

SUMMARY
Expression of an oncogene in a primary cell can, paradoxically, block proliferation by inducing senescence or apoptosis through pathways that remain to be elucidated. Here we perform genome-wide RNA-interference screening to identify 17 genes required for an activated BRAF oncogene (BRAFV600E) to block proliferation of human primary fibroblasts and melanocytes. Surprisingly, we find a secreted protein, IGFBP7, has a central role in BRAFV600E-mediated senescence and apoptosis. Expression of BRAFV600E in primary cells leads to synthesis and secretion of IGFBP7, which acts through autocrine/paracrine pathways to inhibit BRAF-MEK-ERK signaling and induce senescence and apoptosis. Apoptosis results from IGFBP7-mediated upregulation of BNIP3L, a pro-apoptotic BCL2 family protein. Recombinant IGFBP7 (rIGFBP7) induces apoptosis in BRAFV600E-positive human melanoma cell lines, and systemically administered rIGFBP7 markedly suppresses growth of BRAFV600E-positive tumors in xenografted mice. Immunohistochemical analysis of human skin, nevi and melanoma samples implicates loss of IGFBP7 expression as a critical step in melanoma genesis.
INTRODUCTION
When expressed in primary cells, activated oncogenes can block cellular proliferation by inducing senescence or apoptosis (reviewed in Campisi, 2005; Mooi and Peeper, 2006). Oncogene-induced senescence and apoptosis are thought to play important roles in suppressing tumorigenesis by preventing proliferation of cells at risk for neoplastic transformation (Campisi, 2005; Michaloglou et al., 2005).
The BRAF proto-oncogene provides a paradigm for studying oncogene-induced senescence and apoptosis. BRAF is a serine-threonine protein kinase that functions as an immediate downstream effector of RAS (reviewed in Dhomen and Marais, 2007). BRAF activates the MAP kinase extracellular signal regulated kinase (MEK), which in turn phosphorylates and activates extracellular signal-regulated kinases 1 and 2 (ERK1 and ERK2).
Activating BRAF mutations are found at high frequency in human cancers and are particularly prevalent in melanoma where they occur at a frequency of 50-70% (Davies et al., 2002). Approximately 90% of activating BRAF mutations are a glutamic acid to valine substitution at position 600 (V600E; formally identified as V599E) (Davies et al., 2002). This mutation substantially increases protein kinase activity, resulting in constitutive BRAF-MEK-ERK signaling (Davies et al., 2002). In BRAFV600E-positive melanoma cell lines and mouse xenografts, BRAFV600E has been shown to be required for cell viability, proliferation and tumor growth (Hingorani et al., 2003; Hoeflich et al., 2006; Satyamoorthy et al., 2003; Sharma et al., 2005).
Activating BRAF mutations are also present in up to 82% of melanocytic nevi (Pollock et al., 2003), which are benign skin lesions that rarely progress to melanoma (Bennett, 2003; Chin et al., 1998). Nevi are growth-arrested and display classical hallmarks of senescence, including expression of senescence-associated β-galactosidase and the cell cycle inhibitor p16INK4a (Michaloglou et al., 2005; Sparrow et al., 1998; Wang et al., 1996). Expression of BRAFV600E also induces senescence in cultured primary human melanocytes (Michaloglou et al., 2005).
How, then, does an activated BRAF oncogene induce uncontrolled proliferation in melanoma and senescence in benign nevi? One hypothesis is that melanomas contain a second oncogenic lesion that inactivates the BRAF-mediated senescence pathway (reviewed in Campisi, 2005). Although attractive, this hypothesis remains to be proven largely because the downstream factors and signaling pathways involved in BRAF-mediated senescence have not been characterized. Here we perform a genome-wide RNA interference (RNAi) screen to identify factors required for BRAFV600E to block cellular proliferation.
RESULTS
A Genome-Wide shRNA Screen Identifies Factors Required for BRAFV600E-Mediated Senescence and Apoptosis
To identify genes required for BRAFV600E to block proliferation of primary cells, we performed a genome-wide small hairpin RNA (shRNA) screen (Figure 1A). The primary screen was performed in human primary foreskin fibroblasts (PFFs). A human shRNA library comprising ∼62,400 shRNAs directed against ∼28,000 genes was divided into 10 pools, which were packaged into retrovirus particles and used to stably transduce PFFs. The cells were then infected with a retrovirus expressing BRAFV600E under conditions in which all cells were infected. Cells that bypassed the BRAFV600E-mediated cellular proliferation block formed colonies, which were pooled and expanded, and the shRNAs identified by sequence analysis. Positive candidates were confirmed by stable transduction of PFFs with single shRNAs directed against the candidate genes, infection with the BRAFV600E-expressing retrovirus, and quantitation of cellular proliferation. Confirmed candidate shRNAs were then tested in a secondary screen for their ability to bypass the proliferation block in BRAFV600E-expressing primary human melanocytes.
Figure 1. A Genome-Wide shRNA Screen Identifies Factors Required for BRAFV600E-Mediated Senescence and Apoptosis.

(A) Schematic summary of the genome-wide shRNA screen.
(B) Proliferation of the 17 BRAFV600E/PFF KD cell lines. 1×104 PFF fibroblasts stably expressing the indicated shRNA were cultured in 12-well plates, infected with the BRAFV600E-expressing retrovirus and after 14 days stained with crystal violet.
(C) Quantitative proliferation assays of the 17 BRAFV600E/melanocyte KD cell lines. Melanocytes stably expressing the indicated shRNA were infected with the BRAFV600E-expressing retrovirus and after 14 days analyzed by trypan blue exclusion test. Growth of BRAFV600E/melanocytes is expressed relative to that of normal melanocytes. Growth of BRAFV600E/melanocyte KD cell lines is normalized to that of the corresponding melanocyte KD cell line in the absence of BRAFV600E expression. Error bars represent standard error.
(D) DNA replication assays of the 17 BRAFV600E/melanocyte KD cell lines, monitored by BrdU incorporation.
(E) Apoptosis assays of the 17 BRAFV600E/melanocyte KD cell lines, monitored by Annexin V-PE staining.
(F) Immunoblot analysis monitoring induction of p16INK4a and H3K9 acetylation in each of the 17 BRAFV600E/melanocyte KD cell lines. β-ACTIN (ACTB) was monitored as a loading control.
The screen identified 17 genes that, following shRNA-mediated knockdown, enabled BRAFV600E-expressing PFFs (BRAFV600E/PFFs) and melanocytes (BRAFV600E/melanocytes) to proliferate. These genes are listed in Supplemental Table S1 (see Supplemental Data available with this article online) and proliferation assays of the 17 BRAFV600E/PFF knockdown (KD) cell lines are shown in Figure 1B (for quantitation, see Figure S1A). Expression of BRAFV600E in PFFs (data not shown) or in PFFs containing a control non-silencing (NS) shRNA (Figure 1B) efficiently inhibited cellular proliferation. Significantly, however, this block was overcome in all 17 BRAFV600E/PFF KD cell lines. Quantitative real-time RT-PCR (qRT-PCR) confirmed in all cases that expression of the target gene was decreased in the corresponding KD cell lines (Figure S1B). For all 17 genes, a second, unrelated shRNA directed against the same target gene also enabled PFFs to proliferate following BRAFV600E expression (Figure S1C).
As expected from previous studies (Michaloglou et al., 2005), expression of BRAFV600E in primary melanocytes efficiently blocked cellular proliferation (Figure 1C). By contrast, BRAFV600E failed to block cellular proliferation in all 17 melanocyte KD cell lines.
Following expression of BRAFV600E in melanocytes, the majority of cells became senescent (Figure 1D and data not shown), consistent with previous studies (Michaloglou et al., 2005), although we found ∼10% of cells underwent apoptosis (Figure 1E). To determine the role of the 17 genes in these two pathways, apoptosis and senescence assays were performed in each melanocyte KD cell line following BRAFV600E expression. Figure 1E shows that only three of the 17 genes were required for apoptosis: BNIP3L, which encodes a pro-apoptotic BCL2 family protein; SMARCB1, which encodes a component of the SWI/SNF chromatin remodeling complex; and insulin growth factor binding protein 7 (IGFBP7), which encodes a secreted protein with weak homology to IGF binding proteins. By contrast, all but one of the 17 genes, BNIP3L, were required for BRAFV600E to induce growth arrest (Figure 1D) and characteristic markers of senescence (see below). Identical results were obtained using BRAFV600E/melanocyte KD cell lines that had undergone an additional 15 population doublings (Figure S2A) and in PFFs (Figure S2B).
p16INK4a has been proposed to play an important role in replicative and oncogene-induced senescence (reviewed in Ben-Porath and Weinberg, 2005). We were therefore interested in determining whether the genes identified in our screen were required for p16INK4a induction. Figure 1F shows, as expected, that p16INK4a levels increased substantially following BRAFV600E expression in control melanocytes expressing an NS shRNA. Significantly, p16INK4a expression was not induced by BRAFV600E in 16 of the 17 melanocyte KD cell lines. The sole exception was the cell line knocked down for BNIP3L, which, as described above, is specifically involved in apoptosis (Figure 1F). Loss of histone H3 lysine 9 (H3K9) acetylation, another well characterized senescence marker (Narita et al., 2006), also occurred following BRAFV600E expression in control melanocytes but not in any of the melanocyte KD cell lines except for the BNIP3L KD cell line (Figures 1F and S2A).
A Secreted Protein, IGFBP7, Induces Senescence and Apoptosis through Autocrine/Paracrine Pathways
One of the genes required for the induction of both senescence and apoptosis was IGFBP7, which encodes a secreted protein (Wilson et al., 1997), raising the possibility that the BRAFV600E-mediated block to cellular proliferation might occur through an autocrine/paracrine pathway. To determine whether IGFBP7 is secreted and functions extracellularly, we analyzed the ability of conditioned medium (CM) from BRAFV600E/melanocytes to induce senescence. Figure 2A (top panel) shows that following expression of BRAFV600E in melanocytes, the level of IGFBP7 in CM increased substantially. Addition of CM from BRAFV600E/melanocytes to naïve melanocytes blocked cellular proliferation, primarily resulting from the induction of senescence (bottom panel and see Figure 2D below).
Figure 2. A Secreted Protein, IGFBP7, Induces Senescence and Apoptosis through an Autocrine/Paracrine Pathway.

(A) (Top) Immunoblot analysis of IGFBP7 levels in CM from normal melanocytes, BRAFV600E/melanocytes, BRAFV600E/melanocytes stably expressing an IGFBP7 shRNA or in BRAFV600E/melanocyte CM treated with an α-IGFBP7 antibody. (Bottom) Proliferation assays on naïve melanocytes following addition of the different CMs described above. Proliferation was measured and normalized to the growth of untreated melanocytes. Error bars represent standard error.
(B) Coomassie-stained gel of purified, recombinant IGFBP7 (rIGFBP7). Molecular weight markers are shown on the left, in kDa.
(C) Proliferation assay monitoring the effect of rIGFBP7 on the growth of melanocytes 14 days after treatment.
(D) β-galactosidase staining of melanocytes infected with a retrovirus expressing either empty vector or BRAFV600E, or melanocytes treated with CM from BRAFV600E/melanocytes or rIGFBP7. Images are shown at a magnification of 10X, 20X and 40X.
(E) Proliferation assay monitoring growth rates of untreated melanocytes, or melanocytes stably expressing an NS or IGFBP7 shRNA.
Two experiments verified that IGFBP7 activation was downstream of BRAF-MEK-ERK signaling. First, BRAFV600E-mediated induction of IGFBP7 was blocked by addition of a MEK inhibitor (Figure S3A). Second, expression of a constitutively activated ERK mutant (ERK2Q103A or ERK2L73P,S151D) was sufficient to activate IGFBP7 transcription (Figure S3B).
The IGFBP7 promoter contains a consensus binding site for the dimeric AP-1 (JUN/FOS) transcription factor (Figure S4). Significantly, JUN (also known as c-Jun) is activated through RAF-MEK-ERK signaling (Leppa et al., 1998), raising the possibility that AP-1 is involved in BRAFV600E-mediated induction of IGFBP7. Chromatin immunoprecipitation (ChIP) analysis verified that JUN bound to the IGFBP7 promoter in response to BRAFV600E expression (Figure S4A), and siRNA-mediated knockdown of JUN abrogated induction of IGFBP7 transcription in BRAFV600E/melanocytes (Figure S4B).
We next sought to verify that IGFBP7 was the secreted protein responsible for the BRAFV600E-mediated cellular proliferation block. In one experiment, we treated BRAFV600E/melanocytes with an shRNA targeting IGFBP7. Figure 2A shows that IGFBP7 was absent from the CM of BRAFV600E/melanocytes expressing an IGFBP7 shRNA (top panel), and that this CM did not inhibit cellular proliferation of naïve melanocytes (bottom panel). In a second experiment, immunodepletion with an α-IGFBP7 antibody efficiently removed IGFBP7 from CM of BRAFV600E/melanocytes (top panel) and this immunodepleted CM failed to inhibit cellular proliferation of naïve melanocytes (bottom panel).
To confirm that IGFBP7 could block cellular proliferation, we purified recombinant IGFBP7 (rIGFBP7) from baculovirus-infected insect cells. Figure 2B shows that following expression and purification, a polypeptide of ∼33 kDa was detected, the expected size of IGFBP7. Addition of rIGFBP7 blocked proliferation of primary melanocytes in a dose-dependent manner (Figure 2C). The growth-arrested cells had an enlarged flat morphology, characteristic of senescent cells, and stained positively for senescence-associated β-galactosidase (Figure 2D).
The finding that primary melanocytes expressed IGFBP7 (Figure 2A and see below) raised the possibility that under normal conditions IGFBP7 might regulate melanocyte proliferation. To test this idea, we compared the proliferation rates of untreated melanocytes, control melanocytes expressing an NS shRNA, and melanocytes expressing an IGFBP7 shRNA. Figure 2E shows that melanocyte proliferation increased following IGFBP7 knockdown. Thus, normal melanocytes express low levels of IGFBP7, which restrains proliferation. When present at high levels, such as following expression of BRAFV600E, IGFBP7 induces senescence.
Selective Sensitivity of Melanoma Cell Lines Containing an Activating BRAF Mutation to IGFBP7-Mediated Apoptosis
We next analyzed the ability of IGFBP7 to block cellular proliferation in a panel of human melanoma cell lines. The cells contained either an activating BRAF mutation (BRAFV600E; SK-MEL-28, MALME-3M, WM793B, WM39 and WM278), an activating RAS mutation (RASQ61R; SK-MEL-2, SK-MEL-103 and WM1366), or were wild type for both BRAF and RAS (CHL, SK-MEL-31, WM1321 and WM3211). For each cell line, the presence of IGFBP7 in the CM was determined by immunoblot analysis (Figure 3A) and sensitivity to IGFBP7-induced growth inhibition was measured in a proliferation assay (Figure 3B). The results reveal a striking inverse correlation between IGFBP7 expression and sensitivity to IGFBP7-mediated growth inhibition that correlates with the status of BRAF or RAS. Most importantly, melanoma cell lines harboring an activating BRAF mutation fail to express IGFBP7 and are highly sensitive to IGFBP7-mediated growth inhibition. By contrast, cells that are wild type for BRAF and RAS express IGFBP7 and are relatively insensitive to IGFBP7-mediated growth inhibition. Finally, melanoma cell lines containing an activating RAS mutation express low levels of IGFBP7 and are partially sensitive to IGFBP7-mediated growth inhibition. We further analyzed the IGFBP7-mediated cellular proliferation block with regard to apoptosis and senescence. Significantly, in melanoma cell lines harboring an activating BRAF mutation, rIGFBP7 strongly induced apoptosis and surviving senescent cells were undetectable (Figure 3C and data not shown). Thus, IGFBP7 primarily induced senescence in melanocytes and apoptosis in BRAFV600E-positive melanoma cells.
Figure 3. Selective Sensitivity of Melanoma Cell Lines Containing an Activating BRAF Mutation to IGFBP7-Mediated Apoptosis.

(A) (Top) Immunoblot analysis monitoring IGFBP7 levels in the CM from a panel of human melanoma cell lines. (Bottom) Quantitative real-time RT-PCR analysis of IGFBP7 expression. Error bars represent standard error.
(B) Proliferation assays of human melanoma cell lines 24 hrs after rIGFBP7 treatment. Proliferation was normalized to the growth of the corresponding cell line in the absence of rIGFBP7 addition.
(C) Apoptosis assays of human melanoma cell lines treated with rIGFBP7.
(D) (Top), Immunoblot analysis in SK-MEL-28 cells in the presence or absence of rIGFBP7 and stably expressing either an NS, SMARCB1 or BNIP3L shRNA. (Bottom), Schematic summary of the IGFBP7-mediated apoptotic pathway.
(E) ChIP analysis monitoring STAT1 recruitment to the SMARCB1 promoter in SK-MEL-28 cells.
(F) qRT-PCR analysis of SMARCB1 (left) or STAT1 (right) mRNA levels in SK-MEL-28 cells following treatment with an NS or STAT1 siRNA.
(G) ChIP analysis monitoring SMARCB1 (left) or BRG1 (right) recruitment to the BNIP3L promoter in SK-MEL-28 cells.
(H) SK-MEL-28 cells were incubated with rIGFBP7 for 0, 2, 6, 12 or 24 hrs, following which the cells were washed and cultured in medium lacking rIGFBP7 and apoptosis was quantitated.
To understand the basis of this differential response, we analyzed expression of the 17 genes in primary melanocytes and SK-MEL-28 melanoma cells. Figure S5A (left panel) shows that in primary melanocytes, expression of BRAFV600E resulted in the transcriptional upregulation of seven genes, which are involved in apoptosis (BNIP3L, IGFBP7 and SMARCB1) and senescence (PEA15, IGFBP7, MEN1, FBXO31, SMARCB1 and HSPA9). BRAFV600E-mediated induction of all seven genes did not occur following knockdown of IGFBP7 (Figure S5B). Following addition of rIGFBP7 to melanocytes, six of the seven genes were induced, IGFBP7 being the exception (Figure S5A, middle panel). Significantly, following addition of rIGFBP7 to SK-MEL-28 cells, neither IGFBP7 nor PEA15 were upregulated (Figure S5A, right panel). PEA15, a known regulator of BRAF-MEK-ERK signaling (Formstecher et al., 2001), is required for senescence (see Figure 1F). Thus, the lack of PEA15 induction in IGFBP7-treated SK-MEL-28 cells can explain their failure to undergo senescence. We note that BNIP3L is only modestly upregulated in primary melanocytes following expression of BRAFV600E or addition of rIGFBP7, consistent with the relatively low level of apoptosis in IGFBP7-treated melanocytes (see Figure 1E).
IGFBP7 Induces Apoptosis through Upregulation of BNIP3L
As described above, BRAFV600E-mediated apoptosis was dependent upon IGFBP7, SMARCB1 and BNIP3L, raising the possibility that these three proteins were components of a common pathway required for apoptosis. We performed a series of experiments to confirm this idea and establish the order of the pathway. Figure 3D shows that following addition of rIGFBP7 to SK-MEL-28 cells, expression of SMARCB1 and BNIP3L were significantly increased, and apoptosis occurred as evidenced by caspase 3 activation. Expression of a SMARCB1 shRNA blocked induction of BNIP3L and apoptosis. By contrast, expression of a BNIP3L shRNA still resulted in induction of SMARCB1 following rIGFBP7 addition although apoptosis did not occur. Collectively, these results reveal a pathway in which IGFBP7 increases expression of SMARCB1, which in turn leads to increased expression of BNIP3L culminating in apoptosis (Figure 3D, bottom panel).
In BRAFV600E/melanocytes, induction of SMARCB1 and BNIP3L was blocked following IGFBP7 knockdown (Figure S6A). Moreover, addition of CM from BRAFV600E-expressing melanocytes to naïve melanocytes substantially upregulated SMARCB1 and BNIP3L, which did not occur with various control CMs that lacked IGFBP7 (Figure S6B). Thus, in BRAFV600E/melanocytes induction of SMARCB1 and BNIP3L is also dependent upon and downstream of IGFBP7.
We next sought to determine the mechanistic basis for IGFBP7-mediated induction of BNIP3L and SMARCB1. A previous study analyzing genome-wide targets of STAT proteins had provided evidence that STAT1 was involved in certain SMARCB1-inducible transcription responses, and had shown that the SMARCB1 promoter contains a STAT1 binding site located ∼2.4 kb upstream of the transcription start-site (Hartman et al., 2005). We therefore investigated the potential role of STAT1 in IGFBP7-mediated induction of SMARCB1 transcription. ChIP experiments revealed that following addition of rIGFBP7 to SK-MEL-28 cells, STAT1 was recruited to the SMARCB1 promoter (Figure 3E), and shRNA-mediated knockdown experiments confirmed that STAT1 was required for IGFBP7-mediated upregulation of SMARCB1 (Figure 3F).
As described above, SMARCB1 is required for upregulation of BNIP3L by IGFBP7 (Figure 3D). ChIP experiments revealed that following addition of rIGFBP7, SMARCB1 as well as BRG1, an essential subunit of the SWI/SNF complex (Bultman et al., 2000), were recruited to the BNIP3L promoter near the transcription start-site (Figure 3G). Following knockdown of SMARCB1, BRG1 (and, as expected, SMARCB1) failed to associate with the BNIP3L promoter. Collectively, these results indicate that IGFBP7 stimulates BNIP3L transcription, at least in part, by increasing intracellular levels of SMARCB1, leading to formation of a SMARCB1-containing SWI/SNF chromatin-remodeling complex, which is recruited to the BNIP3L promoter and facilitates BNIP3L transcriptional activation.
Finally, we asked whether apoptosis was dependent upon the continual presence of rIGFBP7 or was irreversible following transient exposure to rIGFBP7. SK-MEL-28 cells were incubated with rIGFBP7 for various lengths of time, following which the cells were washed and cultured in medium lacking rIGFBP7, and apoptosis was quantitated after 24 hours. Figure 3H shows that following 6 hours of incubation with rIGFBP7, the cells were irreversibly committed to apoptosis, which occurred even after removal of rIGFBP7.
IGFBP7 Inhibits BRAF-MEK-ERK Signaling
In BRAFV600E-positive melanoma cells BRAF-MEK-ERK signaling is hyper-activated, rendering the cells highly dependent on this pathway. Thus, treatment of BRAFV600E-positive melanoma cells with a BRAF shRNA (Hoeflich et al., 2006) or an inhibitor of BRAF (Sharma et al., 2005) or MEK (Solit et al., 2006) blocks cellular proliferation. We therefore considered the possibility that IGFBP7 blocks cellular proliferation, at least in part, by inhibiting BRAF-MEK-ERK signaling.
To test this idea we added rIGFBP7 to SK-MEL-28 cells and analyzed the levels of total and activated ERK (phospho-ERK). Figure 4A shows that addition of rIGFBP7 resulted in a dose-dependent loss of phospho-ERK. Similarly, expression of BRAFV600E in melanocytes markedly decreased phospho-ERK levels, which did not occur in BRAFV600E/melanocytes expressing an IGFBP7 shRNA (Figure S7A). Moreover, addition of CM from BRAFV600E/melanocytes to naïve melanocytes substantially decreased the levels of phospho-ERK, which did not occur with various control CMs that lacked IGFBP7 (Figure S7B). rIGFBP7 also blocked growth factor-induced ERK activation (Figure S7C). Collectively, these results indicate that IGFBP7 inhibits BRAF-MEK-ERK signaling.
Figure 4. IGFBP7 Blocks BRAF-MEK-ERK Signaling to Activate the Apoptotic Pathway.

(A) Immunoblot analysis in SK-MEL-28 cells treated with increasing concentrations of rIGFBP7 (0.2, 1.0, 2.0, 5.0 or 10 μg/ml) for 24 hrs.
(B) Proliferation assays monitoring sensitivity of SK-MEL-28 cells to rIGFBP7. Cells were transfected with an empty expression vector or a constitutively activated ERK2 or MEK1 mutant. Cell growth was analyzed 24 hrs after treatment with rIGFBP7 and normalized to the growth of the corresponding cell line in the absence of rIGFBP7 addition. Error bars represent standard error.
(C) Immunoblot analysis in SK-MEL-28 cells stably transfected with an empty expression vector or a constitutively activated ERK2 mutant. SK-MEL-28 cells were either untreated or treated with 10 μg/ml of rIGFBP7, as indicated, for 24 hrs prior to harvesting cells.
(D) Immunoblot analysis in SK-MEL-28 cells 24 hrs following treatment with rIGFBP, a MEK inhibitor (MEK-i) or a RAF inhibitor (RAF-i).
Addition of rIGFBP7 to SK-MEL-28 cells resulted in decreased levels of activated MEK1/2, corresponding with the reduced phospho-ERK levels and apoptosis (Figure S8A). Moreover, ectopic expression of a constitutively activated MEK1 mutant (MEK1EE) prevented IGFBP7 from blocking ERK activation (Figure S8B). These results demonstrate that IGFBP7 blocks phosphorylation of MEK by BRAF. Finally, we found that addition of IGFBP7 to SK-MEL-28 cells resulted in upregulation of RAF inhibitory protein (RKIP) (Figure S8A,C), which has been shown to interact with several RAF proteins, including BRAF, and inhibit RAF-mediated phosphorylation of MEK (see, for example, Park et al., 2005). Following knockdown of RKIP in SK-MEL-28 cells, rIGFBP7 failed to block activation of MEK or ERK (Figure S8C). Collectively, these results indicate that IGFBP7 inhibits BRAF-MEK-ERK signaling by inducing RKIP, which prevents BRAF from phosphorylating MEK.
To establish the relationship between inhibition of BRAF-MEK-ERK signaling and the IGFBP7-mediated block to cellular proliferation, we ectopically expressed a constitutively activated ERK2 or MEK1 mutant and analyzed sensitivity to rIGFBP7. Figure 4B shows that expression of either an ERK2 (left) or MEK1 (right) mutant in SK-MEL-28 cells substantially overcame the IGFBP7-mediated cellular proliferation block. Expression of a constitutively activated ERK2 mutant also blocked BRAFV600E- and IGFBP7-induced senescence in melanocytes (Figure S9). In addition, ectopic expression of a constitutively activated ERK2 mutant in SK-MEL-28 cells increased phospho-ERK2 levels and prevented the IGFBP7-mediated upregulation of BNIP3L and induction of apoptosis (Figure 4C).
The above results allowed us to draw two conclusions. First, IGFBP7 blocks cellular proliferation, at least in part, by inhibiting BRAF-MEK-ERK signaling. Second, inhibition of BRAF-MEK-ERK signaling is required for activation of the IGFBP7-mediated apoptotic pathway. This latter observation prompted us to ask whether inhibition of BRAF-MEK-ERK signaling was sufficient to induce apoptosis. Figure 4D shows, as expected, that addition of a MEK or RAF inhibitor blocked BRAF-MEK-ERK signaling. However, unlike rIGFBP7, MEK and RAF inhibitors did not increase BNIP3L levels or efficiently induce apoptosis. Thus, inhibition of BRAF-MEK-ERK signaling is necessary but not sufficient for IGFBP7-mediated upregulation of BNIP3L and induction of apoptosis.
IGFBP7 Suppresses Growth of BRAFV600E-Positive Tumors in Xenografted Mice
The ability of IGFBP7 to inhibit proliferation of BRAFV600E-positive human melanoma cell lines (see Figure 3B) raised the possibility that IGFBP7 could suppress growth of tumors containing an activating BRAF mutation. As a first test of this possibility, human melanoma cells that contained (SK-MEL-28) or lacked (SK-MEL-31) an activating BRAF mutation were injected subcutaneously into the flanks of nude mice. Three, six and nine days later, the mice were injected at the tumor site with either rIGFBP7 or, as a control, PBS. Figure 5A shows that rIGFBP7 substantially suppressed growth of BRAFV600E-positive tumors but had no effect on tumors containing wild type BRAF.
Figure 5. IGFBP7 Suppresses Growth of BRAFV600E-Positive Tumors in Xenografted Mice.
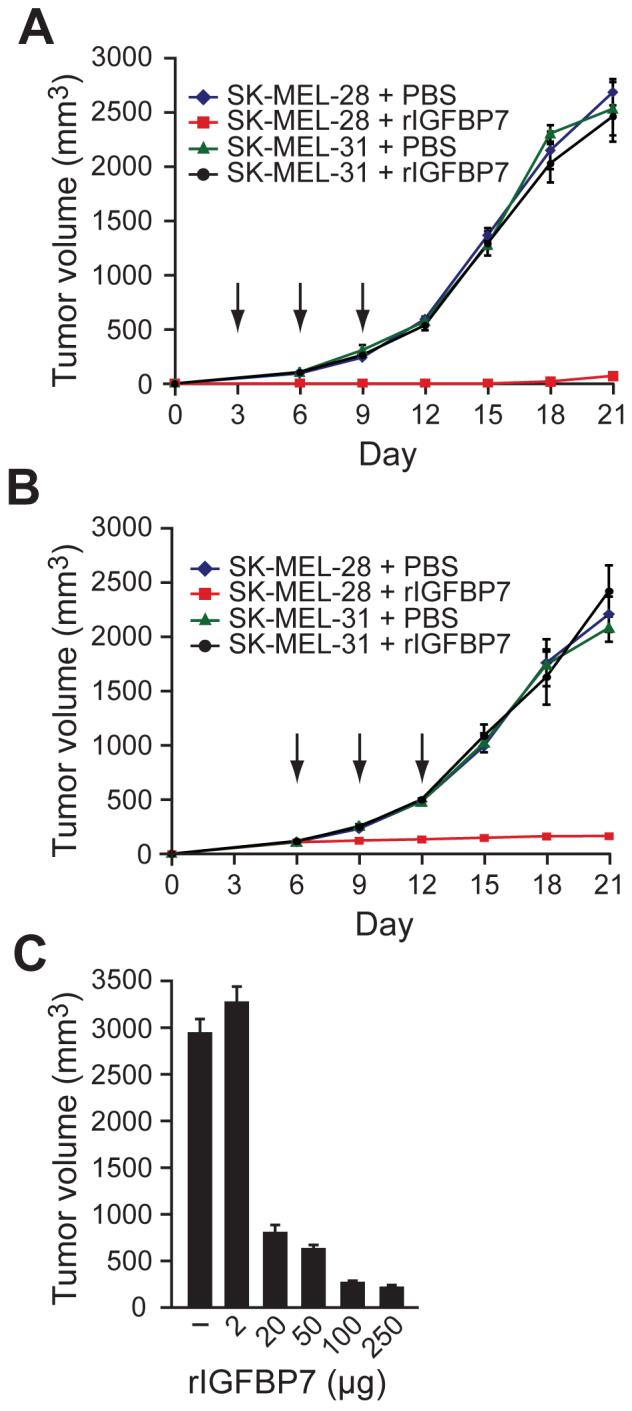
(A) SK-MEL-28 or SK-MEL-31 cells were injected subcutaneously into the flanks of nude mice, and three, six and nine days later (denoted by arrows), the mice were injected at the tumor site with rIGBP7 or, as a control, PBS. Error bars represent standard error.
(B) SK-MEL-28 or SK-MEL-31 cells were injected into the flanks of nude mice. When tumors reached a size of 100 mm3, 100 μg rIGFBP7 was systemically administered by tail vein injection at days 6, 9 and 12 (indicated by arrows).
(C) Dose-dependent suppression of tumor growth by rIGFBP7. SK-MEL-28 cells were injected into the flanks of nude mice as described in (B), following which rIGFBP7 was systemically administered by tail vein injection. Tumor volume was measured at day 21.
We next asked whether tumor growth could also be suppressed by systemic administration of rIGFBP7. SK-MEL-28 or SK-MEL-31 cells were injected into the flanks of nude mice and when tumors reached a size of 100 mm3, 100 μg rIGFBP7 was delivered by tail vein injection at days 6, 9 and 12. Figure 5B shows that systemic administration of rIGFBP7 completely suppressed growth of BRAFV600E-positive tumors, whereas tumors containing wild type BRAF were unaffected. In mice treated with rIGFBP7, BRAFV600E-positive tumors were deoxyuridine triphosphate nick-end labeling (TUNEL)-positive (Figure S10), indicating that suppression of tumor growth resulted from apoptosis. Suppression of tumor growth by systemically administered rIGFBP7 was dose-dependent, and concentrations higher than that required for inhibition of tumor growth could be delivered without apparent adverse effects (Figure 5C and data not shown).
Loss of IGFBP7 Expression is a Critical Step in Development of a BRAFV600E-Positive Melanoma
As shown above, BRAFV600E-positive melanoma cell lines fail to express IGFBP7 and are highly sensitive to IGFBP7-mediated apoptosis. These results raised the possibility that IGFBP7 functions as a tumor suppressor and loss of IGFBP7 might be required for development of BRAFV600E-positive melanoma. To investigate this possibility, we performed immunohistochemical analysis of IGFBP7 expression on a series of human skin, nevi and melanoma samples.
Figure 6 and Table S2 show that normal skin melanocytes expressed low but detectable levels of IGFBP7. BRAFV600E-positive nevi expressed high levels of IGFBP7, consistent with the finding that expression of BRAFV600E in melanocytes increased IGFBP7 levels (Figure 2A). Significantly, expression of IGFBP7 was not detectable in BRAFV600E-positive melanomas. By contrast, IGFBP7 was clearly expressed in melanomas lacking activated BRAF.
Figure 6. Loss of IGFBP7 Expression is a Critical Step in Development of a BRAFV600E-Positive Melanoma.

Immunohistochemical analysis of IGFBP7 expression in human tissue samples. Samples were stained with hematoxylin and eosin (H&E). Arrowheads indicate IGFBP7-positive melanocytes. Images are shown at 2X and/or 20X.
To determine whether loss of IGFBP7 expression was the result of epigenetic silencing, we performed bisulfite sequence analysis. Figure 7A shows that the IGFBP7 promoter was densely hypermethylated in BRAFV600E-positive melanomas but not in BRAFV600E-positive nevi or melanomas lacking activated BRAF. Similar analyses in a panel of melanoma cell lines showed that the IGFBP7 promoter was densely hypermethylated in BRAFV600E-positive melanoma cell lines and modestly hypermethylated in NRASQ61R-positive melanoma cell lines (Figure 7B). Treatment of these cell lines with the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine restored IGFBP7 expression in BRAFV600E- and NRASQ61R-positive cell lines but had no effect in BRAF/RAS-wild type cell lines (Figure 7C). Collectively, these results indicate that loss of IGFBP7 expression in BRAFV600E-positive melanomas and cell lines results from epigenetic silencing involving promoter hypermethylation.
Figure 7. The IGFBP7 Promoter is Hypermethylated in BRAFV600E-Positive Melanoma Cell Lines and Tissues.

(A) Bisulfite sequence analysis of the IGFBP7 promoter in human tissue samples. (Top) Schematic of the IGFBP7 promoter; positions of the CpG dinucleotides are shown to scale by vertical lines. (Bottom) Each circle represents a CpG dinucleotide: open (white) circles denote unmethylated CpG sites and filled (black) circles indicate methylated CpG sites. Each row represents a single clone.
(B) Bisulfite sequence analysis of the IGFBP7 promoter in a panel of melanoma cell lines.
(C) qRT-PCR analysis of IGFBP7 mRNA levels in melanoma cell lines following treatment with the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine (5-aza). Error bars represent standard error.
(D) Schematic summary of BRAFV600E-mediated senescence and melanoma progression. Normal melanocytes (BRAF-wt) express and secrete low levels of IGFBP7, which inhibits BRAF-MEK-ERK signaling through an autocrine/paracrine pathway, thereby restraining proliferation. In BRAFV600E-positive nevi, constitutive activation of the BRAF-MEK-ERK pathway increases expression and secretion of IGFBP7, and the resultant high levels of IGFBP7 inhibit BRAF-MEK-ERK signaling and activate senescence. In a BRAFV600E-positive melanoma, IGFBP7 expression is lost, enabling the cells to escape from senescence and resulting in uncontrolled proliferation. Addition of exogenous IGFBP7 to BRAFV600E-positive melanoma cells inhibits BRAF-MEK-ERK signaling and activates apoptosis.
DISCUSSION
By performing a genome-wide shRNA screen, we have identified 17 genes that are essential for BRAFV600E to induce senescence or apoptosis in primary cells. Unexpectedly, a critical component of the senescence and apoptotic pathways is a secreted protein, IGFBP7. Expression of BRAFV600E in primary cells induces synthesis and secretion of IGFBP7, which functions through autocrine/paracrine pathways to inhibit BRAF-MEK-ERK signaling and induce senescence or apoptosis. Consistent with our findings are previous reports that IGFBP7 (also called IGFBP-rP1 or MAC25) can inhibit proliferation of some cancer cell lines in vitro (see, for example, Hingorani et al., 2003; Mutaguchi et al., 2003; Ruan et al., 2006; Swisshelm et al., 1995; Wilson et al., 2002). Our results provide new insights into how activated BRAF promotes senescence, apoptosis and malignant transformation, which are schematically summarized in Figure 7D and discussed below.
We note that TP53 was one of the genes we identified as required for BRAFV600E-mediated senescence. There have been several previous reports that loss of TP53 does not enable escape from oncogene-induced senescence (see, for example, Beausejour et al., 2003; Narita et al., 2006; Zhu et al., 1998). However, these studies involved either a different cell type or oncogene than those used here.
Activated BRAF-Mediated Senescence Occurs through an Autocrine/Paracrine Pathway that Establishes a Negative Feedback Loop
Previous studies have shown that expression of BRAFV600E in melanocytes results in an initial proliferative burst leading to clonal expansion followed by growth arrest (Michaloglou et al., 2005). Our results explain this biphasic response. In the first phase, the initial expression of BRAFV600E increases BRAF-MEK-ERK signaling, providing a transient proliferative signal. In the second phase, BRAFV600E expression results in the synthesis and secretion of IGFBP7, which acts through an autocrine/paracrine pathway to inhibit BRAF-MEK-ERK signaling and activate a senescence program.
The non-uniform expression of p16INK4a in nevi has prompted speculation that senescent cells secrete a senescence-inducing agent that acts upon neighboring cells (Gray-Schopfer et al., 2007). We find that BRAFV600E-positive melanocytic nevi express high levels of IGFBP7 and propose that secreted IGFBP7 has a central role in the initiation and maintenance of the senescent state. A senescence-inducing secreted protein provides a powerful mechanism for tumor suppression because, following an initial oncogenic event in a single cell, neighboring cells are also protected.
Role for IGFBP7 in Melanoma Development and Therapy
BRAFV600E cannot fully transform human melanocytes, implying the existence of additional, cooperating events required for tumor development (Peeper and Mooi, 2002). The 17 genes we have identified are potential tumor suppressors in malignancies involving an activating BRAF mutation. Unlike nevi, BRAFV600E-positive melanomas do not express IGFBP7. On the basis of this observation and the other results presented in this study, we propose that loss of IGFBP7 expression allows escape from BRAFV600E-mediated senescence and is a critical step in melanoma genesis.
Activated BRAF-positive metastatic melanoma is an aggressive disease that is refractory to conventional chemotherapeutic agents and lacks adequate treatment options (reviewed in Gray-Schopfer et al., 2007). Inhibitors of BRAF have been developed but unfortunately have performed poorly in clinical trials. We have shown that IGFBP7, but not a RAF or MEK inhibitor, efficiently induces apoptosis in BRAFV600E-positive melanoma cell lines. IGFBP7 may be a more efficacious anti-cancer agent than a BRAF or MEK inhibitor because it both inhibits BRAF-MEK-ERK signaling and irreversibly induces apoptosis following transient exposure. The selective sensitivity of BRAFV600E-containing human cancer cell lines to IGFBP7, and the ability of IGFBP7 to suppress BRAFV600E-positive tumor growth in mice, suggests that IGFBP7 may have a role in treating malignancies harboring activating BRAF mutations.
EXPERIMENTAL PROCEDURES
Cell Lines and Culture
Primary foreskin fibroblasts (BJ) and human melanoma cell lines were obtained from ATCC, and human primary melanocytes were obtained from Cascade Biologics; all cells were grown as recommended by the supplier.
Retroviruses and Plasmids
Retroviruses expressing empty vector or BRAFV600E were generated from plasmids pBABE-zeo (Addgene) or pBABE-zeo/BRAFV600E (Michaloglou et al., 2005), respectively. Plasmids expressing ERK2Q103A and ERK2L73P,S151D (Emrick et al., 2006) and MEK1EE (Tournier et al., 1999) have been described.
shRNA Screen
The human shRNAmir library (release 1.20; Open Biosystems) was obtained through the UMass Medical School shRNA library core facility. Retroviral pools were generated and used to transduce PFFs as described (Gazin et al., 2007). Cells were then infected with a retrovirus carrying BRAFV600E at MOI 20. Cells that bypassed the BRAFV600E-induced cellular proliferation block formed colonies, which were pooled and expanded, and the shRNAs identified by sequence analysis as previously described (Gazin et al., 2007). Individual knockdown cell lines were generated by stable transduction of 6×104 cells with a single shRNA followed by infection with the BRAFV600E-expressing retrovirus. Individual shRNAs were either obtained from the Open Biosystems library or synthesized (see Table S3).
Quantitative real-time RT-PCR
Total RNA was isolated and reverse transcription was performed as described (Gazin et al., 2007), followed by quantitative real-time PCR using Platinum SYBR Green qPCR SuperMix-UDG with Rox (Invitrogen). Primer sequences are provided in Table S4.
Quantitative Proliferation Assay
Cell viability was measured by trypan blue exclusion at the time point indicated in the relevant figure legend. The number of viable cells was quantitated, and the values were expressed as percent cell growth, as described in the relevant figure legend. For the assay shown in Figure 2A, CM was replenished every 3 days and proliferation was measured after a total of 14 days of CM treatment. Unless otherwise stated, rIGFBP7 was added to the culture medium at 10 μg/ml.
Apoptosis and DNA Replication Assays
PFFs, melanocytes or shRNA knockdown derivatives (5×105 cells) were infected with BRAFV600E-expressing retrovirus, and 4 days later the total cell population was stained for Annexin V-PE (BD Biosciences). To monitor apoptosis in melanoma cells following rIGFBP7 treatment, 5×105 cells were treated with rIGFBP7 (10 μg/ml) for 24 hrs and stained for Annexin V-PE. For DNA replication assays, cells were treated as described above, except 4 hrs prior to the end of the 4-day retroviral infection, cells were incubated with 20 μM BrdU (Sigma) and processed as previously described (Wajapeyee and Somasundaram, 2003).
Antibodies and Immunoblot Analysis
Cell extracts were prepared by lysis in Laemmli buffer in the presence or absence of a phosphatase inhibitor cocktail (Sigma), as needed. To prepare CM, cells were grown in Opti-MEM (Invitrogen) for 24 hrs and media was harvested and concentrated using Centricon plus 20 tubes (Millipore). CM was normalized to cell number prior to loading the gel. For the experiments shown in Figures 3E and 4C, rIGFBP7 was added to the culture medium at 10 μg/ml. For the experiment of Figure 4D, SK-MEL-28 cells were treated with 2 μg/ml or 10 μg/ml rIGFBP7, 20 μM or 40 μM of the MEK inhibitor PD98054 (Calbiochem), or 5 nM or 10 nM the RAF inhibitor GW5074 (Sigma) for 24 hrs prior to harvesting cells. Blots were probed with the following antibodies: α-p16 (Abcam); α-acetylated H3K9 (Upstate); α-IGFBP7, α-BRAFV600E, cleaved caspase-3 p11 (Santa Cruz), α-SMARCB1 (Abnova), α-BNIP3L (Proscience), α-β-actin (Sigma), α-phospho-ERK or α-ERK (Cell Signaling Technology).
Recombinant IGFBP7 Expression and Purification
The human IGFBP7 expression vector pFASTBAC-1/IGFBP7, expressing a C-terminal Flag-tagged fusion protein (Oh et al., 1996), was used to generate recombinant baculovirus using the Bac-to-Bac Baculovirus Expression System (Invitrogen). The recombinant baculovirus construct was then transfected into Sf9 cells (Invitrogen), and CM was collected and incubated with α-Flag M2 beads (Sigma), and the bound IGFBP7 protein was eluted using an α-Flag peptide (synthesized by CFAR, UMass Medical School, USA).
Senescence-associated β-galactosidase Assay
Melanocytes infected with a retrovirus expressing either vector or BRAFV600E, or melanocytes treated with BRAFV600E/melanocyte CM or rIGFBP7 (10 μg/ml) for 14 days were processed as previously described (Dimri et al., 1995). Cells were visualized on a Zeiss Axiovert 40 CFL microscope. Images were captured using QCapture Pro version 5 software (QImaging Corporation).
ChIP Assays
ChIP assays were performed using extracts prepared 24 hrs following rIGFBP7 treatment. The following antibodies were used: α-BRG1 (a gift from A. Imbalzano, UMass Medical School), α-phospho-STAT1 (Upstate), and α-SMARCB1 and α-STAT1 (Santa Cruz). For ChIP experiments on the SMARCB1 promoter, primers spanning a STAT1 binding site located ∼2.4 kb upstream of the transcription start-site were used. For ChIP experiments on the BNIP3L promoter, a series of primer-pairs that covered ∼2 kb of the BNIP3L promoter were used; following addition of rIGFBP7, SMARCB1 and BRG1 were recruited to the BNIP3L promoter near the transcription start-site. shRNA/siRNA sequences are provided in Table S3, and primer sequences used for amplifying the ChIP products are provided in Table S4. ChIP products were analyzed by quantitative real-time PCR using Platinum SYBR Green qPCR SuperMix-UDG with Rox (Invitrogen). Calculation of fold-differences was done as previously described (Pfaffl, 2001).
Tumor Formation Assays
5×106 SK-MEL-28 or SK-MEL-31 cells were suspended in 100 μl of serum-free DMEM and injected subcutaneously into the right flank of athymic Balb/c (nu/nu) mice (Taconic). At days 3, 6 and 9, mice were injected at the tumor site with either rIGBP7 (20 μg in 100 μl total volume) or, as a control, PBS. Tumor dimensions were measured every three days and tumor volume was calculated using the formula π/6 × (length) × (width)2. For the systemic administration experiments, cells were injected into the flank as described above and when tumors reached a size of 100 mm3, 100 μg rIGFBP7 in a total volume of 100 μl was delivered by tail vein injection at days 6, 9 and 12. Tumor dimensions were measured every three days. Animal experiments were performed in accordance with the Institutional Animal Care and Use Committee (IACUC) guidelines.
Immunohistochemistry
The study was approved by the UMass Medical Center institutional review board (IRB #12543). Archival materials from normal skin (n=5), nevi (n=20) and malignant melanoma (primary (n=7) and metastatic (n=13)) were retrieved from the pathology files of UMass Medical Center, Worcester, MA. The histologic sections of all cases were re-reviewed and the diagnoses confirmed by a dermatopathologist (MM). All patient data were de-identified.
Five μm-thick sections were cut for immunohistochemical studies, which were performed, using standard techniques, with heat-induced epitope retrieval buffer and an α-IGFBP7 antibody (1:20 dilution; Santa Cruz). Appropriate positive and negative controls were included. Positive staining was noted by ascertaining expression of IGFBP7 in the cytoplasm; significant nuclear staining was not noted. All stained slides were reviewed by the dermatopathologist (MM) in a blind fashion with respect to genotype. Genomic DNA was isolated and quantitated, followed by PCR amplification (see Table S4 for primer sequences) and TA cloning (Promega). Multiple clones were sequenced for identifying the V600E mutation (T1796A) in exon 15 of the BRAF gene.
Bisulfite Sequencing
Bisulfite modification was carried out essentially as described (Frommer et al., 1992) except that hydroquinone was used at a concentration of 125 mM during bisulfite treatment (carried out in the dark) and DNA was desalted on Qiaquick columns (Qiagen) after the bisufite reaction. Six clones were sequenced for each cell line or human tissue sample (see Table S4 for primer sequences). For 5-aza-2′-deoxycytidine (5-aza) treatment, melanoma cell lines were treated with 10 μM 5-aza (Calbiochem) for 48 hrs.
Supplementary Material
ACKNOWLEDGMENTS
We thank the UMass Medical School (UMMS) shRNA library core facility; the UMMS Diabetes and Endocrine Research Center (DERC) for immunohistochemical staining; Natalie Ahn, Roger Davis, Anthony Imbalzano, Greg Nolan, Daniel Peeper and Ron Rosenfeld for reagents; Stephane Gobeil and Amy Virbasius for assistance with the shRNA screen; Susan Griggs for assistance with the animal experiments; and Sara Evans for editorial assistance. M.R.G. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003;22:4212–4222. doi: 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Porath I, Weinberg RA. The signals and pathways activating cellular senescence. Int. J. Biochem. Cell Biol. 2005;37:961–976. doi: 10.1016/j.biocel.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Bennett DC. Human melanocyte senescence and melanoma susceptibility genes. Oncogene. 2003;22:3063–3069. doi: 10.1038/sj.onc.1206446. [DOI] [PubMed] [Google Scholar]
- Bultman S, Gebuhr T, Yee D, La Mantia C, Nicholson J, Gilliam A, Randazzo F, Metzger D, Chambon P, Crabtree G, Magnuson T. A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol. Cell. 2000;6:1287–1295. doi: 10.1016/s1097-2765(00)00127-1. [DOI] [PubMed] [Google Scholar]
- Campisi J. Suppressing cancer: the importance of being senescent. Science. 2005;309:886–887. doi: 10.1126/science.1116801. [DOI] [PubMed] [Google Scholar]
- Chin L, Merlino G, DePinho RA. Malignant melanoma: modern black plague and genetic black box. Genes Dev. 1998;12:3467–3481. doi: 10.1101/gad.12.22.3467. [DOI] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Dhomen N, Marais R. New insight into BRAF mutations in cancer. Curr. Opin. Genet. Dev. 2007;17:31–39. doi: 10.1016/j.gde.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emrick MA, Lee T, Starkey PJ, Mumby MC, Resing KA, Ahn NG. The gatekeeper residue controls autoactivation of ERK2 via a pathway of intramolecular connectivity. Proc. Natl. Acad. Sci. USA. 2006;103:18101–18106. doi: 10.1073/pnas.0608849103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formstecher E, Ramos JW, Fauquet M, Calderwood DA, Hsieh JC, Canton B, Nguyen XT, Barnier JV, Camonis J, Ginsberg MH, Chneiweiss H. PEA-15 mediates cytoplasmic sequestration of ERK MAP kinase. Dev. Cell. 2001;1:239–250. doi: 10.1016/s1534-5807(01)00035-1. [DOI] [PubMed] [Google Scholar]
- Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Molloy PL, Paul CL. A genomicsequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazin C, Wajapeyee N, Gobeil S, Virabasius CM, Green MR. An elaborate pathway required for Ras-mediated epigenetic silencing. Nature. 2007;449:1073–1077. doi: 10.1038/nature06251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445:851–857. doi: 10.1038/nature05661. [DOI] [PubMed] [Google Scholar]
- Hartman SE, Bertone P, Nath AK, Royce TE, Gerstein M, Weissman S, Snyder M. Global changes in STAT target selection and transcription regulation upon interferon treatments. Genes Dev. 2005;19:2953–2968. doi: 10.1101/gad.1371305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Jacobetz MA, Robertson GP, Herlyn M, Tuveson DA. Suppression of BRAF(V599E) in human melanoma abrogates transformation. Cancer Res. 2003;63:5198–5202. [PubMed] [Google Scholar]
- Hoeflich KP, Gray DC, Eby MT, Tien JY, Wong L, Bower J, Gogineni A, Zha J, Cole MJ, Stern HM, et al. Oncogenic BRAF is required for tumor growth and maintenance in melanoma models. Cancer Res. 2006;66:999–1006. doi: 10.1158/0008-5472.CAN-05-2720. [DOI] [PubMed] [Google Scholar]
- Leppa S, Saffrich R, Ansorge W, Bohmann D. Differential regulation of c-Jun by ERK and JNK during PC12 cell differentiation. EMBO J. 1998;17:4404–4413. doi: 10.1093/emboj/17.15.4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- Mooi WJ, Peeper DS. Oncogene-induced cell senescence--halting on the road to cancer. N. Engl. J. Med. 2006;355:1037–1046. doi: 10.1056/NEJMra062285. [DOI] [PubMed] [Google Scholar]
- Mutaguchi K, Yasumoto H, Mita K, Matsubara A, Shiina H, Igawa M, Dahiya R, Usui T. Restoration of insulin-like growth factor binding protein-related protein 1 has a tumor-suppressive activity through induction of apoptosis in human prostate cancer. Cancer Res. 2003;63:7717–7723. [PubMed] [Google Scholar]
- Narita M, Krizhanovsky V, Nunez S, Chicas A, Hearn SA, Myers MP, Lowe SW. A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell. 2006;126:503–514. doi: 10.1016/j.cell.2006.05.052. [DOI] [PubMed] [Google Scholar]
- Oh Y, Nagalla SR, Yamanaka Y, Kim HS, Wilson E, Rosenfeld RG. Synthesis and characterization ofinsulin-like growth factor-binding protein (IGFBP)-7. Recombinant human mac25 protein specifically binds IGF-I and - II. J. Biol. Chem. 1996;271:30322–30325. doi: 10.1074/jbc.271.48.30322. [DOI] [PubMed] [Google Scholar]
- Park S, Yeung ML, Beach S, Shields JM, Yeung KC. RKIP downregulates B-Raf kinase activity in melanoma cancer cells. Oncogene. 2005;24:3535–3540. doi: 10.1038/sj.onc.1208435. [DOI] [PubMed] [Google Scholar]
- Peeper DS, Mooi WJ. Pathogenesis of melanocytic naevi: growth arrest linked with cellular senescence? Histopathology. 2002;41:S139–S143. [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock PM, Harper UL, Hansen KS, Yudt LM, Stark M, Robbins CM, Moses TY, Hostetter G, Wagner U, Kakareka J, et al. High frequency of BRAF mutations in nevi. Nat. Genet. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- Ruan WJ, Lin J, Xu EP, Xu FY, Ma Y, Deng H, Huang Q, Lv BJ, Hu H, Cui J, et al. IGFBP7 plays a potential tumor suppressor role against colorectal carcinogenesis with its expression associated with DNA hypomethylation of exon 1. J Zhejiang Univ. Sci. B. 2006;7:929–932. doi: 10.1631/jzus.2006.B0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satyamoorthy K, Li G, Gerrero MR, Brose MS, Volpe P, Weber BL, Van Belle P, Elder DE, Herlyn M. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation. Cancer Res. 2003;63:756–759. [PubMed] [Google Scholar]
- Sharma A, Trivedi NR, Zimmerman MA, Tuveson DA, Smith CD, Robertson GP. Mutant V599EB-Raf regulates growth and vascular development of malignant melanoma tumors. Cancer Res. 2005;65:2412–2421. doi: 10.1158/0008-5472.CAN-04-2423. [DOI] [PubMed] [Google Scholar]
- Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, Ye Q, Lobo JM, She Y, Osman I, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–362. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow LE, Eldon MJ, English DR, Heenan PJ. p16 and p21WAF1 protein expression in melanocytic tumors by immunohistochemistry. Am. J. Dermatopathol. 1998;20:255–261. doi: 10.1097/00000372-199806000-00006. [DOI] [PubMed] [Google Scholar]
- Swisshelm K, Ryan K, Tsuchiya K, Sager R. Enhanced expression of an insulin growth factor-like binding protein (mac25) in senescent human mammary epithelial cells and induced expression with retinoic acid. Proc. Natl. Acad. Sci. USA. 1995;92:4472–4476. doi: 10.1073/pnas.92.10.4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournier C, Whitmarsh AJ, Cavanagh J, Barrett T, Davis RJ. The MKK7 gene encodes a group of c-Jun NH2-terminal kinase kinases. Mol. Cell Biol. 1999;19:1569–1581. doi: 10.1128/mcb.19.2.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajapeyee N, Somasundaram K. Cell cycle arrest and apoptosis induction by activator protein 2alpha (AP-2alpha) and the role of p53 and p21WAF1/CIP1 in AP-2alpha-mediated growth inhibition. J. Biol. Chem. 2003;278:52093–52101. doi: 10.1074/jbc.M305624200. [DOI] [PubMed] [Google Scholar]
- Wang YL, Uhara H, Yamazaki Y, Nikaido T, Saida T. Immunohistochemical detection of CDK4 and p16INK4 proteins in cutaneous malignant melanoma. Br. J. Dermatol. 1996;134:269–275. [PubMed] [Google Scholar]
- Wilson EM, Oh Y, Rosenfeld RG. Generation and characterization of an IGFBP-7 antibody: identification of 31kD IGFBP-7 in human biological fluids and Hs578T human breast cancer conditioned media. J. Clin. Endocrinol. Metab. 1997;82:1301–1303. doi: 10.1210/jcem.82.4.3983. [DOI] [PubMed] [Google Scholar]
- Wilson HM, Birnbaum RS, Poot M, Quinn LS, Swisshelm K. Insulin-like growth factor binding protein-related protein 1 inhibits proliferation of MCF-7 breast cancer cells via a senescence-like mechanism. Cell Growth Differ. 2002;13:205–213. [PubMed] [Google Scholar]
- Zhu J, Woods D, McMahon M, Bishop JM. Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev. 1998;12:2997–3007. doi: 10.1101/gad.12.19.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.