

Abstract
The ATM (Ataxia telangiectasia mutated) gene has been implicated as an early barrier to the growth and progression of incipient solid tumors. Here, we show that germline nullizygosity for the mouse Atm gene significantly increases the proliferative index, net growth rate, and multiplicity of intestinal adenomas in two distinct models of familial colon cancer: ApcMin/+ and Apc1638N/+. These effects of Atm deficiency are quantitatively different from deficiency for either of the genomic stability genes Bloom’s Syndrome helicase (Blm) or DNA ligase IV (Lig4), and the effect of Atm loss on tumor multiplicity is largely independent of the effect of ionizing radiation. Furthermore, the LOH (loss of heterozygosity) rates at the Apc (Adenomatous polyposis coli) locus are unaffected by Atm loss. Taken together, these data implicate the Atmgene product as a barrier to dysplastic growth in the early stages of intestinal tumor progression, independent of its effects on genomic stability.
Keywords: Gastrointestinal cancer, Atm, Apc, Blm, Lig4, modifier
Action of the ATM gene product is highly pleiotropic; its loss of function is associated with a wide range of neurological and aging phenotypes, including a strong predisposition to lymphoid malignancies (Savitsky et al., 1995). Recently, it has been discovered that the Atm pathway suppresses the progression of various solid tumors by triggering DNA damage response mechanisms, including senescence (Gorgoulis et al., 2005; Bartkova et al., 2005; Bartkova et al., 2006). Atm-deficient mice rapidly develop aggressive thymic lymphomas (Barlow et al., 1996), but have not yet shown a predisposition to epithelial neoplasia. We have therefore sought to test these aspects of Atm function using mouse models for epithelial cancer. The intestine provides a powerful experimental platform to ask whether Atm deficiency affects the formation and progression of solid tumors on Apc-mutant backgrounds. ApcMin/+ mice - which have a stop codon at position 850 - develop multiple tumors throughout the intestinal tract, and are widely used to test for the effects of various mutations and environmental factors on tumorigenesis (Taketo, 2006); Apc1638N/+ - which truncates Apc at codon 1638 and decreases protein levels to ~2% (Fodde et al., 1994) - is an independent model that develops 1–2 intestinal tumors with biological and molecular characteristics different from those of ApcMin/+ tumors (Haigis et al., 2004). We compare the results of crosses of Apc-mutant mice to Atm-deficient mice with similar crosses to mice deficient in the genomic stability proteins Bloom’s helicase (Blm) or DNA ligase 4 (Lig4).
We introgressed a null Atm allele (Barlow et al., 1996) onto the C57BL/6J (B6) background before crossing it to B6.Apc+/− mice. Atm−/− animals on both the B6 and 129/S6 (129) backgrounds showed a significant reduction in body mass, accompanied by a consistent shortening of the small intestinal tract by an average of 3 cm (data not shown). B6.Atm−/− mice are also infertile and developed thymic lymphomas at a reduced rate (10/44, 23%) compared to age-matched 129.Atm−/− animals (9/14, 63%; p<0.05, Fisher’s Exact Test). However, the lymphoma phenotype did not correlate with any intestinal phenotypes in Apc+/− derivatives of these Atm−/− genotypes.
To generate the three possible Atm genotypes on an Apc-mutant background, we intercrossed B6.Atm+/−;Apc+/− and B6. Atm+/−;Apc+/+ mice. B6.Atm−/− mice on an ApcMin/+ background developed 2-fold more intestinal tumors than heterozygous Atm or wildtype age-matched controls (Table 1a). To normalize for the length of the intestinal tract, tumors/cm² were measured and also found to be significantly different: Atm+/+, 0.4 ± 0.1; Atm+/−, 0.4 ± 0.1; Atm−/−, 0.9 ± 0.1 (p<0.01, Kruskal-Wallis test). Atm deficiency also produced a significant increase in colon tumor multiplicity and incidence. Notably, Atm+/− heterozygotes did not differ significantly from Atm+/+ homozygous wildtype mice for tumor multiplicity in the small intestine (p>0.1) or colon (p>0.2) or for incidence of colonic tumors (p>0.6). However, the average tumor diameter at 100–109 days of age increased significantly with decreasing allelic dosage of wildtype Atm (Fig. 1). The increase in size was uniform throughout the length of the intestine, with each quartile of the small intestine demonstrating a significant difference (p<0.02, Jonckheere-Terpstra test; data not shown). Preliminary data from four mice indicates that this size increase also holds true at 160–168 days of age (data not shown). We investigated whether the increase in tumor size reflected an enhancement of cellular proliferation. Immunohistochemical staining for the Ki-67 antigen revealed an increase in the percentage of Ki-67-positive cells in Atm−/−;ApcMin/+ tumors and in tumor-associated hyperplasia when compared to Atm+/+; ApcMin/+ ( Fig. 2a–f and Table 2).
Table 1. Effect of the genotype of (a) Atm and (b) Blm on intestinal tumor multiplicity.
The approximate fold-increase refers to Atm−/− or Blmm3/m3 compared to the other two classes. 129/S6 (129).Blmm3/+ mice were a gift from Allan Bradley (Baylor College of Medicine, Houston, TX); 129.Atm+/− mice were purchased from The Jackson Laboratory (Bar Harbor, ME); 129.Lig4+/− mice were a gift from Frederick Alt (Harvard University, Boston, MA). All mice were backcrossed to B6 for at least 11 (Atm), 10 (Blm), or 5 (Lig4) generations. Mouse husbandry and dissection were performed as previously described (Haigis et al., 2002). All mice were dissected between 3 and 5 months of age except for irradiated mice, which became moribund at 2 months. Whole-body IR was delivered using a Mark I 137 Cs irradiator, model 30 (J L Shepard & Assoc., San Fernando, CA), as previously described (Luongo and Dove, 1996). It was noted that Atm−/−;Apc1638N/+ and Blmm3/m3;Apc1638N/+ mice, but no other genotypic combination, developed numerous desmoid fibromas which were not enumerated.
| a | Atm+/+ | Atm+/− | Atm−/− | |||||
|---|---|---|---|---|---|---|---|---|
| Loc. | Tumor count, mean ± SD (incidence) | N | Tumor count, mean ± SD (incidence) | N | Tumor count, mean ± SD (incidence) | N | Fold Increase (95%CI‡) | |
| B6.ApcMin/+ | SI | 31 ± 7 | 47 | 35 ± 12 | 49 | 67 ± 10 | 17 | 2.0 (1.8–2.3)* |
| LI | 0.9 ± 1.5 (47%) | 1.1 ± 1.3 (47%) | 3.8 ± 3.7 (94%*) | 4.6 (2.2–9.3)* | ||||
| B6.ApcMin/+ +5.0Gy IR | SI | 280 ± 70 | 7 | 276 ± 54 | 12 | 267 ± 53 | 3 | 1.0 (0.7–1.3)† |
| LI | 7.7 ± 3.0 (100%) | 10 ± 7.5 (100%) | 22 ± 5.5 (100%†) | 2.4 (1.4–4.2)* | ||||
| B6.Apc1638N/+ | SI | 1.4 ± 1.1 | 13 | 1.5 ± 1.0 | 21 | 26 ± 9.4 | 20 | 16 (11–22)* |
| LI | 0 ± 0 (0%) | 0 ± 0 (0%) | 0.8 ± 0.8 (45%*) | N/A* | ||||
| b | Blm+/+ | Blmm3/+ | Blmm3/m3 | |||||
| Loc. | Tumor count, mean ± SD (incidence) | N | Tumor count, mean ± SD (incidence) | N | Tumor count, mean ± SD (incidence) | N | ~Fold-increase (95%CI‡) | |
| B6.ApcMin/+ | SI | 28 ± 12 | 9 | 25 ± 7 | 15 | 99 ± 16 | 4 | 3.8 (2.9–5.0)* |
| LI | 0.1 ± 0.3 (11%) | 0.4 ± 0.6 (33%) | 1.3 ± 0.5 (100%†) | 4.3 (1.8–21)* | ||||
| B6.Apc1638N/+ | SI | 1.8 ± 0.8 | 5 | 0.9 ± 1.1 | 8 | 6.2 ± 3.2 | 20 | 5.1 (2.8–10)* |
| LI | 0 ± 0 (0%) | 0 ± 0 (0%) | 0.1 ± 0.2 (5%†) | N/A† | ||||
p<0.05, Jonkheere-Terpstra Test or χ² test compared to controls
No significant difference
Bootstrap ratios comparing the homozygous knockouts to the combined heterozygotes and wildtypes; N/A, Not Applicable; SI, small intestine; LI, large intestine.
Figure 1.
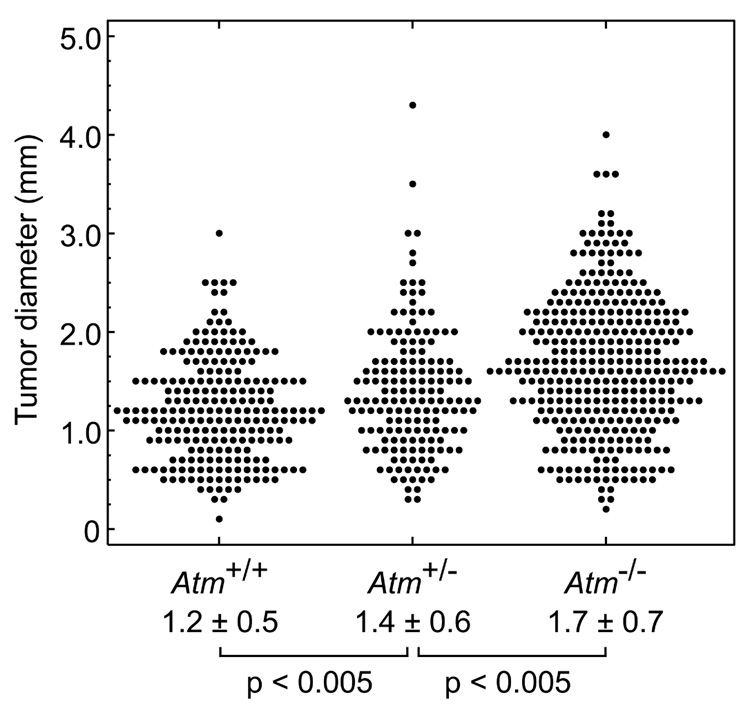
Effect of Atm genotype on the size of small intestinal tumors in 100–109 day old ApcMin/+ mice (Atm+/+: n=7 animals, 227 tumors; Atm+/−: n=6 animals, 164 tumors; Atm−/−: n=5 animals, 353 tumors). Intestinal tumors were measured at the maximal diameter using a dissection microscope with an ocular sizing reticle. Mean ± SD for each set and p-values (Jonckheere-Terpstra test) are shown.
Figure 2.
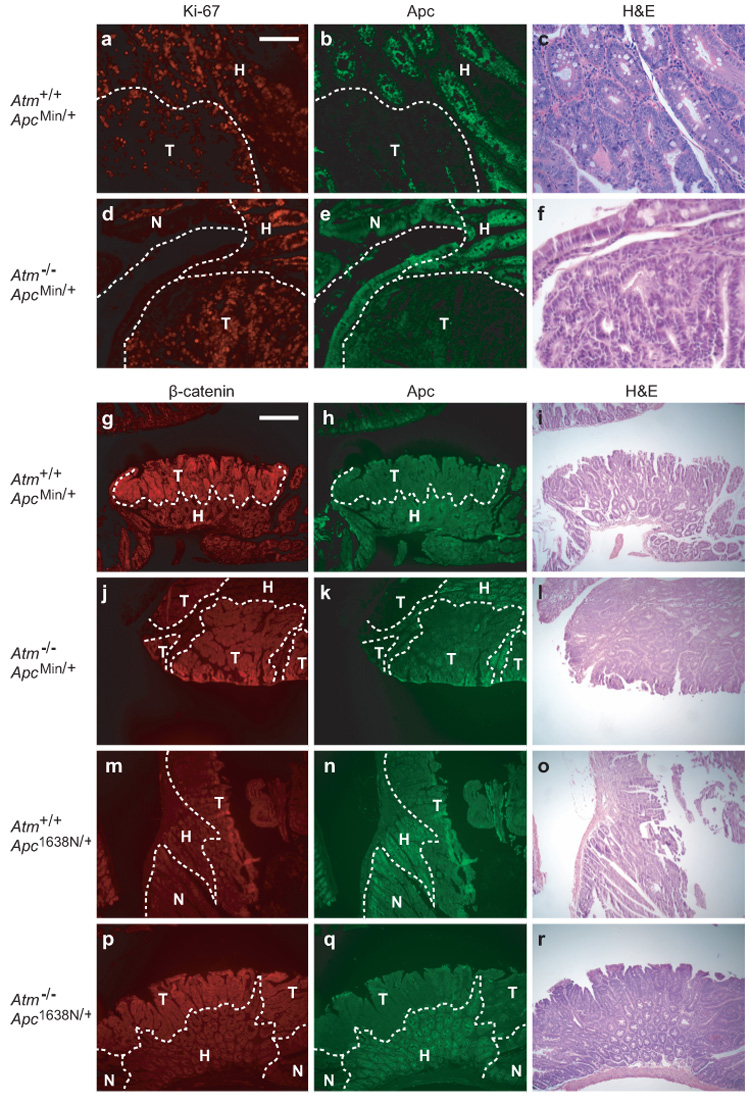
Ki-67, Apc, and β-catenin staining in intestinal tumors. (a–c) Atm+/+;ApcMin/+ and (d–f) Atm−/−;ApcMin/+ tumors stained for Ki-67 (red), Apc (green) and H&E. (g–i) Atm+/+;ApcMin/+, (j–l) Atm−/−;ApcMin/+, (m–o) Atm+/+;Apc1638N/+, and (p–r) Atm−/−;Apc1638N/+ tumors stained for β-catenin (red), Apc (green) and H&E. Histological classification was determined using Apc levels and cellular morphology. Immunofluorescence was performed as described previously (Haigis et al., 2002). All sections shown are representative. T, tumor; H, tumor-associated hyperplasia; N, normal tissue. Scale bars: a–f, 0.1mm; g–r, 0.4mm.
Table 2. The effect of Atm deficiency on the proliferative index of ApcMin/+ tumors.
The signals were counted, with the observer blind to the genotype, from digital photomicrographs displayed on a computer screen. Only crypts that were clearly epithelial by H&E staining on adjacent serial sections were assessed. Apc staining was used to discriminate between tumor (negative staining) and non-tumor (positive staining) tissue. Cells from the poorly characterized “cap” of Apc-positive cells surrounding the lumenal edges of tumors were not included. Six tumors and adjacent hyperplastic tissue were analyzed per genotype. The p-values were calculated using Fisher’s Exact Test.
| Ki-67-positive cells/total (percent positive; 95% CI) |
|||
|---|---|---|---|
| Tissue type | Atm+/+ | Atm−/− | P-value |
| Hyperplastic | 481/2163 | 424/1461 | <0.01 |
| (22.2%; 20.5–24.1) | (29.0%; 27.7–31.4) | ||
| Tumor | 749/2128 | 1175/2338 | <0.01 |
| (35.2%; 33.2–37.3) | (50.3%; 48.2–52.3) | ||
We then asked whether Atm-deficiency affects a different model of intestinal tumorigenesis, Apc1638N/+. First, we confirmed that the molecular mechanisms involved in Apc1638N/+ tumors differed from ApcMin/+ tumors. ApcMin/+ tumors showed a pattern of strong nuclear β-catenin staining in Apc-negative cells - a hallmark of neoplastic ApcMin/+ tissue. This observation was independent of the Atm status (12/14, 86% and 20/22, 91% for Atm+/+ and Atm−/−, respectively; Fig. 2g–1). By contrast, all 28 Apc1638N/+-induced tumors (14 Atm+/+ and 14 Atm−/−) were negative or only weakly positive for nuclear β-catenin, despite the loss of detectable Apc antigen (Fig. 2, compare m–r to g–l). We controlled for differences in staining by processing ApcMin/+ and Apc1638N/+ tumor sections in parallel. A similar result has been reported elsewhere (Janssen et al., 2006), but it remains unclear by what alternative mechanism Apc1638N/+ tumors initiate.
Next, we found that nullizygosity for Atm dramatically increased the multiplicity of Apc1638N/+ tumors in the small intestine, by 16-fold (Table 1a). Atm heterozygotes did not differ from Atm wildtype mice in the multiplicity of Apc1638N/+ tumors in the small intestine (p>0.7). Colonic tumor multiplicity and incidence were also significantly increased by Atm deficiency, as both Atm+/− heterozygous and Atm+/+ wildtype control groups of Apc1638N/+ developed no colonic tumors, while 45% of Atm−/− derivatives developed at least one. These colonic tumors showed a histological appearance indistinguishable from those of ApcMin/+ animals. The low overall tumor multiplicity in controls precluded a rigorous statistical analysis of tumor size. Overall, these results indicate that loss of Atm function enhances intestinal tumorigenesis, regardless of the initial underlying molecular mechanism.
Because the functions of Atm encompass more than cellular growth, we tested the effects of Atm loss on genomic stability. First, we measured rates of LOH at the Apc locus in ApcMin/+ mice. Quantitative Pyrosequencing® assays (Pyrosequencing is a registered trademark of Biotage) for the ApcMin/+ SNP (performed as described in Amos-Landgraf et al., 2007) demonstrated no statistically significant difference in the percentage of LOH-initiated tumors among the three genotypes: Atm+/+, 31/33 (94%); Atm+/−, 29/34 (85%); and Atm−/−, 45/51 (88%), ruling out a strong effect of Atm loss on this mechanism of tumor initiation. Second, we measured the regional localization of intestinal tumors, which is hypothesized to correlate with the mechanism of induction (Haigis et al., 2004). Atm deficiency did not affect the regional distribution of tumors per 10%- or 25% sub-interval of the small intestine, for either ApcMin/+ or Apc1638N/+ mice (data not shown). Finally, we induced DNA double-strand breaks, using whole-body ionizing radiation (IR). Unlike the acute sensitivity of 129.Atm−/− mice to IR, B6.Atm−/− mice with or without Apc mutations survived for over two months after a single 5Gy dose at 10 days of age. Dissections of age-matched mice showed that, after IR, Atm deficiency affected the multiplicity of ApcMin/+-induced tumors in the colon, but not in the small intestine, compared to irradiated Atm+/+;ApcMin/+ controls (Table 1a). However, after adjusting for intestinal length, a significant effect of Atm deficiency was seen for tumors/cm² in the small intestine: Atm+/+, 3.3 ± 0.6; Atm+/−, 3.4 ± 0.5; Atm−/− 4.4 ± 0.4 (p<0.05, Jonkheere-Terpstra Test). Thus, Atm nullizygosity has an effect at least partially independent of the genomic instability caused by IR.
To probe further the effects of genomic stability on intestinal tumors, we contrasted Atm mutants with a mutant for the Blm gene. In agreement with other observations (Luo et al., 2000), homozygosity for a Blm hypomorphic allele (Blmm3) increased ApcMin/+ tumor multiplicity 3.8-fold (Table 1b). Blmm3/+ heterozygotes and homozygous wildtypes did not differ significantly from each other (p>0.3). No studies have yet been reported for Blm and Apc1638N/+; we found that, in contrast to Atm-deficiency, Blm-deficiency had only a 5-fold enhancing effect on B6.Apc1638N/+ tumor multiplicity (Table 1b). Importantly, the 95% CI for the fold-increase caused by Blm-deficiency does not overlap with the larger effect caused by Atm-deficiency. Furthermore, only one of 20 B6.Blmm3/m3;Apc1638N/+ mice developed colonic tumors, which is not statistically different from heterozygotes or wildtype controls (p>0.4) but is significantly different from Atm−/−;Apc1638N/+ (p<0.01). This Blm hypomorph is known to increase ApcMin/+ tumor multiplicity via an increase in somatic recombination proximal to the Apc locus (Luo et al., 2000; Goss et al., 2002). Therefore, our results with the Blmm3 allele argue that the generation of Apc1638N/1638N cells is not sufficient to induce adenomas at a high rate. Thus, although genome-wide somatic hyperrecombination strongly enhances ApcMin/+-induced tumors, it is insufficient to explain the dramatic affect of Atm loss on Apc1638N/+ tumorigenesis.
Finally, we sought further evidence for an effect of Atm deficiency distinct from genomic instability by studying Apc-mutant mice haploinsufficient for Lig4, which encodes a protein involved in maintaining genomic stability and which, unlike some modifiers of ApcMin/+ - Blm (Luo et al., 2000; Goss et al., 2002), Recql4 (Mann et al., 2005), or Bub1R (Rao et al., 2005)- has no known role in cell cycle checkpoints (Frank et al., 2000). Lig4+/− mice have previously been shown to increase the genomic instability and multiplicity of sarcomas in Ink4a/Arf/− mice (Sharpless et al., 2001). By contrast, B6.Lig4+/−;ApcMin/+ or B6.Lig4+/−;Apc1638N/+ mice did not have a significantly different tumor multiplicity (p>0.6; data not shown) or average size (p>0.9) when compared to Lig4+/+ controls. Taken together, these and the preceding results suggest that the effect of Atm deficiency on intestinal tumorigenesis involves functions beyond the maintenance of genomic stability.
Atm encodes a protein with pleiotropic functions involving cell cycle checkpoints, DNA double strand break repair, senescence, and apoptosis. It has recently been proposed that Atm and other checkpoint proteins also serve as barriers to the growth of incipient tumors (Gorgoulis et al., 2005; Bartkova et al., 2005). Various human solid tumors show a sharp increase in an activated form of Atm protein at early stages, signals that are then diminished or lost in later-stage carcinomas. Other recent evidence in human tumors correlates a decrease in colon tumor-specific Atm protein levels (Grabsch et all., 2006) and an increase in Atm promoter hypermethylation (Bai et al., 2004) with progression to carcinoma and decreased survival. Allelic imbalance at the 11q22–23 interval containing Atm is also associated with polyploidy in colorectal carcinomas (Sugai et al., 2001). Inactivation of the Atm and/or related pathways might therefore be required for tumor progression. We have reported here that ablation of the Atm gene significantly sensitizes intestinal epithelia to both tumor multiplicity and growth. These findings, along with the recent studies of Atm knockdown in mouse cell line explants and the inverse correlation between senescence and colorectal tumor progression (Bartkova et al., 2006) lends strong support to the hypothesis that the Atm function can negatively regulate tumor emergence in epithelial tissues. Additionally, heterozygosity for the Atm mutation appeared to affect intestinal tumor size (but not multiplicity); Atm haploinsufficiency has long been controversially associated with human breast cancer (Renwick et al., 2006).
How exactly does Atm control intestinal tumor multiplicity and size via proliferation? The pleiotropy of Atm action makes this a difficult question to answer, and is further complicated by the possibility that Atm could act in a non-cell autonomous fashion, similar to studies implicating stromal p53 loss in mouse models of prostate cancer (Hill et al., 2005). Nevertheless, models of possible pathways can be constructed. For example, p53 protein is phosphorylated directly by the Atm kinase (Westphal et al., 1997); perhaps perturbed p53 signaling, including decreased expression levels of p21, potentiates increased cell survival and/or proliferation. Indeed, we have shown previously that p53 nullizygosity on the B6 background increases ApcMin/+ tumor multiplicity from 32 to 45 tumors (Halberg et al., 2000); however, this is significantly less than the increase from Atm loss (p<0.05). Thus, other pathways are likely involved: for example, Atm also regulates cell cycle checkpoints, lying directly upstream of many key effectors such as Chk2 (Matsuoka et al., 2000). In principle, defective checkpoint signaling can result in the deregulation of cell division. Finally, the increase in proliferation is consistent with a loss of senescence induction, as recently proposed for the knockdown of Atm (Bartkova et al., 2006). Confirmation of this speculation will require analysis of senescence markers.
In summary, we have demonstrated that Atm can negatively regulate tumorigenesis in epithelial tissues by opposing the proliferation of dysplastic cells, acting independently of effects on genomic stability.
Acknowledgements
We thank Caroline Alexander for discussions and Linda Clipson for critical reading and editing of the manuscript. Allan Bradley and Frederick Alt generously provided key mouse strains. This work was supported by the National Cancer Institute (NCI) training grant CA009135 to L.N.K and by NCI grant R37CA63677
Reference List
- Amos-Landgraf JM, Kwong LN, Kendziorski CM, Reichelderfer M, Torrealba J, Weichert J, et al. A target-selected Apc-mutant rat kindred enhances the modeling of familial human colon cancer. Proc Natl Acad Sci U S A. 2007;104:4036–4041. doi: 10.1073/pnas.0611690104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai AHC, Tong JHM, To KF, Chan MWY, Man EPS, Lo KW, et al. Promoter hypermethylation of tumor-related genes in the progression of colorectal neoplasia. Int J Cancer. 2004;112:846–853. doi: 10.1002/ijc.20485. [DOI] [PubMed] [Google Scholar]
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, et al. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1996;86:159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- Fodde R, Edelmann W, Yang K, van Leeuwen C, Carlson C, Renault B, et al. A targeted chain-termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proc Natl Acad Sci U S A. 1994;91:8969–8973. doi: 10.1073/pnas.91.19.8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank KM, Sharpless NE, Gao Y, Sekiguchi JM, Ferguson DO, Zhu C, et al. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol Cell. 2000;5:993–1002. doi: 10.1016/s1097-2765(00)80264-6. [DOI] [PubMed] [Google Scholar]
- Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- Goss KH, Risinger MA, Kordich JJ, Sanz MM, Straughen JE, Slovek LE, et al. Enhanced tumor formation in mice heterozygous for Blm mutation. Science. 2002;297:2051–2053. doi: 10.1126/science.1074340. [DOI] [PubMed] [Google Scholar]
- Grabsch H, Dattani M, Barker L, Maughan N, Maude K, Hansen O, et al. Expression of DNA double-strand break repair proteins ATM and BRCA1 predicts survival in colorectal cancer. Clin Cancer Res. 2006;12:1494–1500. doi: 10.1158/1078-0432.CCR-05-2105. [DOI] [PubMed] [Google Scholar]
- Haigis KM, Caya JG, Reichelderfer M, Dove WF. Intestinal adenomas can develop with a stable karyotype and stable microsatellites. Proc Natl Acad Sci U S A. 2002;99:8927–8931. doi: 10.1073/pnas.132275099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis KM, Hoff PD, White A, Shoemaker AR, Halberg RB, Dove WF. Tumor regionality in the mouse intestine reflects the mechanism of loss of Apc function. Proc Natl Acad Sci U S A. 2004;101:9769–9773. doi: 10.1073/pnas.0403338101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halberg RB, Katzung DS, Hoff PD, Moser AR, Cole CE, Lubet RA, et al. Tumorigenesis in the multiple intestinal neoplasia mouse: redundancy of negative regulators and specificity of modifiers. Proc Natl Acad Sci U S A. 2000;97:3461–3466. doi: 10.1073/pnas.050585597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill R, Song Y, Cardiff RD, Van Dyke T. Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell. 2005;123:1001–1011. doi: 10.1016/j.cell.2005.09.030. [DOI] [PubMed] [Google Scholar]
- Janssen KP, Alberici P, Fsihi H, Gaspar C, Breukel C, Franken P, et al. APC and oncogenic KRAS are synergistic in enhancing Wnt signaling in intestinal tumor formation and progression. Gastroenterology. 2006;131:1096–1109. doi: 10.1053/j.gastro.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Luo G, Santoro IM, McDaniel LD, Nishijima I, Mills M, Youssoufian H, et al. Cancer predisposition caused by elevated mitotic recombination in Bloom mice. Nat Genet. 2000;26:424–429. doi: 10.1038/82548. [DOI] [PubMed] [Google Scholar]
- Luongo C, Dove WF. Somatic genetic events linked to the Apc locus in intestinal adenomas of the Min mouse. Genes Chromosomes Cancer. 1996;17:194–198. doi: 10.1002/1098-2264(199611)17:3<194::aid-gcc2870170302>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Mann MB, Hodges CA, Barnes E, Vogel H, Hassold TJ, Luo G. Defective sister-chromatid cohesion, aneuploidy and cancer predisposition in a mouse model of type II Rothmund-Thomson syndrome. Hum Mol Genet. 2005;14:813–825. doi: 10.1093/hmg/ddi075. [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci U S A. 2000;97:10389–10394. doi: 10.1073/pnas.190030497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao CV, Yang YM, Swamy MV, Liu T, Fang Y, Mahmood R, et al. Colonic tumorigenesis in BubR1+/−ApcMin/+ compound mutant mice is linked to premature separation of sister chromatids and enhanced genomic instability. Proc Natl Acad Sci U S A. 2005;102:4365–4370. doi: 10.1073/pnas.0407822102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006;38:873–875. doi: 10.1038/ng1837. [DOI] [PubMed] [Google Scholar]
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- Sharpless NE, Ferguson DO, O’Hagan RC, Castrillon DH, Lee C, Farazi PA, et al. Impaired nonhomologous end-joining provokes soft tissue sarcomas harboring chromosomal translocations, amplifications, and deletions. Molecular Cell. 2001;8:1187–1196. doi: 10.1016/s1097-2765(01)00425-7. [DOI] [PubMed] [Google Scholar]
- Sugai T, Habano W, Uesugi N, Jiao YF, Nakamura S, Yoshida T, et al. Frequent allelic imbalance at the ATM locus in DNA multiploid colorectal carcinomas. Oncogene. 2001;20:6095–6101. doi: 10.1038/sj.onc.1204731. [DOI] [PubMed] [Google Scholar]
- Taketo MM. Mouse models of gastrointestinal tumors. Cancer Science. 2006;97:355–361. doi: 10.1111/j.1349-7006.2006.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal CH, Rowan S, Schmaltz C, Elson A, Fisher DE, Leder P. atm and p53 cooperate in apoptosis and suppression of tumorigenesis, but not in resistance to acute radiation toxicity. Nat Genet. 1997;16:397–401. doi: 10.1038/ng0897-397. [DOI] [PubMed] [Google Scholar]