

Abstract
We have used flash photolysis of a caged form of nitric oxide (NO), potassium pentachloronitrosylruthenate (K2Ru(NO)Cl5), to apply known concentrations of NO, with a high degree of temporal resolution, to hippocampal slices prepared from juvenile male rats maintained in an interface recording chamber.
Photolytically released NO (1–4.5 μM) from bath applied caged NO reduced the magnitude of long-term potentiation (LTP) in a concentration-dependent manner. This effect was abolished in the presence of the NO scavenger haemoglobin. NO had no effect on pre-established LTP.
Exposure to photolytically released NO had no effect on normal fast synaptic transmission, but did result in depression of N-methyl-D-aspartate (NMDA) receptor-mediated transmission recorded using extracellular electrodes. The onset of NO-induced depression was relatively slow, taking >40 s to manifest itself, and several minutes to achieve maximum depression (t½≈ 70 s). NO-induced depression persisted for more than 2 h after photolysis. The time courses of the action of NO on NMDA receptor-mediated responses and its action on the induction of LTP were similar.
These results suggest that released NO may play a role in determining the subsequent threshold for the induction of LTP at Schaffer-commissural synapses through a reduction in the efficacy of NMDA receptor function when repeated conditioning trains are used.
The role of nitric oxide (NO) in the induction of N-methyl-D-aspartate (NMDA) receptor-dependent long-term potentiation (LTP) remains controversial. Based on observations that pharmacological blockade of nitric oxide synthase (NOS) or sequestration of NO impair the induction of LTP (O'Dell et al. 1991; Schuman & Madison, 1991; Haley et al. 1992) and that exogenous application of NO or NO donors induces potentiation (Bohme et al. 1991; Bon et al. 1992; Zhuo et al. 1993; Arancio et al. 1996), it has been claimed that NO may serve as a retrograde messenger during the induction of LTP. However, this view has been challenged by studies from several laboratories showing that neither blockade of NOS (Kato & Zorumski, 1993; Bannerman et al. 1994; Cummings et al. 1994; Kirkwood & Bear, 1994) nor exogenous application of NO or NO donors (Boulton et al. 1994; Murphy et al. 1994) affects the induction of LTP.
Under certain experimental conditions, namely in slices prepared from young rats (5-7 weeks of age) and maintained at room temperature (∼24°C), we have found it possible to block the induction of LTP with antagonists of NOS. However, if the experiments were repeated in slices maintained at a more physiological temperature (∼30°C), or in slices prepared from adult rats, then blockade of NOS did not affect the induction of LTP (Williams et al. 1993). In further experiments, we used a caged form of NO in an attempt to induce LTP by pairing synaptic activation with photolytic release of NO (Murphy et al. 1994). These experiments were performed under the same conditions (young animals, low temperature) as those in which we had previously shown that NOS blockade impaired the induction of LTP. Pairing tetanic stimulation with exposure to NO, in the presence of the NMDA receptor antagonist D(-)-2-amino-5-phosphonopentanoic acid (D-AP5) did not induce LTP, a result suggesting that even in young animals NO is unlikely to be involved in the induction of LTP (Murphy et al. 1994). However, we noted that exposure to photolytically released NO affected the outcome of subsequent high-frequency trains. Specifically, after washout of AP5 and caged NO, slices previously exposed to concentrations of NO of less than 1 μM expressed normal LTP, whereas those exposed to higher concentrations showed a decrease in the magnitude of potentiation. A similar inhibition of LTP has been reported by Izumi et al. (1992) using an NO donor, nitroprusside.
In this study we have further investigated the effects of exposure to NO on the induction and expression of LTP. We have used caged NO to apply known quantities of NO with millisecond accuracy, an approach not possible using other methods of applying NO. We find that NO has no effect on the maintenance of LTP but markedly affects induction in a time- and concentration-dependent manner. The latter appears to be a consequence of the depressant action of NO on NMDA receptors (Lei et al. 1992; Manzoni et al. 1992; Murphy et al. 1994). Based on these findings, we propose that NO, if endogenously released, may render freshly conditioned synapses refractory to further NMDA receptor-dependent plasticity.
Some of the results presented here have been published in preliminary form (Murphy & Bliss, 1995).
METHODS
Hippocampal slices were prepared from young male Sprague-Dawley rats (5-7 weeks). Animals were killed by stunning followed by cervical dislocation and decapitation. The brain was rapidly removed and the hippocampus dissected out and placed in cold (0-4°C) oxygenated (95% O2, 5% CO2) artificial cerebrospinal fluid (ACSF) of the following composition (mM): 120 NaCl, 3 KCl, 2 MgSO4, 2 CaCl2, 1.2 NaH2PO4, 23 NaHCO3, 11 glucose. Transverse slices (420 μm thick) were cut with a Mcllwain tissue chopper, the CA3 subfield excised and the resulting minislice transferred to an interface-type recording chamber maintained at 24 ± 0.3 or 30 ± 0.3°C and perfused with oxygenated ACSF at a flow rate of 100 μl min−1, or to a holding chamber containing oxygenated ACSF at room temperature.
Extracellular field EPSPs were recorded using glass microelectrodes (filled with 0.5 M sodium acetate and 2% Pontamine Blue; 5-12 MΩ) placed in the stratum radiatum of the CA1 field and evoked by monopolar stimulation of the Schaffer collateral- commissural pathway (0.033 Hz). The test stimulus (constant current, duration 20-40 μS, intensity 50-300 μA) was set to elicit a response with an initial slope of 30-40% of the maximum. The conditioning tetanus consisted of a train of 20 pulses at 100 Hz, repeated 6 times with an inter-train interval of 3 s (stimulus duration increased by 1.5 times). In most experiments responses were elicited by alternate stimulation of two separate afferent pathways (each pathway at 0.033 Hz). The independence of these pathways was verified by ensuring that prior activation of one pathway did not affect the response evoked by stimulation of the second (20-60 ms stimulation interval). NMDA receptor-mediated responses were pharmacologically isolated using modified, nominally magnesium free ACSF containing 10 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; Tocris Cookson, Bristol, UK). In additional experiments, NMDA receptor-mediated potentials were recorded in the presence of 2 mM magnesium in ACSF containing 10 μM CNQX and 20 μM of the GABAA receptor antagonist bicuculline (Sigma). Initial slope measurements were made during the linear phase for each evoked potential with fixed latencies of less than 5 ms for normal (fast) responses and less than 10 ms for NMDA receptor-mediated potentials.
NO was released by flash photolysis of bath applied caged NO (potassium pentachloronitrosylruthenate, K2Ru(NO)Cl5; Johnson Matthey, Royston, Herts, UK) using a 1 ms pulse of near-UV light (fixed energy of ∼70 mJ; photolysing 0.9% of the caged compound) delivered by a xenon flashlamp (Rapp, Hamburg, Germany/Cairn Research Instruments, Faversham, UK) irradiating the whole slice. The approach offers several advantages over traditional chemical methods for applying NO: firstly, the biological inert caged NO can equilibrate within the slice prior to photolysis; secondly, photolysis occurs rapidly (< 1 ms) allowing excellent temporal resolution of NO delivery; and thirdly, the extent of photolysis and therefore the concentration of NO liberated is known (see Murphy et al. (1994) and Bettache et al. (1996) for detailed description of the calibration procedure and characterization of caged NO and Ogden et al. (1990) and Murphy et al. (1997) for details of photolytic efficiency within brain slices).
All statistical data are expressed as means followed by the standard error of the mean. Unless otherwise stated, all P values are derived using Student's two-tailed t test.
RESULTS
Nitric oxide and the induction of LTP
Photolytic release of NO from K2Ru(NO)Cl5 blocked the induction of LTP in a dose-dependent manner without affecting pre-existing LTP (Fig. 1). The ability of slices to support LTP was unaffected by the presence of caged NO. Slices exposed to concentrations as high as 500 μM exhibited LTP of a magnitude comparable with that induced in a set of control slices not exposed to the caged compound (69.8 ± 4.3% (n = 7) and 78.0 ± 17.3% (n = 6), respectively, measured 1 h after induction (P > 0.8); Fig. 1A and B). The action of NO on the induction and maintenance of LTP was examined concurrently. Slices were bathed with varying concentrations of caged NO, and EPSPs evoked by alternate stimulation of two separate and independent afferent pathways. After a 20 min control period a conditioning tetanus was applied to one pathway (P1) and followed 10 min later by application of a single 1 ms pulse of near-UV light, releasing 0-4.5 μM NO from caged NO. Ten minutes after the flash, a conditioning tetanus was then applied to the second pathway (P2). Exposure to NO had no effect on the expression of LTP in P1 nor did it alter the baseline response in P2. In slices exposed to 0, 1, 3 and 4.5 μM NO the degree of potentiation in P1, measured 1 h after induction, was 78.0 ± 17.3% (n = 6), 79.4 ± 11.1% (n = 5), 71.5 ± 4.6% (n = 3) and 69.6 ± 4.3% (n = 7), respectively. However, prior exposure to NO impaired the ability to induce LTP in P2: the magnitude of potentiation measured 40 min after tetanic stimulation was 73.2 ± 13.5% (n = 6), 34.0 ± 6.5% (n = 5), 16.4 ± 11.8% (n = 3) and 5.9 ± 3.8% (n = 7) after exposure to 0, 1, 3 and 4.5 μM NO, respectively. The concentration dependency of the action of NO on the induction of LTP is illustrated in Fig. 1C. Regardless of the concentration of NO applied, LTP in P2 was non-decremental, suggesting that in P2, as in P1, the mechanisms underlying the expression of LTP were largely unaffected. Note that prior exposure to 4.5 μM NO unambiguously abolished the ability to induce LTP. Consequently, this concentration of NO was used in subsequent experiments designed to assess the role of NO in the induction of LTP.
Figure 1. Differential action of photolytically released NO on the induction and maintenance of LTP at CA1 synapses in slices maintained at 24 °C.
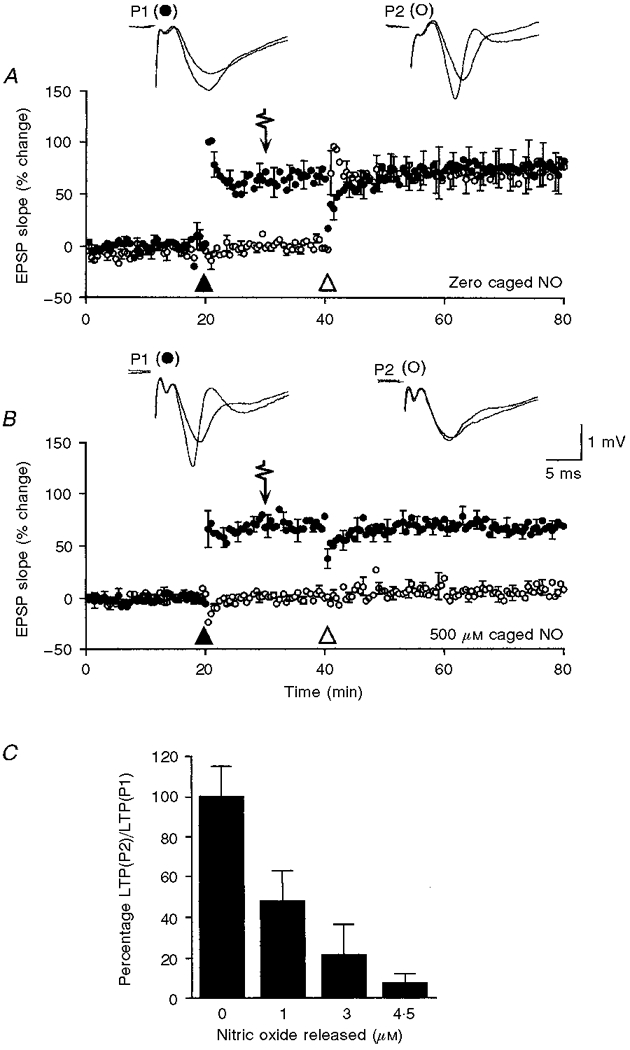
In A and B the normalized percentage change in the slope of the field EPSP is displayed as a function of time; each plot is the mean of pooled data obtained from 6 and 7 slices in A and B, respectively, and normalized with respect to the mean of the control period 10 min prior to the delivery of the conditioning tetanus. Standard error bars are restricted to every fifth mean. In this and the other figures, pathway 1 (P1) is denoted by • and pathway 2 (P2) by ○. A tetanus consisting of 20 pulses at 100 Hz repeated 6 times at an interval of 3 s was first applied to P1 and is denoted by ▴; 20 min later a second conditioning tetanus was applied to P2 and is denoted by ▵. A single 1 ms pulse of near-UV light was applied 10 min before conditioning of the second pathway and is indicated by an arrow. A, slices were bathed with control medium and LTP was sucessfully induced in both pathways. B, slices were bathed with 500 μM caged NO, liberating 4.5 μM NO upon photolysis. Exposure to NO reduced the magnitude of LTP expressed in P2. Superimposed traces of averaged pre- and post-tetanus responses (averages of five consecutive responses; post-tetanus responses collected 40 min after tetanic stimulation). C, histogram showing the extent of LTP expressed in P2 as a percentage of that expressed in P1 for each experiment, after exposure to 0 μM (n = 6), 1 μM (n = 5), 3 μM (n = 3) and 4.5 μM (n = 7) NO. Each data bar is the pooled mean ±s.e.m. Experiments performed at 24 °C.
Haemoglobin abolishes the action of NO on the induction of LTP
To demonstrate that suppression of LTP was attributable to NO, a series of experiments were performed in the presence of the NO scavenger haemoglobin (Martin et al. 1985). We have previously reported (Williams et al. 1993) that haemoglobin can impair the induction of LTP in a temperature-dependent manner: extracellularly applied haemoglobin suppresses the induction of LTP at 24°C but not at 30°C. Consequently, these experiments were performed at 30°C, a temperature at which haemoglobin has no effect on the induction and maintenance of LTP. Figure 2A confirms that the action of NO on the induction of LTP is not dependent on temperature. Slices were bathed with 500 μM caged NO and field EPSPs evoked by alternate stimulation of two independent afferent pathways. LTP was successfully induced in P1 in both the absence and presence of 100 μM haemoglobin (71.4 ± 17.2% (n = 4) and 59.1 ± 10.4% (n = 5), respectively, measured 1 h after induction; Fig. 2). In contrast, the absence (Fig. 2A) or presence (Fig. 2B) of haemoglobin significantly affected the outcome of conditioning in P2 after exposure to 4.5 μM NO. In the absence of the NO scavenger, LTP observed in P2 was negligible (8.9 ± 4.2%, measured 40 min after induction) whereas it was normal in slices bathed with haemoglobin (74.9 ± 31.6%). Expressed as a percentage of LTP in P1, the magnitude of LTP in P2 was 19.5 ± 13.1% (n = 4) in the absence of haemoglobin and 86.9 ± 22.0% (n = 5) in its presence.
Figure 2. Haemoglobin abolishes the action of NO on the induction of LTP at CA1 synapses in slices maintained at 30 °C.
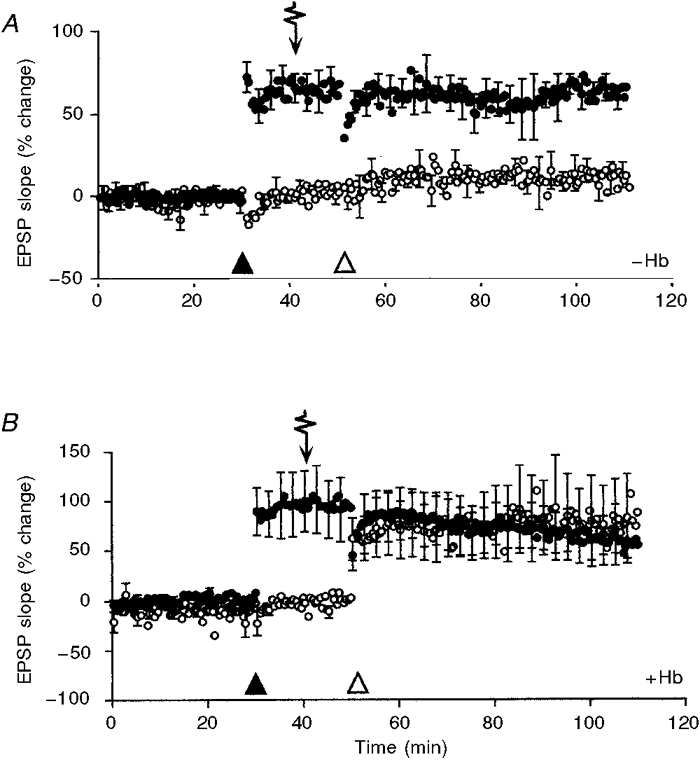
The percentage change in the slope of field EPSPs evoked by stimulation of two independent afferent pathways is displayed as a function of time. The plot is the mean data obtained from 4 slices in A (in the absence of haemoglobin) and 5 slices in B (in the presence of 100 μM haemoglobin), normalized to the mean of the control period prior to delivery of conditioning stimuli. The s.e.m. is plotted for every fifth mean. Slices were perfused with 500 μM caged NO throughout. Conditioning tetani were applied to P1 (•) and P2 (○) as indicated by ▴ and ▵, respectively. A pulse of near-UV light (arrow), releasing 4.5 μM NO from the caged compound, was applied 10 min before the delivery of conditioning stimuli to P2. B, addition of 100 μM haemoglobin to the perfusion medium abolished the depressive action of NO on the induction of LTP in P2.
At both 24 and 30°C brief exposure to 4.5 μM NO prior to tetanic stimulaton reduced the efficacy of the conditioning tetanus, resulting in little or no potentiation. The persistence of this effect is illustrated in Fig. 3; attempts to induce LTP at 70 and 130 min after exposure to 4.5 μM NO at 24°C were unsuccessful. Exposure to a brief pulse of NO rendered the slices refractory to the induction of LTP for more than 2 h, while the expression of established LTP remained unaffected (Fig. 3A).
Figure 3. Brief exposure to NO has long-lasting consequences for both NMDA receptor-mediated transmission and the induction of LTP.

A, the percentage change in the slope of responses evoked by stimulation of two independent afferent pathways (P1, •; P2, ○) is displayed as a function of time. Mean data obtained from 5 slices are plotted, normalized to the mean of the control period prior to delivery of conditioning stimuli. The s.e.m. is plotted for every fifth mean. Slices were perfused with 500 μM caged NO throughout. After a 20 min control period a conditioning tetanus was applied to P1 (▴) followed 10 min later by a 1 ms pulse of near-UV light releasing 4.5 μM NO (arrow). Conditioning tetani ▵ applied to P2 at 10, 70 and 130 min after photolysis failed to induce LTP. B, the mean slope of NMDA receptor-mediated responses is plotted against time (pooled data from 4 slices) and the s.e.m. is plotted for every fifth mean. The slices were bathed with 400 μM caged NO throughout. A single 1 ms pulse of near-UV light (arrow) releasing 3.6 μM NO produced a marked and persistent decrease in the efficacy of NMDA receptor transmission. Bath application of 25 μM D-AP5 abolished the NMDA receptor-mediated potential (horizontal bar). The NMDA receptor-mediated potentials were isolated pharmacologically in nominally magnesium-free ACSF containing 10 μM CNQX. Experiments performed at 24 °C.
How does nitric oxide modulate the induction of LTP?
The induction of LTP at CA1 synapses is dependent upon the activation of NMDA receptors (Collingridge et al. 1983). It is possible that the selective impairment of induction reported here reflects an action of NO on NMDA receptor function. This suggestion is supported by reports that NO donors reduce the efficacy of NMDA receptor function (Lei et al. 1992; Manzoni et al. 1992; Lipton et al. 1993) and by our observation (Murphy et al. 1994) that photolytically released NO reduces NMDA receptor-mediated transmission at hippcampal synapses. In the latter study, the effects of NO on NMDA receptor-mediated transmission were followed for 50 min. In this study we have extended this period up to 180 min (see Fig. 3B). Exposure to 3.6 μM NO induced a 70.5 ± 9.0% (n = 4 slices) reduction in the initial slope of the NMDA receptor-mediated potential (measured 3 min after photolysis of caged NO) and persisted for the duration of the recording showing little signs of recovery (59.6 ± 7.3% and 54.7 ± 6.6% depression at 60 and 120 min, respectively). The long-term effects of NO on the induction of LTP correspond well with its effects on transmission at NMDA receptors. It is likely that failure to induce LTP is directly attributable to a decrease in the efficacy of transmission mediated by NMDA receptors. However, the LTP experiments, unlike the NMDA receptor experiments, were carried out in the presence of 2 mM extracellular magnesium ions. Additional experiments showed the action of NO at NMDA receptors not to be affected by the presence of 2 mM magnesium. The slope of NMDA receptor-mediated potentials recorded in disinhibited slices (20 μM bicuculline) and in the presence of 2 mM magnesium were reduced by 68.1 ± 3.6% (n = 6 inputs, 3 slices) after exposure to ∼4 μM photolytically released NO.
The time course of nitric oxide-induced depression at the NMDA receptor
Earlier studies employing pharmacological techniques to apply NO have lacked the necessary temporal precision to assay the early time course of NO-induced depression of NMDA receptor function. Flash photolysis overcomes these restrictions (see Methods), allowing the delivery of known concentrations of NO with millisecond accuracy. The time course of NO-induced depression of NMDA receptor-mediated transmission is shown in Fig. 4. Synaptic responses were evoked at 0.1 Hz and monitored for a period of 20 min to ensure stability. A pulse of near-UV light was then applied releasing 4.5 μM NO and the responses followed for a further 30 min. The effects of exposure to NO were not immediate; at 30 s the responses were reduced by only 11.0 ± 2.8%, at 60 s by 37.0 ± 8.0% and at 10 min by 81.2 ± 8.1% (n = 6 slices; means represent pooled data taken from single responses corresponding to the elapsed times indictated). Half-maximal depression was reached in 70.0 ± 3.4 s (n = 6 slices). In a further series of experiments, slices exposed to 2.7 μM NO exhibited a slower time course (time taken to reach half-maximal depression: 104.3 ± 5.3 s (n = 6)).
Figure 4. NO impairs the induction of LTP in a time-dependent manner.

A, the time course of the depressive action of photolytically released NO on NMDA receptor-mediated transmission is plotted against time. The plot is pooled data from six experiments and the s.e.m. is plotted for every fifth mean (responses evoked at 0.1 Hz). A single 1 ms pulse of near-UV light was applied at time zero (inverted arrow), releasing 4.5 μM NO. The inset shows the early effects of NO on NMDA receptor-mediated transmission on an expanded time base. The arrows indicate time zero, and elapsed times at 30 s and 60 s after photolysis. B, the mean slope of normal synaptic responses is plotted against time and the s.e.m. is plotted for every fifth mean. Each slice was exposed to 500 μM caged NO throughout. A tetanus was applied at the time indicated (▴). Slices were exposed to photolytically released NO (4.5 μM) either in conjunction with the first shock of the conditioning train (•, n = 5), or 30 s (○, n = 5), 60 s (▪, n = 6), or 10 min (□, n = 6) before tetanic stimulation. Experiments performed at 24 °C.
Nitric oxide impairment of LTP is time dependent
If modulation of NMDA receptors is the primary means by which NO impairs the induction of LTP, then the rate at which NO depresses NMDA receptor transmission should also determine the time dependency of the action of NO on the induction of LTP. NO photolytically released during a tetanus would be expected to have no effect on the magnitude of LTP induced, since the NO-mediated block of NMDA receptor function takes several seconds to develop. Conversely, if an interval of more than 30 s is allowed between the release of NO and the onset of the tetanus, a decrease in the efficacy of the conditioning tetanus should ensue, resulting in poor expression of LTP. Experiments designed to test the above prediction are shown in Fig. 4. Slices were bathed with caged NO and monitored for 30 min prior to the application of a conditioning tetanus. A photolytic pulse of near-UV light was applied either at the start of the conditionng tetanus, or 30 s, 60 s and 10 min before it. The outcome of a tetanus was determined by the interval between exposure to NO and the tetanus. Slices exposed to NO at the onset of the tetanus exhibited LTP (62.5 ± 21.7%; n = 5 slices) which was similar to that induced in a control set of slices not exposed to NO (64.8 ± 19.5% (n = 7 slices), measured 1 h after induction). Slices exposed to NO 30 s prior to conditioning also supported normal LTP; 54.7 ± 7.4% (n = 5). However, LTP was severely depressed in slices exposed to NO 60 s before application of the conditioning tetanus (11.0 ± 8.1% (n = 6)), and abolished when the interval was extended to 10 min (0.37 ± 2.5% (n = 6)).
DISCUSSION
Exposure of hippocampal neurones to NMDA can cause the release of NO into the extracellular medium, presumably via calcium activation of nitric oxide synthase (East & Garthwaite, 1991; Garthwaite, 1991; Luo & Vincent, 1994; Bhardwaj et al. 1995), suggesting a role for NO as an extracellular messenger in NMDA receptor-mediated synaptic plasticity (Schuman & Madison, 1991; Zhuo et al. 1993). It has not been unequivocally established that NO is released at biologically effective concentrations by synaptic activation of NMDA receptors, nor, if it is, whether it behaves as a retrograde messenger, acting on the presynaptic terminal (Meffert et al. 1994; Boulton et al. 1995; Arancio et al. 1996) or whether its action is restricted to the postsynaptic apparatus (Kato & Zorumski, 1993; Williams et al. 1993; Murphy et al. 1994). In this study, we have used a caged form of NO to examine in a time-resolved manner the effects of known concentrations of NO on NMDA receptor-mediated transmission, and on the induction and maintenance of LTP. However, a weakness of this approach is that we do not know the range of biologically effective concentrations achieved at active hippocampal synapses during the induction of LTP and consequently, the range of 1 to 5 μM NO used in this study may not be physiologically relevant. We show that NO depresses NMDA receptor-mediated responses with slow kinetics. Our main finding is that synapses exposed to NO undergo a persistent decrease in their abilility to sustain LTP; this effect has similar kinetics to the NO-induced depression of NMDA receptor-mediated responses, reaching a maximum 1-2 min after the release of NO. Our previous experiments failed to demonstrate a role for NO as a potentiating agent at CA1 synapses (but see Zhuo et al. 1993 for a contrary finding). Our present results suggest instead that NO may act to modulate the threshold for the induction of NMDA receptor-dependent LTP. On this view, NO produced during a brief tetanus will not affect the potentiation produced by that tetanus, but will render activated synapses refractory to further potentiation once the depressive action of NO at the NMDA receptors has become established. Note that any action of endogenous NO is likely to be confined to the immediate neighbourhood of the tetanized synapses, since in the two-pathway experiment illustrated in Fig. 1A, the induction of LTP in P1 did not influence the induction or magnitude of LTP in P2.
The proposed role for NO as a modulator of the threshold for LTP provides a possible explanation for certain forms of NMDA receptor-dependent metaplasticity at hippocampal synapses (Abraham & Bear, 1996). Prior activation of synaptic inputs, in a manner that allows NMDA receptors to participate in but not potentiate synaptic transmission, raises the threshold for the subsequent induction of LTP (Coan et al. 1989; Huang et al. 1992). Furthermore, the effect of this ‘priming’ stimulation is not immediate, taking several minutes to manifest itself (Christie et al. 1995). It is tempting to suggest that both the time course and change in inductive threshold associated with metaplasticity is attributable to the action of NO at the NMDA receptor. It is also possible that NO might determine the final magnitude of potentiation and the rate at which synapses become saturated for LTP during repeated periods of conditioning (Frey et al. 1995). Interestingly, Christie et al. (1995) reported that pharmacological blockade of L-type calcium channels enhanced LTP and slowed down the rate at which synapses became saturated. At the time they could not offer an explanation for such an unexpected result. However, recent evidence demonstrating that L-type channels play a critical role in NMDA receptor-mediated release of NO (Rodriguez-Alvarez et al. 1997) may provide an answer: bockade of L-type channels will reduce the production of NO, thereby preventing an NO-mediated decrease in NMDA receptor efficacy.
If NO is released during the induction of LTP, the predicted outcome would be a decrease in NMDA receptor-mediated transmission in parallel with an increase in the non-NMDA receptor-mediated response. Studies comparing the relative extent of potentiation in these two components have produced inconclusive results; ranging from no change in NMDA receptor transmission (Kauer et al. 1988; Perkel & Nicoll, 1993; Isaac et al. 1995; Liao et al. 1995), to a small (10-15%) (Muller et al. 1988; Muller & Lynch, 1990) or modest (20-30%) increase (Asztely et al. 1992; Muller et al. 1992) or an increase comparable with the non-NMDA receptor-mediated potential (Clark & Collingridge, 1995). However, in one study in which the expression of hippocampal NMDA receptors had been elevated, conditioning stimuli that successfully induced LTP in the non-NMDA receptor component produced a reproducible and persistent depression of ∼40% in the NMDA receptor-mediated response (Bernard & Wheal, 1995). Moreover, whilst potentiation of the non-NMDA receptor-mediated response was apparent immediately after conditioning, depression of the NMDA receptor-mediated potential took a minute or more to establish itself, resembling the slow onset of NO-induced depression at the NMDA receptor reported here.
Other evidence supporting a role for NMDA receptor-mediated NO release in the control of the induction of LTP comes from the observation that application of NMDA directly to slices immediately before tetanic conditioning can prevent the induction of LTP. Furthermore, this inhibitory action of NMDA is reversed by NOS antagonists and mimicked by the NO donor, nitroprusside (Izumi et al. 1992).
In contrast to our experiments, several laboratories have reported that exposure to exogenous NO donors reversibly depresses non-NMDA receptor-mediated synaptic transmission (Bohme et al. 1991; Bon et al. 1992; Boulton et al. 1994). In our experiments, brief exposure to photolytically released NO selectively and irreversibly depressed NMDA receptor-mediated transmission. The multiplicity of actions reported for NO is likely to reflect the redox species involved. Compounds that evolve the free radical (NO.) have very little effect on non-NMDA receptor function, but compounds releasing nitrosonium ions (NO+) reversibly depress synaptic transmission (Pan et al. 1996). We are confident that our caged compound releases the free radical NO., the form of NO produced by the enzymatic action of NOS (Garthwaite, 1991), on the basis of changes in the absorption spectrum of haemoglobin when exposed to photolytically released NO; these changes, reflecting the conversion of deoxyhaemoglobin to nitrosyl-haemoglobin, are stimulated by NO. but not by NO+ (Bettache et al. 1996). Moreover, photolysis of our caged NO affected NMDA but not non-NMDA receptor-mediated responses (see, for example, Fig. 3A and B) as predicted for the free radical (NO.) but not the nitrosonium (NO+) species (Pan et al. 1996).
The mechanism by which NO depresses NMDA receptor function is not known, though redox modification of thiol groups associated with the receptor has been strongly implicated (Moriyoshi et al. 1991; Lei et al. 1992; Manzoni et al. 1992; Lipton et al. 1993). Evidence that the redox state of NMDA receptors can influence synaptic plasticity is provided by the observation that reducing agents, which prevent redox modification of thiol groups, lower the threshold for the induction of LTP (Tauck & Ashbeck, 1990). Likewise, it has been reported that inhibition of NO production also facilitates the induction of LTP, possibly by reversing the oxidative effects of NO at the NMDA receptor (Kato & Zorumski, 1993). One result inconsistent with this interpretation has been reported: application of the oxidizing agent 5,5′-dithiobis-2-nitrobenzoic acid (DNTB), which depresses NMDA receptor function, has very little effect on the induction of LTP expressed by non-NMDA receptors (Bernard et al. 1995). A possible explanation for this difference is that the maximal reduction in the NMDA receptor-mediated potential is only 45%, which is sufficient in our experience to produce a significant, but not a complete block of LTP. Unlike NO, DNTB cannot readily pass through the plasma membrane, suggesting that a cytosolic target on the NMDA receptor may also be involved in the NO-mediated modulaton of NMDA receptor function.
Other possible mechanisms by which NO could reduce NMDA receptor-mediated potentials and block the induction of LTP is by modulation of metabotropic glutamate receptors or GABA-mediated inhibition. Intracerebral perfusion of rat brain with the NO donor Sin-1 causes an increase in extracellular levels of GABA in the hippocampus (Segovia et al. 1994). It is conceivable that an increase in GABA release after exposure to NO might account for some of the effects of NO on synaptic transmission reported here. However, we believe this to be unlikely as NO was still capable of markedly reducing (> 60%) NMDA receptor-mediated responses in slices disinhibited with the GABAA antagonist, bicuculline. Furthermore, the reported action of Sin-1 on extracellular GABA concentration may not have been attributable to a direct action of NO, but rather to the formation of peroxynitrite, a neurotoxic byproduct of Sin-1 decomposition.
Perhaps the most surprising finding reported here is the persistent (> 2 h) depression of NMDA receptor-mediated transmission produced by brief exposure to NO. It is the persistence of this depression that leads us to believe that NO may play a role in conferring a refractory period upon NMDA receptor-dependent plasticity, effectively preventing the further potentiation of recently conditioned synapses. It would be interesting to establish if the refractory period also extends to long-term depression and depotentiation. However, we cannot exclude the possibility that persistent suppression of LTP induction after exposure to NO may be the result of irreversible damage to the induction mechanism independently of the action of NO at the NMDA receptor.
The concentration range of NO delivered by flash photolysis in these experiments was 1-5 μM, which is comparable with that used to induce long-term depression at parallel fibre-Purkinje cell synapses in the cerebellum (Lev Ram et al. 1995). Nevertheless, the physiological relevance of our results will remain difficult to assess until it becomes possible to measure in situ the concentration of NO evolved during stimulus protocols which lead to persistent changes in synaptic efficacy.
References
- Abraham WC, Bear MF. Metaplasticity - the plasticity of synaptic plasticity. Trends in Neurosciences. 1996;19:126–130. doi: 10.1016/s0166-2236(96)80018-x. 10.1016/S0166-2236(96)80018-X. [DOI] [PubMed] [Google Scholar]
- Arancio O, Kiebler M, Lee CJ, Lev Ram V, Tsien R Y, Kandel E R, Hawkins RD. Nitric oxide acts directly in the presynaptic neuron to produce long-term potentiation in cultured hippocampal neurons. Cell. 1996;87:1025–1035. doi: 10.1016/s0092-8674(00)81797-3. [DOI] [PubMed] [Google Scholar]
- Asztely F, Wigstrom H, Gustafsson B. The relative contribution of NMDA receptor channels in the expression of long-term potentiation in the hippocampal CA1 region. European Journal of Neuroscience. 1992;4:681–690. doi: 10.1111/j.1460-9568.1992.tb00177.x. [DOI] [PubMed] [Google Scholar]
- Bannerman DM, Chapman PF, Kelly PA, Butcher SP, Morris RGM. Inhibition of nitric oxide synthase does not prevent the induction of long-term potentiation in vivo. Journal of Neuroscience. 1994;14:7415–7425. doi: 10.1523/JNEUROSCI.14-12-07415.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard C, Hirsch J, BenAri Y. Non-involvement of the redox site of NMDA receptors in bidirectional synaptic plasticity in the CA1 area of the rat hippocampus in vitro. Neuroscience Letters. 1995;193:197–200. doi: 10.1016/0304-3940(95)11702-x. [DOI] [PubMed] [Google Scholar]
- Bernard CL, Wheal HV. Simultaneous expression of long-term depression of NMDA and long-term potentiation of AMPA receptor-mediated synaptic responses in the CA1 area of the kainic acid-lesioned hippocampus. European Journal of Neuroscience. 1995;7:1651–1655. doi: 10.1111/j.1460-9568.1995.tb01160.x. [DOI] [PubMed] [Google Scholar]
- Bettache N, Carter T, Corrie JET, Ogden D, Trentham DR. Photolabile donors of nitric oxide: ruthenium nitrosyl chlorides as caged nitric oxide. Methods in Enzymology. 1996;268:266–281. doi: 10.1016/s0076-6879(96)68029-x. [DOI] [PubMed] [Google Scholar]
- Bhardwaj A, Northington FJ, Koehler RC, Stiefel T, Hanley DF, Traystman RJ. Adenosine modulates N-methyl-D-aspartate-stimulated hippocampal nitric oxide production in vivo. Stroke. 1995;26:1627–1633. doi: 10.1161/01.str.26.9.1627. [DOI] [PubMed] [Google Scholar]
- Bohme GA, Bon C, Stutzmann JM, Doble A, Blanchard JC. Possible involvement of nitric oxide in long-term potentiation. European Journal of Pharmacology. 1991;199:379–381. doi: 10.1016/0014-2999(91)90505-k. [DOI] [PubMed] [Google Scholar]
- Bon C, Bohme GA, Doble A, Stutzmann JM, Blanchard JC. A role for nitric oxide in long-term potentiation. European Journal of Neuroscience. 1992;4:420–424. doi: 10.1111/j.1460-9568.1992.tb00891.x. [DOI] [PubMed] [Google Scholar]
- Boulton CL, Irving AJ, Southam E, Potier B, Garthwaite J, Collingridge GL. The nitric oxide-cyclic GMP pathway and synaptic depression in rat hippocampal slices. European Journal of Neuroscience. 1994;6:1528–1535. doi: 10.1111/j.1460-9568.1994.tb00543.x. [DOI] [PubMed] [Google Scholar]
- Boulton CL, Southam E, Garthwaite J. Nitric oxide-dependent long-term potentiation is blocked by a specific inhibitor of soluble guanylyl cyclase. Neuroscience. 1995;69:699–703. doi: 10.1016/0306-4522(95)00349-n. [DOI] [PubMed] [Google Scholar]
- Christie BR, Stellwagen D, Abraham WC. Reduction of the threshold for long-term potentiation by prior theta-frequency synaptic activity. Hippocampus. 1995;5:52–59. doi: 10.1002/hipo.450050107. [DOI] [PubMed] [Google Scholar]
- Clark KA, Collingridge GL. Synaptic potentiation of dual-component excitatory postsynaptic currents in the rat hippocampus. The Journal of Physiology. 1995;482:39–52. doi: 10.1113/jphysiol.1995.sp020498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coan EJ, Irving AJ, Collingridge GL. Low-frequency activation of the NMDA receptor system can prevent the induction of LTP. Neuroscience Letters. 1989;105:205–210. doi: 10.1016/0304-3940(89)90038-4. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Kehl SJ, McLennan H. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. The Journal of Physiology. 1983;334:33–46. doi: 10.1113/jphysiol.1983.sp014478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings JA, Nicola SM, Malenka RC. Induction in the rat hippocampus of long-term potentiation (LTP) and long-term depression (LTD) in the presence of a nitric-oxide synthase inhibitor. Neuroscience Letters. 1994;176:110–114. doi: 10.1016/0304-3940(94)90883-4. [DOI] [PubMed] [Google Scholar]
- East SJ, Garthwaite J. NMDA receptor activation in rat hippocampus induces cyclic-GMP formation through the L-arginine nitric-oxide pathway. Neuroscience Letters. 1991;123:17–19. doi: 10.1016/0304-3940(91)90147-l. 10.1016/0304-3940(91)90147-L. [DOI] [PubMed] [Google Scholar]
- Frey U, Schollmeier K, Reymann KG, Seidenbecher T. Asymptotic hippocampal long-term potentiation in rats does not preclude additional potentiation at later phases. Neuroscience. 1995;67:799–807. doi: 10.1016/0306-4522(95)00117-2. 10.1016/0306-4522(95)00117-2. [DOI] [PubMed] [Google Scholar]
- Garthwaite J. Glutamate, nitric oxide and cell-cell signalling in the nervous system. Trends in Neurosciences. 1991;14:60–67. doi: 10.1016/0166-2236(91)90022-m. 10.1016/0166-2236(91)90022-M. [DOI] [PubMed] [Google Scholar]
- Haley JE, Wilcox GL, Chapman PF. The role of nitric oxide in hippocampal long-term potentiation. Neuron. 1992;8:211–216. doi: 10.1016/0896-6273(92)90288-o. [DOI] [PubMed] [Google Scholar]
- Huang YY, Colino A, Selig DK, Malenka RC. The influence of prior synaptic activity on the induction of long-term potentiation. Science. 1992;255:730–733. doi: 10.1126/science.1346729. [DOI] [PubMed] [Google Scholar]
- Isaac JT, Nicoll RA, Malenka RC. Evidence for silent synapses: implications for the expression of LTP. Neuron. 1995;15:427–434. doi: 10.1016/0896-6273(95)90046-2. 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Clifford DB, Zorumski CF. Inhibition of long-term potentiation by NMDA-mediated nitric oxide release. Science. 1992;257:1273–1276. doi: 10.1126/science.1519065. [DOI] [PubMed] [Google Scholar]
- Kato K, Zorumski CF. Nitric oxide inhibitors facilitate the induction of hippocampal long-term potentiation by modulating NMDA responses. Journal of Neurophysiology. 1993;70:1260–1263. doi: 10.1152/jn.1993.70.3.1260. [DOI] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC, Nicoll RA. A persistent postsynaptic modification mediates long-term potentiation in the hippocampus. Neuron. 1988;1:911–917. doi: 10.1016/0896-6273(88)90148-1. [DOI] [PubMed] [Google Scholar]
- Kirkwood A, Bear MF. Hebbian synapses in visual cortex. Journal of Neuroscience. 1994;14:1634–1645. doi: 10.1523/JNEUROSCI.14-03-01634.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei SZ, Pan ZH, Aggarwal SK, Chen HSV, Hartman J, Sucher NJ, Lipton SA. Effect of nitric oxide production on the redox modulatory site of the NMDA receptor-channel complex. Neuron. 1992;8:1087–1099. doi: 10.1016/0896-6273(92)90130-6. [DOI] [PubMed] [Google Scholar]
- van Ram V, Makings LR, Keitz PF, Kao JP, Tsien RY. Long-term depression in cerebellar Purkinje neurons results from coincidence of nitric oxide and depolarization-induced Ca2+ transients. Neuron. 1995;15:407–415. doi: 10.1016/0896-6273(95)90044-6. 10.1016/0896-6273(95)90044-6. [DOI] [PubMed] [Google Scholar]
- Liao DZ, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Choi YB, Pan ZH, Lei SZ, Chen HS, Sucher NJ, Loscalzo J, Singel DJ, Stamler JS. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature. 1993;364:626–632. doi: 10.1038/364626a0. 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- Luo D, Vincent SR. NMDA-dependent nitric oxide release in the hippocampus in vivo: interactions with noradrenaline. Neuropharmacology. 1994;33:1345–1350. doi: 10.1016/0028-3908(94)90035-3. 10.1016/0028-3908(94)90035-3. [DOI] [PubMed] [Google Scholar]
- Manzoni O, Prezeau L, Marin P, Deshager S, Bockaert J, Fagni L. Nitric oxide-induced blockade of NMDA receptors. Neuron. 1992;8:653–662. doi: 10.1016/0896-6273(92)90087-t. [DOI] [PubMed] [Google Scholar]
- Martin W, Villani GM, Jothianandan D, Furchgott RF. Selective blockade of endothelium-dependent and glyceryl trinitrate-induced relaxation by hemoglobin and by methylene blue in the rabbit aorta. Journal of Pharmacology and Experimental Therapeutics. 1985;232:708–716. [PubMed] [Google Scholar]
- Meffert MK, Premack BA, Schulman H. Nitric oxide stimulates Ca2+-independent synaptic vesicle release. Neuron. 1994;12:1235–1244. doi: 10.1016/0896-6273(94)90440-5. 10.1016/0896-6273(94)90440-5. [DOI] [PubMed] [Google Scholar]
- Moriyoshi K, Masu M, Ishii T, Shigemoto R, Mizuno N, Nakanishi S. Molecular cloning and characterization of the rat NMDA receptor. Nature. 1991;354:31–36. doi: 10.1038/354031a0. 10.1038/354031a0. [DOI] [PubMed] [Google Scholar]
- Muller D, Arai A, Lynch G. Factors governing the potentiation of NMDA receptor-mediated responses in hippocampus. Hippocampus. 1992;2:29–38. doi: 10.1002/hipo.450020105. [DOI] [PubMed] [Google Scholar]
- Muller D, Joly M, Lynch G. Contributions of quisqualate and NMDA receptors to the induction and expression of LTP. Science. 1988;242:1694–1697. doi: 10.1126/science.2904701. [DOI] [PubMed] [Google Scholar]
- Muller D, Lynch G. Synaptic modulation of N-methyl-D-aspartate receptor mediated responses in hippocampus. Synapse. 1990;5:94–103. doi: 10.1002/syn.890050203. [DOI] [PubMed] [Google Scholar]
- Murphy KPSJ, Bliss TVP. Photolytic release of nitric oxide blocks induction of LTP by depression of NMDA receptor-mediated transmission in the CA1 region of rat hippocampus. Society for Neuroscience Abstracts. 1995;21:1809. [Google Scholar]
- Murphy KPSJ, Reid GP, Trentham DR, Bliss TVP. Activation of NMDA receptors is necessary for the induction of associative long-term potentiation in area CA1 of the rat hippocampal slice. The Journal of Physiology. 1997;504:379–385. doi: 10.1111/j.1469-7793.1997.379be.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy KPSJ, Williams JH, Bettache N, Bliss TVP. Photolytic release of nitric oxide modulates NMDA receptor-mediated transmission but does not induce long-term potentiation at hippocampal synapses. Neuropharmacology. 1994;33:1375–1385. doi: 10.1016/0028-3908(94)90039-6. 10.1016/0028-3908(94)90039-6. [DOI] [PubMed] [Google Scholar]
- O'Dell TJ, Hawkins RD, Kandel ER, Arancio O. Tests of the roles of two diffusible substances in long-term potentiation: evidence for nitric oxide as a possible early retrograde messenger. Proceedings of the National Academy of Sciences of the USA. 1991;88:11285–11289. doi: 10.1073/pnas.88.24.11285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden DC, Capiod T, Walker JW, Trentham DR. Kinetics of the conductance evoked by noradrenaline, inositol trisphosphate or Ca2+ in guinea-pig isolated hepatocytes. The Journal of Physiology. 1990;422:585–602. doi: 10.1113/jphysiol.1990.sp018002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan ZH, Segal MM, Lipton SA. Nitric oxide-related species inhibit evoked neurotransmission but enhance spontaneous miniature synaptic currents in central neuronal cultures. Proceedings of the National Academy of Sciences of the USA. 1996;93:15423–15428. doi: 10.1073/pnas.93.26.15423. 10.1073/pnas.93.26.15423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkel DJ, Nicoll RA. Evidence for all-or-none regulation of neurotransmitter release: Implications for long-term potentiation. The Journal of Physiology. 1993;471:481–500. doi: 10.1113/jphysiol.1993.sp019911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Alvarez J, Lafon-Cazal M, Blanco I, Bockaert J. Different routes of Ca2+ influx in NMDA-mediated generation of nitric oxide and arachidonic acid. European Journal of Neuroscience. 1997;9:867–870. doi: 10.1111/j.1460-9568.1997.tb01437.x. [DOI] [PubMed] [Google Scholar]
- Schuman EM, Madison DV. A requirement for the intercellular messenger nitric oxide in long-term potentiation. Science. 1991;254:1503–1506. doi: 10.1126/science.1720572. [DOI] [PubMed] [Google Scholar]
- Segovia G, Porras A, Mora F. Effects of nitric oxide donor on glutamate and GABA release in the striatum and hippocampus of conscious rat. NeuroReport. 1994;5:1937–1940. doi: 10.1097/00001756-199410000-00024. [DOI] [PubMed] [Google Scholar]
- Tauck DL, Ashbeck GA. Glycine synergistically potentiates the enhancement of LTP induced by a sulfhydryl reducing agent. Brain Research. 1990;519:129–132. doi: 10.1016/0006-8993(90)90070-r. 10.1016/0006-8993(90)90070-R. [DOI] [PubMed] [Google Scholar]
- Williams JH, Li YG, Nayak A, Errington ML, Murphy KPSJ, Bliss TVP. The suppression of long-term potentiation in rat hippocampus by inhibitors of nitric oxide synthase is temperature and age dependent. Neuron. 1993;11:877–884. doi: 10.1016/0896-6273(93)90117-a. 10.1016/0896-6273(93)90117-A. [DOI] [PubMed] [Google Scholar]
- Zhuo M, Small SA, Kandel ER, Hawkins RD. Nitric oxide and carbon monoxide produce activity-dependent long-term synaptic enhancement in hippocampus. Science. 1993;260:1946–1950. doi: 10.1126/science.8100368. [DOI] [PubMed] [Google Scholar]