

Abstract
The major part of the brain’s energy budget (~60%–80%) is devoted to its communication activities. While inhibition is critical to brain function, relatively little attention has been paid to its metabolic costs. Understanding how inhibitory interneurons contribute to brain energy consumption (brain work) is not only of interest in understanding a fundamental aspect of brain function but also in understanding functional brain imaging techniques which rely on measurements related to blood flow and metabolism. Herein we examine issues relevant to an assessment of the work performed by inhibitory interneurons in the service of brain function.
Background
The brain of an average adult human represents about 2% of the body weight yet accounts for approximately 20% of the energy consumed. How this energy consumption is apportioned among the cell types within the brain and the various activities they perform is an area of active research (cf. Fox and Raichle, 2007). This research has been stimulated in large part by the rapidly increasing use of functional imaging techniques like positron emission tomography (PET) and functional magnetic resonance imaging (fMRI), which are based on measurements of brain blood flow and metabolism. Most research, to date, has focused on the role of glutamatergic principal cells and astrocytes.
While glutamatergic principal cells comprise the majority of cortical neurons, the remaining 15%–20% of the population is inhibitory GABAergic interneurons (Sillito, 1984; Hendry et al., 1987; DeFelipe, 1993; Somogyi et al., 1998; Markram et al., 2004). GABAergic interneurons differ from principal cells in many ways, including their dendritic organization, axonal connectivity, intrinsic biophysical properties, firing patterns, network activity, and behavioral correlates (Freund and Buzsáki, 1996; Somogyi et al., 1998; McBain and Fisahn, 2001; Somogyi and Klausberger, 2005; Buzsáki et al., 2004; Markram et al., 2004; Soltesz, 2006).
The dynamic partnership between principal cells and interneurons (Figure 1A) ensures an overall homeostatic regulation of global firing rates of neurons over extended territories of the cerebral cortex yet allows for dramatic changes in local excitability in short time windows, a requirement for processing and sending messages and modifying network connections. Coordinated inhibition insures that excitatory trajectories are properly routed and that competing cell assemblies are functionally segregated. As a result, in response to the same input, a given network can produce different output patterns at different times, depending on the state of inhibition (Figures 1B–1D). Interneuron-supported oscillations provide a temporal context for the content represented by the spatio-temporal discharge patterns of principal cells (cf. Buzsáki and Chrobak, 1995; Salinas and Sejnowski, 2001).
Figure 1. The Relationship between Brain Energy Consumption and Neurophysiology Is Critically Dependent upon the Partnership that Exists between Principal Cells (P) and Interneurons (i).
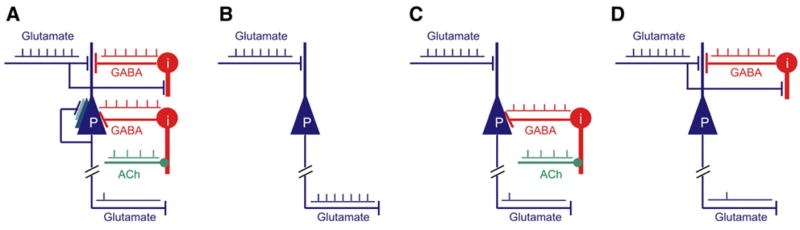
Both “spontaneous” ongoing (intrinsic) activity (A), which accounts for the largest fraction of the brain’s energy budget, and evoked activity are a combination of excitation (glutamate) and inhibition (GABA).
(B) In the simplest (but nonrealistic) example of a change in activity resulting from an increase in glutamate release unopposed by a change in GABA, EPSPs and spike output are well correlated and accompanied by an increase in energy consumption. However, parallel changes in GABA and glutamate induce nonlinear responses in the system. For example, in the presence of an increase in somatic feed-forward inhibition (C) produced by subcortical neurotransmitters (e.g., acetylcholine [Ach]) the same increase in glutamate release produces the same EPSPs as in (B), but the spike output of the principal cell is decreased. The local energy consumption increases as in (B) despite the fact that the output of the principal cell has not changed. An imaging method detecting only glutamate release-related metabolic activity cannot distinguish between (B) and (C) despite the difference in local computation and spike output.
(D) In the presence of dendritic inhibition (D), the quantity of glutamate released locally may be even greater than in (B) or (C), yet the output of the principal cell, in this hypothetical case, is the same as in (C). Energy consumption related to the generation of EPSPs and the processing of glutamate by astrocytes would clearly increase, possibly more than in (B) or (C). These examples serve to illustrate the dependence of brain imaging signals on the energy demanding events associated with the input to both principal cells and interneurons. They also serve to alert researchers to the potential complexity of comparing the output of principal cells (spikes) to changes in brain imaging signals.
While the many functions of both phasic and tonic inhibition are critical to brain operations, the energy they consume has received far less attention than that afforded to the excitatory effects of principal cells (cf. Heeger and Ress, 2002; Attwell and Gibb, 2005; Raichle and Mintun, 2006). Below, we discuss the differences between excitatory and inhibitory networks and how their complex interactions in the working brain may have important consequences for the assessment of the metabolic expenditure of neuronal computation.
In order to provide a proper context in which to understand the role of interneurons in the genesis of the brain’s energy consumption, we begin with a brief review of some aspects of brain energy metabolism and circulation that are relevant to an appraisal of the contribution of interneurons to brain work. As well, we review the limited data available on that portion of the brain’s energy budget attributable to inhibition. Because of the incompleteness of the latter, we then devote the remainder of this review to a consideration of those properties of interneurons and inhibition that should guide future work.
Brain Work
Recent appraisals of overall brain energy consumption using a variety of approaches have consistently indicated that between 50% and 80% of the energy consumption of the brain appears to be devoted to signaling associated with the input and output activity of neurons (Sibson et al., 1997, 1998; Ames, 2000; Attwell and Laughlin, 2001; Lennie, 2003). While the majority of the energy devoted to signaling is committed to the activities of glutamate, a recent NMR spectroscopy study in the rat neocortex suggests that the contribution of GABA to the glutamate/glutamine cycling (GABA can be converted to glutamate in astrocytes) may account for 10%–15% of the total oxidative metabolism (Patel et al., 2005).
Most of the energy needed for signaling is derived from the metabolism of glucose to carbon dioxide and water through a two-stage process beginning with glycolysis and ending with oxidative phosphorylation. Traditionally, these two processes have been viewed as acting in concert with glycolysis providing the necessary substrate for oxidative phosphorylation (i.e., pyruvate). It is the case, however, that despite the presence of adequate brain oxygenation, glycolysis occurs in excess of that needed to provide substrate for oxidative phosphorylation. While known for some time (Gibbs et al., 1942; Raichle et al., 1970; Siesjö, 1978), the importance of this excess glycolysis for normal brain function has been appreciated only recently as the result of its unique association with the metabolic demands incurred with increased cellular activity in the brain (Figure 2; Fox et al., 1988).
Figure 2. Relationship between Neuronal Activity and BOLD.
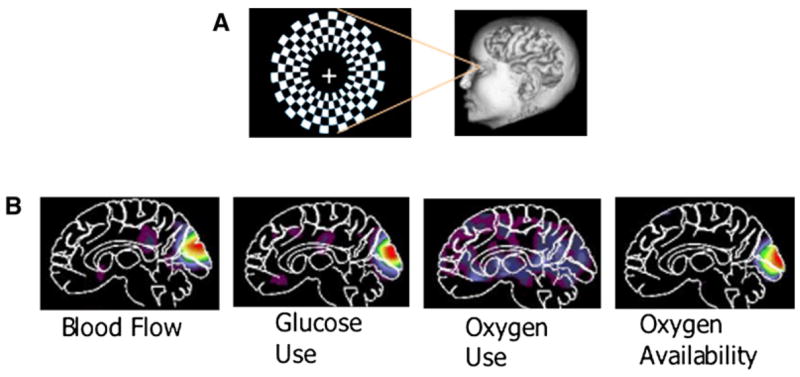
An increase in brain activity involves an increase in blood flow and glucose use, as illustrated when the visual system is stimulated with a reversing annular checkerboard (A) (data adapted from Fox et al., 1988). The degree to which oxygen use increases is variable but always less than the increase in glucose utilization and blood flow. The fMRI BOLD signal emerges as a marker of brain activity because of the resulting increase in oxygen availability and hemoglobin oxygenation, which has a direct effect on the MRI magnet field (Ogawa et al., 1990). If circumstances of increased activity involved equal increases in oxygen consumption, glucose utilization, and blood flow then measurements of glucose utilization with 2-deoxyglucose would reveal a change but the fMRI BOLD signal would not (B). Is it possible that an increase in excitatory neurotransmission involves increases in blood and glucose utilization that are greater than the increase in oxygen consumption but with an increase in inhibitory neurotransmission the three move in parallel? To date, the lack of rigorous experiments prevents one from answering this question.
The identification of glycolysis as a unique component of brain activity (cf. Raichle and Mintun, 2006) has stimulated a reappraisal of the cell biology of brain energy metabolism. Interest has centered on the astrocyte and its role in the uptake of glutamate from the synapse. When astrocytes are exposed to glutamate they exhibit a burst in glycolysis but not when exposed to GABA (Chatton et al., 2003). These data were interpreted to raise doubts about the possibility that GABA-mediated inhibition will be reflected in changes in the fMRI BOLD signal.
While the majority of the brain’s energy budget is devoted to ongoing (“spontaneous”) activity, most research using functional imaging techniques have focused on changes in brain energy consumption (Figure 2). When measured in terms of blood flow or glucose utilization, these changes usually range from 5% to 10% locally. However, the “baseline versus task”-related differences may be even less in terms of local brain energy consumption because glucose utilization (glycolysis) significantly exceeds the changes in oxygen consumption (Figure 2). Glycolysis produces only 6% of the energy realized when glucose is fully metabolized to carbon dioxide and water. It can be estimated that if glucose consumption increases 10% locally in the brain, the increase in energy consumption from its resting baseline can range from 1%, where there is no increase in oxygen consumption, to 10%, where glucose is fully metabolized to carbon dioxide and water. Because the imaging signal of fMRI is dependent upon a local change in the amount of available oxygen (Ogawa et al., 1990), it follows that a change in energy consumption that is fully supported by an increase in oxidative phosphorylation will not be “seen” by fMRI. The implication is that inhibitory functions, which may be supported more by oxidative mechanisms than excitatory signaling (see below), may remain undetected. It is therefore critical to point out that assessing the relative contributions of glycolysis and oxidative phosphorylation requires PET measurements of both oxygen consumption and glucose utilization (Fox et al., 1988).
Independent of the methods used, it is important to understand how energy consumption is distributed in the neuropil. Most studies suggest that the major metabolic cost is in the excitatory input to cells (Schwartz et al., 1979; Mata et al., 1980; Kadekaro et al., 1985; Nudo and Masterton, 1986; Logothetis et al., 2001; Logothetis, 2003; Logothetis and Wandell, 2004; Viswanathan and Freeman, 2007) rather than in their output spikes, and the cost of inhibitory inputs are often ignored. Illustrative in this regard is the discordance between the tonic firing rates of neurons in caudate-putamen versus globus pallidus and the associated metabolism in these two areas. Tonic discharge rates of medium spiny GABAergic neurons in caudate-putamen are approximately 1 Hz, whereas GABAergic neurons in globus pallidus that project mainly to the thalamus exhibit sustained rates up to 60–80 Hz (Mink, 1996; Tepper et al., 2007). These firing-rate differences should be contrasted with the metabolic rates for glucose in these two structures, which showed the opposite pattern in rats, cats, and monkeys, i.e., high in the caudate-putamen and low in the globus pallidus (Sokoloff, 1984), in accordance with the stronger glutamatergic innervation of caudate-putamen.
From the above experiments, one would expect that the cost of the fast firing rate of cells in the globus pallidus to be expressed in the energy consumption of the thalamus where their GABAergic inhibitions are expressed. Consistent with this prediction, variations in the firing rate of inhibitory cells in globus pallidus are positively correlated with resting-state glucose metabolism in the thalamus measured with PET and 18F-fluorodeoxyglucose (Eidelberg et al., 1997). Furthermore, lesions of globus pallidus for the treatment of Parkinson’s disease led to a decrease in glucose metabolism in the thalamus and an increase in the supplementary motor area of the cerebral cortex to which thalamic neurons project (Eidelberg et al., 1996). Using PET measurements of blood flow, Hershey and colleagues (Hershey et al., 2003) furthered the above line of research by examining the effect of subthalamic nucleus (STN) stimulation in patients with implanted electrodes placed for the treatment of Parkinson’s disease. They observed an increase in blood flow in globus pallidus consistent with the excitatory input from STN, accompanied by an increase in blood flow in the thalamus attributed to the inhibitory input from globus pallidus and a decrease in the supplementary motor area.
Experiments in the neocortex also highlight the metabolic demand of interneuron activity. In an early attempt to assess the energy costs of task-evoked GABAergic neurotransmission, Ackerman and colleagues (Ackermann et al., 1984) used 2-deoxyglucose tissue autoradiography (Sokoloff et al., 1977) to measure changes in glucose metabolism and reported increased glucose metabolism upon silencing of principal cells in the hippocampus. They inferred that the stimulation-evoked increase in energy consumption was due mainly to the firing of inter-neurons and associated inhibition (see also Nudo and Masterton, 1986). Because neither GABAergic inhibition nor GABA neuron activity was measured directly in these experiments, a causal role of interneurons and inhibition could not be supported unequivocally. Nevertheless, in a subsequent study, elegantly done with 2-deoxyglucose labeling of immunocytochemically identified GABAergic neurons in the somatosensory cortex of hamsters, glucose metabolism was significantly stronger in GABAergic neurons than in glutamatergic neurons (McCasland and Hibbard, 1997).
Unfortunately, none of the studies reviewed above tell us whether the energy requirements attributed to inhibition are provided by glycolysis alone, oxidative phosphorylation, or some combination of the two. Without measurements of oxygen consumption, the magnitude of changes in brain energy consumption remains unknown.
Summarizing, the energy consumption of the brain is largely devoted to its functional activity with up to 80% devoted to signaling associated with the input and output of neurons. Changes in energy consumption related to evoked activity are small compared to the overall energy budget. A contribution of inhibition to evoked changes in functional brain activity is undoubtedly present, but its magnitude remains to be determined. Because excitatory principal cells and inhibitory interneurons contribute to different aspects of brain function and because the activity patterns of the excitatory and inhibitory systems can dissociate in a task-dependent manner, difference-based metabolic changes can reflect activity shifts in either systems or their complex interactions. A critical step in understanding their respective contributions will require quantitative assessment of glycolysis and oxidative phosphorylation.
Because of the incompleteness of data relating inhibition and brain work, we turn next to a more detailed consideration of the properties of interneurons and the role of inhibition in brain function to highlight the potential mechanisms of energy savings and expenditures of the inhibitory system.
Basic Properties of Inhibitory Interneurons
Anatomical Features
The discussion that follows is mainly based on data obtained from the hippocampus of the rat because quantitative neuroanatomical and physiological data on identified neurons are mainly available in this simple cortical structure. However, the major conclusions should hold for other cortical structures and species as well.
GABAergic interneurons in the hippocampus proper (75,000 in CA1–3 combined) comprise 15%–20% of the neuronal population, approximately a third of which innervate the perisomatic region of pyramidal neurons (Freund and Buzsáki, 1996). The total axon length of individual interneurons is two to four times less than that of CA3 pyramidal cells (Li et al., 1994) and varies substantially across the various groups: perisomatic basket and chandelier cells (40–55 mm; 9,000–12,000 boutons), dendrite-targeting interneurons (80–220 mm; 16,000–80,000 boutons), long-range interneurons (20–100 mm; 20,000–25,000 boutons; Li et al., 1992, 1994; Sik et al., 1994, 1995, 1997; Jinno et al., 2007). Calculating the total length of interneuron axons and the total number of inhibitory terminals is difficult because the proportion of cells in the various interneuron classes is not known (Parra et al., 1998; Somogyi et al., 1998; Buzsáki et al., 2004; Markram et al., 2004). For the estimated 30,000–40,000 basket cells in the rat hippocampus (Ribak et al., 1990) the total axon length is approximately 1.5 km (i.e., 4% of the length of the total axon arbor of CA3 pyramidal cells) with half a billion boutons (i.e., 5%–10% of the total). Assuming similar average values for the remaining interneurons, the hippocampal interneuron population as a whole has to transmit action potentials for over five to eight times less distance than the pyramidal cell population (Li et al., 1994; Wittner et al., 2007).
In addition, dendritic arbors of interneurons are shorter than those of the principal cells. The cumulative length of dendrites in a single CA1 pyramidal cell is approximately 12 mm, over which approximately 30,000 excitatory inputs are received (Megias et al., 2001), whereas interneurons have shorter dendritic lengths (4 mm) and fewer excitatory inputs (5,000–17,000; Gulyás et al., 1999). Basket cells receive the largest number of excitatory inputs (16,000) and a similar ratio of inhibitory and excitatory inputs (6% versus 94%) as pyramidal cells (Megias et al., 2001; Gulyás et al., 1999). Other interneurons have a lower ratio of excitatory inputs (70%–80%; Gulyás et al., 1999).
In summary, because an “average” interneuron receives two to six times less excitatory inputs than an average pyramidal cell, an estimated 3%–10% of all excitatory terminals innervate the GABAergic interneuron population. Although similar quantitative data are not yet available for neocortical principal cells and interneurons, given the comparable percentage representation of interneurons in the neocortex, their similar anatomical divisions and principal cell target domains, the above estimates from the hippocampus are likely similar in the neocortex (Somogyi et al., 1998; Somogyi and Klausberger, 2005; Markram et al., 2004; Douglas and Martin, 2004). The lower fraction of the excitatory inputs onto interneurons, together with the shorter distances through which action potentials should be conducted along axons and dendrites, can be considered energy saving mechanisms in interneurons (Sarpeshkar, 1998; Attwell and Laughlin, 2001; Laughlin and Sejnowski, 2003). However, these anatomical features should be contrasted to the differences in the physiological activity of interneurons and pyramidal cells.
Firing Patterns of Interneurons Are Different from Those of Principal Neurons
The “resting” membrane potential of interneurons is several mV less negative (i.e., closer to spike threshold) than that of the principal cells (Fricker and Miles, 2000; Markram et al., 2004), reflecting further energy savings in interneurons in the nonspiking state. However, this same property will make interneurons respond more effectively to inputs. In addition, the glutamatergic terminals on interneuron dendrites are generally larger than on pyramidal cells (Acsády et al., 1998; Gulyás et al., 1993). This is translated to more effective transmission in several interneuron types: rapid rise of depolarization, larger amplitude EPSPs, and less frequent failures, compared to the glutamatergic synapses on principal cells (Miles, 1990; Gulyás et al., 1993; Jonas et al., 2004; Kraushaar and Jonas, 2000; Losonczy et al., 2004), although considerable variability exists among the various cortical interneuron classes (Reyes et al., 1998; Markram et al., 2004; Thomson and Lamy, 2007). Due to the combination of these factors, the same amount of glutamate released per terminal is translated onto higher frequency spiking output in interneurons. It is this higher frequency output of interneurons that allows for a full control of the complex network operations performed by the principal neurons (Shadlen and Newsome, 1998; Shu et al., 2003; Swadlow, 2003), despite the low share of inhibitory synapses on cortical neurons (6% hippocampus; Megias et al., 2001; estimated 16% in neocortex; Markram et al., 2004).
Elevated postsynaptic potentials and firing rates enhance metabolic costs. The pyramidal cell population in the hippocampus can sustain long-term firing at an average rate of approximately 1.4 Hz, although individual neurons can respond robustly for short periods (O’Keefe and Nadel, 1978; Csicsvari et al., 1999, 2003; Hirase et al., 2001; Dragoi et al., 2003; Wilson and McNaughton, 1994). The referred long-term rate is likely an overestimate because the proportion of sampled but silent neurons in any given behavioral situation is difficult to assess (Henze et al., 2000). In contrast, putative basket and chandelier interneurons, on average, discharge steadily at approximately 15 Hz, and the long-term discharge rates of other interneuron types are also several-fold above the rates of pyramidal cells (Kawaguchi and Kondo, 2002; Csicsvari et al., 1999; Swadlow, 2003; Markram et al., 2004). Therefore, the minority interneuron population with mostly local connectivity in the hippocampus emits as many or more spikes than all cortical principal cells combined with important metabolic consequences.
Similar quantitative comparisons are not available in the neocortex, mainly because of the technical difficulties of identifying the principal cell types and separating them from the inhibitory groups in vivo. Estimates indicate that interneurons, overall, may sustain three times higher firing rates than principal cells (Markram et al., 2004). This ratio is likely an underestimate for the following reasons: in the neocortex, spike cost calculations include 4 Hz sustained firing rates for an average pyramidal cell, a rate based primarily on the activity of layer 5 pyramidal cells (Attwell and Laughlin, 2001). However, averaged over neocortical areas, pyramidal cells in layers 2, 3, and 6 make up 60%–70% of human neocortical principal neurons, whereas large pyramidal cells of layer 5 and stellate cells of layer 4 constitute only about 10%–15% and 15%–25%, respectively (Blinkov and Glezer, 1968). Single-cell recordings are strongly biased toward the much more active layer 4 and 5 neurons and large-scale recordings in various cortical regions provided a mean firing rate of 1.9 Hz in deep layer neurons (Battaglia et al., 2004; Isomura et al., 2006). This latter rate estimate is also biased because it includes unidentified interneurons as well. The few studies that compared laminar-specific rates show that the minority layer 4–5 cells may be two to four times more active than the majority of pyramidal cells in the remaining layers (Swadlow, 1988; Krupa et al., 2004). In this context, it is interesting to note that metabolic activity in primary visual cortex of the monkey, as assessed by 2-DG autoradiography, is highest in layer 4 (Kennedy et al., 1976).
On the basis of these considerations, the ratio of spiking activity between neocortical excitatory principal cells and inhibitory interneurons may be more similar to those in the hippocampus than to the current estimates. In summary, the higher firing rates of the minority interneuron population may match the sparse firing of the majority principal cells. Translated into target activity, an average cortical neuron may experience the same numbers of IPSPs and EPSPs on the time scale of seconds. However, as will be discussed below, the energetic costs of excitation and inhibition do not sum up linearly, and hence they cannot be estimated in isolation.
Powering sustained high levels of cellular activity requires maintained energy supplies. Indeed, the activity of mitochondrial cytochrome-C oxidase, an activator of ATP synthase, is three times higher in GABAergic neurons in the striate cortex of the monkey than in the surrounding pyramidal cells (Nie and Wong-Riley, 1995). Among the GABAergic neuron types in the hippocampus, fast-firing basket cells and long-range interneurons stain most strongly for cytochrome-C (Gulyás et al., 2006). In addition, their beaded dendrites and large axon terminals possess a higher density of mitochondria than in pyramidal neurons, emphasizing the importance of oxidative metabolism in sustained interneuron function.
Direct and Indirect Metabolic Costs of Interneuron Operations
GABAergic control by the interneuron population is achieved by two distinct mechanisms: action potential-dependent or “phasic” inhibition and action potential-independent or “tonic” inhibition (Mody and Pearce, 2004; Semyanov et al., 2004; Farrant and Nusser, 2005; Farrant and Kaila, 2007). GABA, acting through GABAA receptors, increases membrane permeability to Cl− and HCO3− ions (Kaila, 1994). At lower frequencies, IPSPs rarely fail, in contrast to the lower fidelity EPSPs on principal cells (Tamas et al., 1997; Somogyi et al., 1998; Kraushaar and Jonas, 2000). The functional consequence of GABAA receptor activation depends on the polarization level of the postsynaptic membrane. Typically, the activity of chloride-extruding K-Cl co transporters (KCCs) such as KCC2 (Rivera et al., 1999; Payne et al., 2003) creates a transmembrane Cl− gradient that is needed in the generation of hyperpolarizing postsynaptic responses. GABAA channel activation also brings about a decrease in input resistance of the target cell, known as “shunting” inhibition (Farrant and Kaila, 2007; Bartos et al., 2007). In the assessment of the metabolic costs of these “direct” GABA transmission mechanisms, one also needs to consider the indirect effects of interneuron operations in maintaining the dynamic equilibrium of network activity in the in vivo brain.
Tonic Activity Mediated by GABA
Work during the past decade has shown that low (micromolar or nanomolar) concentrations of extracellular GABA can persistently activate GABAA receptors to generate a tonic conductance. The tonic effect is mediated mainly by high-affinity extrasynaptic receptors, containing δ subunits most often coassembled with α6 or α4 subunits (cf. Farrant and Nusser, 2005; Farrant and Kaila, 2007). The significance of the diffuse action of GABA is that the time-integrated GABAergic current, and, consequently, the persistently increased membrane conductance can be several times larger than what is produced by synapse-mediated IPSCs occurring at a frequency of 10 Hz (Mody and Pearce, 2004). The ambient level of GABA, activating the extrasynaptic receptors, is regulated by both neuronal and glial GABA transporters. Nevertheless, the magnitude of the tonic GABA effect is heterogeneous in different types of neurons and is significantly larger in certain hippocampal interneurons than in principal cells (Semyanov et al., 2004). The extrasynaptic δ subunit-containing receptors are also the main targets of neuroactive steroids (Majewska, 1992; Stell et al., 2003; Maguire et al., 2005). Finally, subcortical neurotransmitters often selectively target certain classes of interneurons. For example, serotonergic terminals typically avoid parvalbumin-immunoreactive basket cells and innervate mainly dendrite-targeting interneurons and CCK-immunoreactive basket cells, by which mechanism the activity of a given subcortical input can engage specific subgroups of interneurons and, in turn, target GABA receptors with specific subunit composition (cf. Freund, 2003). GABA released into the extrasynaptic space is taken up by both neurons and astrocytes by the membrane-bound GABA transporter (Semyanov et al., 2004). In summary, tonic inhibition is slow and diffuse, yet multiple mechanisms, especially GABA uptake (Richerson and Wu, 2003), can regulate its effects both temporally and spatially.
Indirect Effects of Inhibition on the Activity of Principal Cells
Although further research is needed to assess the direct metabolic costs of inhibition (Lauritzen, 2005), the indirect effects of inhibition, expressed through changing the firing rates and patterns of the principal cells, are easier to illustrate. Inhibition exerts a gain control on principal cell firing by both phasic (Figure 1) and tonic mechanisms. Inhibition can alter the membrane conductance and time constant and, therefore, the temporal window over which excitatory synaptic integration occurs (Pouille and Scanziani, 2001). This is not simply a linear (additive) operation by shifting the input-output excitability curve of principal cells to the left or right but a change in the slope of the input-output relationship because neuronal excitability depends on the variability of input conductance (Semyanov et al., 2004). The result is a hard-to-predict nonlinear relationship between afferent excitation (glutamate release and EPSPs) and output spiking. Oftentimes information is not represented by firing changes of the neuronal population but by changing the membership in cell assemblies, which represent a constant fraction of the population over time. For example, despite the robust changes in firing rates of both principal cells and interneurons at the ten milliseconds scale, the total firing rate of the hippocampal CA1 pyramidal cell population and several groups of interneurons at the time scale of seconds remains stable across the sleep-wake cycle, learning conditions, network state changes, and even after long-term potentiation (Csicsvari et al., 1999; Hirase et al., 2001; Dragoi et al., 2003). What does change is the membership of neurons in temporally defined cell assemblies and the spatiotemporal combinatorics of assembly sequences (Harris et al., 2003). On the other hand, subgroups of interneurons, located in the CA1 stratum oriens-alveus, can show as much as 50% increase in discharge rate during theta frequency oscillations of the network (Csicsvari et al., 1999), a state associated with increased attention, arousal, exploration, and locomotion (cf. Buzsáki, 2002). Despite the relatively constant firing rate of the principal cell population across various conditions and states, fMRI investigation can detect BOLD changes in the hippocampus in various tasks (Maguire et al., 1998; Zeineh et al., 2003), indicating that neuroimaging signals and firing rates of principal cells are not simply interchangeable. The lack of task-related firing-rate changes of the principal cells has been reported in other systems as well. In the V2 cortex of primates, robust attention-mediated increases were detected in the activity of putative interneurons but only negligible changes in firing rates of principal cells (Mitchell et al., 2007). The implication of these findings is that while increased spiking of the principal cells correlates with both the amount of released glutamate at the terminals and the frequency of EPSPs at their targets, the reverse may not be always true. Increased afferent excitation/glutamate release and associated dendritic depolarization, reflected by quantitative changes of the EEG, may result in increased output spiking of the principal cells, no change in mean firing rate or even decreased discharge output, depending on the state of inhibition (Figure 1).
Coactive Inhibition and Excitation of Single Neurons: Effect on Ion Fluxes
From the above summary, it follows that in the assessment of the metabolic expenditure of inhibition, it is important to distinguish between the direct or immediate ionicenergetic consequences of GABA-mediated mechanisms and the functional consequences of inhibitory transmission in intact neurons and networks. Synaptic transmission leads to dissipation of plasmalemmal ion gradients, and their restoration by active transport mechanisms is generally thought to be a major factor in the energy consumption of the brain (Siesjö, 1978; Attwell and Laughlin, 2001; Erecinska and Silver, 1989). However, as will be evident from the discussion below, it is not (either theoretically or empirically) possible to estimate the energetic costs of inhibition or excitation in isolation.
To illustrate our point, let us consider the total ion flux associated with a given glutamatergic postsynaptic event that activates a fixed number of ionotropic glutamate receptors in a target neuron. In the absence of inhibition, a certain number of Na+ ions will enter the cell across the glutamate receptor channels, and the target neuron will be depolarized. Now, if exactly the same glutamatergic conductance is evoked in the presence of GABAA receptor-mediated inhibition, a seemingly paradoxical consequence is that while the total flux of Na+ across glutamate-gated channels will be enhanced, there is a smaller depolarizing effect on membrane potential. The key to this “paradox” is simple: the driving force for Na+ will be larger in the presence of the coincident inhibition. It should be noted that the situation is analogous if we consider a fixed postsynaptic GABAA conductance in isolation and in the presence of coincident excitation: in the latter case, the target neuron will accumulate much more Cl− than in the former. An important conclusion from these elementary considerations based on ionic driving forces is that the total ion fluxes and hence, the cost of both excitation and inhibition at the level of a single target neuron is increased if there is temporal overlap between the two types of synaptic events. With coactive inhibition and excitation, most of the total ion flux is electrically neutral (based on the mutually neutralizing net influx of Na+ and Cl−). This will be discussed in more quantitative terms below.
In an isolated neuron, where a single synaptic input acts on the background of high input resistance to generate an IPSP, GABAA receptor-mediated net influx of Cl− is negligible, amounting to a net gain in the micromolar range of Cl− that is needed to charge up the membrane capacitance close to the chloride equilibrium potential, ECl (Figure 3; see Kaila, 1994; Farrant and Kaila, 2007; Plonsey and Barr, 2000). For a neuron in a real network, which receives inhibitory and excitatory signals simultaneously, most of the coincident influx of Cl− and Na+ will have an electrically neutralizing action on each other (see above). Hence, the total net ion fluxes will be orders of magnitude higher than the capacitive current. The metabolic consequences of simultaneous ionotropic excitation and inhibition can be best approached by examining the charge transfer mediated by inhibition (Qi) and excitation (Qe), by Cl− and Na+, respectively. Because capacitive currents are negligible and bulk electroneutrality must prevail (i.e., Qi = Qe), the postsynaptic energy cost of inhibition is dependent on coincident excitatory current mediated by ion-otropic glutamate receptors. At the same time, the magnitude of the postsynaptic energy cost of glutamate receptor activation is larger in the presence of inhibition. Based on the equivalent circuit shown in Figure 3, a quantitative expression for the energy cost (W) of coincident inhibition (mediated by Cl−) and excitation (mediated by Na+) can be obtained from
Figure 3. Coactive Inhibition and Excitation Increases the Amount of Net Influx of Cl− and Na+ and the Consequent Energetic Cost of Both Kinds of Postsynaptic Actions.
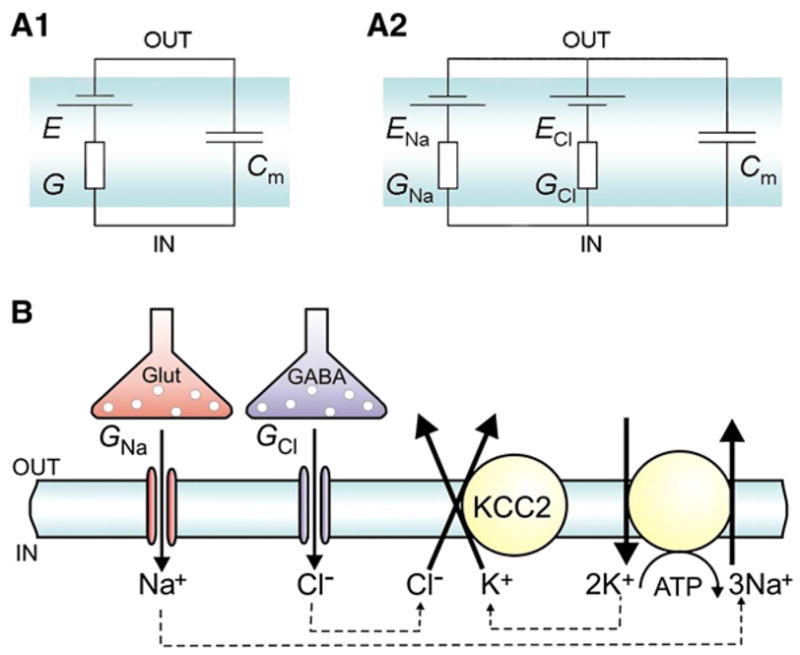
(A1) When channels that are permeable for a single ion species only are activated (depicted as conductance, G), the current that is needed to charge the membrane capacitance Cm to the equilibrium potential of this ion (E) is negligible. Hence, the net ion flux across the membrane is very small. (A2) During coactive excitation and inhibition, glutamate-and GABA-gated ion channels (GNa and GCl, respectively) are activated. A simultaneous influx of the oppositely charged Na+ and Cl− ions, driven by the ionic electrochemical gradients (Vm−ENa and Vm−ECl), takes place. Therefore, most of the net influx of the Na+ and Cl− ions is electrically neutral (see Equation 1).
(B) Simplified scheme depicting channel and transporter mediated ionic fluxes at excitatory and inhibitory synapses, where the net influx of Cl− and Na+ ions across glutamate- and GABA-gated ion channels is counteracted by the K-Cl cotransporter KCC2 (or some other KCC isoform) and by the Na-K ATPase. K+ accumulation by the Na-K AT-Pase leads typically to values of EK of about −100 mV, which is a fundamental requirement for hyperpolarizing GABAergic inhibition: the K+ electrochemical gradient sets the driving force for Cl− extrusion by KCC2. Because the Na-K ATPase takes up two K+ ions for one cycle of ATP hydrolysis, two Cl− ions are extruded at the expense of one ATP. Hence, one cycle of the Na-K pump that consumes one ATP molecule can offset the influx of two Cl− ions and three Na+ ions. “Out” and “in” refer to the extracellular and intracellular compartments, respectively. In (A2), the Hodgkin-Huxley convention of setting the polarities of ENa and ECl was adopted.
| (1) |
where W refers to the work required to re-establish the ion gradients that have been dissipated by the channel-mediated conductive net fluxes (I = current, G = conductance, E = equilibrium potential, and the subscripts refer to the ionic species). It should be re-emphasized that Equation 1 gives the current that flows in the conductive loop at steady state: the total ionic current I is not the net transmembrane current that affects Vm.
The above considerations imply that, in neurons embedded in an active network, quantification of the energy cost of inhibition or excitation is not meaningful in isolation. Because the “direct” or immediate energetic cost of a given GABAergic event is context-dependent, attempts to design experiments to estimate the cost of inhibition by pharmacological blockade of GABA (or glutamate) transmission is not a satisfactory approach. As already noted above, there is an exactly similar kind of context-dependence of the metabolic costs of excitation. It is worth emphasizing here that a pharmacological separation of the above kind is an invalid approach even at the level of a single postsynaptic neuron—this is a fundamental point conveyed by Equation 1.
On the basis of Equation 1, one might predict that enhancing tonic GABAA receptor-mediated conductance (see Farrant and Kaila, 2007) should lead to an increase in the energy consumption as long as excitatory transmission is not severely damped. Here, it is interesting to note that observations in normal human subjects using PET have shown that administration of the specific GABAA receptor agonist, 4, 5, 6, 7-tetrahydroisoxazolo[5,4-c]pyridin-3-ol (THIP; an agonist known to induce a tonic GABA conductance) leads to an increase in glucose metabolism, even though clinical and electroencephalographic monitoring showed a sedative effect and sleepiness after drug administration (Peyron et al., 1994a). In temporal lobe epilepsy patients, the THIP-induced increase of glucose metabolism was highest in the electrically hypoactive focus (Peyron et al., 1994b).
It appears that the main K-Cl cotransporter expressed in cortical neurons, KCC2, that plays a key role in the maintenance of the Cl− electrochemical gradient in cortical principal neurons (Rivera et al., 1999), is optimized for minimizing neuronal energy consumption. The electro-neutrality of K-Cl cotransport is notable here, because (by definition) an electroneutral transporter does not produce changes in the membrane potential and consequent voltage-driven ion fluxes. In particular, because the KCC2 cotransporter operates at near-equilibrium (Payne et al., 2003), it permits maximization of the energy harvested from the K+ gradient to fuel the extrusion of Cl−. At the transporter-stochiometry level, extruding one Cl− ion by neuronal K-Cl cotransporters requires the “energy” of one K+ ion (Figure 3). Here, it is also worth noting that the substantial efflux of bicarbonate across GABAA receptors (Kaila, 1994) will add a further component to the electrically silent net influx of Cl− and hence to the total energy expenditure related to inhibition. This is because the depolarizing driving force of HCO3− is generated by plasmalemmal ion transporters that control intracellular pH (Kaila and Voipio, 1987), which is maintained at a more alkaline steady-state level than what is predicted on the basis of a passive distribution of HCO3− (Chesler and Kaila, 1992).
The present discussion focuses on ionotropic GABAergic and glutamatergic transmission, but mutually neutralizing ion fluxes can arise from the coincident activation of a number of different types of channels (or from a non-ideal selectivity of a given channel type). Indeed, the fact that large electrically silent ion fluxes exist in brain is evident from the large (mM range) activity-dependent shifts of monovalent ions that can be detected under various conditions (Nicholson, 1993; Voipio and Kaila, 2000; Payne et al., 2003).
Further Interneuron Operations
There are other interneuron operations that should be considered in the present context. Some dendrite-targeting interneurons may not modify the firing rates of pyramidal cells significantly. Instead, they affect local computation by altering voltage- and NMDA receptor-mediated Ca2+ influxes (Miles et al., 1996; Buzsáki et al., 1996; Schiller and Schiller, 2001; Larkum et al., 1999). The metabolic costs of intracellular Ca2+ surges and the absorption of extracellular Ca2+ by glia are not well understood (Attwell and Laughlin, 2001) but may be important (Lauritzen, 2005). It has been specifically suggested that synaptic inhibition interferes with Ca2+-dependent production of vasoactive substances in principal cells, such as nitric oxide (NO), prostaglandins, and epoxyeicosatrienoic acid (Lauritzen, 2005). However, the validity of this hypothesis depends on the assumption that these substances are present in pyramidal cells and can be released in sufficient concentration to exert a vascular effect.
In addition to synaptic transmission, interneurons can also communicate through electrical gap junctions (Bennett and Zukin, 2004; Connors and Long, 2004; Traub et al., 2004; Hestrin and Galarreta, 2005). In developing brains, the roles of interneurons and the effects of GABA are less understood. GABA can exert a depolarizing effect on immature principal cell targets through GABAA receptors (Ben-Ari, 2002; Farrant and Kaila, 2007). It is noteworthy that during certain developmental time windows, neurons may show high expression levels of transporters that extrude (KCC2; Rivera et al., 1999) and accumulate (NKCC1; Sipilä et al., 2006) chloride. While this may seem like a costly operation, it is necessary for a strict control of the “set-point” of intracellular Cl−. In addition, the postsynaptic GABAergic currents have a much longer duration in immature than in mature neurons (e.g., Vicini et al., 2001). Although very little is understood about the consequences of such mechanisms for the imaged signal, they can be important in the interpretation of images in the developing brain (Volpe et al., 1983; Fair et al., 2007; Colonnese et al., 2007).
Interneuron Control of Local Circulation
Even though increases in blood flow appear not to be needed to deliver additional oxygen and glucose during increases in brain activity (Mintun et al., 2001; Powers et al., 1996; Raichle and Mintun, 2006), several neuromodulators have been shown to directly affect local circulation (Figure 4; cf. Iadecola, 2002; Hamel, 2006). Many of these vasoactive neuromodulators are supplied by interneurons, including vasoactive intestinal peptide (VIP), somatostatin, calcitonin generelated peptide (CGRP), and neuropeptide Y (NPY; cf. Freund and Buzsáki, 1996). Several interneuron species contain nicotinamide adenine dinucleotide phosphate (NADPH)-diaphorase (Hamel, 2006), the synthesizing enzyme of nitric oxide (NO; Vincent and Kimura, 1992). A characteristic feature of some of these interneurons is the presence “drum stick”-like axon appendages, often found in the proximity of microvessels and separated from the endothelium only by the basal lamina and the thin processes of pericytes (Sik et al., 1995; Estrada and DeFelipe, 1998; Iadecola, 2002; Iadecola et al., 1993; Vaucher et al., 2000). If elevated discharge rate of interneurons is a prerequisite for the release of these substances, local increase of blood flow may occur irrespective of the direction of discharge frequency in principal cells. An added complexity is that interneuron-released substances exert opposite effects on blood vessels, depending on the peptide content of the active interneuron species.
Figure 4. Along with Substances Released from Principal Cells and Astrocytes, Interneurons Are Known to Exert a Direct Control on Local Blood Supply.
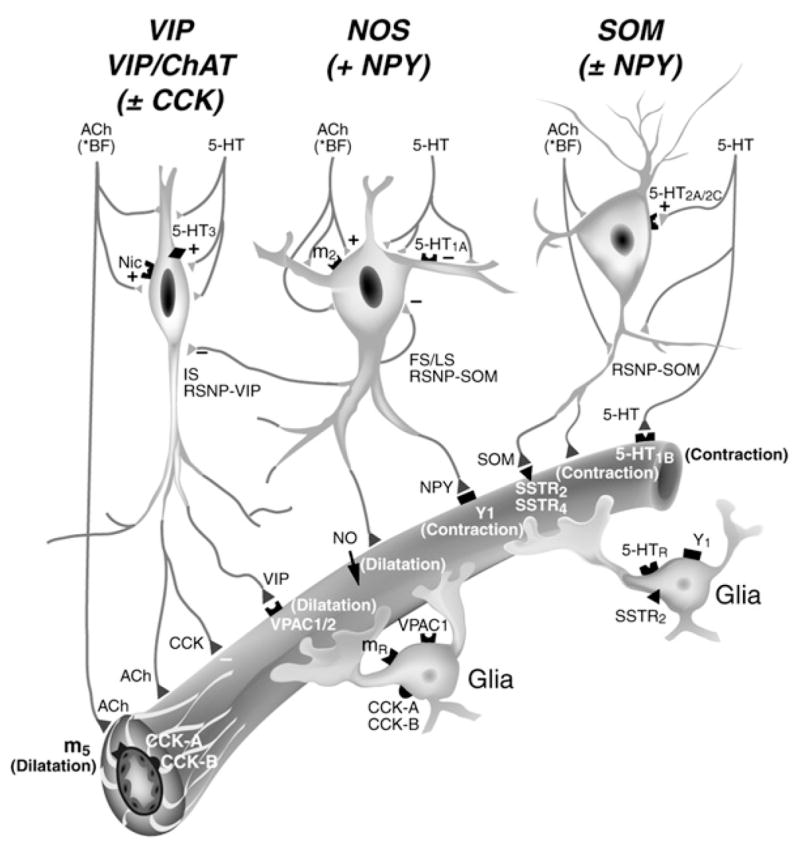
Their direct vasomotor effects are thought to be mediated by m5 muscarinic ACh receptor (dilatation) or 5-HT1B receptor (constriction). Local microvessels are endowed with subtype-specific receptors for vasoactive neuropeptides (VIP, NPY, or SOM) and NO (not shown) coreleased from GABAergic interneurons. Reprinted with permission from Cauli et al. (2004), copyright 2004 by the Society for Neuroscience.
The neurogliaform class of interneurons (Khazipov et al., 1995; Kawaguchi, 1995; Hestrin and Armstrong, 1996; Vida et al., 1998; Tamas et al., 2003) may be particularly well suited for local blood flow regulation. These interneurons, found in the hippocampus and in all layers of the neocortex, give rise to an especially dense local axon tree with thin branches. The branches have relatively few boutons and, importantly, vesicles are found not only in terminal boutons but also in axons far away from synaptic specializations, suggesting nonsynaptic release of neurotransmitters and modulators. In addition to GABA, they synthesize and release NPY and NO (Price et al., 2005). Neurogliaform neurons are therefore ideally positioned to affect local perfusion by integrating local excitatory inputs and releasing vasoactive substances.
In accordance with the above hypothesis, evoked discharge of single interneurons in whole-cell recordings was sufficient to either dilate or constrict neighboring micro-vessels. The interneurons that induced vasodilatation were subsequently characterized by single-cell polymerase chain reaction (PCR) and revealed to express of VIP or NO synthase, whereas interneurons that induced vasocontriction contained genes for SOM and NPY. Constriction appeared spatially restricted, maximal at the level of neurite apposition, and was associated with contraction of surrounding smooth muscle cells, providing direct evidence for interneural regulation of vascular sphincters (Cauli et al., 2004). The mechanisms by which interneurons regulate the caliber of local microvessels is not well understood (Zonta et al., 2003; Mulligan and MacVicar, 2004; Hirase et al., 2004). One mechanism may be due to the enhanced Ca2+ levels in astrocytes in response to elevated GABA levels (Kang et al., 1998). Furthermore, VIP can amplify nor-epinephrine-induced glycogenolysis in astrocytes, providing a local means of increasing glucose availability in a cell that uses glycolysis to provide the energy for glutamate removal from excitatory synapses (Magistretti et al., 1981).
In summary, because interneurons integrate the spiking output of large numbers of principal cells and release vasoactive substances in an activity-dependent manner, they are ideally situated to exert local and differential control on microcirculation, and the metabolic responses to changes in neuronal activity, especially excitatory neurotransmission.
Conclusions
Research over the past several decades has revealed a rather complex relationship between firing patterns of neurons, metabolic activity, and the imaged signal. The goal of our review has been to emphasize the distinct morphological, connection, integration, and firing properties of the cortical principal cell and the GABAergic interneuron populations and their intricate relationship to each other and to brain energetics. Because principal cells and interneurons contribute to information processing in fundamentally different ways, assessing their respective share of brain work and contribution in brain images is desirable.
Functional imaging continues to be the leading tool for the investigation of systems level functions of the human brain. As a measure of collective behavior of neuronal populations, it provides essential information about the modes of operations in distinct networks. However, these signals should not be regarded as a surrogate measure of neuronal spiking or a substitute of cellular resolution neurophysiological methods. Rather, they provide important complementary information. Furthermore, glucose consumption and blood flow alone cannot distinguish between nonoxidative glycolysis and mitochondrium-driven phosphorylation. A measurement of oxygen consumption must also be made to obtain a complete picture of the metabolic consequences of a change in brain activity. By knowing the contribution of principal cell and interneuron activity and their interactions to these distinct energy generating mechanisms one can begin to design paradigms to assess quantitatively the share of the GABAergic interneuron system and its context-dependent effects.
Acknowledgments
We would like to thank M. Farrant, T.F. Freund, A. Gulyás, B. Gulyás, H. Hirase, S. Laughlin, N. Logothetis, E. Nimchinski, S. Ogawa, A. Thomson, and J. Voipio for their comments on earlier versions of the manuscript. This work was supported by National Institutes of Health (to G.B. and M.R.) and by the Academy of Finland (to K.K.).
References
- Ackermann RF, Finch DM, Babb TL, Engel J., Jr Increased glucose metabolism during long-duration recurrent inhibition of hippocampal pyramidal cells. J Neurosci. 1984;4:251–264. doi: 10.1523/JNEUROSCI.04-01-00251.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acsády L, Kamondi A, Sik A, Freund T, Buzsáki G. GABAergic cells are the major postsynaptic targets of mossy fibers in the rat hippocampus. J Neurosci. 1998;18:3386–3403. doi: 10.1523/JNEUROSCI.18-09-03386.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames A., III CNS energy metabolism as related to function. Brain Res Brain Res Rev. 2000;34:42–68. doi: 10.1016/s0165-0173(00)00038-2. [DOI] [PubMed] [Google Scholar]
- Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. 2001;21:1133–1145. doi: 10.1097/00004647-200110000-00001. [DOI] [PubMed] [Google Scholar]
- Attwell D, Gibb A. Neuroenergetics and the kinetic design of excitatory synapses. Nat Rev Neurosci. 2005;6:841–849. doi: 10.1038/nrn1784. [DOI] [PubMed] [Google Scholar]
- Bartos M, Vida I, Jonas J. Synaptic mechanisms of synchroized gamma oscillations in inhibitory interneuron networks. Nat Rev Neurosci. 2007;8:45–56. doi: 10.1038/nrn2044. [DOI] [PubMed] [Google Scholar]
- Battaglia FP, Sutherland GR, McNaughton BL. Hippocampal sharp wave bursts coincide with neocortical “up-state”. Learn Mem. 2004;11:697–704. doi: 10.1101/lm.73504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y. Excitatory actions of gaba during development: the nature of the nurture. Nat Rev Neurosci. 2002;3:728–739. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- Bennett MV, Zukin RS. Electrical coupling and neuronal synchronization in the mammalian brain. Neuron. 2004;41:495–511. doi: 10.1016/s0896-6273(04)00043-1. [DOI] [PubMed] [Google Scholar]
- Blinkov SM, Glezer II. Das Zentralnervensystem in Zahlen und Tabellen. Jena, Germany: Fischer; 1968. [Google Scholar]
- Buzsáki G. Theta oscillations in the hippocampus. Neuron. 2002;33:325–340. doi: 10.1016/s0896-6273(02)00586-x. [DOI] [PubMed] [Google Scholar]
- Buzsáki G, Chrobak JJ. Temporal structure in spatially organized neuronal ensembles: a role for interneuronal networks. Curr Opin Neurobiol. 1995;5:504–510. doi: 10.1016/0959-4388(95)80012-3. [DOI] [PubMed] [Google Scholar]
- Buzsáki G, Penttonen M, Nádasdy Z, Bragin A. Pattern and inhibition-dependent invasion of pyramidal cell dendrites by fast spikes in the hippocampus in vivo. Proc Natl Acad Sci USA. 1996;93:9921–9925. doi: 10.1073/pnas.93.18.9921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G, Geisler C, Henze DA, Wang XJ. Interneuron diversity series: circuit complexity and axon wiring economy of cortical interneurons. Trends Neurosci. 2004;27:186–193. doi: 10.1016/j.tins.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Cauli B, Tong XK, Rancillac A, Serluca N, Lambolez B, Rossier J, Hamel E. Cortical GABA interneurons in neurovascular coupling: relays for subcortical vasoactive pathways. J Neurosci. 2004;24:8940–8949. doi: 10.1523/JNEUROSCI.3065-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatton JY, Pellerin L, Magistretti PJ. GABA uptake into astrocytes is not associated with significant cost implication for brain imaging of inhibition. Proc Natl Acad Sci USA. 2003;100:12456–12461. doi: 10.1073/pnas.2132096100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler M, Kaila K. Modulation of pH by neuronal activity. Trends Neurosci. 1992;15:396–402. doi: 10.1016/0166-2236(92)90191-a. [DOI] [PubMed] [Google Scholar]
- Colonnese MT, Phillips MA, Constantine-Paton M, Kaila K, Jasanoff A. Emergence of hemodynamic responses and functional connectivity during development of rat somatosensory cortex. Nat Neurosci. 2007 doi: 10.1038/nn2017. in press. Published online November 25, 2007. [DOI] [PubMed] [Google Scholar]
- Connors BW, Long MA. Electrical synapses in the mammalian brain. Annu Rev Neurosci. 2004;27:393–418. doi: 10.1146/annurev.neuro.26.041002.131128. [DOI] [PubMed] [Google Scholar]
- Csicsvari J, Hirase H, Czurko A, Mamiya A, Buzsáki G. Oscillatory coupling of hippocampal pyramidal cells and interneurons in the behaving rat. J Neurosci. 1999;19:274–287. doi: 10.1523/JNEUROSCI.19-01-00274.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csicsvari J, Jamieson B, Wise KD, Buzsáki G. Mechanisms of gamma oscillations in the hippocampus of the behaving rat. Neuron. 2003;37:311–322. doi: 10.1016/s0896-6273(02)01169-8. [DOI] [PubMed] [Google Scholar]
- DeFelipe J. Neocortical neuronal diversity: chemical heterogeneity revealed by colocalization studies of classic neurotransmitters, neuropeptides, calcium-binding proteins, and cell surface molecules. Cereb Cortex. 1993;3:273–289. doi: 10.1093/cercor/3.4.273. [DOI] [PubMed] [Google Scholar]
- Douglas RJ, Martin KA. Neuronal circuits of the neocortex. Annu Rev Neurosci. 2004;27:419–451. doi: 10.1146/annurev.neuro.27.070203.144152. [DOI] [PubMed] [Google Scholar]
- Dragoi G, Harris KD, Buzsáki G. Place representation within hippocampal networks is modified by long-term potentiation. Neuron. 2003;39:843–853. doi: 10.1016/s0896-6273(03)00465-3. [DOI] [PubMed] [Google Scholar]
- Eidelberg D, Moeller JR, Ishikawa T, Dhawan V, Spetsieris P, Sibersweig D, Stern E, Woods RP, Fazzini E, Dogali M, Beric A. regional metabolic correlates of surgical outcome following unilateral pallidotomy for Parkinson’s disease. Ann Neurol. 1996;39:450–459. doi: 10.1002/ana.410390407. [DOI] [PubMed] [Google Scholar]
- Eidelberg D, Moeller JR, Kazumata K, Antonini A, Sterio D, Dhawan V, Spetsieris P, Alterman R, Kelly PJ, Dogali M, et al. Metabolic correlates of pallidal neuronal activity in Parkinson’s disease. Brain. 1997;120:1315–1324. doi: 10.1093/brain/120.8.1315. [DOI] [PubMed] [Google Scholar]
- Erecinska M, Silver IA. ATP and brain function. J Cereb Blood Flow Metab. 1989;9:2–19. doi: 10.1038/jcbfm.1989.2. [DOI] [PubMed] [Google Scholar]
- Estrada C, DeFelipe J. Nitric oxide-producing neurons in the neocortex: morphological and functional relationship with intraparenchymal microvasculature. Cereb Cortex. 1998;8:193–203. doi: 10.1093/cercor/8.3.193. [DOI] [PubMed] [Google Scholar]
- Fair DA, Dosenbach NU, Church JA, Cohen AL, Brahmbhatt S, Miezin FM, Barch DM, Raichle ME, Petersen SE, Schlaggar BL. Development of distinct control networks through segregation and integration. Proc Natl Acad Sci USA. 2007;104:13507–13512. doi: 10.1073/pnas.0705843104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat Rev Neurosci. 2005;6:215–229. doi: 10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- Farrant M, Kaila K. The cellular, molecular and ionic basis of GABA(A) receptor signalling. Prog Brain Res. 2007;160:59–87. doi: 10.1016/S0079-6123(06)60005-8. [DOI] [PubMed] [Google Scholar]
- Fox MD, Raichle M. Spontaneous fluctuations in brain activity observed with fMRI. Nat Rev Neurosci. 2007;8:700–711. doi: 10.1038/nrn2201. [DOI] [PubMed] [Google Scholar]
- Fox PT, Raichle ME, Mintun MA, Dence C. Nonoxidative glucose consumption during focal physiologic neural activity. Science. 1988;241:462–464. doi: 10.1126/science.3260686. [DOI] [PubMed] [Google Scholar]
- Freund TF. Interneuron diversity series: rhythm and mood in perisomatic inhibition. Trends Neurosci. 2003;26:489–495. doi: 10.1016/S0166-2236(03)00227-3. [DOI] [PubMed] [Google Scholar]
- Freund TF, Buzsáki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Fricker D, Miles R. Differences between EPSP—spike transduction in pyramidal and inhibitory cells of the hippocampus. Neuron. 2000;28:559–569. [Google Scholar]
- Gibbs EL, Lennox WG, Nims LF, Gibbs FZ. Arterial and venous cerebral blood arterial-venous differences in man. J Biol Chem. 1942;18:325–332. [Google Scholar]
- Gulyás AI, Miles R, Sik A, Toth K, Tamamaki N, Freund TF. Hippocampal pyramidal cells excite inhibitory neurons through a single release site. Nature. 1993;366:683–687. doi: 10.1038/366683a0. [DOI] [PubMed] [Google Scholar]
- Gulyás AI, Megias M, Emri Z, Freund TF. Total number and ratio of excitatory and inhibitory synapses converging onto single interneurons of different types in the CA1 area of the rat hippocampus. J Neurosci. 1999;19:10082–10097. doi: 10.1523/JNEUROSCI.19-22-10082.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyás AI, Buzsaki G, Freund TF, Hirase H. Populations of hippocampal inhibitory neurons express different levels of cytochrome c. Eur J Neurosci. 2006;23:2581–2594. doi: 10.1111/j.1460-9568.2006.04814.x. [DOI] [PubMed] [Google Scholar]
- Hamel E. Perivascular nerves and the regulation of cerebrovascular tone. J Appl Physiol. 2006;100:1059–1064. doi: 10.1152/japplphysiol.00954.2005. [DOI] [PubMed] [Google Scholar]
- Harris KD, Csicsvari J, Hirase H, Dragoi G, Buzsáki G. Organization of cell assemblies in the hippocampus. Nature. 2003;424:552–556. doi: 10.1038/nature01834. [DOI] [PubMed] [Google Scholar]
- Heeger DJ, Ress D. What does fMRI tell us about neuronal activity? Nat Rev Neurosci. 2002;3:142–151. doi: 10.1038/nrn730. [DOI] [PubMed] [Google Scholar]
- Hendry SH, Schwark HD, Jones EG, Yan J. Numbers and proportions of GABA-immunoreactive neurons in different areas of monkey cerebral cortex. J Neurosci. 1987;7:1503–1519. doi: 10.1523/JNEUROSCI.07-05-01503.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henze DA, Borhegyi Z, Csicsvari J, Mamiya A, Harris KD, Buzsáki G. Intracellular features predicted by extracellular recordings in the hippocampus in vivo. J Neurophysiol. 2000;84:390–400. doi: 10.1152/jn.2000.84.1.390. [DOI] [PubMed] [Google Scholar]
- Hershey T, Revilla FJ, Wernle AR, McGee-Minnich L, Antenor JV, Videen TO, Dowling JL, Mink JW, Perlmutter JS. Cortical and subcortical blood flow effects of subthalamic nucleus stimulation in PD. Neurology. 2003;61:816–821. doi: 10.1212/01.wnl.0000083991.81859.73. [DOI] [PubMed] [Google Scholar]
- Hestrin S, Armstrong WE. Morphology and physiology of cortical neurons in layer I. J Neurosci. 1996;16:5290–5300. doi: 10.1523/JNEUROSCI.16-17-05290.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hestrin S, Galarreta M. Electrical synapses define networks of neocortical GABAergic neurons. Trends Neurosci. 2005;28:304–309. doi: 10.1016/j.tins.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Hirase H, Leinekugel X, Czurkó A, Csicsvari J, Buzsáki G. Firing rates of hippocampal neurons are preserved during subsequent sleep episodes and modified by novel awake experience. Proc Natl Acad Sci USA. 2001;98:9386–9390. doi: 10.1073/pnas.161274398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirase H, Qian L, Bartho P, Buzsaki G. Calcium dynamics of cortical astrocytic network in vivo. PloS Biol. 2004;2:e96. doi: 10.1371/journal.pbio.0020096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C. Intrinsic signals and functional brain mapping: caution, blood vessels at work. Cereb Cortex. 2002;12:223–224. doi: 10.1093/cercor/12.3.223. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Beitz AJ, Renno W, Xu X, Mayer B, Zhang F. Nitric oxide synthase-containing neural processes on large cerebral arteries and cerebral microvessels. Brain Res. 1993;606:148–155. doi: 10.1016/0006-8993(93)91583-e. [DOI] [PubMed] [Google Scholar]
- Isomura Y, Sirota A, Ozen S, Montgomery S, Mizuseki K, Henze DA, Buzsaki G. Integration and segregation of activity in entorhinal-hippocampal subregions by neocortical slow oscillations. Neuron. 2006;52:871–882. doi: 10.1016/j.neuron.2006.10.023. [DOI] [PubMed] [Google Scholar]
- Jinno S, Klausberger T, Marton TF, Dalezios Y, Roberts JDB, Fuentealba P, Bushong A, Henze DA, Buzsáki G, Somogyi P. Neuronal diversity in GABAergic long-range projections from the hippocampus. J Neurosci. 2007;27:8790–8804. doi: 10.1523/JNEUROSCI.1847-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas P, Bischofberger J, Fricker D, Miles R. Fast in, fast out—temporal and spatial signal processing in hippocampal interneurons. Trends Neurosci. 2004;27:30–40. doi: 10.1016/j.tins.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Kadekaro M, Crane AM, Sokoloff L. Differential effects of electrical stimulation of sciatic nerve on metabolic activity in spinal cord and dorsal root ganglion in the rat. Proc Natl Acad Sci USA. 1985;82:6010–6013. doi: 10.1073/pnas.82.17.6010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila K. Ionic basis of GABAA receptor channel function in the nervous system. Prog Neurobiol. 1994;42:489–537. doi: 10.1016/0301-0082(94)90049-3. [DOI] [PubMed] [Google Scholar]
- Kaila K, Voipio J. Postsynaptic fall in intracellular pH induced by GABA-activated bicarbonate conductance. Nature. 1987;330:163–165. doi: 10.1038/330163a0. [DOI] [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman SA, Nedergaard M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci. 1998;1:683–692. doi: 10.1038/3684. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y. Physiological subgroups of nonpyramidal cells with specific morphological characteristics in layer II/III of rat frontal cortex. J Neurosci. 1995;15:2638–2655. doi: 10.1523/JNEUROSCI.15-04-02638.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Kondo S. Parvalbumin, somatostatin and cholecystokinin as chemical markers for specific GABAergic interneuron types in the rat frontal cortex. J Neurocytol. 2002;31:277–287. doi: 10.1023/a:1024126110356. [DOI] [PubMed] [Google Scholar]
- Kennedy C, Rosiers MHD, Sakurada O, Shinohara MM, Reivich JW, Jehle L, Sokoloff L. Metabolic mapping of the primary visual system of the monkey by means of the autoradiographic [14C]deoxyglucose technique. Proc Natl Acad Sci USA. 1976;76:4203–4204. doi: 10.1073/pnas.73.11.4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khazipov R, Congar P, Ben-Ari Y. Hippocampal CA1 lacunosum-moleculare interneurons: modulation of monosynaptic GABAergic IPSCs by presynaptic GABAB receptors. J Neurophysiol. 1995;74:2126–2137. doi: 10.1152/jn.1995.74.5.2126. [DOI] [PubMed] [Google Scholar]
- Kraushaar U, Jonas P. Efficacy and stability of quantal GABA release at a hippocampal interneuron-principal neuron synapse. J Neurosci. 2000;20:5594–5607. doi: 10.1523/JNEUROSCI.20-15-05594.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupa DJ, Wiest MC, Shuler MG, Laubach M, Nicolelis MA. Layer-specific somatosensory cortical activation during active tactile discrimination. Science. 2004;304:1989–1992. doi: 10.1126/science.1093318. [DOI] [PubMed] [Google Scholar]
- Larkum ME, Kaiser KM, Sakmann B. Calcium electro-genesis in distal apical dendrites of layer 5 pyramidal cells at a critical frequency of back-propagating action potentials. Proc Natl Acad Sci USA. 1999;96:14600–14604. doi: 10.1073/pnas.96.25.14600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughlin SB, Sejnowski TJ. Communication in neuronal networks. Science. 2003;301:1870–1874. doi: 10.1126/science.1089662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritzen M. Opinion: reading vascular changes in brain imaging: is dendritic calcium the key? Nat Rev Neurosci. 2005;6:77–85. doi: 10.1038/nrn1589. [DOI] [PubMed] [Google Scholar]
- Lennie P. The cost of cortical computation. Curr Biol. 2003;13:493–497. doi: 10.1016/s0960-9822(03)00135-0. [DOI] [PubMed] [Google Scholar]
- Li XG, Somogyi P, Tepper JM, Buzsáki G. Axonal and dendritic arborization of an intracellularly labeled chandelier cell in the CA1 region of rat hippocampus. Exp Brain Res. 1992;90:519–525. doi: 10.1007/BF00230934. [DOI] [PubMed] [Google Scholar]
- Li XG, Somogyi P, Ylinen A, Buzsáki G. The hippocampal CA3 network: an in vivo intracellular labeling study. J Comp Neurol. 1994;339:181–208. doi: 10.1002/cne.903390204. [DOI] [PubMed] [Google Scholar]
- Logothetis NK. The underpinnings of the BOLD functional magnetic resonance imaging signal. J Neurosci. 2003;23:3963–3971. doi: 10.1523/JNEUROSCI.23-10-03963.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logothetis NK, Wandell BA. Interpreting the BOLD signal. Annu Rev Physiol. 2004;66:735–769. doi: 10.1146/annurev.physiol.66.082602.092845. [DOI] [PubMed] [Google Scholar]
- Logothetis NK, Pauls J, Augath M, Trinath T, Oeltermann A. Neurophysiological investigation of the basis of the fMRI signal. Nature. 2001;412:150–157. doi: 10.1038/35084005. [DOI] [PubMed] [Google Scholar]
- Losonczy A, Biro AA, Nusser Z. Persistently active cannabinoid receptors mute a subpopulation of hippocampal interneurons. Proc Natl Acad Sci USA. 2004;101:1362–1367. doi: 10.1073/pnas.0304752101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti PJ, Morrison JH, Shoemaker WJ, Sapin V, Bloom FE. Vasoactive intestinal polypeptide induces glycogenolysis in mouse cortical slices: a possible regulatory mechanism for the local control of energy metabolism. Proc Natl Acad Sci USA. 1981;78:6535–6539. doi: 10.1073/pnas.78.10.6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire EA, Burgess N, Donnett JG, Frackowiak RS, Frith CD, O’Keefe J. Knowing where and getting there: a human navigation network. Science. 1998;280:921–924. doi: 10.1126/science.280.5365.921. [DOI] [PubMed] [Google Scholar]
- Maguire JL, Stell BM, Rafizadeh M, Mody I. Ovarian cycle-linked changes in GABAA receptors mediating tonic inhibition alter seizure susceptibility and anxiety. Nat Neurosci. 2005;8:797–804. doi: 10.1038/nn1469. [DOI] [PubMed] [Google Scholar]
- Majewska MD. Neurosteroids: endogenous bimodal modulators of the GABAA receptor. Mechanism of action and physiological significance. Prog Neurobiol. 1992;38:379–395. doi: 10.1016/0301-0082(92)90025-a. [DOI] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 2004;5:793–807. doi: 10.1038/nrn1519. [DOI] [PubMed] [Google Scholar]
- Mata M, Fink DJ, Gainer H, Smith CB, Davidsen L, Savaki H, Schwartz WJ, Sokoloff L. Activity-dependent energy metabolism in rat posterior pituitary premarily reflects sodium pump activity. J Neurochem. 1980;34:213–215. doi: 10.1111/j.1471-4159.1980.tb04643.x. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Fisahn A. Interneurons unbound. Nat Rev Neurosci. 2001;2:11–23. doi: 10.1038/35049047. [DOI] [PubMed] [Google Scholar]
- McCasland JS, Hibbard LS. GABAergic neurons in barrel cortex show strong, whisker-dependent metabolic activation during normal behavior. J Neurosci. 1997;17:5509–5527. doi: 10.1523/JNEUROSCI.17-14-05509.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megias M, Emri Z, Freund TF, Gulyás AI. Total number and distribution of inhibitory and excitatory synapses on hippocampal CA1 pyramidal cells. Neuroscience. 2001;102:527–540. doi: 10.1016/s0306-4522(00)00496-6. [DOI] [PubMed] [Google Scholar]
- Miles R. Variation in strength of inhibitory synapses in the CA3 region of guinea-pig hippocampus in vitro. J Physiol. 1990;431:659–676. doi: 10.1113/jphysiol.1990.sp018353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles R, Toth K, Gulyás AI, Hajos N, Freund TF. Differences between somatic and dendritic inhibition in the hippocampus. Neuron. 1996;16:815–823. doi: 10.1016/s0896-6273(00)80101-4. [DOI] [PubMed] [Google Scholar]
- Mink JW. The basal ganglia: focused selection and inhibition of competing motor programs. Prog Neurobiol. 1996;50:381–425. doi: 10.1016/s0301-0082(96)00042-1. [DOI] [PubMed] [Google Scholar]
- Mintun MA, Lundstrom BN, Snyder AZ, Vlassenko AG, Shulman GL, Raichle ME. Blood flow and oxygen delivery to human brain during functional activity: theoretical modeling and experimental data. Proc Natl Acad Sci USA. 2001;98:6859–6864. doi: 10.1073/pnas.111164398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JF, Sundberg K, Reynolds J. Differential attention-dependent response modulation across cell classes in macaque visual area V4. Neuron. 2007;55:131–141. doi: 10.1016/j.neuron.2007.06.018. [DOI] [PubMed] [Google Scholar]
- Mody I, Pearce RA. Diversity of inhibitory neurotransmission through GABA(A) receptors. Trends Neurosci. 2004;27:569–575. doi: 10.1016/j.tins.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Mulligan SJ, MacVicar BA. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature. 2004;431:195–199. doi: 10.1038/nature02827. [DOI] [PubMed] [Google Scholar]
- Nicholson C. Ion-selective microelectrodes and diffusion measurements as tools to explore the brain cell microenvironment. J Neurosci Methods. 1993;48:199–213. doi: 10.1016/0165-0270(93)90092-6. [DOI] [PubMed] [Google Scholar]
- Nie F, Wong-Riley MT. Double labeling of GABA and cytochrome oxidase in the macaque visual cortex: quantitative EM analysis. J Comp Neurol. 1995;356:115–131. doi: 10.1002/cne.903560108. [DOI] [PubMed] [Google Scholar]
- Nudo RJ, Masterton RB. Stimulation-induced [14C]2-deoxyglucose labeling of synaptic activity in the central auditory system. J Comp Neurol. 1986;245:553–565. doi: 10.1002/cne.902450410. [DOI] [PubMed] [Google Scholar]
- Ogawa S, Lee TM, Kay AR, Tank DW. Brain magnetic resonance imaging with contrast dependent on blood oxygenation. Proc Natl Acad Sci USA. 1990;87:9868–9872. doi: 10.1073/pnas.87.24.9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Keefe J, Nadel L. The Hippocampus as a Cognitive Map. Oxford: Oxford University Press; 1978. [Google Scholar]
- Parra P, Gulyás AI, Miles R. How many subtypes of inhibitory cells in the hippocampus? Neuron. 1998;20:983–993. doi: 10.1016/s0896-6273(00)80479-1. [DOI] [PubMed] [Google Scholar]
- Patel AB, de Graaf RA, Mason GF, Rothman DL, Shulman RG, Behar KL. The contribution of GABA to glutamate/glutamine cycling and energy metabolism in the rat cortex in vivo. Proc Natl Acad Sci USA. 2005;102:5588–5593. doi: 10.1073/pnas.0501703102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne JA, Rivera C, Voipio J, Kaila K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003;26:199–206. doi: 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- Peyron R, Le Bars D, Cinotti L, Garcia-Larrea L, Galy G, Landais P, Millet P, Lavenne F, Froment JC, Krogsgaard-Larsen P, et al. Effects of GABAA receptors activation on brain glucose metabolism in normal subjects and temporal lobe epilepsy (TLE) patients. A positron emission tomography (PET) study Part I: Brain glucose metabolism is increased after GABAA receptors activation. Epilepsy Res. 1994a;19:45–54. doi: 10.1016/0920-1211(94)90087-6. [DOI] [PubMed] [Google Scholar]
- Peyron R, Cinotti L, Le Bars D, Garcia-Larrea L, Galy G, Landais P, Millet P, Lavenne F, Froment JC, Krogsgaard-Larsen P, et al. Effects of GABAA receptors activation on brain glucose metabolism in normal subjects and temporal lobe epilepsy (TLE) patients. A positron emission tomography (PET) study Part II: The focal hypometabolism is reactive to GABAA agonist administration in TLE. Epilepsy Res. 1994b;19:55–62. doi: 10.1016/0920-1211(94)90088-4. [DOI] [PubMed] [Google Scholar]
- Plonsey R, Barr RC. A Quantitative Approach. 2. New York: Kluwer Academic/Plenum Publishers; 2000. Bioelectricity. [Google Scholar]
- Pouille F, Scanziani M. Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science. 2001;293:1159–1163. doi: 10.1126/science.1060342. [DOI] [PubMed] [Google Scholar]
- Powers WJ, Hirsch IB, Cryer PE. Effect of stepped hypoglycemia on regional cerebral blood flow response to physiological brain activation. Am J Physiol. 1996;270:H554–H559. doi: 10.1152/ajpheart.1996.270.2.H554. [DOI] [PubMed] [Google Scholar]
- Price CJ, Cauli B, Kovacs ER, Kulik A, Lambolez B, Shigemoto R, Capogna M. Neurogliaform neurons form a novel inhibitory network in the hippocampal CA1 area. J Neurosci. 2005;25:6775–6786. doi: 10.1523/JNEUROSCI.1135-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raichle ME, Mintun MA. Brain work and brain imaging. Annu Rev Neurosci. 2006;29:449–476. doi: 10.1146/annurev.neuro.29.051605.112819. [DOI] [PubMed] [Google Scholar]
- Raichle ME, Posner JB, Plum F. Cerebral blood flow during and after hyperventilation. Arch Neurol. 1970;23:394–403. doi: 10.1001/archneur.1970.00480290014002. [DOI] [PubMed] [Google Scholar]
- Reyes A, Lujan R, Rozov A, Burnashev N, Somogyi P, Sakmann B. Target-cell-specific facilitation and depression in neocortical circuits. Nat Neurosci. 1998;1:279–285. doi: 10.1038/1092. [DOI] [PubMed] [Google Scholar]
- Ribak CE, Nitsch R, Seress L. Proportion of parvalbumin-positive basket cells in the GABAergic innervation of pyramidal and granule cells of the rat hippocampal formation. J Comp Neurol. 1990;300:449–461. doi: 10.1002/cne.903000402. [DOI] [PubMed] [Google Scholar]
- Richerson GB, Wu YM. Dynamic equilibrium of neurotransmitter transporters: not just for reuptake anymore. J Neurophysiol. 2003;90:1363–1374. doi: 10.1152/jn.00317.2003. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- Salinas E, Sejnowski TJ. Correlated neuronal activity and the flow of neural information. Nat Rev Neurosci. 2001;2:539–550. doi: 10.1038/35086012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarpeshkar R. Analog versus digital: extrapolating from electronics to neurobiology. Neural Comput. 1998;10:1601–1638. doi: 10.1162/089976698300017052. [DOI] [PubMed] [Google Scholar]
- Schiller J, Schiller Y. NMDA receptor-mediated dendritic spikes and coincident signal amplification. Curr Opin Neurobiol. 2001;11:343–348. doi: 10.1016/s0959-4388(00)00217-8. [DOI] [PubMed] [Google Scholar]
- Schwartz WJ, Smith CB, Davidsen L, Savaki H, Sokoloff L, Mata M, Fink DJ, Gainer H. Metabolic mapping of functional activity in the hypothalamo-neurohypophysial system of the rat. Science. 1979;205:723–725. doi: 10.1126/science.462184. [DOI] [PubMed] [Google Scholar]
- Semyanov A, Walker MC, Kullmann DM, Silver RA. Tonically active GABA A receptors: modulating gain and maintaining the tone. Trends Neurosci. 2004;27:262–269. doi: 10.1016/j.tins.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Shadlen MN, Newsome WT. The variable discharge of cortical neurons: implications for connectivity, computation, and information coding. J Neurosci. 1998;18:3870–3896. doi: 10.1523/JNEUROSCI.18-10-03870.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y, Hasenstaub A, McCormick DA. Turning on and off recurrent balanced cortical activity. Nature. 2003;423:288–293. doi: 10.1038/nature01616. [DOI] [PubMed] [Google Scholar]
- Sibson NR, Dhankhar A, Mason GF, Behar KL, Rothman DL, Shulman RG. In vivo 13C NMR measurements of cerebral glutamine synthesis as evidence for glutamate-glutamine cycling. Proc Natl Acad Sci USA. 1997;94:2699–2704. doi: 10.1073/pnas.94.6.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibson NR, Dhankhar A, Mason GF, Rothman DL, Behar KL, Shulman RG. Stoichiometric coupling of brain glucose metabolism and glutamatergic neuronal activity. Proc Natl Acad Sci USA. 1998;95:316–321. doi: 10.1073/pnas.95.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siesjö B. Brain energy metabolism. New York: Wiley; 1978. [Google Scholar]
- Sik A, Ylinen A, Penttonen M, Buzsáki G. Inhibitory CA1–CA3-hilar region feedback in the hippocampus. Science. 1994;265:1722–1724. doi: 10.1126/science.8085161. [DOI] [PubMed] [Google Scholar]
- Sik A, Penttonen M, Ylinen A, Buzsáki G. Hippocampal CA1 interneurons: an in vivo intracellular labeling study. J Neurosci. 1995;15:6651–6665. doi: 10.1523/JNEUROSCI.15-10-06651.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sik A, Penttonen M, Buzsáki G. Interneurons in the hippocampal dentate gyrus: an in vivo intracellular study. Eur J Neurosci. 1997;9:573–588. doi: 10.1111/j.1460-9568.1997.tb01634.x. [DOI] [PubMed] [Google Scholar]
- Sillito AM. Functional considerations of the operation of GABAergic inhibitory processes in the visual cortex. In: Jones EG, Peters A, editors. Cerebral Cortex, Volume 2: Functional Properties of Cortical Cells. New York: Plenum Press; 1984. pp. 91–117. [Google Scholar]
- Sipilä ST, Schuchmann S, Voipio J, Yamada J, Kaila K. The cation-chloride cotransporter NKCC1 promotes sharp waves in the neonatal rat hippocampus. J Physiol. 2006;573:765–773. doi: 10.1113/jphysiol.2006.107086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokoloff L. Metabolic Probes of Central Nervous System Activity in Experiment Animals and Man. I. Sunderland, MA: Sinauer Associates; 1984. [Google Scholar]
- Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD, Sakurada O, Shinohara M. The [14C]de-oxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem. 1977;28:897–916. doi: 10.1111/j.1471-4159.1977.tb10649.x. [DOI] [PubMed] [Google Scholar]
- Soltesz I. Diversity in the Neuronal Machine. Order and Variability in Interneuronal Microcircuits. Oxford: Oxford Univesity Press; 2006. [Google Scholar]
- Somogyi P, Klausberger T. Defined types of cortical interneuron structure space and spike timing in the hippocampus. J Physiol. 2005;562:9–26. doi: 10.1113/jphysiol.2004.078915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi P, Tamas G, Lujan R, Buhl EH. Salient features of synaptic organisation in the cerebral cortex. Brain Res Brain Res Rev. 1998;26:113–135. doi: 10.1016/s0165-0173(97)00061-1. [DOI] [PubMed] [Google Scholar]
- Stell BM, Brickley SG, Tang CY, Farrant M, Mody I. Neuroactive steroids reduce neuronal excitability by selectively enhancing tonic inhibition mediated by delta subunit-containing GABAA receptors. Proc Natl Acad Sci USA. 2003;100:14439–14444. doi: 10.1073/pnas.2435457100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swadlow HA. Efferent neurons and suspected interneurons in binocular visual cortex of the awake rabbit: receptive fields and binocular properties. J Neurophysiol. 1988;59:1162–1187. doi: 10.1152/jn.1988.59.4.1162. [DOI] [PubMed] [Google Scholar]
- Swadlow HA. Fast-spike interneurons and feedforward inhibition in awake sensory neocortex. Cereb Cortex. 2003;13:25–32. doi: 10.1093/cercor/13.1.25. [DOI] [PubMed] [Google Scholar]
- Tamas G, Buhl EH, Somogyi P. Fast IPSPs elicited via multiple synaptic release sites by different types of GABAergic neurone in the cat visual cortex. J Physiol. 1997;500:715–738. doi: 10.1113/jphysiol.1997.sp022054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamas G, Lorincz A, Simon A, Szabadics J. Identified sources and targets of slow inhibition in the neocortex. Science. 2003;299:1902–1905. doi: 10.1126/science.1082053. [DOI] [PubMed] [Google Scholar]
- Tepper JM, Abercrombie ED, Bolam JP. Progress in Brain Research. Vol. 160. Amsterdam: Elsevier; 2007. Gaba and the Basal Ganglia—from Molecules to Systems. [DOI] [PubMed] [Google Scholar]
- Traub RD, Bibbig A, LeBeau FE, Buhl EH, Whittington MA. Cellular mechanisms of neuronal population oscillations in the hippocampus in vitro. Annu Rev Neurosci. 2004;27:247–278. doi: 10.1146/annurev.neuro.27.070203.144303. [DOI] [PubMed] [Google Scholar]
- Thomson AM, Lamy C. Functional maps of neocortical local circuitry. Fontiers Neurosci. 2007;1:19–42. doi: 10.3389/neuro.01.1.1.002.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaucher E, Linville D, Hamel E. GABA neurons provide a rich input to microvessels but not nitric oxide neurons in the rat cerebral cortex: a means for direct regulation of local cerebral blood flow. J Comp Neurol. 2000;421:161–171. [PubMed] [Google Scholar]
- Vicini S, Ferguson C, Prybylowski K, Kralic J, Morrow AL, Homanics GE. GABAA receptor alpha1 subunit deletion prevents developmental changes of inhibitory synaptic currents in cerebellar neurons. J Neurosci. 2001;21:3009–3016. doi: 10.1523/JNEUROSCI.21-09-03009.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vida I, Halasy K, Szinyei C, Somogyi P, Buhl EH. Unitary IPSPs evoked by interneurons at the stratum radiatum-stratum lacunosum-moleculare border in the CA1 area of the rat hippocampus in vitro. J Physiol. 1998;506:755–773. doi: 10.1111/j.1469-7793.1998.755bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent SR, Kimura H. Histochemical mapping of nitric oxide synthase in the rat brain. Neuroscience. 1992;46:755–784. doi: 10.1016/0306-4522(92)90184-4. [DOI] [PubMed] [Google Scholar]
- Viswanathan A, Freeman RD. Neuometabolic coupling in cerebral cortex reflects synaptic more than spiking activity. Nat Neurosci. 2007 doi: 10.1038/nn1977. in press. [DOI] [PubMed] [Google Scholar]
- Voipio J, Kaila K. GABAergic excitation and K+-mediated volume transmission in the hippocampus. Prog Brain Res. 2000;125:329–338. doi: 10.1016/S0079-6123(00)25022-X. [DOI] [PubMed] [Google Scholar]
- Volpe JJ, Herscovitch P, Perlman JM, Raichle ME. Positron emission tomography in the newborn: extensive impairment of regional cerebral blood flow with intraventricular hemorrhage and hemorrhagic intracerebral involvement. Pediatrics. 1983;72:589–601. [PubMed] [Google Scholar]
- Wilson MA, McNaughton BL. Reactivation of hippocampal ensemble memories during sleep. Science. 1994;265:676–679. doi: 10.1126/science.8036517. [DOI] [PubMed] [Google Scholar]
- Wittner L, Henze DL, Záborszky L, Buzsáki G. Three-dimensional reconstruction of the axon arbor of a CA3 pyramidal cell recorded and filled in vivo. Brain Struct Funct. 2007;212:75–83. doi: 10.1007/s00429-007-0148-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeineh MM, Engel SA, Thompson PM, Bookheimer SY. Dynamics of the hippocampus during encoding and retrieval of face-name pairs. Science. 2003;299:577–580. doi: 10.1126/science.1077775. [DOI] [PubMed] [Google Scholar]
- Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, Carmignoto G. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci. 2003;6:43–50. doi: 10.1038/nn980. [DOI] [PubMed] [Google Scholar]