

Abstract
Genome activity and nuclear metabolism clearly depend on accessibility, but it is not known whether and to what extent nuclear structures limit the mobility and access of individual molecules. We used fluorescently labeled streptavidin with a nuclear localization signal as an average-sized, inert protein to probe the nuclear environment. The protein was injected into the cytoplasm of mouse cells, and single molecules were tracked in the nucleus with high-speed fluorescence microscopy. We analyzed and compared the mobility of single streptavidin molecules in structurally and functionally distinct nuclear compartments of living cells. Our results indicated that all nuclear subcompartments were easily and similarly accessible for such an average-sized protein, and even condensed heterochromatin neither excluded single molecules nor impeded their passage. The only significant difference was a higher frequency of transient trappings in heterochromatin, which lasted only tens of milliseconds. The streptavidin molecules, however, did not accumulate in heterochromatin, suggesting comparatively less free volume. Interestingly, the nucleolus seemed to exclude streptavidin, as it did many other nuclear proteins, when visualized by conventional fluorescence microscopy. The tracking of single molecules, nonetheless, showed no evidence for repulsion at the border but relatively unimpeded passage through the nucleolus. These results clearly show that single-molecule tracking can provide novel insights into mobility of proteins in the nucleus that cannot be obtained by conventional fluorescence microscopy. Our results suggest that nuclear processes may not be regulated at the level of physical accessibility but rather by local concentration of reactants and availability of binding sites.
INTRODUCTION
Although the nucleus is the hallmark of all eukaryotic cells, remarkably little is known of its internal structure and function. The development of antibodies and, more recently, the ability to tag proteins fluorescently have revealed a complex structure with multiple discrete subcompartments involved in RNA and DNA metabolism (1,2). Unlike cytoplasmic organelles, however, subnuclear compartments are not surrounded by membranes. This raises the question of how they arise and whether they impose constraints on the accessibility and mobility of other molecules. Such constraints would impact and thereby control nuclear functions; e.g., restricted access to chromatin or subsets thereof would have as a consequence their transcriptional silencing. Furthermore, such general restricted access to a particular subnuclear compartment in combination with sequestration of subsets of factors to the same compartment mediated by protein or protein–nucleic acid interactions would increase not only the speed of individual nuclear reactions but also their specificity (3). It is therefore necessary to elucidate whether and how the physical structure of the nucleus affects the dynamics and access of proteins.
Earlier studies with fluorescent dextrans showed that diffusion in the cell is four to eight times slower than in aqueous solutions (4,5). Moreover, diffusion measurements of macromolecules in cells using fluorescence photobleaching, correlation microscopy, and time-resolved anisotropy revealed unexpectedly high mobilities (6). From such studies, a view of the cell's interior as a watery but crowded environment rather than a homogeneous viscous gel has emerged.
Recent studies have now combined fluorescent macromolecules with subnuclear compartment labels. Labeling whole chromatin with either fluorescent histones or with DNA dyes and measuring the intranuclear steady-state distribution of injected dextrans relative to chromatin density provided evidence for a high degree of penetration of the probes into chromatin (7–9). The facts that dextrans do not have a rigid shape and that the polymer preparations are not always homogeneous in size introduce some variability in the results from different reports. Nevertheless, gradual exclusion from chromatin was observed for dextrans of 77 kDa or larger (10).
Kinetic studies have also been performed investigating RNA movement through nuclear subcompartments using fluorescence photobleaching or uncaging as well as single-molecule video microscopy (11–15). The outcome of these studies indicated that most regions of the nucleus are accessible to RNA particles, although some results pointed to a preferential movement through the interchromatin space (12), and the reports differed on its energy dependence (11,13).
A profusion of kinetics studies in recent years (16) have measured the mobility of fluorescently tagged nuclear proteins, mostly employing fluorescence photobleaching/activation and in some cases also fluorescence correlation microscopy. Such analyses provided a highly dynamic view of the nuclear interior with proteins diffusing rapidly within the nucleus and most often showing a fast exchange at their binding sites. The experimental limitations of such photobleaching/activation measurements and corresponding data analysis have recently been discussed (17,18) and include a relatively low temporal and spatial resolution as well as the difficulty in extracting accurate physicochemical parameters (e.g., residence times). Because most of the factors measured have endogenous binding partners, probing the structural accessibility of the nuclear interior independent of their specific interactions was not feasible. Although in other reports GFP§§ was used as an inert tracer protein to probe the nuclear interior, no correlation with the different subnuclear compartments was made.
In this study, we set out to measure how a single protein travels through the nucleus and to determine whether its movement is controlled by subnuclear compartmentalization, in particular whether dense chromatin regions or the nucleolus restricts its access and/or mobility.
MATERIALS AND METHODS
Cell culture and transfection
Mouse C2C12 myoblasts were cultured as described previously (19). For live cell analysis, cells were seeded onto either one-well LabTek chambers (Nalge Nunc International, Rochester, NY) or glass-bottom dishes (MatTek, Ashland, MA). Transfection using the CaPO4-DNA coprecipitation method (20) and microinjection were performed 1 and 2 days later, respectively. Mouse RAW264.7 macrophage cells were grown in RPMI-1640 medium. The human Sk-N-SH neuroblastoma cell line was grown in Minimum Essential Medium supplemented with fetal bovine serum.
Plasmids
As reference label for the interchromatin space (“speckled compartment”) and for pericentric heterochromatin, we transfected cells with plasmids coding for fusion proteins of ASF/SF2-GFP (21) or MeCP2-GFP (22), respectively. To monitor the cell cycle stage, cells were additionally transfected with RFP-PCNA (23). Only non-S-phase cells were selected for measurements.
Probe preparation and microinjection
SAv-Cy5 (20 μM; Amersham Life Sciences, Arlington Heights, IL; labeling ratio four to five molecules per protein) and biotin-NLS (1 μM) were mixed 1:5 ∼30 min before the injection and, after 15 min of incubation at room temperature, centrifuged at 13,000 rpm for 15 min. For the SMT experiments, the solution was further diluted 1:10 with PBS before centrifugation. Microinjection was carried out with an Eppendorf injection and micromanipulation setup. The parameters for cytoplasmic microinjection were set to 0.7-s injection time with 20-fPa injection pressure and 15-fPa holding pressure. Imaging and measurements were started 60 min after microinjection to allow cells to recover, which was ascertained by examining the cellular morphology.
Confocal microscopy and fluorescence photobleaching
Steady-state distributions for NLS-SAv-Cy5 complexes were analyzed by live cell microscopy using a confocal laser scanning microscope LSM510Meta (Carl Zeiss, Jena, Germany) equipped with a 63× planapochromat phase-contrast oil-immersion objective, NA 1.4, heated to 37°C. GFP was excited with the 488-nm line of an argon ion laser, whereas Cy5 was excited at 633-nm light from a HeNe laser. The main beam splitter for both settings was a UV/488/543/633 filter, and the secondary beam splitter was NFT545. For the detection of GFP fluorescence, we used a band-pass filter BP500-530, and for Cy5 detection, a long-pass filter LP650. The constitutive heterochromatin domains were marked by the MeCP2-GFP label, and the nucleoli were identified according to the phase-contrast image. In these domains, the respective mean fluorescence intensities were calculated in the red channel and normalized to the maximum values. The time series and FLIP experiments were done on the same equipment using 100% power of the 633-nm HeNe laser for photobleaching. The FLIP was performed by bleaching a spot of 3.6-μm diameter for 554-ms duration followed by one imaging scan of 0.8-s duration and repeated 200 times.
Single-molecule microscopy
Single-molecule experiments were performed at room temperature using an inverted wide-field microscope equipped with a 63× NA 1.4 objective lens employing three fluorescence channels (24,25). For identification of the pericentric heterochromatin, cells were transfected with MeCP2-GFP. In these cells, the pericentric heterochromatin and nucleoplasmic trajectories were recorded. The nucleoplasm was defined as the nuclear space excluding the MeCP2 domains. Therefore, the nucleoplasmic trajectories also contained a small fraction of traces occurring in the nucleoli. Because the nucleoli displayed a low concentration of SAv-Cy5 molecules (42%, see Table S1) and occupied on average only 35% of the non-MeCP2-GFP regions within the nuclei, this gives an approximate contribution of 15%. Single-molecule experiments aimed at analyzing the trajectories within the nucleoli were performed using C2C12 cells transfected with ASF/SF2-GFP. In these experiments, nucleoli were identified according to their exclusion of ASF/SF2-GFP. For all measurements, non-S-phase cells were selected according to their RFP-PCNA subnuclear distribution (26), and the RFP fluorescence was bleached completely before microinjection of NLS-SAv-Cy5 into the cytoplasm. After imaging of the green channel with an Axiocam (Zeiss), movies were recorded in the red channel and illustrated the intranuclear motion of SAv-Cy5 molecules after their nuclear import. Movies comprising 2000 frames were acquired at a frame rate of 191 Hz using an image integration time of 5 ms. A total of 22 cells were examined yielding more than 40 single movies. The green and red fluorescence channels were scaled and aligned to each other as described (27).
Image processing of video images
Before data analysis, all images were filtered using a FFT band-pass filter (high frequency threshold, 2 pixels; low frequency threshold, 20 pixels) using ImageJ (W. S. Rasband, ImageJ, U.S. National Institutes of Health, Bethesda, MD, http://rsb.info.nih.gov/ij/,1997–2005) to facilitate the automatic detection of single-molecule signals. The effect of the filter can be noted in Fig. 1 C (lower panels). Identification and tracking of the single-molecule signals were performed using Diatrack 3.0 (Semasopht, Chavannes, Switzerland), a commercial software package developed specifically to identify and localize single-particle signals and also to detect single-particle tracks. A maximal displacement of 15 pixels from frame to frame was allowed, and a point spread function of 1.7 pixels corresponding to 325 nm was assumed. The application of an automated scheme to our data was not straightforward because the single-molecule data often featured low SNR ratios. For this reason, we visually verified each detected single-molecule track identified by Diatrack in the original, unprocessed data. The analysis of trajectories was performed by means of user-written macros in Origin 7.5 (Microcal, Northampton, MA). The tracks were assigned to the following compartments: pericentric heterochromatin, nucleoplasm, nucleoli, and cytoplasm. Each compartment was marked in a specific color using IPLab, and this false color reference image was used for compartment assignment of the individual tracks. All tracks in the cytoplasm and a 10-pixel border region at the nuclear envelope were discarded to avoid evaluation of molecules being imported into the nucleus. Furthermore, all tracks within a distance of eight pixels from the image border were discarded.
FIGURE 1.

NLS-SAv-Cy5 complexes in living cells and their nuclear import. (A) Scheme of the experimental approach for confocal imaging and photobleaching experiments and for single-molecule detection. Mouse myoblast C2C12 cells transfected with plasmids coding for MeCP2-GFP (green) were microinjected into the cytoplasm with preformed NLS-SAv-Cy5 complexes as tracer molecules. Nuclear import was followed by confocal microscopy (CLSM), and after its completion, photobleaching experiments were performed. In a separate experimental setup, single molecules were tracked using a widefield fluorescence microscope (SMT). (B, left panel) Phase contrast (PC) images of a nucleus, time points of the image sequence as indicated. (B, middle panel) Green channel showing the distinct MeCP2-GFP labeling of the pericentric heterochromatin. (B, right panel) confocal time lapse images acquired in the red channel after the cytoplasmic microinjection of NLS-SAv-Cy5 complexes, demonstrating efficient nuclear import. (C) In the single-molecule setup, single SAv-Cy5 molecules could be observed while entering into the nucleus (white arrows) across the nuclear envelope (red dotted line). Further molecules could be identified and tracked within cytoplasm or nucleus (black arrows). In the upper panel unprocessed data are shown; in the lower panel, the effect of bandpass filtering used for further processing is demonstrated. Scale bars, 5 μm.
Trajectory analysis
Identification and tracking of the single-molecule signals and analysis of the trajectories were performed as described elsewhere (27). The probe molecules moved in all three spatial directions; therefore, most molecules were observed for only a few frames because our focal depth was limited to less than ±400 nm (28). Such tracks were too short for an analysis based on an approach to analyze stochastic motion, namely the plot of the mean-square displacement of single molecules against time (29). A jump distance analysis of the trajectories was performed. In this type of analysis, the probability that a particle starting at a specific position will be encountered within a shell of radius r and width dr at time t from that position is considered. For a single species diffusing in two dimensions (30),
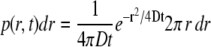 |
(1) |
if we identify the starting position with the origin. Experimentally, this probability distribution can be approximated by a frequency distribution, which is obtained by counting the jump distances within respective intervals [r, r + dr] traveled by single particles after a given time. For each compartment, the displacements within 2 to 20 frames were calculated and drawn in a histogram to visualize the global displacement distribution as a function of time. In cases of particles with multiple diffusive species, the jump distance distributions cannot satisfactorily be fitted by Eq. 1, assuming a single diffusion coefficient. Such different mobility populations can be detected and quantified by curve fitting taking several diffusion terms into account (27).
Three-dimensional movement and tracking
If single molecules are tracked, all three degrees of freedom have to be considered. In case of free and isotropic diffusion in the three-dimensional space, all degrees of freedom are equivalent, and the observation of the movement in respect to a single axis is sufficient to characterize the movement of the probe in the three-dimensional space. Therefore, a time sequence of images is a valid two-dimensional representation of the three-dimensional movement of the molecule. For the limited focal depth of the SMT setup, it was considered that the assumption above holds.
For tracking of particles within the nucleolus or the heterochromatin region, there was the possibility that the molecule traveled below or above the compartment but within the focal depth of the microscope. To reduce false-positive observations, we restricted the analysis for the nucleolus and the chromatin region to signals presenting a higher SNR than accepted in the nucleoplasm. A high SNR is most likely a result of the molecule being central in the focal plane rather than at the border of the depth of field. Effectively such a SNR-based filter reduces the depth of field below the ±400 nm we estimate for the nucleoplasm. The z extension of a nucleus in a living fibroblast cell is in the range of several micrometers. The focal plane was adjusted to the center of the labeled compartment. Although the nucleolus can span the entire nucleus, the pericentric heterochromatin compartments are more likely restricted to less than 2 μm.
Electron spectroscopic imaging
Electron spectroscopic imaging (ESI) was performed as previously described (31,32). Nitrogen and phosphorus maps were collected using a transmission electron microscope (Tecnai 20 (FEI)) fitted with an electron imaging spectrometer (Gatan). Cryopreservation of the macrophage cells was performed by freeze-slamming and cryosectioning (Leica). Frozen sections were transferred under liquid nitrogen (Gatan) to the column of the electron microscope (Tecnai20 (FEI)), where they were freeze-dried before imaging.
RESULTS AND DISCUSSION
Distribution of SAv within the cell nucleus
In a first series of experiments, the overall distribution and potential interactions of SAv-Cy5 within the cell nucleus were examined by live-cell confocal laser scanning microscopy and fluorescence photobleaching experiments. SAv-Cy5 was chosen as a mobility probe because it represents an average-size protein (60 kDa) and has no known binding sites within the nuclear interior. SAv-Cy5 was preincubated with a biotinylated nuclear localization sequence peptide (NLS-biotin), and this complex was microinjected into the cytoplasm of living mouse myoblast C2C12 cells (Fig. 1 A), which expressed GFP-tagged MeCP2 (22). The latter recognizes 5-methylcytosines, which are very abundant in mouse constitutive heterochromatin around centromeres, and thus, pericentric heterochromatin was very selectively marked by green fluorescence (Fig. 1 B, central panel). The complex was imported into cell nuclei within a few minutes (Fig. 1 B, right panel). In the absence of NLS-biotin, no detectable import of SAv-Cy5 into the nucleus was detected over 30 min. Careful examination of the confocal images did not show signs of aggregation or accumulation processes of SAv-Cy5 within specific nuclear domains (Fig. 1 and Fig. S1). Rather, the probe fluorescence was homogeneously distributed with a slight reduction within the pericentric heterochromatin and unambiguous reduction within nucleolar domains, which was already apparent when the first nuclear SAv-Cy5 fluorescence was detected (Fig. 1 B, right panel, 60 s). A quantitative analysis revealed that the concentration of the probe molecules was reduced by only 4% within the pericentric heterochromatin domains, whereas it was significantly reduced within the nucleoli to 42% of the nucleoplasmic level (Table S1). Clearly, the pericentric heterochromatin was not packed densely enough to produce a significant exclusion of proteins of the size of SAv, which is similar to measurements of the distributions of fluorescently labeled sugars (7,9).
To more rigorously control for possible interactions of our probe molecules with intranuclear structures, we performed FLIP experiments. On repeated bleaching of selected spots within nuclei, the fluorescence intensity inside the remaining nuclear space decreased homogeneously (Fig. S2). Altogether, the confocal and photobleaching data clearly indicated that SAv-Cy5 did not bind noticeably within any nuclear domain and suggested that compartment borders presented no major hindrances for the molecules to enter and leave distinct nuclear compartments.
Single-molecule tracking allows real-time imaging of intracellular molecular motions
We recently demonstrated that single SAv-Cy5 molecules in aqueous buffer solution may be visualized by high-speed single-molecule microscopy at a frame rate of 340 Hz (20). Because it is generally assumed that the intracellular viscosity is at least four times higher than in aqueous solution (6), we were confident that movies recorded at frame rates of 200 Hz would produce realistic real-time movies of the intracellular motion of SAv-Cy5 molecules. SAv-Cy5 coupled to NLS-biotin was microinjected at a low concentration so that the diffraction-limited signals originating from single probe molecules within the cell nuclei could be singled out. One hour after the microinjection, single cell nuclei were imaged in the green fluorescence channel to identify the nucleoplasmic, pericentric heterochromatin and nucleolar compartments. Then, SAv-Cy5 fluorescence was excited with 633-nm laser light, and single-molecule signals were recorded with high temporal and spatial resolution for ∼10 s with a frame rate close to 200 Hz (see Movie S1). In Fig. 1, three sample frames from one of the movies are shown, where, e.g., the entry of a single probe molecule into a cell nucleus can be observed (white arrows). Further mobile single molecules within the nucleus or the cytoplasm (black arrows) can be recognized as diffraction-limited bright spots.
Such movies showing the intracellular motion of single molecules contain a wealth of information. To extract this information we identified and localized the single-particle signals and the corresponding single-molecule trajectories. Because we had taken the GFP reference images for all nuclei, we could exactly locate each track within the context of the different intranuclear domains (Fig. S1).
Individual trajectories reveal predominantly highly mobile probe molecules
A plot of all probe molecule trajectories observed within a 10-s time window within the corresponding green reference image provided a direct first impression of the intranuclear probe molecule mobility (Fig. 2). It was immediately obvious that the great majority of the molecules were mobile. At first glance, the trajectories within the pericentric heterochromatin showed no obvious difference from those in the nucleoplasm. Most importantly, it appeared as if the probe molecules could jump into and roam through the pericentric heterochromatin with apparently no hindrance (Movie S1). Most trajectories were relatively short with a mean length of 6.2 frames (median four frames) for this cell, which supported the existence of a high mobility because highly mobile molecules tend to leave the focal plane very rapidly (28).
FIGURE 2.

Individual SAv trajectories within a MeCP2-GFP-labeled cell nucleus. Trajectories of single NLS-SAv-Cy5 (Fig. 1 C and Movie S1) showing different mobilities and located in different nuclear subcompartments were located and identified with Diatrack 3.0. The MeCP2-GFP reference image was recorded before the acquisition of the single-molecule movie and inserted as a background for the trajectory plot. The figure shows all trajectories identified in the data of Movie S1. The dark gray lines show the borders of the MeCP2 domains (light gray), and the white line indicates the nuclear region considered for the evaluation. The overlaid single-molecule tracks are shown in different colors to better distinguish tracks that appear in the same nuclear region. In total, 63 tracks were identified at this measurement, and each 10 consecutive tracks are drawn in the same color. The radius of the spots (100 nm) corresponds to 2σ with σ designating the experimental localization precision of ±50 nm. Hence, movements beyond that radius represent true diffusional steps with a probability of 95%. Scale bar, 1 μm.
In the next step, we distinguished two main classes of traces: whereas most molecules traversed larger distances, others stayed in confined regions (Fig. 2). The individual positions of tracks belonging to the second class were typically scattered in small regions with diameters of ∼100 nm (Fig. 2). This size corresponded to the localization precision of the instrument, which is mainly limited by the SNR of the single-molecule signals (33). Therefore, either these molecules were completely immobile, or their restricted motion within the 100-nm-sized region could not be further resolved. Binding processes can lead to the occurrence of this behavior, as was observed in the case of intranuclear splicing factor mobility (34,27). For example, for single uridine-rich small nuclear ribonucleoprotein particle (U1snRNP) we observed immobilization in ∼75% of all trajectories analyzed, and measured local dwell times often longer than 150 ms in live cells. However, such a behavior was neither expected nor observed for a nonfunctional, ectopic protein such as SAv, and our photobleaching results (Fig. S2) directly argued against binding of SAv to intracellular structures. Thus, we concluded that the virtually motionless molecules were actually trapped in local structures with dimensions smaller than the ±50-nm localization precision of our instrument. The intranuclear space is filled with an extremely complex and intricate network of chromatin fibers, which presumably form all types of structures including caves and mazes. Mobile protein molecules encountering such structures may be caught and transiently trapped there until their own stochastic motion—or chromatin movements—lead to liberation.
The analysis of individual molecule trajectories provided further insights into the intranuclear probe dynamics. An example is given in Fig. 3, where the last frame represents a maximum-intensity projection of the frames shown. The trajectory revealed a molecule that was obviously moving from one trap to another. Although trapping of the molecule leads to an accumulation of signal, the mobile part of the trajectory presents a fading signal in the time projection because of the short time the molecule is observed at any one position. Hence, trapping was a reversible process and sometimes persisted long enough that such traces could be identified.
FIGURE 3.

Repeated trapping of a single SAv molecule. An example of a molecule switching between different modes of motion is shown in a magnified view. (A) The raw data sequence, filtered with an FFT band pass (see Material and Methods), is shown. The last image shows a maximum projection of the trace. Scale bar, 1 μm. Individual frames are marked in a color corresponding to the fitted positions shown in B. Although the molecule was seen in a fixed state at the beginning (red positions, state 1), it was released after 12 frames (blue, state 2) but was trapped again for eight more frames (cyan, state 3). (B) The positions of the molecule observed in A were fitted and plotted on a nanometer scale.
The existence of structural traps would be further supported if trapping would occur repeatedly in the same spatial intranuclear regions. SAv distributed homogeneously within the nucleus and showed no specific binding sites. Therefore, the observation of two different molecules at exactly the same site by pure chance would be exceptionally improbable considering the extremely low concentration used in our experiments, which argues for the existence of stable structural traps in the nucleus. But indeed, such events were observed (Fig. S3).
Different modes of mobility were present within the cell nucleus, but probe molecules were extremely mobile within all three compartments
To obtain a general picture of the SAv-Cy5 mobility, we analyzed the distances covered by the probe molecules between subsequent frames, the so-called jump distances. Because the probe molecules moved in all three spatial directions and our focal depth was limited to less than ±400 nm (28), most molecules were observed for only a few frames. Fig. 4, A, C, and E shows the jump distance distributions for SAv-Cy5 molecules that were observed within nucleoplasm, pericentric heterochromatin, and nucleoli, respectively, with the largest number observed within the nucleoplasm. All three distributions could not satisfactorily be fitted assuming a single diffusion coefficient (Eq. 1), in contrast to jump distance distributions of molecules moving in free solution (35,33). An advantage of the jump distance analysis is, nonetheless, that different mobility populations can be detected and quantified by curve fitting (27). For the SAv-Cy5 data, a sum of three diffusion terms was required, and the fitting results were indicated by the red lines in Fig. 4, A, C, and E. Two fractions, f2 and f3, corresponded to fast motion with diffusion coefficients of D2 = 0.8 μm2/s and D3 = 5 μm2/s, whereas the first fraction f1 corresponded to molecules that appeared motionless on the time scale of the measurements (Table 1). This latter fraction corresponded to those molecules that were interpreted above as trapped.
FIGURE 4.

Global distributions of jump distances in the nuclear subcompartments. The distributions of jump distances occurring between subsequent frames (within 5.22 ms) are shown in panels A, C, and E for the three compartments. The distributions were fitted with a three-component model using the diffusion coefficients D1 = 0.15 μm2/s (for the fixed fraction), D2 = 0.8 μm2/s, and D3 = 5 μm2/s. The data could not be fitted satisfactorily assuming only one or two diffusing components. The fraction of molecules in each mobility class varies strongly between compartments (see Table 1). In panels B, D, and F, the loss of observations over time was monitored for the different compartments. The jumps were binned into two classes: displacements <100 nm (corresponding to the twofold localization precision) and >100 nm. The limited compartment size of pericentric heterochromatin and the nucleolus makes the observation of large jumps less probable, and hence, very long jumps >700 nm were not considered.
TABLE 1.
Fraction of molecules in each mobility class derived from the analysis of jump distances occurring within 5.22 ms
| Nuclear domain | f1*†D = 0.15 μm2/s | f2†D = 0.8 μm2/s | f3†D = 5 μm2/s | f2 + f3 |
|---|---|---|---|---|
| Nucleoplasm | 25 ± 1 | 32 ± 1.5 | 43 ± 2.5 | 75 |
| Pericentric heterochromatin | 49 ± 1 | 34 ± 2 | 17 ± 2 | 51 |
| Nucleoli | 10 ± 1 | 57 ± 1 | 33 ± 2.5 | 90 |
The first fraction with D = 0.15 μm2/s corresponded to the immobile fraction (because σp = 50 nm ≈ 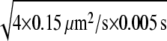 and f2 + f3 to the total mobile fraction).
and f2 + f3 to the total mobile fraction).
These fits were performed with the diffusion coefficients held constant to obtain results for the amplitudes that were directly comparable. Free fits yielded similar results with only slight deviations from the given parameters.
The data in Table 1 suggested differences between compartments in the ratios of shorter to longer jumps, which were only apparent and partly caused by differences in the subcompartment size and geometry. Therefore, we conclude that the general mobility pattern in all three nuclear compartments was comparable.
Single-molecule tracking revealed short-lived trapping of the probe molecules
SAv-Cy5 molecules of fraction f1 (Table 1) did not move beyond the average localization precision of our system, namely 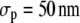 (
(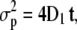 with t = 5 ms). This fraction corresponding to trapped molecules occurred predominantly within the pericentric heterochromatin (49% in f1), less within the nucleoplasm (25% in f1), and least within the nucleoli (10% in f1). The latter value was especially striking because there was still a 2.5-fold reduction between nucleoplasm and nucleoli. The fact that a noticeably smaller fraction of trapped molecules was observed here than in the pericentric heterochromatin suggested that the nucleolar environment contained fewer traps for SAv-size molecules than pericentric heterochromatin.
with t = 5 ms). This fraction corresponding to trapped molecules occurred predominantly within the pericentric heterochromatin (49% in f1), less within the nucleoplasm (25% in f1), and least within the nucleoli (10% in f1). The latter value was especially striking because there was still a 2.5-fold reduction between nucleoplasm and nucleoli. The fact that a noticeably smaller fraction of trapped molecules was observed here than in the pericentric heterochromatin suggested that the nucleolar environment contained fewer traps for SAv-size molecules than pericentric heterochromatin.
To highlight the distinct properties of the three compartments, we divided the jumps into two fractions and examined their time dependence. The first fraction contained jumps of trapped molecules, with jump distances smaller than 2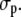 The second fraction contained the mobile molecules performing jumps between 100 and 700 nm. Fig. 4, B, D, and F, shows the time dependence of these fractions. It was obvious that the number of observations for both fractions decreased very rapidly with time. Although this was expected for mobile molecules because they frequently and rapidly jumped out of the focal plane, it was not so for trapped molecules. For these, the decay time, rather, reflected the time span for which they were motionless. The number of the total trapped events detected decayed rapidly with a kinetics that could be described by a biexponential decay with an averaged time constant of 22–25 ms for the pericentric chromatin domains and nucleoplasm and even faster (7 ms) for the nucleoli. Clearly, the immobilization or trapping incidents were short-lived in comparison to functionally relevant binding events, as in the case of splicing factors, which displayed a threefold larger immobile fraction with average binding times of >60 ms (34). The short trapping times of the SAv-Cy5 explain why they could not be measured in the bulk photobleaching experiments, which were performed at much longer time scales (Fig. S2).
The second fraction contained the mobile molecules performing jumps between 100 and 700 nm. Fig. 4, B, D, and F, shows the time dependence of these fractions. It was obvious that the number of observations for both fractions decreased very rapidly with time. Although this was expected for mobile molecules because they frequently and rapidly jumped out of the focal plane, it was not so for trapped molecules. For these, the decay time, rather, reflected the time span for which they were motionless. The number of the total trapped events detected decayed rapidly with a kinetics that could be described by a biexponential decay with an averaged time constant of 22–25 ms for the pericentric chromatin domains and nucleoplasm and even faster (7 ms) for the nucleoli. Clearly, the immobilization or trapping incidents were short-lived in comparison to functionally relevant binding events, as in the case of splicing factors, which displayed a threefold larger immobile fraction with average binding times of >60 ms (34). The short trapping times of the SAv-Cy5 explain why they could not be measured in the bulk photobleaching experiments, which were performed at much longer time scales (Fig. S2).
Altogether, we conclude that on a time scale >100 ms, SAv was freely mobile in all nuclear compartments. However, on the millisecond timescale, many more molecules were trapped within the pericentric heterochromatin than within the nucleoplasm and especially the nucleoli. The different trapping abilities could in part be related to SAv complexation with the NLS peptide.
Compartment borders did not form barriers
In contrast to cytoplasmic organelles, functional compartments in cell nuclei are not separated from each other by membranes. The question of how nuclear subcompartments are maintained is therefore a matter of debate. In principle, even molecules that, at equilibrium, are equally concentrated in two distinct domains may encounter hindrance in crossing the domain borders. Thus, we analyzed in detail the jumps performed by molecules that crossed between nucleoplasm and pericentric heterochromatin and between nucleoplasm and nucleoli, respectively. The distance distributions of these jumps are shown in Fig. S4. For both types of compartment crossings, broad distributions of jump distances were found. Very short jumps were underrepresented compared with the distribution within the compartments (compare to Fig. 4) because of the selection criterion that a border had to be crossed. Still, it was remarkable that all jump distances up to 800 nm occurred, indicating that the border region of neither compartment represented a significant hindrance for the SAv-Cy5 probe. This view was supported by the visual impressions from the movies, where one could see in several cases that the probe molecules changed from the nucleoplasm into the perichromatin domains with no apparent hindrance or delay. Finally, it could be clearly seen that, for jumps crossing the pericentric heterochromatin, the ratio of short jumps to longer jumps was significantly higher than for the nucleolus, strengthening the result of a higher mobility in the nucleolus (Fig. 4 and Fig. S4).
Different environments in the nucleus
On the basis of the almost 60% lower accumulation of SAv-Cy5 in the nucleolus (Table S1), we postulated that there is less free volume available than in the surrounding nucleoplasm. This would be in accord with measurements of protein concentrations in different nuclear bodies performed in Xenopus egg nuclei (36). The lower percentage of immobilized molecules (Fig. 4) and the fact that the nucleolar border did not impede the access of SAv-Cy5 molecules (Fig. S4) nevertheless reflect a highly permeable compartment. This is surprising because this nuclear subcompartment is thought to be a mass-dense structure comprised of rDNA, RNA polymerase I transcription/splicing apparatus, rRNA transcripts, preribosomes, and many proteins that are not involved in ribosome biogenesis. To address this question, it is important to consider whether the viscosity of the nucleolus is high because of a high concentration of nucleic acid and protein or whether molecules experience a reduced free volume but have unimpeded access. Therefore, we measured the relative mass density of the nucleolus and nucleoplasm from sections of conventionally fixed cells by ESI and from freeze-dried cryosections of cryofixed cells.
With ESI, the biological structure is visible without heavy atom contrast agents (e.g., uranium salts) because of energy loss events detected by an imaging electron spectrometer. Background mass contributions from the embedding resin can be removed in the analysis. Intensity values in an image recorded at 120 eV, before the carbon K edge, provide a direct measure of the mass density. Relative mass densities of various regions of interest can also be obtained. A region corresponding to a nucleolus and a region corresponding to the nucleoplasm are shown (Fig. 5 A). The ratio of the mean intensity of these two regions was 112/58 = 1.93. The mean ratio of nucleolus mass density to nucleoplasm mass density, measured for eight nucleoli in six cells, was 1.64 ± 0.34 and varied from 1.20 to 2.17. The variation between measurements may be related to the degree of extraction of soluble protein from the nucleoplasm during the fixation, dehydration, and embedding procedure. It is possible that the degree of extraction is greater from the nucleoplasm than from the nucleolus. To avoid this complication, and to obtain a ratio of mass density between the nucleolus and nucleoplasm that accurately reflects that of an intact cell, we cryofixed a pellet of RAW cells (macrophages) by freeze-slamming. The cell morphology indicated good cryopreservation with a minimal degree of ice crystal damage. The mass density of the nucleolus relative to that of the nucleoplasm in the regions indicated (Fig. 5 B) was 62/47 = 1.32. The average ratio measured from 12 nucleoli in 12 cells was 1.55 ± 0.32. These measurements clearly indicate substantially less free space within the nucleolus in relation to the surrounding nucleoplasm, which likely is a major reason for the lower concentration of proteins such as the SAv-Cy5 with no binding sites in this compartment.
FIGURE 5.

ESI mass-density measurements of the subnuclear compartments. (A) ESI of an Sk-N-SH neuroblastoma cell nucleus. The image was recorded at 120 eV energy loss. Sample preparation involved fixation in 2% paraformaldehyde for 30 min, followed by fixation in 0.5% glutaraldehyde overnight. Cells were dehydrated in ethanol and embedded in Qetol resin. Then 70-nm sections were picked up on 400-mesh electron microscopy grids, which were subsequently covered with a 3-nm carbon film picked up from the surface of water. The ratio (nucleolus divided by nucleoplasm) of the average intensity values of the two regions indicated is 1.93. (B) Bright-field image of a macrophage cell (RAW) cryopreserved by slam-freezing. The frozen hydrated sections were picked up onto a copper grid coated with a 3-nm carbon film. The grid was transferred to the electron microscope at liquid nitrogen temperature. The section was freeze-dried in the microscope column before imaging. The removal of water and the absence of a resin embedding material allow for very high contrast without the need for heavy atom contrast agents. The images, therefore, provide mass-density information. The ratio (nucleolus divided by nucleoplasm) of the average intensity values of the two regions indicated is 1.32. (C) Low-magnification mass-sensitive image of an Sk-N-SH cell recorded at 120 eV. (D) Net phosphorus image of a field from the nucleus presented in C. The arrows in C and D provide a fiduciary for each image and indicate the slight rotation between the images recorded at different magnifications. (E) The net phosphorus image was subtracted from the net nitrogen image (the resulting signal was represented in shades of blue). The net phosphorus signal (yellow) was superimposed on the nitrogen-phosphorus signal. GC, DFC and Ch represent granular component, dense fibrillar component, and chromatin, respectively. (F) High magnification of a field in E. Chromatin fibers can be visualized in the nucleoplasm, bordering the nucleolus, and within the nucleolus (referred to as NuChromatin in G). (G) Identification of different regions and structures associated with nucleolus. Scale bar represents 4 μm in A–C, 1 μm in D and E, and 280 nm in F.
Although the nucleolus showed a higher mass density, the probe molecules had no hindrance in penetrating the nucleolus and were highly mobile within it. Therefore, we further analyzed the ultrastructure of this compartment by acquiring high-resolution electron spectroscopic images of the nucleolus. Nitrogen and phosphorus mapping by ESI can be used to delineate protein- from nucleic acid-based structures and provide structural detail without the use of heavy atom contrast agents. The phosphorus map provides contrast predominantly of nucleic acid. Chromatin fibers, for example, were readily apparent (Fig. 5, C–G, yellow fibers in the region labeled “Nucleoplasm/Chromatin”). If the phosphorus signal is subtracted from the nitrogen signal, protein structures that do not overlap with chromatin can be visualized (blue signal in Fig. 5, E–G). Features of nucleolar structure could be identified with this approach, such as the chromatin on the periphery (Ch), chromatin within the nucleolus (NuChromatin), the granular compartment (GC), and the dense fibrillar component (DFC). It is apparent that the nucleolus could be permeable to proteins, such as SAv-Cy5 (5.3-nm × 4.5-nm barrel-shaped structure). The structural features that support this are:
Channels through the interior of the nucleolus (Fig. 5 G, arrowhead).
Gaps of 10–30 nm between granules (preribosome subunits) in the granular component.
Spaces between RNA fibers in the dense fibrillar component (DFC). Although there is also a high concentration of protein in this subcompartment, it may reflect protein that became fixed in place during fixation and dehydration steps. It should be possible for small molecules to diffuse through this provided they do not bind to the RNA or the RNA-associated proteins. Molecules from the nucleoplasm could easily penetrate the nucleolus through regions where the peripheral chromatin density was not extreme, such as that seen in Fig. 5 D, or through the granular component, which is typically observed on the periphery of the nucleolus.
Our main conclusions are summarized in Fig. 6. Nucleoplasm and pericentric heterochromatin are dominated by the presence of chromatin fibers, albeit to different densities, whereas the nucleolus is composed largely of ribonucleoprotein particles, i.e., ribosomes. It should be noted that the nucleoplasm is a very heterogeneous compartment, and, for simplicity, splicing speckles, Cajal bodies, and other nuclear bodies were not considered. Because pericentric heterochromatin in the mouse constitutes 10% of the total genome and occupies a volume of ∼5% of the nucleus (data not shown), this results in a twofold higher chromatin concentration on average in the pericentric heterochromatin subcompartment and consequently less free space. This agrees with measurements of the relative concentration of GFP-tagged histone distribution (37). In the case of the nucleolus, based on the almost 60% lower accumulation of SAv and the 1.5 times higher mass density, we conclude that there is less free volume available than in the surrounding nucleoplasm. In general, the nuclear subcompartments were easily accessible to a small protein such as SAv, and their borders did not form barriers because molecules could move unimpeded in and out of each compartment. Obviously, the pericentric heterochromatin was not packed densely enough to produce significant exclusion of proteins of the size of SAv, and, hence, reduced heterochromatin physical accessibility per se should not be a major determinant of genome silencing. Different mobility distributions were detected on a millisecond time scale by SMT with short-term trapped molecules in all compartments. In detail, the percentage of trapped SAv molecules was higher in the pericentric heterochromatin and lowest in the nucleolus; i.e., it correlated inversely with the amount of chromatin. Also, the retention or decay times of immobile molecules in heterochromatin and nucleoplasm were longer (>20 ms) than in the nucleolus (∼7 ms). These observations suggested that molecular trapping is possibly a result of short-term nonspecific chromatin interactions.
FIGURE 6.

A view of different environments in the nucleus. Nucleoplasm and pericentric heterochromatin are dominated by the presence of chromatin fibers, whereas the nucleolus is composed largely of ribonucleoprotein particles. The green and red arrows summarize the increase and decrease, respectively, of trapping sites and available space by comparison to the nucleoplasm. See text for details.
An alternative explanation for the very large differences detected in local mobility (Fig. 3) would be strongly differing local viscosities. However, this possibility could clearly be ruled out by our data. Small local domains of higher viscosity would not be stable over time but would rapidly be smeared out and vanish. However, we observed that the structural properties leading to the immobilization or trapping were stable over time (Fig. S3). We, therefore, conclude that short-term trapping of proteins is responsible for their slower diffusion within the cell nucleus.
In summary, our results indicate that nuclear structure affects the mobility and local concentration of proteins and may thus contribute to the regulation of nuclear processes. The transient, short-time trapping of proteins at chromatin may be a feature of the functional architecture of the nucleus and a general strategy to scan the genome for potential binding sites. An additional function of the cationic NLS could also be to enhance such binding. The observation that single molecules move freely in and out of the nucleolus sheds new light on the accessibility of this most prominent structure of the nucleus. Although wide-field epifluorescence images suggest a blocked access, the single-molecule tracings and ESI presented here rather argue for a reduced free volume in combination with a lower ability to trap molecules, i.e., lower retention. This free accessibility of the nucleolus has obvious consequences for possible modes of regulation and underscores the importance of SMT in studying the access and interaction of proteins with subcellular compartments.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Acknowledgments
We thank J. Sleeman (University of Dundee, UK) and M. Elbaum (The Weizmann Institute of Science, Israel) for the generous gift of ASF/SF2-GFP plasmid and NLS-biotin, respectively. U.K. is indebted to R. Peters for using numerous experimental facilities in his laboratory. We thank R. Temkin and R. Li for the electron microscopy.
ESI was funded by an operating grant to D.P.B.-J. from the Natural Sciences and Engineering Research Council of Canada. D.P.B.-J. holds a Canada Research Chair in Molecular and Cellular Imaging. M.C.C., H.L., and U.K. gratefully acknowledge financial support by the Volkswagen Foundation and the German Research Council.
Ulrich Kubitscheck and M. Cristina Cardoso contributed equally to this work.
David Grünwald's present address is Dept. of Anatomy and Structural Biology, Albert Einstein College of Medicine, Bronx, NY 10461.
Volker Buschmann's present address is PicoQuant GmbH, 12489 Berlin, Germany.
Abbreviations used: GFP, green fluorescent protein; ASF/SF2, alternative splicing factor/splicing factor 2; ESI, electron spectroscopic imaging; FLIP, fluorescence loss in photobleaching; MeCP2, methyl cytosine binding protein 2; NLS, nuclear localization sequence; SAv-Cy5, streptavidin-Cy5; SMT, single-molecule tracking; SNR, signal/noise ratio.
Editor: Thomas Schmidt.
References
- 1.Handwerger, K. E., and J. G. Gall. 2006. Subnuclear organelles: new insights into form and function. Trends Cell Biol. 16:19–26. [DOI] [PubMed] [Google Scholar]
- 2.Spector, D. L. 2001. Nuclear domains. J. Cell Sci. 114:2891–2893. [DOI] [PubMed] [Google Scholar]
- 3.Leonhardt, H., H. P. Rahn, and M. C. Cardoso. 1999. Functional links between nuclear structure, gene expression, DNA replication, and methylation. Crit. Rev. Eukaryot. Gene Expr. 9:345–351. [DOI] [PubMed] [Google Scholar]
- 4.Lang, I., M. Scholz, and R. Peters. 1986. Molecular mobility and nucleocytoplasmic flux in hepatoma cells. J. Cell Biol. 102:1183–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seksek, O., J. Biwersi, and A. S. Verkman. 1997. Translational diffusion of macromolecule-sized solutes in cytoplasm and nucleus. J. Cell Biol. 138:131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verkman, A. S. 2002. Solute and macromolecule diffusion in cellular aqueous compartments. Trends Biochem. Sci. 27:27–33. [DOI] [PubMed] [Google Scholar]
- 7.Görisch, S. M., K. Richter, M. O. Scheuermann, H. Herrmann, and P. Lichter. 2003. Diffusion-limited compartmentalization of mammalian cell nuclei assessed by microinjected macromolecules. Exp. Cell Res. 289:282–294. [DOI] [PubMed] [Google Scholar]
- 8.Görisch, S. M., M. Wachsmuth, K. F. Toth, P. Lichter, and K. Rippe. 2005. Histone acetylation increases chromatin accessibility. J. Cell Sci. 118:5825–5834. [DOI] [PubMed] [Google Scholar]
- 9.Verschure, P. J., I. van der Kraan, E. M. Manders, D. Hoogstraten, A. B. Houtsmuller, and R. van Driel. 2003. Condensed chromatin domains in the mammalian nucleus are accessible to large macromolecules. EMBO Rep. 4:861–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Görisch, S. M., P. Lichter, and K. Rippe. 2005. Mobility of multi-subunit complexes in the nucleus: accessibility and dynamics of chromatin subcompartments. Histochem. Cell Biol. 123:217–228. [DOI] [PubMed] [Google Scholar]
- 11.Molenaar, C., A. Abdulle, A. Gena, H. J. Tanke, and R. W. Dirks. 2004. Poly(A)+ RNAs roam the cell nucleus and pass through speckle domains in transcriptionally active and inactive cells. J. Cell Biol. 165:191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Politz, J. C., R. A. Tuft, T. Pederson, and R. H. Singer. 1999. Movement of nuclear poly(A) RNA throughout the interchromatin space in living cells. Curr. Biol. 9:285–291. [DOI] [PubMed] [Google Scholar]
- 13.Politz, J. C., R. A. Tuft, K. V. Prasanth, N. Baudendistel, K. E. Fogarty, L. M. Lifshitz, J. Langowski, D. L. Spector, and T. Pederson. 2006. Rapid, diffusional shuttling of poly(A) RNA between nuclear speckles and the nucleoplasm. Mol. Biol. Cell. 17:1239–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shav-Tal, Y., X. Darzacq, S. M. Shenoy, D. Fusco, S. M. Janicki, D. L. Spector, and R. H. Singer. 2004. Dynamics of single mRNPs in nuclei of living cells. Science. 304:1797–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vargas, D. Y., A. Raj, S. A. Marras, F. R. Kramer, and S. Tyagi. 2005. Mechanism of mRNA transport in the nucleus. Proc. Natl. Acad. Sci. USA. 102:17008–17013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gorski, S. A., M. Dundr, and T. Misteli. 2006. The road much traveled: trafficking in the cell nucleus. Curr. Opin. Cell Biol. 18:284–290. [DOI] [PubMed] [Google Scholar]
- 17.Beaudouin, J., F. Mora-Bermudez, T. Klee, N. Daigle, and J. Ellenberg. 2006. Dissecting the contribution of diffusion and interactions to the mobility of nuclear proteins. Biophys. J. 90:1878–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Braga, J., J. M. Desterro, and M. Carmo-Fonseca. 2004. Intracellular macromolecular mobility measured by fluorescence recovery after photobleaching with confocal laser scanning microscopes. Mol. Biol. Cell. 15:4749–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cardoso, M. C., H. Leonhardt, and B. Nadal-Ginard. 1993. Reversal of terminal differentiation and control of DNA replication: cyclin A and Cdk2 specifically localize at subnuclear sites of DNA replication. Cell. 74:979–992. [DOI] [PubMed] [Google Scholar]
- 20.Cardoso, M. C., C. Joseph, H. P. Rahn, R. Reusch, B. Nadal-Ginard, and H. Leonhardt. 1997. Mapping and use of a sequence that targets DNA ligase I to sites of DNA replication in vivo. J. Cell Biol. 139:579–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sleeman, J., C. E. Lyon, M. Platani, J. P. Kreivi, and A. I. Lamond. 1998. Dynamic interactions between splicing snRNPs, coiled bodies and nucleoli revealed using snRNP protein fusions to the green fluorescent protein. Exp. Cell Res. 243:290–304. [DOI] [PubMed] [Google Scholar]
- 22.Brero, A., H. P. Easwaran, D. Nowak, I. Grunewald, T. Cremer, H. Leonhardt, and M. C. Cardoso. 2005. Methyl CpG-binding proteins induce large-scale chromatin reorganization during terminal differentiation. J. Cell Biol. 169:733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sporbert, A., P. Domaing, H. Leonhardt, and M. C. Cardoso. 2005. PCNA acts as a stationary loading platform for transiently interacting Okazaki fragment maturation proteins. Nucleic Acids Res. 33:3521–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kubitscheck, U., D. Grünwald, A. Hoekstra, D. Rohleder, T. Kues, J. P. Siebrasse, and R. Peters. 2005. Nuclear transport of single molecules: dwell times at the nuclear pore complex. J. Cell Biol. 168:233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siebrasse, J. P., D. Grünwald, and U. Kubitscheck. 2007. Single-molecule tracking in eukaryotic cell nuclei. Anal. Bioanal. Chem. 387:41–44. [DOI] [PubMed] [Google Scholar]
- 26.Leonhardt, H., H. P. Rahn, P. Weinzierl, A. Sporbert, T. Cremer, D. Zink, and M. C. Cardoso. 2000. Dynamics of DNA replication factories in living cells. J. Cell Biol. 149:271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kues, T., A. Dickmanns, R. Lührmann, R. Peters, and U. Kubitscheck. 2001. High intranuclear mobility and dynamic clustering of the splicing factor U1 snRNP observed by single particle tracking. Proc. Natl. Acad. Sci. USA. 98:12021–12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kues, T., and U. Kubitscheck. 2002. Single molecule motion perpendicular to the focal plane of a microscope: application to splicing factor dynamics within the cell nucleus. Single Mol. 3:218–224. [Google Scholar]
- 29.Saxton, M. J., and K. Jacobson. 1997. Single-particle tracking: applications to membrane dynamics. Annu. Rev. Biophys. Biomol. Struct. 26:373–399. [DOI] [PubMed] [Google Scholar]
- 30.Crank, J. 1975. The Mathematics of Diffusion. Clarendon Press, Oxford.
- 31.Eskiw, C. H., G. Dellaire, J. S. Mymryk, and D. P. Bazett-Jones. 2003. Size, position and dynamic behavior of PML nuclear bodies following cell stress as a paradigm for supramolecular trafficking and assembly. J. Cell Sci. 116:4455–4466. [DOI] [PubMed] [Google Scholar]
- 32.Dellaire, G., R. Nisman, and D. P. Bazett-Jones. 2004. Correlative light and electron spectroscopic imaging of chromatin in situ. Methods Enzymol. 375:456–478. [DOI] [PubMed] [Google Scholar]
- 33.Kubitscheck, U., O. Kückmann, T. Kues, and R. Peters. 2000. Imaging and tracking of single GFP molecules in solution. Biophys. J. 78:2170–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grünwald, D., B. Spottke, V. Buschmann, and U. Kubitscheck. 2006. Intranuclear binding kinetics and mobility of single U1 snRNP particles in living cells. Mol. Biol. Cell. 17:5017–5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grünwald, D., A. Hoekstra, T. Dange, V. Buschmann, and U. Kubitscheck. 2006. Direct observation of single protein molecules in aqueous solution. ChemPhysChem. 7:812–815. [DOI] [PubMed] [Google Scholar]
- 36.Handwerger, K. E., J. A. Cordero, and J. G. Gall. 2005. Cajal bodies, nucleoli, and speckles in the Xenopus oocyte nucleus have a low-density, sponge-like structure. Mol. Biol. Cell. 16:202–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weidemann, T., M. Wachsmuth, T. A. Knoch, G. Muller, W. Waldeck, and J. Langowski. 2003. Counting nucleosomes in living cells with a combination of fluorescence correlation spectroscopy and confocal imaging. J. Mol. Biol. 334:229–240. [DOI] [PubMed] [Google Scholar]