

Abstract
Paramyotonia congenita is a temperature-sensitive skeletal muscle disorder caused by missense mutations that occur in the adult skeletal muscle voltage-gated sodium channel. We report here the identification of a new genetic mutation in a family with the paramyotonia congenita phenotype.
Single-strand conformation polymorphism analysis and DNA sequencing showed that the defect was linked to a single nucleotide substitution causing an amino acid change from an arginine to a serine at position 1448 in the human sodium channel α-subunit.
Expression of the altered protein in human embryonic kidney (HEK) 293 cells revealed several defects in channel function: (i) the rate of fast inactivation was slower in the mutant channel compared with wild-type, (ii) steady-state fast inactivation was shifted towards hyperpolarizing potentials, (iii) the R1448S channels deactivated much more slowly, and (iv) the mutant channels recovered from the fast inactivated state more rapidly.
By contrast, the activation curve, steady-state slow inactivation and the rate of onset and recovery from slow inactivation were not altered by the R1448S mutation.
These data show that the defects observed in the sodium channel function could well explain the onset of the paramyotonia congenita in this family and emphasize the role of segment S4 of domain IV in sodium channel inactivation.
Voltage-gated sodium channels are prominent transmembrane proteins in excitable tissues and are responsible for the ascending phase of the action potential. The channel molecule consists of four homologous domains, and each domain possesses six hydrophobic putative transmembrane segments (S1-S6) (Noda et al. 1984). Positively charged arginine and lysine residues embedded within the S4 segments presumably confer voltage sensitivity to the channel. Five muscle diseases are linked to human skeletal and cardiac muscle sodium channels: hyperkalaemic periodic paralysis (HyperKPP) (Fontaine et al. 1990; Ptácek et al. 1991a), paramyotonia congenita (PC) (Ptácek et al. 1991b), potassium-aggravated myotonia (PAM) (Lerche et al. 1993; Ptácek et al. 1994), long QT syndrome (Wang et al. 1995b) and Brugada's syndrome (Chen et al. 1998).
HyperKPP, PC and PAM are autosomal dominant disorders caused by single amino acid mutations occurring in the voltage-gated skeletal muscle sodium channel (hSkM1) (for review see Ptácek & Griggs, 1996; Bulman, 1997). Eight amino acid substitutions have been identified to date in families with the PC phenotype. Substitutions of arginine 1448, located at the outermost portion of the S4 segment of domain IV, have been described in several families with PC (Ptácek et al. 1992; Wang et al. 1995a). This positively charged residue was naturally mutated to proline, cysteine, or histidine, causing a decreasing degree of severity of the disease from proline to histidine, respectively (Wang et al. 1995a). In expression studies, channels with these mutations exhibited a slow rate of current inactivation (Chahine et al. 1994; Yang et al. 1994), a shift in the steady-state inactivation curve (Chahine et al. 1994; Yang et al. 1994; Richmond et al. 1997a; Featherstone et al. 1998), a slower deactivation rate (Ji et al. 1996; Featherstone et al. 1998), and a faster recovery from fast inactivation (Chahine et al. 1994; Yang et al. 1994; Ji et al. 1996; Richmond et al. 1997a; Featherstone et al. 1998).
In the present study, we report a new mutation in a previously unstudied family with the PC phenotype and carrying a nucleotide substitution at position C4419A of the human skeletal muscle sodium channel gene SCN4A (George et al. 1992), predicting an amino acid mutation of arginine 1448 to serine. This mutation is of interest because R1448 is located in segment S4 of domain IV in the human skeletal muscle channel α-subunit, a region of the channel that has been shown to play a crucial role in the coupling of activation and inactivation (Chahine et al. 1994). The arginine to serine substitution is a moderate amino acid change described in PC families at position 1448, and therefore, compared with the other substitutions (proline or cysteine) occurring at the same position, the serine substitution may provide additional insights into the disease onset and severity.
METHODS
Clinical evaluation
Both the proband (III-2) and his father (II-2) (Fig. 1A) were evaluated by one of the authors (H. K.). Routine phlebotomy was performed from the right or left anticubital vein. Clinical studies and sampling of family members were conducted after subjects signed a consent form approved by the Institutional Review Board of the University of Utah Health Sciences Center. Evaluation included general medical and neurological examinations. The latter were performed with particular attention to the muscle exam. The amplitude of the evoked compound muscle action potential (CMAP) was recorded from abductor digiti minimi after stimulation of the ulnar nerve in the wrist. The effects of cooling and short exercise tests on the CMAP amplitude were also studied (Streib, 1987; Jackson et al. 1994). The hand and forearm were cooled to 20°C by immersion in a 16°C water bath. Provocative tests in the warm (physical exercise, potassium load) were also performed, with clinical evaluation only.
Figure 1. A SCN4A missense mutation co-segregates with PC.
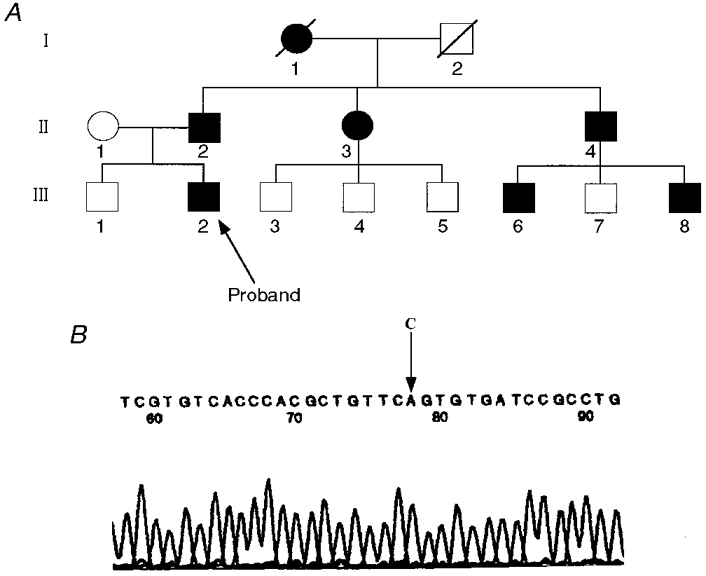
A, pedigree structure of kindred 3403. Affected individuals are represented by filled circles (females) and filled squares (males). Open symbols represent unaffected individuals. B, chromatograph and the corresponding DNA sequence of the SCN4A mutation associated with PC in family K3403. DNA sequence analysis demonstrates a transition from C in the normal DNA to an A in the DNA of the affected individuals at nucleotide 4419, which causes the R1448S substitution in the S4 segment of domain IV of the human skeletal muscle sodium channel. Only the sequence from the aberrant bands is shown.
PCR amplification, single-strand conformation polymorphism analysis and sequencing
DNA was obtained directly from peripheral blood by phenol- chloroform extraction. DNA samples were amplified for single-strand conformation polymorphism (SSCP) analysis using PCR as described previously (Ptácek et al. 1992).
Cell culture and transfection
Human embryonic kidney (HEK) 293 cells (ATCC, USA) were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco BRL) supplemented with 100 u ml−1 penicillin, 100 μg ml−1 streptomycin and 10 % fetal calf serum (Gibco BRL), and maintained at 37°C with 5 % CO2. The calcium-phosphate precipitation technique (Graham & Van Der Eb, 1973) was employed for transfection. Approximately 15 h after transfection, the medium was changed. Stably expressing cell lines were produced as described previously (Bendahhou et al. 1995) using the expression vector pRc-CMV with an insert encoding either wild-type (WT) hSkM1 or the corresponding R1448S mutant construct.
Construction of hSkM1-R1448S
Arginine 1448 was altered by the megaprimer PCR method of site-directed mutagenesis (Sarkar & Sommer, 1990). The reaction was primed using oligonucleotides 5′-GGCTCAATGTCAAGGTCAAC-3′ (forward) and 5′-GGATCACACTGAACAGCGT-3′ (reverse). The second megaprimer set was generated using oligonucleotides 5′-CACGCTGTTCAGTGTGATCC-3′ (forward) and 5′-TGGGCAAGTCCAGTGTGATG-3′ (reverse). The primer contained the nucleotide substitution 4419 C→A (indicated above in bold). The protocol for the first round of PCR was: 2 min at 94°C, one cycle; 20 s at 94°C, 20 s at 50°C, 1 min at 75°C, 30 cycles; 5 min at 72°C, one cycle.
The second round of PCR was primed using first round products (megaprimers) and the primers 5′-GGCTCAATGTCAAGGTCAAC-3′ (forward) and 5′-TGGGCAAGTCCAGTGTGATG-3′ (reverse). The following protocol was applied: 2 min at 94°C, one cycle; 20 s at 94°C, 20 s at 54°C, 90 s at 72°C, 30 cycles; 5 min at 72°C, one cycle. A 1·3 kb product was isolated and ligated to an appropriately cut fragment of WT hSkM1 to construct the full length mutated channel hSkM1-R1448S. The mutant construct was sequenced to ensure that the mutation was present and that the integrity of the cDNA in the region encompassing the mutation was preserved.
Electrophysiology
Recordings were conducted at room temperature (22°C) in the whole-cell configuration (Hamill et al. 1981) and as described previously (Bendahhou et al. 1997), using an Axopatch 200B amplifier (Axon Instruments Inc.) and an EPC-9 amplifier. Data acquisition and analysis were performed with pCLAMP 6 (Axon Instruments Inc.) or Pulse and Pulsefit (HEKA Electronic, Germany), and Igor (Wavemetric Inc.) software.
Twenty-four hours after plating stable lines or 36 h after transfection, the medium was replaced with bathing solution comprising (mM): 140 NaCl, 4 MgCl2, 2 CaCl2, and 10 Na-Hepes, pH 7·3. The internal pipette solution comprised (mM): 130 CsCl, 4 MgCl2, 2·5 EGTA, 5 NaCl, and 10 Hepes, pH 7·3.
Values represent means ±s.e.m.; n, number of cells tested. Statistical data were obtained using Student's t test.
RESULTS
Evaluation of patient
The proband presented at age 15 years with the complaint of ‘muscle stiffness’. He was otherwise healthy. The stiffness was not worsened by any particular food or by cold temperature. The patient has had rare attacks of paralysis, following vigorous exercise. Examination of muscle was only remarkable for action myotonia and percussion myotonia. Electromyographic examination showed diffuse myotonia which was worsened by muscle cooling. The CMAPs were normal as were nerve conduction velocities when studied at room temperature. However, with the routine muscle cooling and exercise test, the CMAP amplitude decreased by 40 % (data not shown). Both patients experienced several episodes of weakness, usually early in the day. An oral potassium load did not induce weakness.
Identification of nucleotide alterations in PC patient DNA
DNA from the proband was amplified using primers from the first part of exon 24. An aberrant SSCP conformer was noted in this patient. The aberrant pattern in this patient was not seen in 106 unrelated, normal individuals whose DNA had been evaluated by SSCP (Ptácek et al. 1992). Sequence analysis of the aberrant conformer revealed a C4419A mutation (Fig. 1B) of the gene encoding hSkM1 that predicts an R1448S change in the sodium channel α-subunit.
Kinetics of fast inactivation
Whole-cell current traces, obtained during voltage clamp experiments, from R1448S mutant channels exhibited altered inactivation compared with WT (Fig. 2A-C). The kinetics of fast inactivation were assessed by fitting the trace after the peak current with a single-exponential function. Figure 2D shows that mutant channel currents decayed more slowly than the WT channel currents over the voltage range tested. This behaviour is similar to that reported previously for R1448C, R1448H and R1448P mutant channels (Chahine et al. 1994; Yang et al. 1994; Richmond et al. 1997a; Featherstone et al. 1998).
Figure 2. Effect of R1448S substitution on fast inactivation.
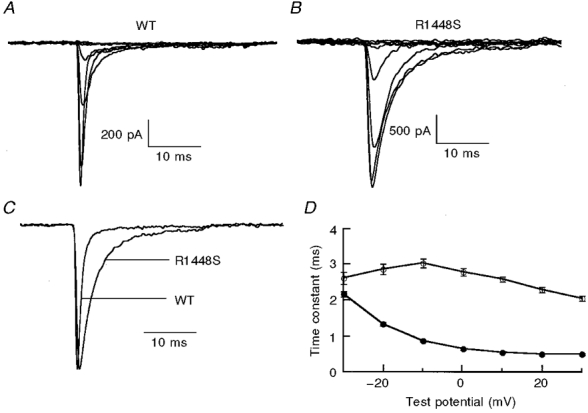
A and B, sodium current traces recorded in the whole-cell configuration for WT channels (A) and R1448S mutant channels (B). Cells were held at -120 mV, then depolarized to a test pulse ranging from -80 to 0 mV in 10 mV increments for 25 ms. In C, two normalized traces (at -10 mV) from WT and R1448S are shown to better visualize the effect of the mutation on inactivation decay. D, time constant of fast inactivation. Fast inactivation phase decays for WT (•, n= 39) and R1448S (○, n= 27) were fitted with a single-exponential function for voltages from -30 to +30 mV, for a -120 mV holding potential.
Steady-state inactivation and activation
The mid-point of steady-state fast inactivation was significantly (P < 0·001) shifted towards more hyperpolarizing potentials for the mutant channels (Fig. 3B) (WT: V0·5= -62·5 ± 4·9 mV, n= 43; R1448S: V0·5= -70·5 ± 7·5 mV, n= 21). This shift in the mid-point of fast inactivation was accompanied by a significant (P < 0·001) change in the slope (WT: z= -3·9 ± 0·7, n= 26; R1448S: z= -2·3 ± 0·5, n= 27). By contrast to steady-state inactivation, the current-voltage relationship and the activation curve that was derived from it showed no significant (P= 0·8) change between WT and mutant channels (Fig. 3A and B) (WT: V0·5= -17·8 ± 3·0 mV, n= 44; R1448S: V0·5= -18·6 ± 3·4 mV, n= 30).
Figure 3. Effect of R1448S on steady-state activation and fast inactivation.
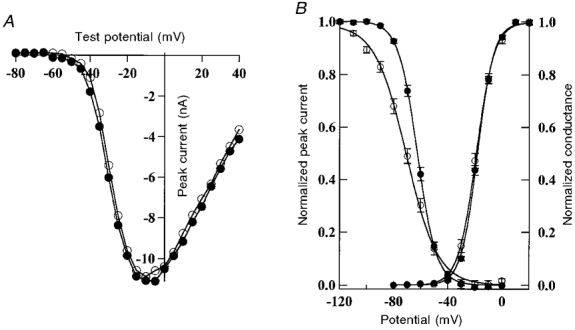
A, current-voltage relationships for a cell held at -120 mV, then depolarized to a test pulse ranging from -80 to 40 mV for WT (•) and R1448S mutant (○) channels. B, steady-state activation and inactivation. Steady-state fast inactivation for WT and R1448S: cells were held at prepulse potentials from -120 to 20 mV for 200 ms, and were then subjected to a 0 mV test pulse for 25 ms. Normalized peak currents were plotted versus prepulse potentials, and curves were fitted by a Boltzmann function: I/Imax= (1 + exp [ze(V - V0·5)/kT])−1, where Imax is the current recorded at the most hyperpolarizing potential (-120 mV), V0·5 is the potential for half-maximal inactivation, k is the Boltzmann constant, z is the apparent gating charge, T is absolute temperature, and kT/e= 25 mV at 22 °C. Steady-state activation: peak sodium conductance (GNa) was measured during a 25 ms test pulse to various test potentials from a holding voltage of -120 mV. GNa was calculated from GNa=INa/(E - Erev), where INa is the peak sodium current during the test depolarization (E) and Erev is the sodium current reversal potential. Data were normalized to maximum peak conductance (Gmax) and fitted with a two-state Boltzmann distribution: GNa/Gmax= (1 + exp[ze(V - V0·5)/kT])−1, where V0·5 is the test potential for half-maximal activation and k, T, z and e are the same as described above. Each value represents the mean ±s.e.m.
R1448S deactivation
A sodium channel defect in deactivation was suggested to explain the muscle hyperexcitability in PC for both the R1448C and the R1448P mutations (Featherstone et al. 1998). We assayed the effect of the R1448S mutation on deactivation by measuring the tail current at a range of potentials after briefly activating the channels at 40 mV. Our data show a clear defect in the deactivation of mutant channels (Fig. 4). Indeed, R1448S tail currents decayed more slowly at potentials ranging from -80 to 0 mV, suggesting that, in addition to slowed fast inactivation, deactivation is also slowed. This result corroborates findings reported earlier for other amino acid substitutions at the same position (Ji et al. 1996; Featherstone et al. 1998).
Figure 4. Deactivation and fast inactivation.
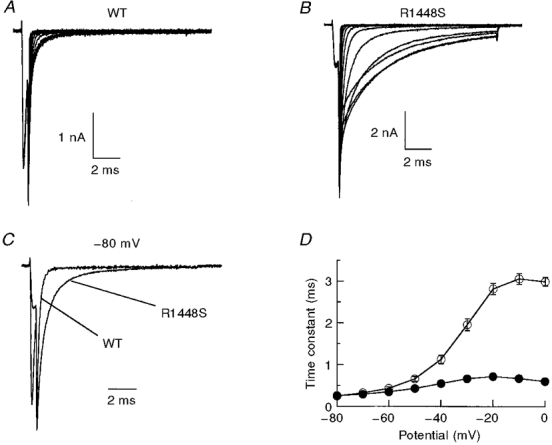
In A and B, traces show tail currents recorded in HEK 293 cells for WT channels (A) and R1448S mutant channels (B). Current was measured using the following pulse protocol: cells were held at -120 mV, depolarized to +40 mV for 0·5 ms, then different voltage steps were applied (-120 to 0 mV). C, normalized tail current traces recorded at -80 mV showing a slower rate of deactivation for the R1448S mutant channels than for the WT channels. D, tail currents were fitted with a single-exponential function and plotted versus voltage steps for WT (•, n= 20) and R1448S (○, n= 25).
Development of and recovery from fast inactivation
Kuo & Bean (1994) have proposed that recovery from fast inactivation and channel deactivation are coupled. Because R1448S channels deactivate more slowly than WT channels, we expected that recovery from inactivation would also be slower for the R1448S channels. We therefore examined the kinetics of recovery from fast inactivation and found that this was not the case. Figure 5 shows that the mutant channels recovered faster from fast inactivation than the WT channels at all voltages tested (from -140 to -70 mV). We also examined the rate of development of fast inactivation at voltages ranging from -80 to -50 mV. Inactivation developed much more rapidly for R1448S than for WT channels at these voltages (Fig. 5). These results suggest that transitions between the closed states and inactivated states are more favourable for R1448S channels than for WT channels.
Figure 5. Development of and recovery from fast inactivation.
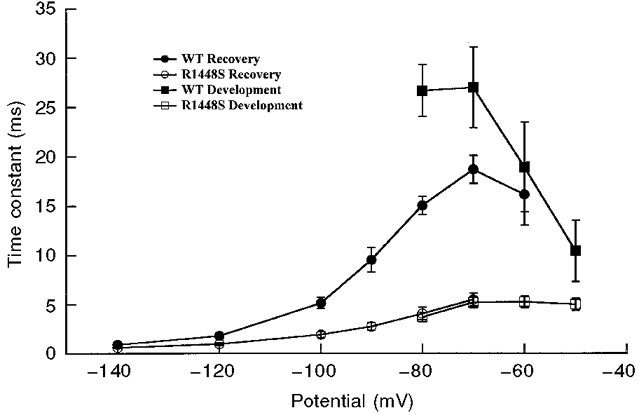
Development of fast inactivation and recovery from fast inactivation. The time constants for development of fast inactivation (squares) and recovery from fast inactivation (circles) are plotted as a function of voltage. Development of inactivation: cells were held at -100 mV, followed by a prepulse ranging from -80 to -50 mV for increasing durations, then stepped to -20 mV to determine the fraction of current inactivated during the prepulse for WT (▪) and R1448S (□). Recovery from fast inactivation: cells were prepulsed to -20 mV for 20 ms to inactivate all of the current, then brought back to potentials from -140 to -60 mV for increasing recovery durations prior to the pulse to -20 mV to assay the fraction of current recovered, for WT (•) and R1448S (○). The time constants were determined by fitting a single-exponential function to plots of peak amplitude during the test pulse versus prepulse duration. The maximum pulse rate was 0·5 Hz.
Slow inactivation
Although defective fast inactivation could underlie hyperexcitability and contribute to myotonia, impaired slow inactivation could also alter excitability. Therefore, we investigated the slow inactivation properties of both the WT and R1448S mutant channels. Figure 6 shows that there was no significant (P= 0·35) change in steady-state slow inactivation (WT: V0·5= -63·8 ± 9·9 mV, n= 11; R1448S: V0·5= -60·2 ± 8·0 mV, n= 8), nor was there any change in the rate of recovery from slow inactivation. Our data indicate that slow inactivation does not contribute to a channel malfunction with the R1448S mutation.
Figure 6. hSkM1 WT and R1448S mutant channel slow inactivation.
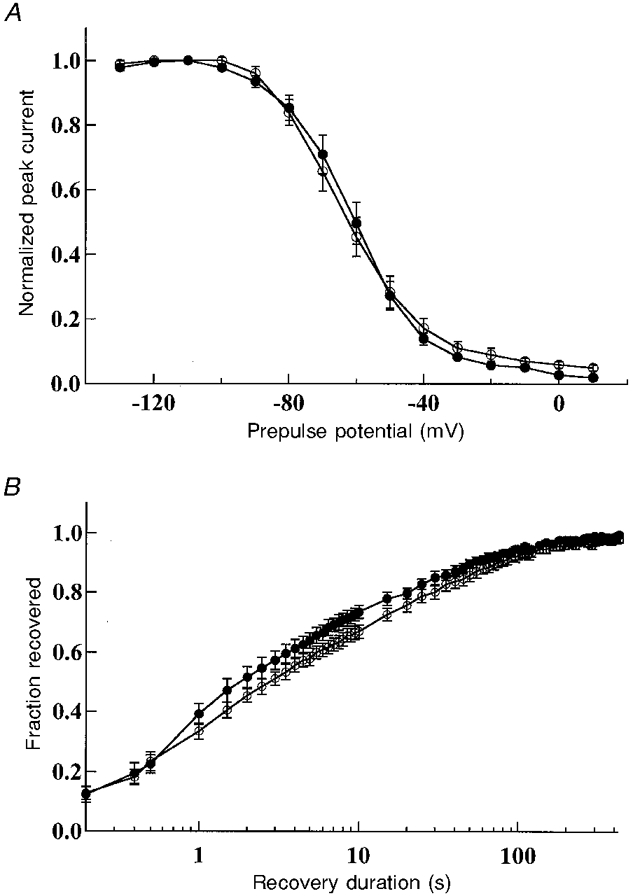
A, steady-state slow inactivation. Cells were held at potentials starting from -130 mV and increasing to 10 mV in 10 mV steps. After 50 s at each holding potential, a 30 ms recovery pulse to -100 mV and a 20 ms test pulse to -10 mV were given before the holding potential was incremented. The peak current elicited by the test pulse to -10 mV is plotted as a fraction of the maximum current for WT channels (•, n= 11) and R1448S channels (○, n= 8). B, slow inactivation recovery. The pulse protocol used has been described previously (Cummins & Sigworth, 1996). Data are means ±s.e.m. (WT: •, n= 9; R1448S: ○, n= 8).
DISCUSSION
Three substitutions of amino acid 1448, located in segment S4 in domain IV, have been described to occur, changing the arginine to proline, cysteine or histidine. These substitutions affected patient phenotype in a distinct manner. Here we have identified a new substitution occurring in a family with the PC phenotype involving a new amino acid mutation from an arginine to a serine at position 1448 in the human skeletal muscle sodium channel α-subunit. Although there was no complaint of clinical worsening with cooling, these patients demonstrated a cold-sensitive phenotype manifested by a decrement of the CMAP with exercise and muscle cooling. Patients with the R1448P mutation have a severe phenotype in which the myotonia interferes with ambulation (Wang et al. 1995b). Patients with the R1448C mutation have some disability from stiffness and frequent attacks of weakness (Ptácek et al. 1992). In the family studied here, stiffness is annoying but not debilitating and attacks of weakness are rare.
The introduction of a serine residue instead of an arginine greatly slowed sodium channel inactivation, which can result in a prolonged sodium influx. A similar effect has also been reported for most of the disease-causing sodium channel mutations (PC or HyperKPP). It was also shown to occur in patches from muscle biopsy of PC patients carrying the R1448P mutation (Lerche et al. 1996). This effect could in itself explain the occurrence of myotonia in the affected families. We further investigated the influence of this substitution on other channel parameters. Steady-state fast inactivation was shifted towards negative potentials, again strongly suggesting that the arginine at position 1448 is located in a critical region that governs channel inactivation. Despite the charge neutralization (due to the substitution) and despite the critical location of the arginine in the voltage sensor S4 segment of domain IV, the activation curve was not affected when compared with that of WT channels, nor was there any change in the slope of the curve. As shown for other mutations located at the same position, inactivation parameters were primarily affected. This agrees with the role of segment S4 of domain IV in sodium channel inactivation.
It has been shown that channel deactivation is also altered in PC mutants in general and in two of the three substitutions previously reported at arginine 1448 (Featherstone et al. 1998), and it was proposed that this may, in part, account for the onset of the disease. Our study shows that introduction of a serine residue at this position also slowed the rate of channel deactivation. Since recovery from inactivation and channel deactivation are postulated to be coupled (Kuo & Bean, 1994), and since the kinetics of recovery from inactivation are important for a cell to fire action potentials, we investigated how altered deactivation in R1448S may affect development and recovery from inactivation. The mutant channels in our expression system exhibited a faster rate of onset and recovery from fast inactivation compared with WT channels. This behaviour was observed among PC mutants expressed in heterologous systems in which a faster recovery from fast inactivation was reported, by contrast to a study carried out with biopsied muscle of PC patients (Lerche et al. 1996) where a slightly slower recovery from fast inactivation was observed. A possible reason for this discrepancy could be the expression background. In the studies of Lerche et al. (1996), channels were studied in vitro but from the native tissue. In contrast, the expression studies reported here were performed in a non-native cell line that normally has a low level of sodium channel expression. The channels may exhibit different properties from one expression system to another. Our data show a faster recovery from inactivation for R1448S, a behaviour similar to that reported for the R1448P, C or H mutations in expression studies. However, our data and those from expression studies do not agree with the Kuo & Bean (1994) model which predicts that the rate of recovery from inactivation should follow the kinetics of deactivation.
Slow inactivation has been shown to be impaired in HyperKPP mutant channels (Cummins & Sigworth, 1996; Hayward et al. 1997) but was not altered in PC mutant channels (Richmond et al. 1997a, b; Hayward et al. 1997). Our study shows that steady-state slow inactivation was not altered in the R1448S channels and that mutant channels recovered from slow inactivation with a rate similar to that of WT. Defective slow inactivation may be a hallmark of the HyperKPP disease.
The alterations in sodium channel gating observed with the R1448S mutation were similar to those described for the other three substitutions at the same amino acid position. All previous studies and the present study were primarily conducted at room temperature (22°C). Chahine et al. (1994) have shown that the rate of fast inactivation is only slightly more temperature dependent in the R1448C mutation than in WT, and found no difference in the temperature sensitivity of fast inactivation with the R1448H mutation. Featherstone et al. (1998) proposed that the effect of temperature on excitability in PC is simply a result of the normal slowing of channel kinetics with cooling, and is not the consequence of any alteration in the temperature sensitivity of sodium channel properties. It should be noted, however, that the majority of data on PC mutant channels has been collected at room temperature, and the few studies that have examined the effects of temperature have focused primarily on the temperature sensitivity of open-state fast inactivation. Therefore it is possible that the PC mutations alter the temperature dependence of other kinetic processes, such as deactivation, which may contribute to the cold-induced hyperexcitability in PC patients. To definitively address this question it may be necessary to examine the temperature sensitivity of all the sodium channel properties at clinically relevant temperatures.
In conclusion, we have identified a new mutation, R1448S, associated with PC, and we have shown that this mutation predominantly alters fast inactivation. Patients with a proline, cysteine or histidine substitution have different degrees of clinical severity of the disease. When proline and cysteine substitutions were compared in oocytes, Featherstone et al. (1998) noticed that the only major difference between the two mutant channels resided in the rate of deactivation, thus suggesting that a slower rate of deactivation may underlie the cold sensitivity in PC patients. Without any direct comparison, it would be difficult to distinguish between the subtle differences in channels carrying proline, cysteine, histidine or serine substitutions. However, it is interesting to note that the shift in the steady-state inactivation curve (compared with WT) is larger for the proline (-15 mV; Featherstone et al. 1998) and cysteine (-16 mV; Yang et al. 1994) substitutions than for the histidine (-7 mV; Yang et al. 1994) and serine (-8 mV; this study) substitutions. In the absence of any direct comparison and based on available data, we hypothesize that a moderate amino acid (not charged or not an α-helix breaker) at position 1448 may lead to a moderate disease phenotype.
Acknowledgments
The authors would like to thank Dr David Featherstone for critical reading of the manuscript and for providing macros for data analysis. We also thank the DNA Sequencing Facility at the University of Utah, supported in part by NCI grant no. 5P30CA42014. This work was supported by a Muscular Dystrophy Association grant (L. J. P.) and by the Paralysed Veterans of America and Eastern Paralysed Veterans Association.
References
- Bendahhou S, Cummins TR, Agnew WS. Mechanism of modulation of the voltage-gated skeletal and cardiac muscle sodium channels by fatty acids. American Journal of Physiology. 1997;272:C592–600. doi: 10.1152/ajpcell.1997.272.2.C592. [DOI] [PubMed] [Google Scholar]
- Bendahhou S, Cummins TR, Potts JF, Tong J, Agnew WS. Serine-1321-independent regulation of the μ1 adult skeletal muscle Na+ channel by protein kinase C. Proceedings of the National Academy of Sciences of the USA. 1995;92:12003–12007. doi: 10.1073/pnas.92.26.12003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulman DE. Phenotype variation and newcomers in ion channel disorders. Human Molecular Genetics. 1997;10:1679–1685. doi: 10.1093/hmg/6.10.1679. [DOI] [PubMed] [Google Scholar]
- Chahine M, George AL, Jr, Zhou M, Ji S, Sun WJ, Barchi RL, Horn R. Sodium channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron. 1994;12:281–294. doi: 10.1016/0896-6273(94)90271-2. [DOI] [PubMed] [Google Scholar]
- Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G, Ortiz-Lopez R, Wang Z, Antzelevitch C, O'Brien RE, Schulze-Bahr E, Keating MT, Towbin JA, Wang Q. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–296. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- Cummins TR, Sigworth FJ. Impaired slow inactivation in mutant sodium channels. Biophysical Journal. 1996;71:227–236. doi: 10.1016/S0006-3495(96)79219-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Featherstone DE, Fujimoto E, Ruben PC. A defect in skeletal muscle sodium channel deactivation exacerbates hyperexcitability in human paramyotonia congenita. The Journal of Physiology. 1998;506:627–638. doi: 10.1111/j.1469-7793.1998.627bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine B, Khurana TS, Hoffman EP, Bruns GAP, Haines JL, Trofatter JA, Hanson MP, Rich J, McFarlane H, Yasek DM, Romano D, Gusella JF, Brown RH. Hyperkalemic periodic paralysis and the adult muscle sodium channel α-subunit gene. Science. 1990;250:1000–1002. doi: 10.1126/science.2173143. [DOI] [PubMed] [Google Scholar]
- George AL, Jr, Komisarof J, Kallen RG, Barchi RL. Primary structure of the adult human skeletal muscle voltage-dependent sodium channel. Annals of Neurology. 1992;31:131–137. doi: 10.1002/ana.410310203. [DOI] [PubMed] [Google Scholar]
- Graham FL, Van Der Eb AJ. A new technique for assay for infectivity of human adenovirus 5 DNA. Virology. 1973;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hayward LJ, Brown RH, Jr, Cannon SC. Slow inactivation differs among mutant Na channels associated with myotonia and periodic paralysis. Biophysical Journal. 1997;72:1204–1219. doi: 10.1016/S0006-3495(97)78768-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson CE, Barohn RJ, Ptácek LJ. Paramyotonia congenita: abnormal short exercise test, and improvement after mexiletine therapy. Muscle and Nerve. 1994;17:763–768. doi: 10.1002/mus.880170710. [DOI] [PubMed] [Google Scholar]
- Ji S, George AL, Jr, Horn R, Barchi RL. Paramyotonia congenita mutations reveal different roles for segments S3 and S4 of domain D4 in hSkM1 sodium channel gating. Journal of General Physiology. 1996;107:183–194. doi: 10.1085/jgp.107.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CC, Bean BP. Na+ channels must deactivate to recover from inactivation. Neuron. 1994;12:819–829. doi: 10.1016/0896-6273(94)90335-2. [DOI] [PubMed] [Google Scholar]
- Lerche H, Heine R, Pika U, George AL, Jr, Mitrovic N, Browatzki M, Weiß T, Rivet-Bastide M, Franke C, Lomonaco M, Ricker K, Lehmann-Horn F. Human sodium channel myotonia: slowed channel inactivation due to substitutions for a glycine within the III-IV linker. The Journal of Physiology. 1993;470:13–22. doi: 10.1113/jphysiol.1993.sp019843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerche H, Mitrovic N, Dubowitz V, Lehmann-Horn F. Paramyotonia congenita: the R1448P Na+ channel mutation in adult human skeletal muscle. Annals of Neurology. 1996;39:599–608. doi: 10.1002/ana.410390509. [DOI] [PubMed] [Google Scholar]
- Noda M, Shimizu S, Tanabe T, Takai T, Kayano T, Ikeda T, Takahashi H, Nakayama H, Kanaoka Y, Minamino N, Kangawa K, Matsuo H, Raftery MA, Hirose T, Inayama S, Hayashida H, Miyata T, Numa S. Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature. 1984;312:121–127. doi: 10.1038/312121a0. [DOI] [PubMed] [Google Scholar]
- Ptácek LJ, George AL, Jr, Barchi RL, Griggs RC, Riggs JE, Robertson M, Leppert MF. Mutations in an S4 segment of the adult skeletal muscle sodium channel cause paramyotonia congenita. Neuron. 1992;8:891–897. doi: 10.1016/0896-6273(92)90203-p. [DOI] [PubMed] [Google Scholar]
- Ptácek LJ, George AL, Jr, Griggs RC, Tawil R, Kallen RG, Barchi RL, Robertson M, Leppert MF. Identification of a mutation in the gene causing hyperkalemic periodic paralysis. Cell. 1991a;67:1021–1027. doi: 10.1016/0092-8674(91)90374-8. [DOI] [PubMed] [Google Scholar]
- Ptácek LJ, Griggs RC. Familial periodic paralysis. In: Schultz SG, editor. Molecular Biology of Membrane Transport Disorders. New York: Plenum Press; 1996. pp. 625–642. [Google Scholar]
- Ptácek LJ, Tawil R, Griggs RC, Meola G, McManis P, Barohn RJ, Mendell JR, Harris C, Spitzer R, Santiago F, Leppert MF. Sodium channel mutations in acetazolamide-responsive myotonia congenita, paramyotonia congenita, and hyperkalemic periodic paralysis. Neurology. 1994;44:1500–1503. doi: 10.1212/wnl.44.8.1500. [DOI] [PubMed] [Google Scholar]
- Ptácek LJ, Trimmer JS, Agnew WS, Roberts JW, Petajan JH, Leppert MF. Paramyotonia congenita and hyperkalemic periodic paralysis map to the same sodium-channel gene locus. American Journal of Human Genetics. 1991b;49:851–854. [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, Featherstone DE, Ruben PC. Human Na+ channel fast and slow inactivation in paramyotonia congenita mutants expressed in Xenopus laevis oocytes. The Journal of Physiology. 1997a;499:589–600. doi: 10.1113/jphysiol.1997.sp021952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, VanDeCarr D, Featherstone DE, George AL, Jr, Ruben PC. Defective fast inactivation recovery and deactivation account for sodium channel myotonia in the I1160V mutant. Biophysical Journal. 1997b;73:1896–1903. doi: 10.1016/S0006-3495(97)78220-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar G, Sommer SS. The ‘megaprimer’ method of site-directed mutagenesis. Biotechniques. 1990;8:404–407. [PubMed] [Google Scholar]
- Streib EW. AAEE minimonograph no. 27: differential diagnosis of myotonic syndromes. Muscle and Nerve. 1987;10:603–615. doi: 10.1002/mus.880100704. [DOI] [PubMed] [Google Scholar]
- Wang J, Dubowitz V, Lehmann-Horn F, Ricker K, Ptácek LJ, Hoffman EP. In vivo sodium channel structure/function studies: consecutive Arg1448 changes to Cys, His, and Pro at the extracellular surface of IVS4. In: Dawson DC, Frizzell RA, editors. Ion Channels and Genetic Diseases. New York: Rockfeller University Press; 1995a. pp. 77–88. [Google Scholar]
- Wang Q, Shen JX, Splawski I, Atkinson D, Li ZZ, Robinson JL, Moss AJ, Towbin JA, Keating MT. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995b;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- Yang N, Ji S, Zhou M, Ptácek LJ, Barchi RL, Horn R, George AL., Jr Sodium channel mutations in paramyotonia congenita exhibit similar biophysical phenotypes in vitro. Proceedings of the National Academy of Sciences of the USA. 1994;91:12785–12789. doi: 10.1073/pnas.91.26.12785. [DOI] [PMC free article] [PubMed] [Google Scholar]