 of ∼15 min (Fig. 1 D). The same redistribution of SAC1 occurred in cells treated with cycloheximide to block protein synthesis before serum stimulation, thus indicating that a preexisting pool of SAC1 translocated from the Golgi to the ER in a growth-dependent fashion (Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.20070109/DC1).
Figure 1.

Growth-dependent localization of SAC1 in fibroblasts. (A) Confocal immunofluorescence microscopy of human fibroblasts grown in the presence of 15% FCS. The cells were fixed, permeabilized, and costained with polyclonal anti-SAC1 (green) and monoclonal anti-PDI or anti-TGN46 antibodies (red). Bar, 15 μm. (B) Human fibroblasts were cultivated for 24 h in the presence of 0.5% FCS to induce quiescence. The quiescent cells were subsequently stimulated with 15% FCS for another 24 h. Cells were processed for confocal immunofluorescence microscopy as in A. Bar, 15 μm. (C) NIH3T3 cells were grown in 10% NBS and then starved in the presence of 0.5% serum for the indicated times. SAC1 was visualized by immunofluorescence microscopy. The ratio of mean fluorescence intensity in Golgi regions (FGolgi) to the mean fluorescence intensity in ER structures (FER) was calculated at different time points and used as a measure of SAC1 distribution. Values are means ± SD from individual cells (n = 7–12). (D) NIH3T3 cells were starved in 0.5% serum for 48 h and then stimulated with 10% serum. SAC1 was visualized by immunofluorescence analysis. The ratio between mean fluorescence intensity in the Golgi regions (FGolgi) and mean fluorescence intensity in ER-structures (FER) was calculated at different time points and used as a measure of SAC1 distribution. Values are means ± SD from individual cells (n = 10–15).
Oligomerization of SAC1 is required for translocation to the Golgi
Although human SAC1 contains a C-terminal ER retrieval signal (Fig. 2 A), this motif failed to prevent Golgi accumulation of SAC1 after serum starvation. Previous studies have shown that oligomerization of membrane cargo, such as plasma membrane receptors, can be linked to ER exit (Letourneur et al., 1995). To investigate whether SAC1 forms oligomers in response to growth conditions, we coexpressed a flag epitope-tagged version of SAC1 along with GFP-SAC1 in COS7 cells and probed for self-association. Very little GFP-SAC1 could be coimmunoprecipitated with flag-SAC1 when cells were grown in the presence of high serum, whereas the extent of this association was significantly higher in quiescent cells (Fig. 2 B). This data therefore indicates that SAC1 forms oligomers induced by serum starvation.
Figure 2.

Growth-dependent oligomerization of SAC1 regulates its ER–Golgi distribution. (A) Sequence motifs in SAC1 orthologues. Leucine residues within the putative LZ motif and lysine residues within the C-terminal ER retrieval motif are highlighted in magenta. (B and C) COS7 cells transiently expressing flag-SAC1 mutants and/or GFP-SAC1 were cultured either in the presence (10%) or absence (0.5%) of serum for 24 h and lysed, and flag-SAC1 was immunoprecipitated with M2 agarose beads. The bound proteins were separated by SDS-PAGE and analyzed by immunoblotting with anti-GFP, β-COP, or SEC23 antibodies. Black lines indicate that intervening lanes have been spliced out. (D and E) COS7 cells transiently expressing GFP-SAC1-K2A (D, green) or GFP-SAC1-LZ (E, green) were cultured either in the presence (10%) or absence (0.5%) of serum for 24 h and then subjected to immunofluorescence microscopy using either polyclonal anti-TGN46 (D, red) or anti-Sec61β (E, red). Bars, 15 μm. (F) COS7 cells were infected with adenoviruses to express flag-tagged versions of SAC1, SAC1-LZ, and phosphatase-dead SAC1-C/S. Cells were then lysed and SAC1 was collected on M2 agarose beads. After the elution with 200 μg/ml of M2 peptide, 2 μg of each purified SAC1 protein was assayed for phosphatase activity using dioctanoyl-PI(4)P as a substrate. Data are from three independent phosphatase measurements. Data represent means ± SD from three independent experiments.
Serum deprivation, which promoted GFP-SAC1/flag-SAC1 association, also allowed for coimmunoprecipitation of the COP-II subunit SEC23 (Fig. 2 B). These results suggest that only SAC1 oligomers interact with COP-II and that oligomerization of SAC1 is indeed a prerequisite for ER exit. A SAC1 mutant containing lysine to alanine substitutions within the C-terminal ER retrieval motif (SAC1-K2A; Rohde et al., 2003) displayed efficient serum-dependent oligomerization but failed to interact with COP-I under any condition (Fig. 2 B). Consequently, GFP-SAC1-K2A resided in the Golgi even in the presence of serum (Fig. 2 C). Interaction of flag-SAC1 with COP-II after serum starvation was also observed after treatment with the protein synthesis inhibitor cycloheximide (Fig. S1 B). These results demonstrate that SAC1 and SAC1-K2A continuously shuttle between the ER and Golgi and that the steady-state distribution of these proteins is determined by the efficiency of their recruitment into COP-I or -II vesicles.
An N-terminal leucine zipper (LZ) motif in SAC1 is required for oligomerization
To identify the critical regions within SAC1 required for its translocation to the Golgi, we generated GFP-tagged SAC1 mutants. A truncated GFP-SAC1 that lacked the entire N-terminal cytosolic domain (GFP-SAC1ΔN) failed to accumulate at the Golgi upon serum starvation (Fig. S1 C), indicating that the N-terminal domain of SAC1 is necessary for SAC1 redistribution to the Golgi. Mammalian SAC1 homologues contain a potential LZ motif within their N-terminal cytosolic domain (Fig. 2 A). LZs form coiled-coil secondary structures implicated in assembly of protein oligomers (Lupas, 1996). To examine whether growth-dependent ER-to-Golgi shuttling of SAC1 is regulated by LZ-dependent oligomerization, we generated the mutant SAC1-LZ, in which this motif was mutated. Alanine substitutions of the four leucines within the LZ abolished the ability of flag-SAC1 to form oligomers, bind to the COP-II complex, or shuttle to the Golgi under any growth conditions (Fig. 2, D and E). Measurements of phosphatase activity of affinity-purified flag-SAC1-LZ showed that the catalytic activity was nearly identical to that determined for flag-SAC1 (Fig. 2 F). This result therefore precludes that oligomerization of SAC1 is a prerequisite for phosphatase activity.
Mitogen activation of quiescent cells triggers Golgi-to-ER translocation of SAC1
Because the intracellular distribution of SAC1 changed significantly in response to serum levels, we surmised that serum constituents might be involved in controlling SAC1 trafficking. Serum growth factors are among the most important stimuli that trigger cell growth and survival. We therefore tested individual growth factors for their ability to induce SAC1 shuttling from the Golgi to the ER in quiescent NIH3T3 cells. Stimulation of quiescent cells with a combination of PDGF, FGF, and EGF caused the same prompt translocation of SAC1 to the ER as was observed after stimulation with serum (Fig. 3 A). When we analyzed individual growth factors for their ability to stimulate SAC1 redistribution from the Golgi to the ER in quiescent cells, we found that FGF and PDGF were equally potent in promoting SAC1 shuttling, whereas EGF was less effective (Fig. 3 B). This finding suggests that Golgi-to-ER shuttling of SAC1 is controlled by growth factor signaling.
Figure 3.

Role for MAPKs in growth-dependent SAC1 translocation to the ER. (A–C) Localization of the endogenous SAC1 in NIH3T3 cells was analyzed by immunofluorescence microscopy using polyclonal anti-SAC1 antibodies. Quiescent cells were stimulated by the addition of 10% serum (C) or a mix of EGF/basic FGF/PDGF (A) or individual growth factors (B) for 1 h at 37°C. (C) Quiescent cells were incubated with 10 μM of the MEK1/2 inhibitor UO126, 10 μM of the JNK MAPK inhibitor SP600125, or 10 μM p38 MAPK before stimulation with 10% serum. Bar, 15 μm.
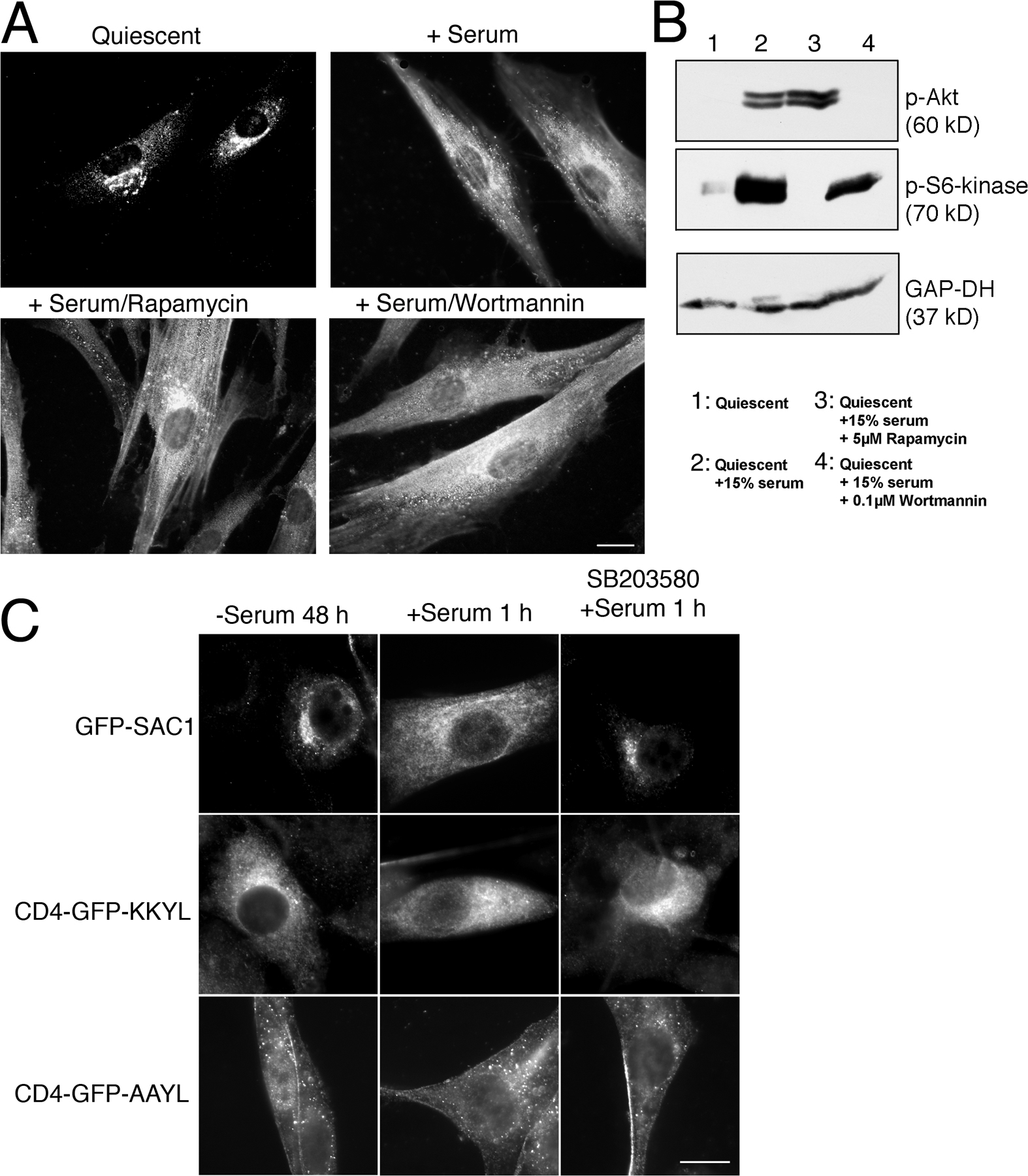
To determine which of the signaling pathways activated by growth factors induce Golgi-to-ER translocation of SAC1, we tested a variety of specific inhibitors in the SAC1 shuttling assay. Inhibition of PI3 kinase by wortmannin or blocking of mTOR by rapamycin did not affect growth-dependent redistribution of SAC1 (Fig. S2, A and B, available at http://www.jcb.org/cgi/content/full/jcb.20070109/DC1). However, inhibition of p38 MAPK by the specific inhibitor SB203580 was highly effective in blocking SAC1 translocation to the ER after serum stimulation (Fig. 3 C). Inhibition of MAPK/extracellular signal-regulated kinase (ERK) kinase (MEK) 1/2 by UO126, which selectively inactivates the downstream ERK1/2 MAPK modules (Johnson and Lapadat, 2002), also reduced Golgi-to-ER redistribution of SAC1, albeit to a lesser extent (Fig. 3 C). In contrast, the JNK MAPK inhibitor SP600125 (English and Cobb, 2002) had no effect (Fig. 3 C). Treatment with SB203580 did not prevent the ER localization of a GFP-CD4 chimera containing a KKXX ER retrieval motif (Zerangue et al., 2001), thus indicating that COP-I–mediated retrieval of cargo proteins to the ER was not generally blocked by p38 inhibition (Fig. S2 C). These results suggest that p38 MAPK selectively regulates Golgi-to-ER shuttling of SAC1. Addition of MEK1/2 or p38 MAPK inhibitors during normal growth conditions neither altered SAC1 localization (Fig. S3 A) nor affected Golgi morphology (Fig. S3 B). Consequently, mitogen-dependent Golgi-to-ER shuttling of SAC1 relies on MAPK activation, yet the steady-state distribution of SAC1 in proliferating cells does not require ongoing MAPK signaling.
Activation of the p38 MAPK pathway triggers dissociation of SAC1 oligomers and is required for mitogen-induced constitutive secretion
Our results show that p38 MAPK activity is necessary for SAC1 redistribution to the ER after mitogen stimulation. However, it remained possible that signaling through the p38 MAPK pathway plays merely a permissive role in this process. To address this question, we expressed dominant active alleles of the upstream kinases MKK3 and 6 (flag-MKK3(Glu) and flag-MKK6(Glu); Raingeaud et al., 1996) in starved COS7 cells. MKK3 and 6 have been shown to specifically activate p38 MAPK without stimulating any of the other MAPKs (Raingeaud et al., 1996). Expression of either flag-MKK3(Glu) or flag-MKK6(Glu) triggered a significant accumulation of SAC1 at the ER in starved cells, thus bypassing the requirement for additional stimulation by serum growth factors (Fig. 4 A). This result shows that activation of the p38 MAPK pathway is sufficient for triggering SAC1 trafficking.
Figure 4.

Effect of p38 MAPK on SAC1 oligomerization and constitutive secretion. (A) Expression of constitutively active alleles of MKK3 and 6 induces ER localization of SAC1 in serum-starved cells. COS7 cells were transfected with plasmids for expressing GFP-SAC1 (green) and flag-MKK6(Glu) (blue) or flag-MKK3(Glu) (blue). Cells were fixed, permeabilized, and costained with M2 antibodies (blue) and polyconal anti-TGN46 (red). Merged images show colocalization of GFP-SAC1 (green) and TGN46 (red). Bar, 15 μm. (B) COS7 cells transiently expressing flag-SAC1 and GFP-SAC1 were starved in the presence of 0.5% serum for 24 h and then stimulated with 10% serum with or without 10 μM SB203580. Cells were lysed and flag-SAC1 was immunoprecipitated with M2 agarose beads. Proteins were separated by SDS-PAGE followed by immunoblotting with anti-GFP or M2 antibodies. (C) COS7 cells transiently expressing flag-SAC1 and GFP-SAC1 were starved in the presence of 0.5% serum for 24 h and then stimulated with 10% serum in the presence of increasing concentrations of SB503280 (0.3–3 μM). Cells were lysed and flag-SAC1 was immunoprecipitated with M2 agarose beads. Proteins were separated by SDS-PAGE followed by immunoblotting with anti-GFP or M2 antibodies. GFP-SAC1 bands were quantified and compared with the relative levels of precipitated GFP-SAC1 from starved nonstimulated cells. Data represent means ± SD from three independent experiments. (D) NIH3T3 cells were grown as indicated. Pulse-chase analysis of protein secretion was conducted as described in Materials and methods. Data represent means ± SD from three independent experiments. (E) COS7 cells were transfected with a control plasmid without insert or with a plasmid for expressing flag-MKK3(Glu). Cells were serum-starved for 24 h and pulse-chase analysis of protein secretion was conducted as described in Materials and methods. Data represent means ± SD from three independent experiments.
Because oligomerization was an important determinant for SAC1 localization to the Golgi, we hypothesized that mitogens may directly regulate this process via downstream p38 MAPK signaling. Indeed, SAC1 oligomers could no longer be recovered after quiescent cells were stimulated with serum (Fig. 4 B). More importantly, blocking p38 activity with SB203580 showed dose-dependent inhibition of serum-induced dissociation of SAC1 oligomers (Fig. 4, B and C). This finding confirms that the oligomeric state of SAC1 is intimately linked to its intracellular localization and suggests that the p38 MAPK pathway controls SAC1 levels in the Golgi by regulating the proportion of SAC1 oligomers. Despite the critical role of p38 MAPK in mitogen-induced SAC1 shuttling, SAC1 is not phosphorylated via the p38 MAPK pathway (unpublished data) and the direct downstream target for p38 MAPK remains to be identified.
Several studies in yeast and mammalian cells have shown that PI(4)P is enriched at Golgi membranes and promotes Golgi trafficking (Stefan et al., 2002; Godi et al., 2004). Because PI(4)P is the major substrate for SAC1, p38 MAPK-mediated control of SAC1 localization may be a mechanism to regulate PI(4)P-dependent anterograde trafficking out of the Golgi. To examine the role of p38 MAPK in stimulating anterograde trafficking after mitogen stimulation, we analyzed constitutive trafficking out of the Golgi. To this end, we measured the discharge of newly synthesized 35S-labeled proteins into the extracellular medium in pulse-chase experiments. Serum stimulation of quiescent NIH3T3 cells resulted in a substantial increase in secretion compared with mock-treated cells (Fig. 4 D). This increase was significantly attenuated when p38 was inhibited by SB203580 (Fig. 4 D). Moreover, expression of the dominant active flag-MKK3(Glu) stimulated constitutive secretion in serum-starved COS7 cells (Fig. 4 E), further supporting a direct role for p38 in mitogen-dependent regulation of secretion.
Starvation-induced Golgi accumulation of SAC1 down-regulates PI(4)P and slows anterograde traffic
Metabolism of Golgi PIs is regulated by lipid kinases and phosphatases. To determine whether SAC1-mediated turnover of PI(4)P in the Golgi affects anterograde trafficking during starvation, we eliminated SAC1 in starved COS7 cells using RNAi. Depletion of SAC1 with specific siRNAs led to a substantial increase in constitutive secretion in serum-starved cells as compared with the basal secretion level in cells transfected with control siRNAs (Fig. 5 A). Knockdown of SAC1 in COS7 cells did not affect secretion rates measured after serum stimulation but largely abolished the effect of p38 inhibition on serum-stimulated constitutive secretion (Fig. 5 B). Moreover, RNAi-mediated knockdown of SAC1 in serum-starved COS7 cells led to a stabilization of the Golgi PI(4)P pool (Fig. 5 C). When both siRNAs against SAC1 and a RNAi-resistant phosphatase-dead version of SAC1 (GFP-SAC1-C/S*) were coexpressed in starved cells, Golgi PI(4)P remained prominent and was partially mislocalized to other membranes (Fig. 5 C). In contrast, Golgi PI(4)P was largely reduced when an RNAi-resistant version of wild-type SAC1 (GFP-SAC1*) was coexpressed with siRNAs against SAC1 (Fig. 5 C). Collectively, these results provide direct evidence that both activation of p38 MAPK signaling and SAC1 depletion up-regulate constitutive secretion in starved cells.
Figure 5.

RNAi-mediated depletion of SAC1 increases constitutive secretion and affects PI(4)P distribution in serum-starved cells. (A) COS7 cells were transfected with siRNAs directed against SAC1 or with mutated control siRNAs. Knockdown efficiency was determined by immunoblotting using anti-SAC1 and anti–glyceraldehyde-3-phosphate dehydrogenase (GAP-DH) antibodies. Cells were serum-starved for 24 h before pulse-chase analysis of protein secretion was conducted as described in Materials and methods. Data represent means ± SD from three independent experiments. (B) COS7 cells were transfected with siRNAs directed against SAC1 or with control siRNAs. Knockdown efficiency was determined by immunoblotting using anti-SAC1 and anti–glyceraldehyde-3-phosphate dehydrogenase antibodies. The cells were then serum-starved for 24 h and incubated in the presence or absence of 10 μM SB203580 for 30 min and then stimulated with serum for another 20 min followed by pulse-chase analysis of protein secretion as described in Materials and methods. Data represent means ± SD from three independent experiments. (C) COS7 cells were cotransfected with siRNAs directed against SAC1 and with plasmids for expressing either RNAi-resistant GFP-SAC1 (GFP-SAC1*, green) or an RNAi-resistant phosphatase-dead SAC1 mutant (GFP-SAC1-C/S*, green). Cells were serum starved for 24 h before being subjected to immunofluorescence microscopy using monoclonal anti-PI(4)P antibodies (red). Bar, 15 μm.
To further examine how starvation-induced redistribution of SAC1 to the Golgi affects Golgi PI(4)P and anterograde traffic, we used the SAC1-K2A mutant, which accumulates at the Golgi regardless of growth conditions (Fig. 2 C). Inhibition of p38 by SB203580 greatly reduced the release of 35S-labeled secretory proteins into the culture medium in mitogen-stimulated NIH3T3 cells expressing flag-SAC1 (Fig. 6 A). However, addition of growth factors to quiescent cells expressing flag-SAC1-K2A failed to stimulate secretion (Fig. 6 A). Furthermore, the reduced rate of secretion observed in flag-SAC1-K2A–expressing cells was not further diminished by SB203580-mediated inhibition of p38 (Fig. 6 A). This result indicates that the inability of SAC1 to shuttle out of the Golgi upon growth factor stimulation abolished p38-dependent stimulation of constitutive secretion.
Figure 6.

Localization of SAC1 to the Golgi down-regulates Golgi PI(4)P and slows anterograde traffic. (A) Quiescent NIH3T3 expressing flag-SAC1 or flag-SAC1-K2A was stimulated with 10% serum in the presence or absence of 10 μM SB203580. Pulse-chase analysis of protein secretion was conducted as described in Materials and methods. Data are means ± SD from three independent experiments. (B) COS7 cells infected with adenoviruses expressing either empty vector, flag-SAC1, flag-SAC1-K2A, or flag-SAC1-LZ were labeled with [3H]myo-inositol, and total cellular PIs were analyzed and quantified by HPLC analysis. Data are means ± SD from four to six independent experiments. *, P < 0.5 × 10−3. (C and D) Human fibroblasts infected with adenoviruses expressing either GFP-SAC1 (C, green) or GFP-SAC1-K2A (D, green) were grown in the presence of 15% (+serum) or 0.5% serum (quiescent) for 48 h. The cells were fixed, permeabilized, costained with anti-PI(4)P antibodies (red), and analyzed by immunofluorescence microcopy. Bars, 20 μm.
Expression of flag-SAC1-K2A in COS7 cells also led to a modest but significant reduction of total cellular PI(4)P (Fig. 6 B). To examine which pool of PI(4)P is down-regulated by SAC1 during serum starvation, we localized intracellular PI(4)P by immunofluorescence microscopy using a PI(4)P-specific antiserum. Proliferating human fibroblasts grown in the presence of high serum showed localization of PI(4)P in Golgi-specific regions (Fig. 6 C). Under these conditions, overexpressed GFP-SAC1 was largely confined to the ER (Fig. 6 C). In contrast, quiescent fibroblasts showed accumulation of GFP-SAC1 at the Golgi, which resulted in a dramatic elimination of PI(4)P from these regions (Fig. 6 C). In addition, tubular or filamentous PI(4)P-positive structures of unknown nature became visible. Overexpression of Golgi-confined GFP-SAC1-K2A led to elimination of Golgi PI(4)P even when cells were grown in the presence of high serum (Fig. 6 D), demonstrating that Golgi accumulation of SAC1 is sufficient to cause depletion of PI(4)P at this organelle.
Discussion
In this paper, we provide direct evidence linking growth factor signaling to lipid signaling at the Golgi. This mechanism involves the strictly controlled redistribution of the lipid phosphatase SAC1 between the ER and Golgi. Our data suggest that growth factor–dependent shuttling of SAC1 regulates the rate of anterograde trafficking out of the Golgi. It is well established that growth factor stimulation induces production of phosphorylated lipids at the plasma membrane. PI3 kinase isoforms generate PIs, such as PI(3,4,5)P3, which act as crucial second messengers in the recruitment of protein complexes involved in regulating cell proliferation, survival, and cytoskeletal arrangement (Vanhaesebroeck et al., 2005; Wymann and Marone, 2005). However, how signals for cell growth are transmitted to regulate membrane traffic is not well defined. Clearly, membrane dynamics within the secretory pathway must be coordinated with the rate of cell proliferation because cell growth and cell division rely heavily on ongoing delivery of proteins and lipids to the cell periphery. Our results define a novel regulatory element directly involved in controlling such secretory organellar traffic in response to growth factor signaling. Interestingly, this regulation does not appear to involve PI3 kinase but instead depends on p38 MAPK and to a lesser extent on ERK1/2 MAPK activities.
ERK signaling has also been implicated in cell growth–dependent regulation of Golgi dynamics during mitosis (Acharya et al., 1998; Shaul and Seger, 2006). Basal signaling through the yeast p38 MAPK homologue Hog1p was previously shown to be required for proper localization of a Golgi glycosyl transferase (Reynolds et al., 1998). In addition, stress-induced p38 MAPK signaling was shown to regulate endocytic trafficking (Cavalli et al., 2001). However, a specific function of p38 MAPK in regulating secretion rates has not been previously reported.
Several studies have documented that PI(4)P is highly concentrated at the Golgi and plays an essential role in secretion (for review see De Matteis et al., 2005). PI4 kinases, which generate this PI, are crucial for promoting anterograde trafficking of cargo out of the trans-Golgi network (Wang et al., 2003; Godi et al., 2004). The cellular function of lipid phosphatases that dephosphorylate PI(4)P has been less clear. SAC1 is the major PI(4)P phosphatase in secretory organelles in yeast and mammals (Foti et al., 2001; Schorr et al., 2001; Rohde et al., 2003). One primary function of SAC1 was thought to be in preventing the random dispersion of PI(4)P at intracellular membranes, thus maintaining compartmentalization of PI(4)P pools (Roy and Levine, 2004; Tahirovic et al., 2005). Our results establish a novel growth factor–dependent role for SAC1 that directly links control of Golgi PIs and secretion to cell proliferation (Fig. 7). In the course of serum starvation, SAC1 redistributes to the Golgi and down-regulates the Golgi-specific PI(4)P pool, which in turn lowers the rate of Golgi traffic. SAC1 appears to continuously cycle between the ER and Golgi, and its distribution between these two organelles largely depends on its oligomerization status and on selective recruitment by COP-II or -I complexes. Although our results establish SAC1 as the first enzyme playing a direct role in growth-dependent regulation of Golgi PIs, other components of the lipid signaling machinery may also respond to mitogens. Recent work in yeast has shown that Golgi localization of the PI4 kinase Pik1p is controlled by nutrients and cell proliferation (Faulhammer et al., 2007).
Figure 7.

Mitogen-dependent regulation of SAC1 and PI(4)P signaling at the Golgi. Schematic illustration of cell growth–dependent control of PI(4)P signaling and Golgi trafficking by SAC1 lipid phosphatase. In quiescent cells, SAC1 oligomerizes and accumulates in the Golgi, which in turn down-regulates Golgi PI(4)P and constitutive secretion. After growth factor stimulation, p38 MAPK activity is required for dissociation of SAC1 complexes, which triggers retrograde traffic and redistribution of SAC1 to the ER. Reduction of SAC1 levels at the Golgi allows for elevated concentration of PI(4)P at this organelle, thus accelerating constitutive secretion.
Most cells in multicellular organisms differentiate to perform specialized functions and then stop dividing. Under certain circumstances, such quiescent cells may be recruited to reenter the cell cycle. This return to growth is strictly controlled by mitogens and can lead to severe human disease if misregulated (Zeisberg et al., 2000; Hedin et al., 2004; Blagosklonny, 2006). MAPK family members are common elements of mitogenic signaling cascades that regulate gene expression, mitosis, movement, and metabolism (Johnson and Lapadat, 2002). We now show that activation of the p38 MAPK is sufficient for triggering Golgi-to-ER translocation of SAC1 in starved cells, which indicates that the p38 MAPK pathway plays a direct role in modulating ER and Golgi PIs upon mitogen stimulation (Fig. 7). p38 MAPK activity is required for dissociation of SAC1 oligomers at the Golgi, which is a prerequisite for rapid COP-I–dependent retrograde trafficking of SAC1 to the ER. Collectively, our results describe elements of a novel pathway for transduction of cell growth signals to the machinery of the secretory pathway. In addition, this study suggests that prompt up-regulation of secretion after growth factor stimulation may constitute a crucial early step in quiescent cell reactivation.
Materials and methods
Antibodies and reagents
Rabbit polyclonal anti-SAC1 serum (clone #69) was generated as described previously (Rohde et al., 2003). Antisera against phospho-Akt and phospho-S6 kinase were a gift from M. Iordanov (Oregon Health and Science University, Portland, OR). Sources for other antibodies are the following: mouse monoclonal anti-flag M2 (Sigma-Aldrich), rabbit polyclonal anti-GFP (Sigma-Aldrich), mouse monoclonal anti-PI(4)P (Echelon), mouse monoclonal anti–β-COP (clone mAD; Abcam), rabbit polyclonal anti-SEC23 (COP-II; Abcam), mouse monoclonal anti-PDI (clone RL90; Affinity BioReagents), goat polyclonal anti-Sec61α (Santa Cruz Biotechnology, Inc.), and sheep polyclonal anti-TGN46 (Serotec). Secondary antibodies conjugated either with HRP, TRITC, or FITC were obtained from Jackson ImmunoResearch Laboratories. Basic FGF, PDGF-AB, and mouse EGF were purchased from PeproTech. The MEK1/2 inhibitor UO126 and p38 MAPK inhibitor SB203580 were purchased from InvivoGen. The JNK inhibitor SP600125 was purchased from BIOMOL International, L.P. Rapamycin and wortmannin were purchased from EMD. Phosphatase inhibitor cocktail 1 was purchased from Sigma-Aldrich and Complete Mini EDTA-free protease inhibitor cocktail was purchased from Roche. [35S]-EXPRESS Easy Tag protein labeling mix, [32P]orthophosphate, and [3H]myo-inositol were purchased from PerkinElmer. All cell culture media were purchased from Thermo Fisher Scientific, FCS was purchased from Hyclone, and newborn bovine serum (NBS) was purchased from Sigma-Aldrich.
DNA constructs and recombinant adenoviruses
The plasmids for expressing flag-MKK3(Glu) and flag-MKK6(Glu) were a gift from R. Davis (University of Massachussetts Medical School, Worcester, MA). The plasmids for expressing CD4-GFP-KKYL and CD4-GFP-AAYL chimeras were obtained from B. Schwappach (University of Manchester, Manchester, UK). cDNAs encoding flag- and GFP-tagged human SAC1 and SAC1-K2A were generated as previously described (Rohde et al., 2003). The cDNA encoding SAC1-LZ containing four leucine-to-alanine substitutions (L105A, L112A, L119A, and L126A) was generated by site-directed mutagenesis using the primers sac1-1 (5′-GACAATGGCGCACTTAACTGATATTCAGGCACAAGATA-3′), sac1-2 (5′-TATCTTGTGCCTGAATATCAGTTAAGTGCGCCATTGTC-3′), sac1-3 (5′-CCTTCGCAGCGATGCTAAACCATGTCGCGAATGTGGA-3′), and sac1-4 (5′-TCCACATTCGCGACATGGTTTAGCATCGCTGCGAAGG-3′). The cDNA encoding GFP-SAC1ΔN was constructed by PCR using primers sac1-5 (5′-TGCAGATCTTAAGTGTTCCAAGGGACTGG-3′) and sac1-6 (5′-GGACTGCAGTCAGTCTATCTTTTCTTTCTGG-3′). The adenovirus system was obtained from K. Früh (Oregon Health and Science University). Recombinant adenoviruses expressing GFP-tagged SAC1, SAC1-K2A, and SAC1-LZ were produced as previously described (Hitt et al., 1997).
Cell lines, mitogen stimulation, transfections, and infections
Monkey kidney COS7 cells were grown as previously described (Rohde et al., 2003). Mouse embryonic fibroblast NIH3T3 cells (CRL-1658; American Type Culture Collection) and primary human fibroblasts (GM00442; Coriell Cell Repositories) were cultured according to the manufacturer's instructions. All cells were grown at 37°C in a humidified atmosphere of 5% CO2. To generate a quiescent cell population, cells were plated and cultured using reduced serum concentrations (0.3–0.5%) for 24–48 h. Stimulation of quiescent cells was performed with either 10 or 15% calf serum or with growth factors (5 ng/ml basic FGF, 3 ng/ml PDGF-AB, or 5 ng/ml EGF). Cells were transfected by electroporation essentially as previously described (Norcott et al., 1996) or infected with transactivator (MOI = 5) and recombinant adenoviruses for 24 (MOI = 10) or 48 h (MOI = 3).
Immunofluorescence microscopy and image analysis
Immunofluorescent staining was performed using paraformaldehyde-fixed and saponin-permeabilized cells as previously described (Blagoveshchenskaya et al., 2002). The mounting medium used was Prolong Antifade (Invitrogen). The fluorochromes were FITC, TRITC, and Alexa Fluor 350. Samples were viewed at 25°C using a microscope (Eclipse E800; Nikon) equipped with a camera (CoolSNAP HQ; Photometrics) and a 60× oil objective (1.45 NA). Confocal micrographs were obtained using a microscope (BX51; Olympus) and a 60× oil objective (1.45 NA; Plan Apo; Olympus). Image acquisition was performed using Metamorph Image software (7.0; MDS Analytical Technologies). Images were analyzed using Image J software (National Institutes of Health) and Photoshop CS (version 8.0; Adobe).
Quantification of the time course of SAC1 trafficking
Images of endogenous SAC1 immunofluorescence were captured at 25°C using a spinning disk confocal microscope (Eclipse TE2000-E; Nikon) equipped with a camera (Orca-ER; Hamamatsu) and a 60× oil objective (1.45 NA; Plan Apo). Image acquisition was performed using QED InVivo (Media Cybernetics, Inc.). All image processing was done with Metamorph software. Because the ER and Golgi occupy different z planes of the cell, images for quantification were made by condensing individual z series (15 planes, 0.5 μm apart) to a single plane by maximum intensity projection. The ratio of mean Golgi to ER fluorescence was used as a measure of SAC1 accumulation in the Golgi. Before analysis, the mean background from areas of the coverslip devoid of cells was subtracted from each image. For each cell, the fluorescence intensity along a 10-pixel-wide line that intersected both the ER and Golgi was used to measure the mean fluorescence intensity in both the Golgi and ER regions (using the linescan function in Metamorph). The boundaries of the Golgi and ER were determined morphologically from the SAC1 signal. The Golgi to ER ratios for each construct were calculated from a minimum of seven cells at each time point.
RNAi
Cells were transfected with 0.2 μM siRNA directed against SAC1 (5′-GGCGUGUUCCGAAGCAAUU-3′) using Lipofectamine 2000 (Invitrogen). Control siRNAs were generated by changing three nucleotides in the siRNA sequence (5′-AGCGUGGUCCGAAGCCAUU-3′). To express RNAi-resistant SAC1 proteins, five silent mutations were introduced into the region of recombinant SAC1 that is targeted by siRNAs. Transfected cells were assayed 72 h after transfection.
PI analysis
After 24 h of labeling with 10 μCi/ml [3H]myo-inositol in inositol-free MEM (ICN) supplemented with 10% dialyzed NBS, cells were washed with ice-cold PBS. Subsequently, 1 ml of ice-cold 0.5-M perchloric acid was added and the cells were scraped into an eppendorf tube. The resulting pellet was washed once with ice-cold 0.5-M perchloric acid. To solubilize lipids, 750 μl methanol/chloroform/HCl (80:40:1 vol/vol) was added to the pellet and mixed briefly. After incubation at room temperature for 30 min, 250 μl chloroform and 450 μl of 0.1-M HCl were added. After 1 min of centrifugation, the lower organic phase was transferred to a new tube and the upper aqueous phase was neutralized with 50 μl of 1-M NH4OH/methanol. The aqueous phase was reextracted twice with 225 μl methanol/chloroform/HCl (80:40:1 vol/vol), 75 μl chloroform, and 135 μl of 0.1-M HCl. The pooled organic phases were washed with 5 vol of 2-M KCl and dried in a SpeedVac (Savant Instruments).
Dried lipid pellet was deacylated by adding 200 μl methlyamine reagent and incubated in a sealed tube at 53°C for 30 min. The mixture was dried in a SpeedVac and the dried lipid pellet was redissolved in 500 μl of distilled water and 600 μl 1-butanol/petroleum ether/ethyl formate (H2O-saturated; 20:4:1 vol/vol), vortexed, and centrifuged for 1 min. The upper phase was discarded and this step was repeated once more with the lower phase, followed by final extraction with petroleum ether/ethyl formate (4:1, vol/vol). The lower phase was dried in a SpeedVac and the pellet was resuspended with 160 μl of 10-mM (NH4)4HPO4, pH 3.8.
HPLC analysis of glycerophosphoinositols was performed on a HPLC system (Jasco) equipped with an LB 508 Radioflow detector (Berthold Technologies). The following gradient for elution of the HPLC column was used: buffer A (distilled water) and buffer B (1 M (NH4)4HPO4, pH 3.8). The gradient was run at 0% buffer B for 10 min and increased to 25% buffer B over 60 min and then 20 min at 0% buffer B. The flow rate was 1 ml/min. Data from at least four independent experiments were used to calculate the respective mean values. To test for the statistical significance of difference between two percentages, t test analysis was performed.
Phosphatase assay
COS7 cells were infected with adenoviruses to express flag-tagged versions of SAC1, SAC1-LZ, or phosphatase-dead SAC1-C/S (Rohde et al., 2003). 2 d after infection, the cells were washed once with PBS and harvested in modified RIPA buffer (1% NP-40, 1% sodium deoxycholate, 150 mM NaCl, 50 mM Tris-HCl, and protease inhibitors, pH 8.0). After centrifugation at 13,000 g for 15 min, flag-tagged SAC1 proteins were collected on M2 agarose beads. The SAC1 proteins were eluted from the beads with 200 μg/ml M2 peptide in TBS for 2 h at 4°C. For measuring phosphatase activity, a modified version of a published protocol was used (Maehama et al., 2000). 24 μl of reaction buffer (200 mM sodium acetate, 100 mM Bis-Tris, 100 mM Tris, pH 6.0, 0.002% porcine gelatin, and 4 mM DTT), 1.4 μl of 1-M dioctanoyl-PI(4)P in water (Sigma-Aldrich) and 25 μl of eluate containing 2 μg of recombinant SAC1 protein was mixed and incubated at 37°C for various times. Reactions were stopped by the addition of an equal amount of 100 mM NEM, and 25 μl of each supernatant was pipetted into 96-well plates. 50 μl of Malachite green solution (1 vol of 4.2% ammonium molybdate in 4 M HCl and 3 vol of 0.045% Malachite green and 0.01% Tween 20) was added to each well. After incubation for 20 min at room temperature the OD620 was measured.
Secretion assay
NIH3T3 or COS7 cells were starved in DME supplemented with 0.5% serum or without serum, respectively, for 24 h to induce quiescence. Cells were either pretreated with 10 μM SB203580 or mock treated and then stimulated or not with 10% serum. Subsequently, cells were labeled with [35S]-EXPRESS for 15 min at 37°C. The cells were then washed and chased for various times at 37°C in the corresponding unlabeled DME. To determine the kinetics of secretion, the incubation medium was collected at different times and cells were lysed in modified RIPA buffer. 35S-labeled proteins were precipitated with 3% TCA from both media and lysates, collected on filters (GF/C; Whatman), and quantified by scintillation counting. For controls, cells were left on ice before supernatants were collected and analyzed.
Immunoprecipitation
COS7 cells were electroporated with 5 μg cDNAs encoding for SAC1 mutants. 2 d after transfection, the cells were washed once with PBS and harvested in colitose (coIP) buffer (PBS, pH 7.5, 1% NP-40, and protease inhibitor cocktail). After centrifugation at 13,000 g for 15 min at 4°C, the supernatants were precleared with agarose beads for 1 h at 4°C. The resulting supernatants were incubated with M2 agarose for 2 h at 4°C. The beads were collected by centrifugation and washed once with ice-cold coIP buffer, twice with coIP buffer containing 0.5 M NaCl, and once with PBS. The proteins were eluted in Laemmli buffer and analyzed by immunoblotting.
Online supplemental material.
Fig. S1 shows that SAC1 shuttling is not sensitive to cycloheximide. This figure also displays shuttling defects of an N-terminally truncated GFP-SAC1 mutant. Fig. S2 shows that Golgi-to-ER shuttling of SAC1 is not inhibited by wortmannin and rapamycin. This figure also shows that inhibition of p38 does not block general Golgi-to-ER traffic. Fig. S3 shows that MAPK inhibition does not affect steady-state distribution of SAC1 or Golgi morphology in proliferating cells. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200708109/DC1.
Supplementary Material
Acknowledgments
We thank Klaus Frueh, Roger Davis, and Blanche Schwappach for reagents. We also thank Michail Iordanov for reagents and helpful advice.
This research was supported by a grant from the National Institutes of Health/National Institute of General Medical Sciences to P. Mayinger (GM071569).
Abbreviations used in this paper: COP, coat protein; ERK, extracellular signal-regulated kinase; LZ, leucine zipper; MEK, MAPK/ERK kinase; PDI, protein disulfide isomerase; PI, phosphoinositide; PI(4)P, phosphatidylinositol-4-phosphate.
References
- Acharya, U., A. Mallabiabarrena, J.K. Acharya, and V. Malhotra. 1998. Signaling via mitogen-activated protein-kinase kinase (MEK 1) is required for Golgi fragmentation during mitosis. Cell. 92:183–192. [DOI] [PubMed] [Google Scholar]
- Blagosklonny, M.V. 2006. Target for cancer therapy: proliferating cells or stem cells. Leukemia. 20:385–391. [DOI] [PubMed] [Google Scholar]
- Blagoveshchenskaya, A.D., M.J. Hannah, S. Allen, and D.F. Cutler. 2002. Selective and signal-dependent recruitment of membrane proteins to secretory granules formed by heterologously expressed von Willebrand factor. Mol. Biol. Cell. 13:1582–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli, V., F. Vilbois, M. Corti, M.J. Marcote, K. Tamura, M. Karin, S. Arkinstall, and J. Gruenberg. 2001. The stress-induced MAP kinase p38 regulates endocytic trafficking via the GDI:Rab5 complex. Mol. Cell. 7:421–432. [DOI] [PubMed] [Google Scholar]
- De Matteis, M.A., A. Di Campli, and A. Godi. 2005. The role of the phosphoinositides at the Golgi complex. Biochim. Biophys. Acta. 1744:396–405. [DOI] [PubMed] [Google Scholar]
- Di Paolo, G., and P. De Camilli. 2006. Phosphoinositides in cell regulation and membrane dynamics. Nature. 443:651–657. [DOI] [PubMed] [Google Scholar]
- English, J.M., and M.H. Cobb. 2002. Pharmacological inhibitors of MAPK pathways. Trends Pharmacol. Sci. 23:40–45. [DOI] [PubMed] [Google Scholar]
- Faulhammer, F., G. Konrad, B. Brankatschk, S. Tahirovic, A. Knodler, and P. Mayinger. 2005. Cell growth–dependent coordination of lipid signaling and glycosylation is mediated by interactions between Sac1p and Dpm1p. J. Cell Biol. 168:185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulhammer, F., S. Kanjilal-Kolar, A. Knodler, J. Lo, Y. Lee, G. Konrad, and P. Mayinger. 2007. Growth control of Golgi phosphoinositides by reciprocal localization of sac1 lipid phosphatase and pik1 4-kinase. Traffic. 8:1554–1567. [DOI] [PubMed] [Google Scholar]
- Foti, M., A. Audhya, and S.D. Emr. 2001. Sac1 lipid phosphatase and stt4 phosphatidylinositol 4-kinase regulate a pool of phosphatidylinositol 4-phosphate that functions in the control of the actin cytoskeleton and vacuole morphology. Mol. Biol. Cell. 12:2396–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godi, A., P. Pertile, R. Meyers, P. Marra, G. Di Tullio, C. Iurisci, A. Luini, D. Corda, and M.A. De Matteis. 1999. ARF mediates recruitment of PtdIns-4-OH kinase-beta and stimulates synthesis of PtdIns(4,5)P2 on the Golgi complex. Nat. Cell Biol. 1:280–287. [DOI] [PubMed] [Google Scholar]
- Godi, A., A.D. Campli, A. Konstantakopoulos, G.D. Tullio, D.R. Alessi, G.S. Kular, T. Daniele, P. Marra, J.M. Lucocq, and M.A. Matteis. 2004. FAPPs control Golgi-to-cell-surface membrane traffic by binding to ARF and PtdIns(4)P. Nat. Cell Biol. 6:393–404. [DOI] [PubMed] [Google Scholar]
- Hedin, U., J. Roy, and P.K. Tran. 2004. Control of smooth muscle cell proliferation in vascular disease. Curr. Opin. Lipidol. 15:559–565. [DOI] [PubMed] [Google Scholar]
- Hitt, M.M., C.L. Addison, and F.L. Graham. 1997. Human adenovirus vectors for gene transfer into mammalian cells. Adv. Pharmacol. 40:137–206. [DOI] [PubMed] [Google Scholar]
- Johnson, G.L., and R. Lapadat. 2002. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 298:1911–1912. [DOI] [PubMed] [Google Scholar]
- Larsson, O., A. Zetterberg, and W. Engstrom. 1985. Cell-cycle-specific induction of quiescence achieved by limited inhibition of protein synthesis: counteractive effect of addition of purified growth factors. J. Cell Sci. 73:375–387. [DOI] [PubMed] [Google Scholar]
- Letourneur, F., S. Hennecke, C. Demolliere, and P. Cosson. 1995. Steric masking of a dilysine endoplasmic reticulum retention motif during assembly of the human high affinity receptor for immunoglobulin E. J. Cell Biol. 129:971–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupas, A. 1996. Coiled coils: new structures and new functions. Trends Biochem. Sci. 21:375–382. [PubMed] [Google Scholar]
- Maehama, T., G.S. Taylor, J.T. Slama, and J.E. Dixon. 2000. A sensitive assay for phosphoinositide phosphatases. Anal. Biochem. 279:248–250. [DOI] [PubMed] [Google Scholar]
- Norcott, J.P., R. Solari, and D.F. Cutler. 1996. Targeting of P-selectin to two regulated secretory organelles in PC12 cells. J. Cell Biol. 134:1229–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raingeaud, J., A.J. Whitmarsh, T. Barrett, B. Derijard, and R.J. Davis. 1996. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol. Cell. Biol. 16:1247–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds, T.B., B.D. Hopkins, M.R. Lyons, and T.R. Graham. 1998. The high osmolarity glycerol response (HOG) MAP kinase pathway controls localization of a yeast golgi glycosyltransferase. J. Cell Biol. 143:935–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde, H.M., F.Y. Cheong, G. Konrad, K. Paiha, P. Mayinger, and G. Boehmelt. 2003. The human phosphatidylinositol phosphatase SAC1 interacts with the coatomer I complex. J. Biol. Chem. 278:52689–52699. [DOI] [PubMed] [Google Scholar]
- Roy, A., and T.P. Levine. 2004. Multiple pools of phosphatidylinositol 4-phosphate detected using the pleckstrin homology domain of Osh2p. J. Biol. Chem. 279:44683–44689. [DOI] [PubMed] [Google Scholar]
- Schorr, M., A. Then, S. Tahirovic, N. Hug, and P. Mayinger. 2001. The phosphoinositide phosphatase Sac1p controls trafficking of the yeast Chs3p chitin synthase. Curr. Biol. 11:1421–1426. [DOI] [PubMed] [Google Scholar]
- Shaul, Y.D., and R. Seger. 2006. ERK1c regulates Golgi fragmentation during mitosis. J. Cell Biol. 172:885–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefan, C.J., A. Audhya, and S.D. Emr. 2002. The yeast synaptojanin-like proteins control the cellular distribution of phosphatidylinositol (4,5)-bisphosphate. Mol. Biol. Cell. 13:542–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahirovic, S., M. Schorr, and P. Mayinger. 2005. Regulation of intracellular phosphatidylinositol-4-phosphate by the Sac1 lipid phosphatase. Traffic. 6:116–130. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck, B., K. Ali, A. Bilancio, B. Geering, and L.C. Foukas. 2005. Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biochem. Sci. 30:194–204. [DOI] [PubMed] [Google Scholar]
- Walch-Solimena, C., and P. Novick. 1999. The yeast phosphatidylinositol-4-OH kinase pik1 regulates secretion at the Golgi. Nat. Cell Biol. 1:523–525. [DOI] [PubMed] [Google Scholar]
- Wang, Y.J., J. Wang, H.Q. Sun, M. Martinez, Y.X. Sun, E. Macia, T. Kirchhausen, J.P. Albanesi, M.G. Roth, and H.L. Yin. 2003. Phosphatidylinositol 4 phosphate regulates targeting of clathrin adaptor AP-1 complexes to the Golgi. Cell. 114:299–310. [DOI] [PubMed] [Google Scholar]
- Weixel, K.M., A. Blumental-Perry, S.C. Watkins, M. Aridor, and O.A. Weisz. 2005. Distinct Golgi populations of phosphatidylinositol 4-phosphate regulated by phosphatidylinositol 4-kinases. J. Biol. Chem. 280:10501–10508. [DOI] [PubMed] [Google Scholar]
- Wymann, M.P., and R. Marone. 2005. Phosphoinositide 3-kinase in disease: timing, location, and scaffolding. Curr. Opin. Cell Biol. 17:141–149. [DOI] [PubMed] [Google Scholar]
- Zeisberg, M., F. Strutz, and G.A. Muller. 2000. Role of fibroblast activation in inducing interstitial fibrosis. J. Nephrol. 13:S111–S120. [PubMed] [Google Scholar]
- Zerangue, N., M.J. Malan, S.R. Fried, P.F. Dazin, Y.N. Jan, L.Y. Jan, and B. Schwappach. 2001. Analysis of endoplasmic reticulum trafficking signals by combinatorial screening in mammalian cells. Proc. Natl. Acad. Sci. USA. 98:2431–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}