

Summary
We have developed a mouse model for ovarian carcinoma by using an avian retroviral gene delivery technique for the introduction of multiple genes into somatic ovarian cells of adult mice. Ovarian cells from transgenic mice engineered to express the gene encoding the avian receptor TVA were efficiently infected in vitro with multiple vectors carrying coding sequences for oncogenes and marker genes. When target cells were derived from TVA transgenic mice deficient for p53, the addition of any two of the oncogenes c-myc, K-ras, and Akt were sufficient to induce ovarian tumor formation when infected cells were injected at subcutaneous, intraperitoneal, or ovarian sites. We demonstrated that the ovarian surface epithelium is the precursor tissue for these ovarian carcinomas, and that introduction of oncogenes causes phenotypic changes in the ovarian surface epithelial cells. The induced ovarian tumors in mice resembled human ovarian carcinomas in their rapid progression and intraperitoneal metastatic spread.
Introduction
Ovarian carcinoma is the fifth most common cause of cancer among women in the United States, with more than 23,000 new cases diagnosed and approximately 14,000 deaths each year (Greenlee et al., 2000). When diagnosed at an early stage, ovarian carcinoma has a survival rate approaching 90%. However, due to the asymptomatic nature of early stages of the disease, more than two-thirds of cases are not diagnosed until the disease has spread beyond the ovaries. For these patients, the five-year survival rate is significantly lower. Despite recent advances in cytoreductive surgery and combination chemotherapy, improvement in long-term survival for ovarian cancer patients has been slight.
The causes of ovarian carcinoma are not known, although the majority of evidence suggests that reproductive factors and heredity may play roles in the origin of this disease (Holschneider and Berek, 2000). Studies of human ovarian cancer specimens have revealed several types of genetic alterations. Mutation of the p53 tumor suppressor gene is the most frequently identified genetic alteration in serous and poorly differentiated epithelial ovarian carcinomas, affecting more than 50% of advanced and early-stage carcinomas (reviewed in Aunoble et al., 2000). Proto-oncogenes such as c-myc, K-ras, and Akt, whose products are involved in the control of growth stimulatory and cell death pathways, are often amplified or mutated in ovarian cancer. Amplification of c-myc has been reported in approximately 30% of ovarian tumors (Baker et al., 1990; Wang et al., 1999). Point mutations at codon 12 of the K-ras gene have been detected in 30%–50% of mucinous adenocarcinomas and tumors of low malignant potential, and in 10%–20% of serous adenocarcinomas (Mok et al., 1993; Teneriello et al., 1993; Cuatrecasas et al., 1997; Caduff et al., 1999; Dokianakis et al., 1999; Morita et al., 2000; Suzuki et al., 2000). Activation of the phosphatidylinositol 3-kinase (PI3-kinase)/Akt pathway has been detected in several ovarian cancer cell lines and in approximately 30% of ovarian cancer specimens (Cheng et al., 1992; Bellacosa et al., 1995; Shayesteh et al., 1999; Yuan et al., 2000; Sun et al., 2001). Most of the evidence that these oncogenes and tumor suppressor genes are involved in ovarian carcinogenesis is based on immunohistological examination of tumors. However, the molecular mechanisms by which these genes contribute to initiation and development of ovarian cancer are still poorly understood.
SIGNIFICANCE
The study of ovarian carcinogenesis has been limited by the lack of appropriate tumor models. We have developed and characterized a mouse model system for recapitulating human ovarian carcinoma development and progression. The system was designed to be used for the evaluation of multiple genetic lesions, individually and in combination. We have demonstrated that combinations of genetic lesions that are commonly present in human ovarian carcinomas can induce ovarian carcinomas in mouse ovarian cells. Our findings support the theory that the ovarian surface epithelium is the precursor tissue for ovarian carcinomas. We expect that this model will be useful for studying the basic biology of ovarian carcinoma initiation and progression, identifying markers of ovarian tumor progression, and establishing parameters for distinguishing a variety of biological behaviors such as metastatic ability, invasiveness, and sensitivity of ovarian tumors to different therapeutic interventions that target specific molecular pathways altered in ovarian carcinoma.
Advancement in understanding the initiation and progression of ovarian carcinoma has been slow, mainly due to the lack of an appropriate experimental model. Even the tissue of origin in ovarian cancer is not completely understood. Most human ovarian carcinomas are thought to arise from the ovarian surface epithelium (reviewed in Feeley and Wells, 2001). However, it is difficult to explain the diversity of ovarian carcinomas that arise from this relatively homogenous tissue. Very little is known about cellular phenotypic changes in the ovarian surface epithelium as it becomes cancerous or metastatic (Auersperg et al., 2001). Despite its clinical importance, the biology of this tissue is poorly understood, and evidence for its role in ovarian carcinogenesis is based almost entirely on morphologic and histologic examination of clinical tumor specimens and immortalized ovarian cancer cell lines.
The direct progression of benign ovarian lesions to clinical carcinoma has not been clearly demonstrated for ovarian cancer (reviewed in Feeley and Wells, 2001). Since early stage malignancy is infrequently detected in patients, the morphologic and genetic changes that occur as the benign epithelium becomes malignant are not well defined. At present, there is little evidence for a genetic model of multi-step tumor progression in ovarian cancer, and there is speculation that ovarian carcinoma occurs de novo, without any intermediate precursor lesion (Bell and Scully, 1994). A suitable animal model, in which ovarian cancer could be predictably induced with defined genetic changes, may be crucial in resolving this controversy.
Here we report the development of a mouse model for ovarian carcinoma with defined genetic lesions. We used an avian retroviral gene delivery technique to introduce multiple genetic lesions in mouse ovarian cells engineered to produce the avian virus receptor, TVA. Combinations of c-myc, K-ras, and Akt oncogenes introduced into primary mouse ovarian cells from p53-deficient mice were able to induce phenotypic changes in mouse ovarian surface epithelial cells in culture and form tumors in vivo, showing that these cells can serve as precursors to ovarian carcinomas and that the system represents a useful tool for understanding the molecular basis of ovarian cancer.
Results
Efficient in vitro gene delivery to a restricted subset of somatic cells in an adult mouse ovary
We used an avian retroviral gene delivery system to introduce multiple oncogenes and marker genes into the somatic cells of an adult mouse ovary. This system relies on ectopic expression of the avian retroviral receptor, TVA, on the surface of mammalian cells, and on infection with a replication-competent avian leukosis virus-derived vector, RCAS (Hughes et al., 1987; Bates et al., 1993; Young et al., 1993). The presence of the TVA receptor renders mouse cells susceptible to infection with subgroup A RCAS viruses. RCAS vectors can carry coding domains of oncogenes or marker genes, which are delivered to the TVA receptor-expressing cells upon infection. After entry into mammalian cells, a newly synthesized DNA copy of the viral genome integrates into the host DNA, allowing long-term expression of the introduced gene. Because mammalian cells do not produce detectable levels of infectious viral particles, avian retroviruses do not spread to surrounding cells and the receptors on the cell surface remain unoccupied, allowing repeated infection with RCAS vectors that can carry different gene inserts.
Tissue specificity of RCAS-mediated delivery is usually obtained by designing transgenic animals that express TVA under the control of tissue-specific promoters (reviewed in Fisher et al., 1999). Since there are no well-characterized promoters specific for ovary epithelium and stroma, we used transgenic mice that express TVA in the ovaries and other tissues, and we ensured ovary-specific gene delivery by removing the ovaries from mice and infecting ovarian cells in vitro (Figure 1). Propagation of ovarian cells in culture facilitated infection by RCAS viruses, which, like all oncoretroviruses, require a round of cell division for the integration of viral DNA into the host genome. The ovarian explants were repeatedly exposed to supernatants from RCAS-producing cultures of the chicken fibroblast cell line, DF-1 (Himly et al., 1998; Schaefer-Klein et al., 1998). The stocks of viruses carrying different oncogenes or marker genes were added individually or in combination, as detailed in the Experimental Procedures section. The infected ovarian cells were examined in culture and also reintroduced into recipient mice by subcutaneous or intraperitoneal injection or by transplantation under the ovarian bursa.
Figure 1.
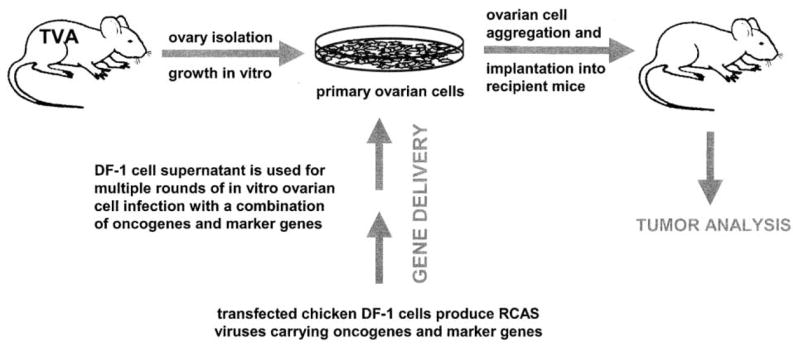
Strategy for viral gene delivery to ovarian cells in vitro and introduction of infected cells into recipient mice
Ovaries are isolated from a TVA transgenic mouse to restrict infection to ovarian cells. DF-1 chicken cells transfected with an RCAS vector carrying a gene of interest are used to obtain high titer virus stock. In vitro propagation of primary ovarian cells facilitates multiple rounds of infection with a combination of RCAS viruses carrying different oncogenes and marker genes. After several days of infection in vitro, ovarian cells are aggregated and implanted into recipient mice.
Two TVA transgenic lines were used to develop ovarian tumor models: a β-actin-TVA line (Federspiel et al., 1996) in which the TVA receptor is expressed in all cells in the ovary under the control of the murine β-actin promoter, and a keratin 5-TVA line in which expression of the TVA receptor is restricted to the ovarian surface epithelium (Figure 2A). Since both of these promoters are active in a variety of tissues other than ovarian, we isolated the ovaries and exposed them to the RCAS viruses in vitro. To determine the cell type that is receptive to infection with the RCAS virus, we infected ovarian cells from each line with a viral supernatant carrying DNA encoding the histological marker gene AP, which we subsequently detected with histochemical stain for alkaline phosphatase (Figure 2B). Ovarian cells cultivated from both transgenic lines are efficiently infected with RCAS-AP vectors (Figure 2B). All ovarian cells from the β-actin-TVA line can be infected with RCAS-AP. On the other hand, the keratin 5-TVA line has the advantage of allowing infection only of the cells derived from the surface epithelium of the ovary, the presumptive precursor of human ovarian carcinoma. We crossed the TVA transgenic mice with p53−/− mice (Jacks et al., 1994) to obtain the β-actin-TVA/p53−/− and the keratin 5-TVA/p53−/− lines. The infection efficiency was the same regardless of the p53 status of ovarian cells (data not shown).
Figure 2.
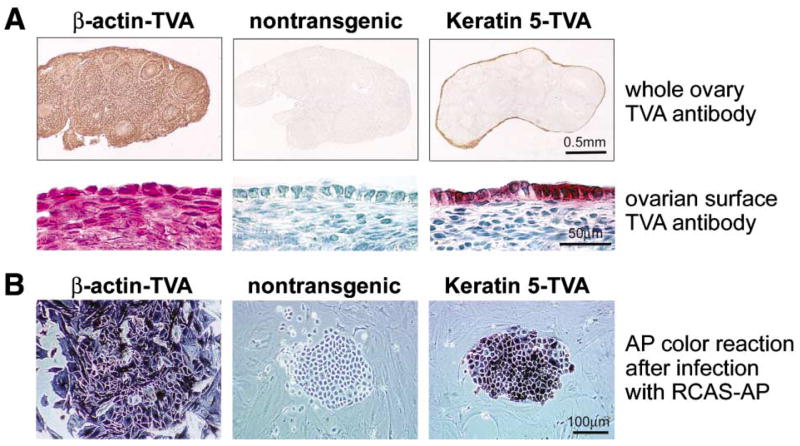
Expression of TVA and susceptibility of ovarian cells from TVA transgenic mice to RCAS infection
A: Immunohistochemical detection of the TVA receptor with the TVA antibody. Ovaries isolated from β-actin-TVA transgenic mice express the TVA receptor in both stromal cells and the ovarian surface epithelium. In ovaries isolated from keratin 5-TVA transgenic mice, expression of the TVA receptor is restricted to the cells of the ovarian surface epithelium.
B: Detection of RCAS-AP-infected cells by AP color reaction. Ovarian cells from each transgenic line were infected with the RCAS-AP virus in culture. Endogenous AP was heat-inactivated, and the infected cells that produce AP were detected by the AP color reaction. Both stromal and epithelial cells from β-actin-TVA ovaries are susceptible to RCAS-AP infection. In ovaries from keratin 5-TVA mice, only the ovarian surface epithelial cells are susceptible to RCAS-AP infection, while stromal cells are AP-negative.
We next wanted to determine if infection of ovarian cells with RCAS viruses carrying proto-oncogenes results in detectable expression of oncoprotein. The ovaries from the β-actin-TVA/p53−/− mice were introduced into culture and infected in vitro with a combination of RCAS viruses carrying coding sequences for human c-myc (RCAS-c-myc), mouse K-ras with the G12D activating mutation (RCAS-K-ras), and myristoylated mouse Akt1 (RCAS-Akt). The expression of c-myc, K-ras, and Akt onco-proteins in RCAS-infected ovarian cells from β-actin-TVA/p53−/− mice was determined by Western blotting (Figure 3) with antibody against human c-myc and the anti-HA antibody that recognizes the HA tag on the virally introduced Akt and K-ras proteins. The level of individual oncoprotein expression did not differ if the cells were infected with a single RCAS virus or a combination of two RCAS viruses (Figure 3).
Figure 3.
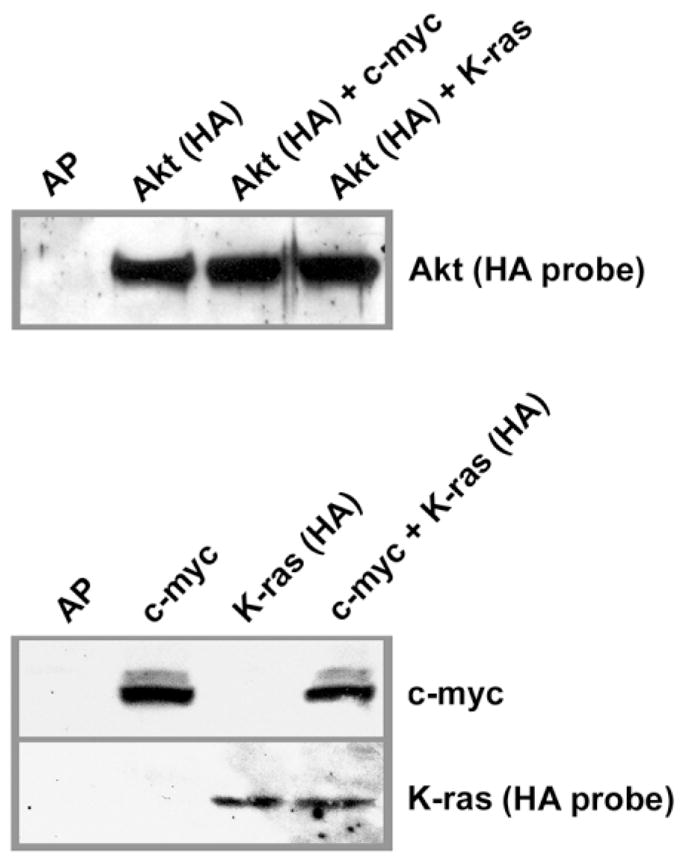
Expression of oncogenic proteins in ovarian cells after in vitro infection with RCAS viruses
Western blot of β-actin-TVA/p53−/− ovarian cells infected with RCAS viruses carrying AP, c-myc, K-ras, or Akt, individually or in combinations of two RCAS viruses. The same amount of cell lysate was loaded into each lane. HA-tagged Akt and K-ras proteins were detected with the anti-HA probe. The expression of c-myc was determined with an antibody against human c-myc.
Induction of oncogenic lesions in ovarian cells alters their growth properties in culture
We asked if the introduction of c-myc and K-ras into p53−/− ovarian cells could induce phenotypic changes in ovarian cells in culture (Figure 4). We first compared in vitro growth characteristics of ovarian cells from β-actin-TVA/p53−/− mice that were infected with RCAS-c-myc or RCAS-K-ras, or with a combination of the two viruses. The number of cells in individual cultures was followed for 8 days (Figure 4A). Infection of ovarian cells with RCAS-K-ras or with RCAS-c-myc increased their proliferation rate in comparison to the ovarian cells infected with RCAS-AP. However, the proliferation rate increased even more dramatically when ovarian cells were infected with the combination of RCAS-K-ras and RCAS-c-myc.
Figure 4.
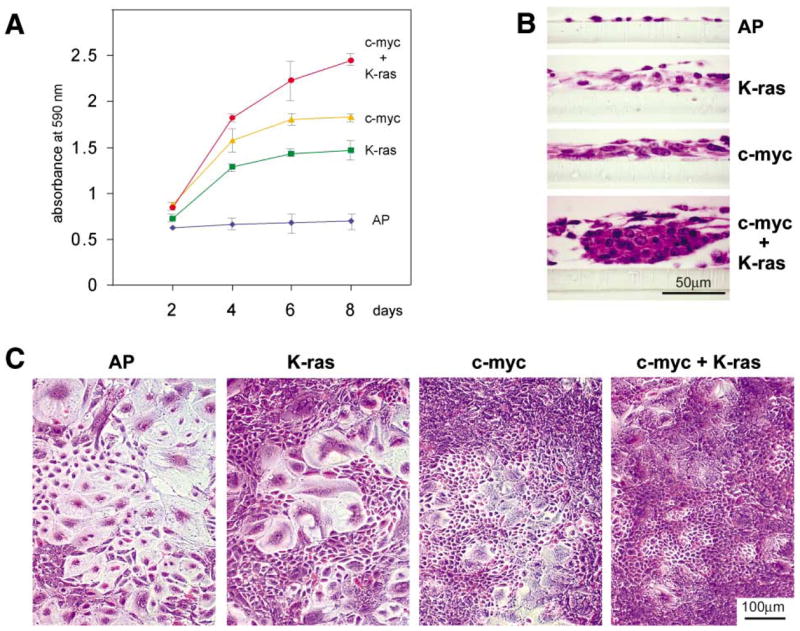
Oncogenic c-myc and K-ras collaborate in inducing proliferation of epithelial ovarian cells
A: Proliferation curves of ovarian cells from β-actin-TVA/p53−/− mice that were infected with RCAS viruses carrying AP, c-myc, K-ras, or with a combination of RCAS viruses carrying c-myc and K-ras. Five days after infection with RCAS viruses, each culture of the virally infected cells was split into 12 separate cultures that were maintained for 8 days. The cell proliferation rates were determined by fixing triplicate cultures every 2 days, staining them with crystal violet dye, and measuring the light absorbance of the extracted dye at 590 nm. This procedure allowed us to count the proliferating cells, as well as the viable terminally differentiated cells that would normally be lost if the cells were continually passaged. Proliferation curves were plotted as a ratio of the light absorbance and the time. The error bars indicate the extreme values of light absorbance in three separate cultures. Proliferation of ovarian cells from β-actin-TVA/p53−/− mice was induced by oncogenic change in either c-myc or K-ras, but the effect was further amplified in ovarian cells with genetic changes in both genes.
B: Morphological appearance of the cell populations described in A at day 6. Cultured ovarian cells infected with RCAS-AP acquire features of normal terminal differentiation and senescence. Introduction of oncogenic lesions induces marked changes to ovarian cell morphology and differentiation, which is especially pronounced in the ovarian epithelial cells.
C: Representative cross-sections of ovarian cell populations in B.
To examine the cellular responses to oncogenic lesions in primary ovarian cells in culture, we visually examined the appearance of cell cultures on day 6. The majority of cells that were infected with RCAS-AP acquired features of normal terminal differentiation, such as enlarged cells with flat morphology (Figure 4B). A subset of cells in the ovarian cultures that were infected with RCAS-K-ras or RCAS-c-myc viruses continued to proliferate. The epithelial-to-stromal cell ratio increased in these cultures as judged by the cobblestone appearance of the proliferating cells. The proliferation of the epithelial cell population was most prominent in cultures infected with the combination of RCAS viruses carrying c-myc and K-ras (Figure 4B). The representative cross-sections of the cell cultures indicate that the cells that were infected with RCAS viruses carrying oncogenes grow in multiple layers (Figure 4C).
Tumor induction in ovarian cells requires a combination of several genetic changes
Mutation or overexpression of genes such as p53, c-myc, K-ras and Akt are frequently detected in human ovarian carcinoma specimens (Aunoble et al., 2000), although it is not known whether these genetic changes must occur in combination to induce tumor growth. Therefore, we asked how many genetic changes are necessary to induce ovarian carcinoma in mouse ovarian cells. Ovarian cells from β-actin-TVA/p53+/+, β-actin-TVA/p53−/−, keratin 5-TVA/p53+/+, and keratin 5-TVA/p53−/− mice were propagated in culture for 7 days, during which they were infected with RCAS-c-myc, RCAS-K-ras and RCAS-Akt viruses, individually or in various combinations. After infection, ovarian cells were trypsinized and 105 or 106 cells were injected subcutaneously into nude mice, and the mice were examined regularly for tumor formation (Table 1). No tumors were produced from ovarian cells infected with an RCAS vector carrying an AP marker gene or only one oncogene (c-myc, K-ras or Akt), regardless of the p53 status or the promoter driving the TVA transgene. Ovarian cells from mice that were wild-type for the p53 gene did not produce tumors even when transduced with any combination of two or all three oncogenes. However, ovarian cells from p53−/− mice resulted in tumors when transduced with any combination of two or three oncogenes, regardless of whether the cells were derived from β-actin-TVA or keratin 5-TVA mice. Therefore, genetic changes in the p53 tumor suppressor gene and at least two oncogenes (c-myc and K-ras, K-ras and Akt, or Akt and c-myc) are required to induce tumors in ovarian cells in this assay (Table 1).
Table 1.
Combinations of changes in oncogenes and tumor suppressor genes required for tumor formation by mouse ovarian cells injected subcutaneously into nude mice
| TVA transgenic | p53 background | Oncogene combination | Tumor |
|---|---|---|---|
| wt | AP marker | − | |
| wt | c-myc | − | |
| wt | K-ras | − | |
| β-actin-TVA ovarian cells | wt | Akt | − |
| wt | c-myc + K-ras | − | |
| wt | K-ras + Akt | − | |
| wt | Akt + c-myc | − | |
| or | wt | Akt + c-myc + K-ras | − |
| p53−/− | AP marker | − | |
| p53−/− | c-myc | − | |
| keratin 5-TVA ovarian cells | p53−/− | K-ras | − |
| p53−/− | Akt | − | |
| p53−/− | c-myc + K-ras | + | |
| p53−/− | K-ras + Akt | + | |
| p53−/− | Akt + c-myc | + | |
| p53−/− | Akt + c-myc + K-ras | + |
Ovaries from β-actin-TVA or from keratin 5-TVA mice, in wild type or p53−/− backgrounds, were isolated and infected in vitro with RCAS vectors carrying AP, c-myc, K-ras, or Akt, individually or in combination. 105 or 106 cells were injected subcutaneously into nude mice. Mice were observed weekly for tumor growth for 8 weeks. Tumors that reached 1 cm in diameter were counted as positive. At least two nude mice were injected with ovarian cells that were infected with each combination of viruses, and the experiment was done at least twice with each combination.
Although individual genetic lesions in ovarian cells are not sufficient for tumor formation, they may induce changes that predispose to tumor development. We monitored the mice for tumor formation for six months to allow development of slow-growing tumors, or tumors that require secondary mutations. Ovarian cells from β-actin-TVA/p53−/− and keratin 5-TVA/p53−/− mice that were infected with only one RCAS virus stock carrying c-myc, K-ras, or Akt, developed tumors with latency varying from 12 weeks to 6 months (data not shown). Thus, ovarian cells with genetic changes that predispose to tumor development most likely acquire additional changes with time.
Ovarian surface epithelium is the precursor tissue for ovarian epithelial cancer
Most human ovarian tumors are believed to arise from the single layer of epithelial cells that covers the ovarian surface. We used histological markers to determine the tissue of origin for the tumors that developed after subcutaneous injection of infected ovarian cells into nude mice. Subcutaneous tumors derived from keratin 5-TVA ovaries consisted mostly of undifferentiated epithelial sheets and stromal cells (Figure 5). Staining with the antibody against TVA (Figure 5A) revealed that the epithelial sheets in the tumor were derived from the ovarian surface epithelium since TVA is present exclusively in the ovarian surface epithelium in the keratin 5-TVA ovaries (Figure 5A, inset). The identity and epithelial characteristics of these cells were confirmed with the TROMA-1 antibody (Figure 5B) which specifically labels the cells of the ovarian surface epithelium by recognizing Keratin 8 (Figure 5B, inset). The same cells in the tumor were reactive to an antibody that recognizes the HA epitope tag on the Akt protein (Figure 5C) which was expressed in tumor cells as a result of infection with the RCAS-Akt virus. The HA epitope, as expected, is not present in the uninfected ovary (Figure 5C, inset). From these results, we concluded that the epithelial component of the tumor is derived from the surface epithelial cells of the ovary from the keratin 5-TVA transgenic mouse. The stromal component of the tumor could represent either cells recruited from the nude mouse subcutaneous stroma or the residual stromal cells from the keratin 5-TVA transgenic ovary.
Figure 5.
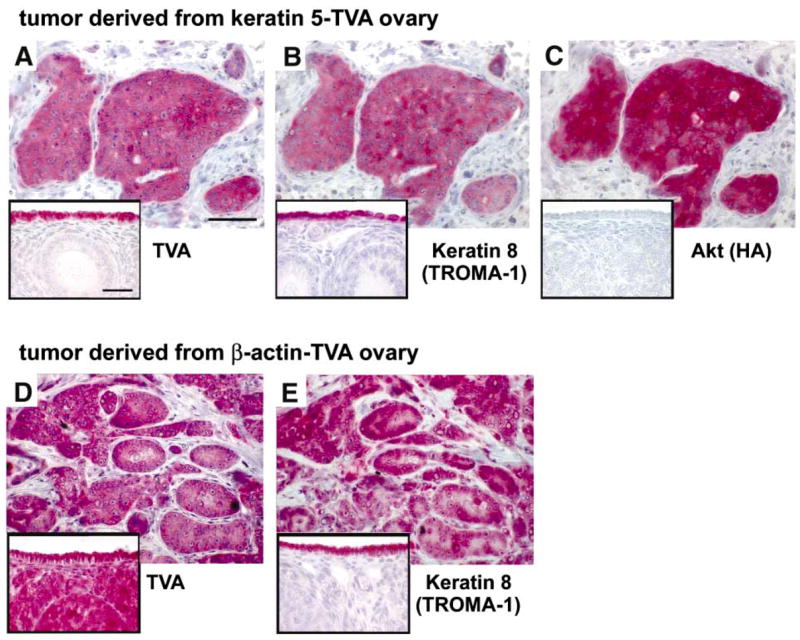
Ovarian surface epithelium is the precursor tissue for ovarian carcinoma
A–C: Sections of a subcutaneous tumor in a nude mouse derived from the keratin 5-TVA/p53−/− ovarian cells infected with a combination of RCAS-c-myc and RCAS-Akt viruses. The consecutive sections were probed with antibodies that recognize TVA, Keratin 8 and the HA-tagged Akt protein. Insets show a normal ovary from an unin-fected keratin 5-TVA mouse probed with the same antibodies. A: The TVA-positive cells in the tumor originate from the ovarian surface epithelium because this is the only cell type that expresses TVA in the ovaries from keratin 5-TVA transgenic mice (A, inset). B: The same TVA-positive cells in the tumor are also positive for the Keratin 8 marker, indicating that they maintain their epithelial characteristics and resemble the Keratin 8-positive cells of the ovarian surface epithelium (B, inset). C: The TVA-positive cells in the tumor also express virally introduced Akt oncogene. The HA-tagged Akt is not present in the uninfected ovary (C, inset).
D and E: Consecutive sections of a subcutaneous tumor in a nude mouse derived from the β-actin-TVA/p53−/− ovarian cells infected with a combination of RCAS-c-myc and RCAS-K-ras viruses. The sections were probed with antibodies that recognize TVA receptor and Keratin 8. Insets show a normal ovary from uninfected β-actin-TVA mouse probed with the same antibodies as the tumor sections. D: The tumor consists of TVA-positive and TVA-negative cells. The TVA-positive cells originate from the ovary of the β-actin-TVA transgenic mouse (D, inset). Since all cells from β-actin-TVA ovaries express TVA, the TVA-negative stromal cells in the tumor must be recruited from the host tissue and not from ovarian stroma. E: The TVA-positive cells in the tumor are epithelial as determined by the expression of the Keratin 8 marker which is present exclusively in the epithelial cells in the ovary (E, inset). Each bar represents 50 μm.
The origin of the stromal component of the tumor can be more accurately determined in the subcutaneous tumors derived from the β-actin-TVA ovaries. In ovaries from the β-actin-TVA mice, both ovarian surface epithelium and the ovarian stroma can be labeled with the antibody against TVA (Figure 5D, inset). However, the stromal cells in the tumor were not reactive with the antibody against TVA, indicating that they were not derived from the β-actin-TVA ovary (Figure 5D). Instead, the stromal cells must have been recruited from the nude mouse stroma. Since all cells in the β-actin-TVA ovaries are susceptible to infection with viruses (Figure 2), it might have been expected that the tumors would be derived from the surface epithelial cells as well as from the cells of the ovarian stroma. Remarkably, only ovarian epithelial cells gave rise to the tumors as determined by staining with the TROMA-1 antibody (Figures 5E and 5E, inset).
Ovarian tumors induced in the mouse model resemble development and metastatic spread of human ovarian carcinoma
The subcutaneous tumor model in nude mice was convenient to use as a rapid method to determine the genetic changes that are necessary for the induction of tumors in ovarian cells. However, ovarian tumors develop in the peritoneal cavity and rarely spread outside the peritoneum. In order to monitor ovarian tumor development in its natural environment, we developed a technique for implanting in vitro-infected ovarian cells into their orthotopic site, under the ovarian bursa (Experimental Procedures section). Ovarian cells isolated from β-actin-TVA/p53−/− mice or from keratin 5-TVA/p53−/− mice were infected in culture with RCAS viruses and implanted under the ovarian bursa of the left ovary of a nude mouse. The first sign of tumor development was a palpable solid tumor of the left ovary. The tumor was confined to the ovary in mice that were sacrificed within the first 2 weeks of ovarian cell implantation (data not shown). Intraperitoneal tumor growths at other sites were palpable within 4 weeks after detection of primary tumor. Metastatic spread was indicated by abdominal bloating and the presence of bloody ascites (Figure 6A). The tumors spread from the left ovary to the intraperitoneal and retroperitoneal organs (Figure 6B). Meta-static sites included the contralateral ovary, intestines, liver, pancreas, kidneys (Figure 6C, arrows), mesothelial lining of the peritoneum, omentum, and the diaphragm (not shown). Most of the metastases grew on the surfaces of organs (Figure 6D, arrows), but some penetrated deep into the tissue as illustrated with sections of liver and spleen that were immunostained with the antibody against the TVA receptor (Figure 6D, arrowheads).
Figure 6.
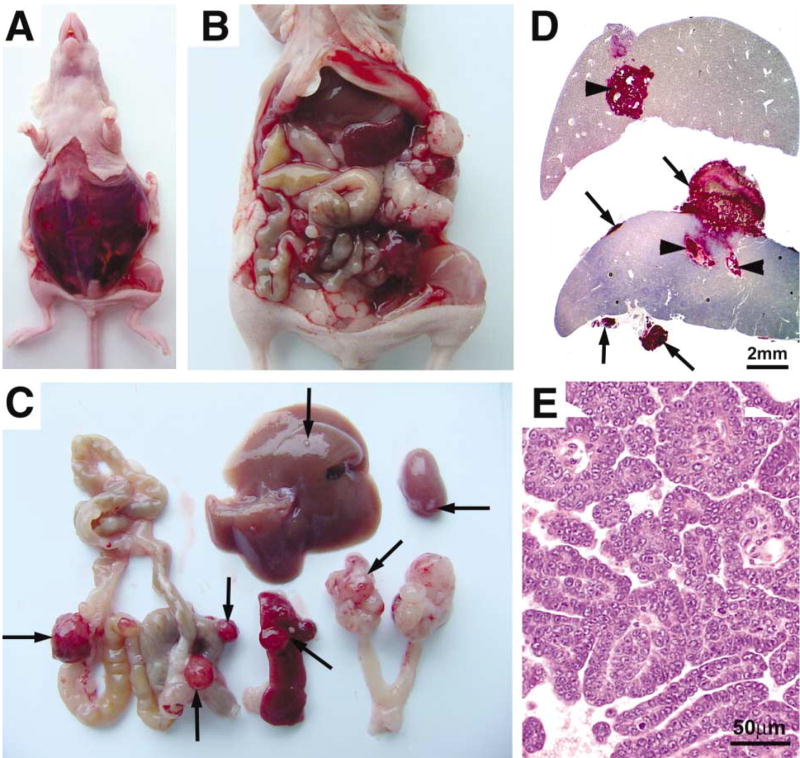
Intraperitoneal tumor spread in nude mice after implantation of ovarian cells with induced genetic lesions under the ovarian bursa
A–E: Tumors derived from β-actin-TVA/p53−/− ovarian cells that were infected with a combination of RCAS viruses carrying c-myc and K-ras oncogenes and implanted under the ovarian bursa of a nude mouse. A: Abdominal bloating and the presence of bloody ascites were the typical signs of the intraperitoneal tumor spread. B: Ovarian tumor spread in the peritoneal cavity. C: Ovarian tumor spread to the intraperitoneal and retroperitoneal organs. Arrows indicate ovarian tumor spread to the spleen, liver, intestine, kidney, and the contralateral ovary. D: Sections of the liver and the spleen probed with antibody against TVA to identify the ovarian tumor cell metastases. The tumor cells metastasize to the surfaces of organs (arrows) and deep into the tissue (arrowheads). E: Papillary organization of metastatic ovarian tumor resembles ovarian papillary carcinoma.
The induced tumors were histologically diagnosed as carcinomas. Primary ovarian tumors consisted mostly of sheets of poorly differentiated epithelial cells (data not shown). The metastases ranged from sheets of poorly differentiated cells (not shown) to organized papillary projections (Figure 6E) that resemble ovarian papillary serous carcinoma.
Induction of ovarian carcinoma in immunocompetent mice
The immune system has been shown to play a role in cancer development in mice (Shankaran et al., 2001). Thus, nude mice may not be the ideal model system in which to study ovarian cancer development and metastatic spread. We attempted to induce ovarian tumors in immunocompetent mice by implanting RCAS-infected ovarian cells under the ovarian bursa. Ovarian cells from the β-actin-TVA/p53−/− mouse or from the keratin 5-TVA/p53−/− mouse were infected in vitro with a combination of RCAS viruses carrying c-myc, K-ras, and Akt. The cells were then implanted under the ovarian bursa of the contralateral ovary of the same mouse. Ovarian tumors developed at the implanted site in both keratin 5-TVA/p53−/− mice, and in β-actin-TVA/p53−/− mice (Figure 7A and data not shown) three months after implantation of the ovarian cells. The histological appearance of ovarian tumors induced in immunocompetent mice (Figure 7B) resembled that of undifferentiated epithelial tumors.
Figure 7.
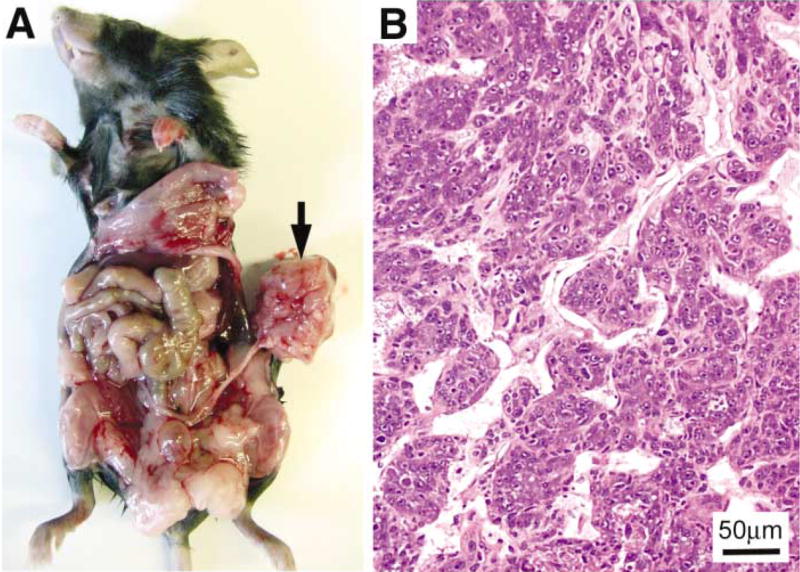
Ovarian tumor induction in immunocompetent mice
A: Ovarian cells from the right ovary of the β-actin-TVA/p53−/− mouse were transduced with c-myc, K-ras, and Akt oncogenes and implanted under the ovarian bursa of the left ovary. The arrow points to the tumor that developed in the left ovary three months after intrabursal implantation of the RCAS-infected ovarian cells. B: H&E staining of the tumor section reveals an undifferentiated epithelial neoplasm, arranged in nests.
Discussion
Our goal was to develop a suitable system for recapitulating human ovarian carcinoma development and progression. The system was designed such that tumors are induced in adult animals, similar to the occurrence in the majority of naturally developing human tumors. Furthermore, the system was designed to be used for the evaluation of multiple genetic lesions, individually and in combination thus increasing the ability to simulate a wide spectrum of human ovarian tumors without extensive breeding protocols.
We used the TVA receptor-dependent avian RCAS retroviral delivery technique to introduce defined multiple genetic lesions into mouse ovarian cells. This delivery technique has recently been adapted for transferring genes into specific mouse cells from transgenic mice programmed to express the avian TVA receptor in various tissues (Federspiel et al., 1994, 1996; Holland et al., 1998, 2000; Holland and Varmus, 1998; Doetsch et al., 1999; Murphy and Leavitt, 1999). We demonstrated that the RCAS-TVA system allows efficient gene transfer to cultured primary ovarian cells. The genes can be transduced to ovarian cells individually or in combination. The ovarian cells that are susceptible to infection with RCAS viruses can be limited by cell-specific expression of the TVA receptor. β-actin-TVA mice (Federspiel et al., 1996) were used to infect both ovarian epithelium and stroma, while keratin 5-TVA mice were used to restrict the infection to the cells of the ovarian surface epithelium. The major drawback of this system is the necessity for propagation of ovarian cells in culture in order to assure efficient infection with multiple RCAS vectors. However, since the cells are propagated in culture for only a short period of time it is unlikely that spontaneous mutations would occur. Another deviation from naturally occurring tumors that arise through clonal expansion is that multiple cells in this system are potential tumor precursors. Although there is an experimental advantage to inducing rapid tumor proliferation in a large number of cells with the same genetic lesions, this system does not entirely reflect the natural development of tumors.
Ovarian carcinomas are thought to arise from the epithelial lining that covers the surface of the ovary. Alternatively, ovarian carcinomas may arise from other cell types within the ovary, or from the components of the secondary Müllerian system that are not part of the ovary (Dubeau, 1999). Our mouse model system is well suited to identify the tissue of origin for the ovarian carcinomas we study. First, we were able to induce genetic lesions in isolated ovaries, thus ensuring that only ovarian cells contribute to tumor formation in our model. Second, we were able to use the keratin 5-TVA system to induce oncogenic changes specifically in the cells of the ovarian surface epithelium. Using the TVA receptor as a marker for the ovarian surface epithelial cells, we have demonstrated that the ovarian tumors in the keratin 5-TVA model system arise from the ovarian surface epithelial cells. Surprisingly, ovarian stromal cells that were transduced with oncogenes after infection of cells from β-actin-TVA transgenic mice did not contribute to the tumor (Figures 5D and 5E). There has been much speculation as to why the surface epithelial cells are the ovarian cells that most frequently give rise to ovarian tumors (Auersperg et al., 2001). Our results with cells propagated in vitro indicate that p53−/− ovarian epithelial cells proliferate more slowly than stromal cells. However, in the presence of oncogenic lesions, the proliferation of epithelial cells is more rapid than the proliferation of stromal cells. It is possible that the combination of oncogenic lesions that we used in this study specifically induces tumorigenic changes in the surface epithelial cells and not in the cells of the ovarian stroma. Because some residual epithelial components of the secondary Müllerian system may exist within the ovarian hilum and medulla (Dubeau, 1999), we cannot completely rule out the possibility that the tumors in mice are derived from the secondary Müllerian epithelium instead of the ovarian surface epithelium. However, this is very unlikely since the oncogene-induced proliferation of the morphologically distinct ovarian surface epithelial cells is already apparent in vitro.
Several studies indicate that ovarian tumors are the end result of a complex pathway involving multiple oncogenes and tumor suppressor genes, which include c-myc, K-ras, Akt, Her-2/neu, p53, and BRCA1/2 (reviewed in Gallion et al., 1995; Berchuck and Carney, 1997; Lynch et al., 1998; Aunoble et al., 2000). In our study, we introduced c-myc, K-ras and Akt oncogenes into cells from p53+/+ and p53−/− mice in an attempt to model the genetic aberrations that characterize many human ovarian carcinomas. A combination of three oncogenes (c-myc, K-ras, and Akt) is not sufficient to induce a tumorigenic state in ovarian cells from p53+/+ mice, indicating the importance of the p53 lesion. Our results suggest that the absence of p53 predisposes ovarian epithelial cells to tumor formation while in the presence of another initiating event, such as a growth signal produced by a mutant or an overexpressed oncogene. We determined that at least two oncogenic changes are required for rapid tumor formation in the p53-deficient ovarian cells (Table 1). Our preliminary results suggest that deletion of other tumor suppressor genes, such as INK4a/ARF, also contribute to ovarian tumor development when combined with oncogenic lesions in c-myc, K-ras, or Akt (S.O., unpublished). Thus, the necessity for a proliferative signal balanced with an anti-apoptotic signal may be a prerequisite for tumor formation in the ovarian cells.
Although changes in the function of p53 are commonly seen in ovarian carcinomas, patients with Li-Fraumeni syndrome rarely develop ovarian carcinomas, suggesting that a mutation in p53 is probably an essential step in ovarian carcinogenesis, but not the initial event. Although we have not addressed the temporal sequence of genetic events in ovarian carcinoma initiation and progression, the retroviral gene delivery system allows for multiple sequential infections with different RCAS vectors. Thus, multiple oncogenes, dominant-negative tumor suppressor genes, or the gene encoding the Cre recombinase, could be sequentially introduced into ovarian cells. Additionally, the use of inducible promoters in the RCAS system could provide the means to study the dependence of tumor growth on sustained production of a tumor-inducing oncogene.
Thus far, we have employed this model system to improve our understanding of the genetic changes leading to the initiation of ovarian cancer and to identify cooperating events in malignancy by using several different combinations of genes that are thought to contribute to cancer development. Unlike other existing mouse models that utilize mouse ovarian cell lines that are spontaneously transformed during prolonged growth in culture (Adams and Auersperg, 1981; Kido and Shibuya, 1998; Roby et al., 2000), our model system uses genetically defined combinations of lesions. We have focused on p53, c-myc, K-ras, and Akt genes because lesions in these genes are commonly present in human ovarian carcinoma samples. However, we have not tested other genes, such as HER-2/neu, FGFs, TGF-β, met, and BRCA 1 and 2, that have been shown to play a role in ovarian tumorigenesis. The system that we established can also be used to test the oncogenic potential of many other genes, in order to identify those that might contribute to the initiation and progression of ovarian cancer.
Experimental procedures
Mouse strains
β-actin-TVA mice (Federspiel et al., 1996) and p53−/− mice (Jacks et al., 1994) have been described. For the generation of keratin 5-TVA mice, a bluescript KS(−) plasmid containing a 5.2 kb fragment of the bovine keratin 5 promoter, a 0.6 kb fragment of the rabbit β-globin intron, a polylinker including a NheI site which was replaced for a NotI site, and a 3′ poly A sequence (Ramirez et al., 1994) were used to insert the pg800 tva cDNA (Bates et al., 1993) as a NotI fragment. The transgene was isolated after digestion by SalI. Transgenic mice were generated in FVB/N mice using standard techniques, and identified by Southern blot using the tva cDNA as a probe. The female nude mice (nu/nu) were purchased from the Charles River Laboratories. The TVA transgenic mice were crossed with the p53−/− mice to obtain TVA/p53−/− mice. The resulting crosses were on a mixed genetic background. Genotypes were determined by PCR with tail DNA.
Viral constructs and virus production
RCAS-AP encodes the heat-resistant placental alkaline phosphatase (Holland and Varmus, 1998). RCAS-K-ras carries the mutant G12D activated K-ras insert obtained from Dave Tuveson and Tyler Jacks, MIT (Johnson et al., 2001). RCAS-Akt carries myristoylated and HA-tagged mouse Akt1 that is constitutively activated. RCAS-c-myc carries the human c-myc (obtained from Brian Lewis, MSKCC). The DF-1 cell line, an immortalized line of chicken cells (Himly et al., 1998; Schaefer-Klein et al., 1998), was used for the production of viruses. The cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal calf serum (FCS), 0.2 mM L-glutamine and 1% penicillin/streptomycin. Plasmid forms of RCAS vectors were transfected into DF-1 cells, resulting in viral spread and high titer production of viruses. Viral supernatant was passed through an 0.8 μm syringe filter to obtain cell-free viruses for ovarian cell infection.
Ovary isolation
Three- to five-week-old female mice were anesthetized with Avertin and the right ovary was isolated using standard aseptic surgical procedures. The ovarian bursa was cut with microsurgical scissors and pushed aside to expose the ovarian surface. The ovary was separated from the bursa and transferred to a tissue culture dish containing DMEM.
Growth of ovary explants, infection of ovarian cells in culture, and ovarian cell aggregation
Each ovary was cut into 5–10 pieces and the explants were allowed to grow in a CO2 incubator for 48 hr to allow for cell attachment and growth. The medium was then replaced with fresh viral supernatant every 12 hr for 5 days. The transduced explants at day 7 were washed with Phosphate Buffer Saline (PBS), trypsinized, and briefly centrifuged to obtain a tight aggregate of cells.
Ovary implantation
Prior to ovary implantation, the intact ovary on the left side was exposed as described above. The bursa was opened with microsurgical scissors at one end, keeping most of the membrane intact, and the entire ovary was removed. While holding the bursa open with tweezers, the replacement ovarian cell aggregate was pushed into the bursa. A very small amount of surgical glue was applied to close the bursa and to keep the ovarian cell aggregate in place. The implanted ovary was placed inside the body wall and the skin was closed with wound clips.
Ovarian cell injection into mice
Infected ovarian cells were washed three times in PBS, trypsinized, and collected by brief centrifugation. The pelleted cells were washed in PBS. The mice were injected subcutaneously with 105 or 106 cells in a 100–200 μl volume of PBS.
Alkaline phosphatase (AP) staining
The AP was detected as described (Holland et al., 1998) with minor modifications. Briefly, cells infected with RCAS-AP were washed two times with PBS, fixed with methanol for 5 min, and washed three times with PBS. AP buffer (100 mM Tris, pH 9.5; 100 mM NaCl; 50 mM MgCl2) was added to the cells. Endogenous AP was heat-inactivated by floating the Petri dish in a water bath at 65°C for 45 min. After cooling the cells to room temperature, the AP buffer was replaced with the AP staining solution (5 ml AP buffer; 100 μl of 100 mM levamisole; 10 μl BCIP; 10 μl NBT). The reaction was left to develop overnight in the dark and stopped the next morning by rinsing out the staining solution with distilled water.
Determining efficiency of in vitro infection with RCAS-AP
Ovarian cells from the β-actin-TVA line or the keratin 5-TVA line were exposed to fresh viral supernatant twice a day for 5 days. After development of AP color reaction, four representative frames were photographed and the APpositive and AP-negative cells were counted in all four photographs. Surface epithelial cells were recognizable by their tightly packed cobblestone appearance; all other cell types were counted as stromal cells.
Measurement of cell proliferation
Ovaries from 12 β-actin-TVA/p53−/− mice were isolated and transferred into culture. The ovarian explants were replated two times to obtain a homogenous culture of ovarian cells. The cells were then split into four separate cultures and each culture was infected with viruses carrying the following genes: AP, c-myc, K-ras, or a combination of c-myc and K-ras. After 5 days of infection with viruses, each culture was separated into 12 cultures in a six-well dish, 1 × 103 cells per well. In order to monitor cell proliferation, triplicate cultures from each viral infection were sacrificed every second day. The cells were fixed with 10% buffered formalin, rinsed with water, and stained with a 0.1% solution of crystal violet (Sigma) for 6 hr. The cells were then washed extensively under running water and dried. Cell-associated dye was extracted with 2 ml of acetic acid. The aliquots were diluted 1:4 with water, and the optical density was determined at 590 nm.
Western blot analysis
Cells were lysed with PBS containing 0.1% Triton X-100 and a protease inhibitor mix (CompleteTM, Boehringer Mannheim), scraped from the culture dish, and cleared with centrifugation in a microfuge for 5 min at 4°C. Equal amounts of total cell protein were boiled in Laemmli buffer for 5 min, separated by 10% SDS-polyacrylamide gel and transferred to nitrocellulose. Filters were blocked for 1 hr at room temperature in 5% dry milk in Tris Buffered Saline + 0.1% TritonX-100 (TBST), washed in TBST, and incubated with primary antibodies in 5% dry milk in TBST at 4°C overnight. Anti-human c-myc (9E10; Santa Cruz) and anti-HA probe (Y-11; Santa Cruz), were used at a 1:100 dilution. After several washes, secondary peroxidase conjugated antibodies (Boehringer) were used at a 1:5,000 dilution. The membrane was washed in TBST and the proteins were detected using an enhanced chemiluminescence (ECL) detection system (Amersham) following the manufacturer’s recommendation.
Immunohistochemistry
Tissues and ovarian cells grown on Millipore filters were fixed in Carnoy’s fixative (absolute ethanol: chloroform: acetic acid; 6:3:1) for 6 hr at 4°C, after which they were transferred to 70% ethanol and embedded in paraffin. Paraffin-embedded sections were cleared in a graded xylene/ethanol series and used for immunohistochemistry with an ABC antibody staining kit (Vector Laboratories) according to manufacturer’s instructions. After color development, slides were counterstained with hematoxylin (Sigma) and mounted with water-based mounting medium (Triangle Biomedical Sciences). Partially purified supernatant of TROMA-1 antibody (Brulet et al., 1980) was obtained from the Developmental Studies Hybridoma Bank at The University of Iowa and used at a 1:25 dilution. Affinity-purified rabbit polyclonal anti-TVA antibody (Bates et al., 1993) was obtained from Andrew Leavitt (UCSF) and used at a concentration of 1 μg/ml. Anti-HA antibody (Y-11) was used at a concentration of 2 μg/ml.
Acknowledgments
We thank Steve Hughes (FCRC, NCI, NIH) for the β-actin-TVA mice; Doug Foster for the DF-1 cells; Brian Lewis (MSKCC) for the human c-myc cDNA; Alfonso Bellacosa (FCCC) for the Akt1 insert; Eric Holland (MSKCC) for the RCAS-AP vector; Dave Tuveson and Tyler Jacks (MIT) for the mutant K-ras cDNA; Andrew Leavitt (UCSF) for the TVA antibody; Galen Fisher for the HA-tagged K-ras virus; Jennifer Doherty for mouse genotyping and immunohistochemical staining; Amy Chen (NHGRI, NIH) for the initial assistance with survival surgery; Wendy Hively, Jennifer Doherty, and Mary Barrett for technical assistance and mouse husbandry; Kristy Daniels for assistance in preparation of the manuscript; David Spriggs, Jeff Boyd, and members of the Varmus lab for useful discussions. This work was supported by the Susan G. Komen Fellowship to S.O. and the Department of Defense Breast Cancer Fellowship to Y.L.
References
- Adams AT, Auersperg N. Transformation of cultured rat ovarian surface epithelial cells by Kirsten murine sarcoma virus. Cancer Res. 1981;41:2063–2072. [PubMed] [Google Scholar]
- Auersperg N, Wong AS, Choi KC, Kang SK, Leung PC. Ovarian surface epithelium: biology, endocrinology, and pathology. Endocr Rev. 2001;22:255–288. doi: 10.1210/edrv.22.2.0422. [DOI] [PubMed] [Google Scholar]
- Aunoble B, Sanches R, Didier E, Bignon YJ. Major oncogenes and tumor suppressor genes involved in epithelial ovarian cancer (review) Int J Oncol. 2000;16:567–576. doi: 10.3892/ijo.16.3.567. [DOI] [PubMed] [Google Scholar]
- Baker VV, Borst MP, Dixon D, Hatch KD, Shingleton HM, Miller D. c-myc amplification in ovarian cancer. Gynecol Oncol. 1990;38:340–342. doi: 10.1016/0090-8258(90)90069-w. [DOI] [PubMed] [Google Scholar]
- Bates P, Young JA, Varmus HE. A receptor for subgroup A Rous sarcoma virus is related to the low density lipoprotein receptor. Cell. 1993;74:1043–1051. doi: 10.1016/0092-8674(93)90726-7. [DOI] [PubMed] [Google Scholar]
- Bell DA, Scully RE. Early de novo ovarian carcinoma. A study of fourteen cases. Cancer. 1994;73:1859–1864. doi: 10.1002/1097-0142(19940401)73:7<1859::aid-cncr2820730714>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Bellacosa A, de Feo D, Godwin AK, Bell DW, Cheng JQ, Altomare DA, Wan M, Dubeau L, Scambia G, Masciullo V. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int J Cancer. 1995;64:280–285. doi: 10.1002/ijc.2910640412. [DOI] [PubMed] [Google Scholar]
- Berchuck A, Carney M. Human ovarian cancer of the surface epithelium. Biochem Pharmacol. 1997;54:541–544. doi: 10.1016/s0006-2952(97)00061-0. [DOI] [PubMed] [Google Scholar]
- Brulet P, Babinet C, Kemler R, Jacob F. Monoclonal antibodies against trophectoderm-specific markers during mouse blastocyst formation. Proc Natl Acad Sci USA. 1980;77:4113–4117. doi: 10.1073/pnas.77.7.4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caduff RF, Svoboda-Newman SM, Ferguson AW, Johnston CM, Frank TS. Comparison of mutations of Ki-RAS and p53 immunoreactivity in borderline and malignant epithelial ovarian tumors. Am J Surg Pathol. 1999;23:323–328. doi: 10.1097/00000478-199903000-00012. [DOI] [PubMed] [Google Scholar]
- Cheng JQ, Godwin AK, Bellacosa A, Taguchi T, Franke TF, Hamilton TC, Tsichlis PN, Testa JR. AKT2, a putative oncogene encoding a member of a subfamily of protein-serine/threonine kinases, is amplified in human ovarian carcinomas. Proc Natl Acad Sci USA. 1992;89:9267–9271. doi: 10.1073/pnas.89.19.9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuatrecasas M, Villanueva A, Matias-Guiu X, Prat J. K-ras mutations in mucinous ovarian tumors: a clinicopathologic and molecular study of 95 cases. Cancer. 1997;79:1581–1586. doi: 10.1002/(sici)1097-0142(19970415)79:8<1581::aid-cncr21>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- Dokianakis DN, Varras MN, Papaefthimiou M, Apostolopoulou J, Simiakaki H, Diakomanolis E, Spandidos DA. Ras gene activation in malignant cells of human ovarian carcinoma peritoneal fluids. Clin Exp Metastasis. 1999;17:293–297. doi: 10.1023/a:1006611220434. [DOI] [PubMed] [Google Scholar]
- Dubeau L. The cell of origin of ovarian epithelial tumors and the ovarian surface epithelium dogma: does the emperor have no clothes? Gynecol Oncol. 1999;72:437–442. doi: 10.1006/gyno.1998.5275. [DOI] [PubMed] [Google Scholar]
- Federspiel MJ, Bates P, Young JA, Varmus HE, Hughes SH. A system for tissue-specific gene targeting: transgenic mice susceptible to subgroup A avian leukosis virus-based retroviral vectors. Proc Natl Acad Sci USA. 1994;91:11241–11245. doi: 10.1073/pnas.91.23.11241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federspiel MJ, Swing DA, Eagleson B, Reid SW, Hughes SH. Expression of transduced genes in mice generated by infecting blastocysts with avian leukosis virus-based retroviral vectors. Proc Natl Acad Sci USA. 1996;93:4931–4936. doi: 10.1073/pnas.93.10.4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feeley KM, Wells M. Precursor lesions of ovarian epithelial malignancy. Histopathology. 2001;38:87–95. doi: 10.1046/j.1365-2559.2001.01042.x. [DOI] [PubMed] [Google Scholar]
- Fisher GH, Orsulic S, Holland E, Hively WP, Li Y, Lewis BC, Williams BO, Varmus HE. Development of a flexible and specific gene delivery system for production of murine tumor models. Oncogene. 1999;18:5253–5260. doi: 10.1038/sj.onc.1203087. [DOI] [PubMed] [Google Scholar]
- Gallion HH, Pieretti M, DePriest PD, van Nagell JR. The molecular basis of ovarian cancer. Cancer. 1995;76:1992–1997. doi: 10.1002/1097-0142(19951115)76:10+<1992::aid-cncr2820761315>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Greenlee RT, Murray T, Bolden S, Wingo PA. Cancer statistics, 2000. CA Cancer J Clin. 2000;50:7–33. doi: 10.3322/canjclin.50.1.7. [DOI] [PubMed] [Google Scholar]
- Himly M, Foster DN, Bottoli I, Iacovoni JS, Vogt PK. The DF-1 chicken fibroblast cell line: transformation induced by diverse oncogenes and cell death resulting from infection by avian leukosis viruses. Virology. 1998;248:295–304. doi: 10.1006/viro.1998.9290. [DOI] [PubMed] [Google Scholar]
- Holland EC, Varmus HE. Basic fibroblast growth factor induces cell migration and proliferation after glia-specific gene transfer in mice. Proc Natl Acad Sci USA. 1998;95:1218–1223. doi: 10.1073/pnas.95.3.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland EC, Hively WP, DePinho RA, Varmus HE. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev. 1998;12:3675–3685. doi: 10.1101/gad.12.23.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet. 2000;25:55–57. doi: 10.1038/75596. [DOI] [PubMed] [Google Scholar]
- Holschneider CH, Berek JS. Ovarian cancer: epidemiology, biology, and prognostic factors. Semin Surg Oncol. 2000;19:3–10. doi: 10.1002/1098-2388(200007/08)19:1<3::aid-ssu2>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Hughes SH, Greenhouse JJ, Petropoulos CJ, Sutrave P. Adaptor plasmids simplify the insertion of foreign DNA into helper-independent retroviral vectors. J Virol. 1987;61:3004–3012. doi: 10.1128/jvi.61.10.3004-3012.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, Jacks T. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- Kido M, Shibuya M. Isolation and characterization of mouse ovarian surface epithelial cell lines. Pathol Res Pract. 1998;194:725–730. doi: 10.1016/S0344-0338(98)80133-7. [DOI] [PubMed] [Google Scholar]
- Lynch HT, Casey MJ, Lynch J, White TE, Godwin AK. Genetics and ovarian carcinoma. Semin Oncol. 1998;25:265–280. [PubMed] [Google Scholar]
- Mok SC, Bell DA, Knapp RC, Fishbaugh PM, Welch WR, Muto MG, Berkowitz RS, Tsao SW. Mutation of K-ras protooncogene in human ovarian epithelial tumors of borderline malignancy. Cancer Res. 1993;53:1489–1492. [PubMed] [Google Scholar]
- Morita K, Ono Y, Fukui H, Tomita S, Ueda Y, Terano A, Fujimori T. Incidence of P53 and K-ras alterations in ovarian mucinous and serous tumors. Pathol Int. 2000;50:219–223. doi: 10.1046/j.1440-1827.2000.01028.x. [DOI] [PubMed] [Google Scholar]
- Murphy GJ, Leavitt AD. A model for studying megakaryocyte development and biology. Proc Natl Acad Sci USA. 1999;96:3065–3070. doi: 10.1073/pnas.96.6.3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez A, Bravo A, Jorcano JL, Vidal M. Sequences 5′ of the bovine keratin 5 gene direct tissue- and cell-type-specific expression of a lacZ gene in the adult and during development. Differentiation. 1994;58:53–64. doi: 10.1046/j.1432-0436.1994.5810053.x. [DOI] [PubMed] [Google Scholar]
- Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik O, Persons DL, Smith PG, Terranova PF. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis. 2000;21:585–591. doi: 10.1093/carcin/21.4.585. [DOI] [PubMed] [Google Scholar]
- Schaefer-Klein J, Givol I, Barsov EV, Whitcomb JM, VanBrocklin M, Foster DN, Federspiel MJ, Hughes SH. The EV-O-derived cell line DF-1 supports the efficient replication of avian leukosis-sarcoma viruses and vectors. Virology. 1998;248:305–311. doi: 10.1006/viro.1998.9291. [DOI] [PubMed] [Google Scholar]
- Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, Schreiber RD. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- Shayesteh L, Lu Y, Kuo WL, Baldocchi R, Godfrey T, Collins C, Pinkel D, Powell B, Mills GB, Gray JW. PIK3CA is implicated as an oncogene in ovarian cancer. Nat Genet. 1999;21:99–102. doi: 10.1038/5042. [DOI] [PubMed] [Google Scholar]
- Sun M, Wang G, Paciga JE, Feldman RI, Yuan ZQ, Ma XL, Shelley SA, Jove R, Tsichlis PN, Nicosia SV, Cheng JQ. AKT1/PKBalpha kinase is frequently elevated in human cancers and its constitutive activation is required for oncogenic transformation in NIH3T3 cells. Am J Pathol. 2001;159:431–437. doi: 10.1016/s0002-9440(10)61714-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Saito S, Saga Y, Ohwada M, Sato I. Mutation of K-RAS protooncogene and loss of heterozygosity on 6q27 in serous and mucinous ovarian carcinomas. Cancer Genet Cytogenet. 2000;118:132–135. doi: 10.1016/s0165-4608(99)00192-2. [DOI] [PubMed] [Google Scholar]
- Teneriello MG, Ebina M, Linnoila RI, Henry M, Nash JD, Park RC, Birrer MJ. p53 and Ki-ras gene mutations in epithelial ovarian neoplasms. Cancer Res. 1993;53:3103–3108. [PubMed] [Google Scholar]
- Wang ZR, Liu W, Smith ST, Parrish RS, Young SR. c-myc and chromosome 8 centromere studies of ovarian cancer by interphase FISH. Exp Mol Pathol. 1999;66:140–148. doi: 10.1006/exmp.1999.2259. [DOI] [PubMed] [Google Scholar]
- Young JA, Bates P, Varmus HE. Isolation of a chicken gene that confers susceptibility to infection by subgroup A avian leukosis and sarcoma viruses. J Virol. 1993;67:1811–1816. doi: 10.1128/jvi.67.4.1811-1816.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan ZQ, Sun M, Feldman RI, Wang G, Ma X, Jiang C, Coppola D, Nicosia SV, Cheng JQ. Frequent activation of AKT2 and induction of apoptosis by inhibition of phosphoinositide-3-OH kinase/Akt pathway in human ovarian cancer. Oncogene. 2000;19:2324–2330. doi: 10.1038/sj.onc.1203598. [DOI] [PubMed] [Google Scholar]