

Abstract
A degenerate polymerase chain reaction (PCR) homology screening procedure was applied to rat brain cDNA in order to identify novel genes belonging to the amiloride-sensitive Na+ channel and degenerin (NaC/DEG) family of ion channels. A single gene was identified that encodes a protein related to but clearly different from the already cloned members of the family (18-30 % amino acid sequence identity). Phylogenetic analysis linked this protein to the group of ligand-gated channels that includes the mammalian acid-sensing ion channels and the Phe-Met-Arg-Phe-amide (FMRFamide)-activated Na+ channel.
Expression of gain-of-function mutants after cRNA injection into Xenopus laevis oocytes or transient transfection of COS cells induced large constitutive currents. The activated channel was amiloride sensitive (IC50, 1.31 μm) and displayed a low conductance (9-10 pS) and a high selectivity for Na+ over K+ (ratio of the respective permeabilities, PNa+/PK+≥ 10), all of which are characteristic of NaC/DEG channel behaviour.
Northern blot and reverse transcriptase-polymerase chain reaction (RT-PCR) analysis revealed a predominant expression of its mRNA in the small intestine, the liver (including hepatocytes) and the brain. This channel has been called the brain-liver-intestine amiloride-sensitive Na+ channel (BLINaC).
Corresponding gain-of-function mutations in Caenorhabditis elegans degenerins are responsible for inherited neurodegeneration in the nematode. Besides the BLINaC physiological function that remains to be established, mutations in this novel mammalian degenerin-like channel might be of pathophysiological importance in inherited neurodegeneration and liver or intestinal pathologies.
The amiloride-sensitive Na+ channel and degenerin family (NaC/DEG) is an expanding family of cationic channels associated with very diverse functions in many organisms (Barbry & Hofman, 1997; Horisberger, 1998). Despite this functional diversity, all these channels share common properties including permeability to Na+, inhibition by the diuretic amiloride and voltage-independent gating. The NaC/DEG family includes channels that are constitutively active, like the epithelial Na+ channel (ENaC) involved in taste perception and Na+ homeostasis (Garty & Palmer, 1997) or the Drosophila gonad-specific amiloride-sensitive Na+ channel (dGNaC1) (Darboux et al. 1998a), also named ripped pocket (RPK) (Adams et al. 1998a), suggested to participate in gametogenesis or early embryonic development.
The NaC/DEG family also comprises ligand-gated channels like the Phe-Met-Arg-Phe-NH2 (FMRFamide) peptide-gated Na+ channel (FaNaC) cloned from the snail Helix aspersa nervous system (Lingueglia et al. 1995; Cottrell, 1997; Zhainazarov & Cottrell, 1998) where it participates in neuromodulation or the mammalian acid-sensing ion channels (ASICs) recently characterized from mammalian brain and sensory neurons (Waldmann et al. 1997b; Waldmann & Lazdunski, 1998) where they are thought to play an important role in the pain accompanying tissue acidosis.
Stretch is another stimulus that has been proposed to activate members of this ion channel family cloned from the nematode Caenorhabditis elegans and from Drosophila melanogaster (Adams et al. 1998a; Darboux et al. 1998b). MEC-4 (Driscoll & Chalfie, 1991) and MEC-10 (Huang & Chalfie, 1994) are expressed in the mechanosensory neurons of Caenorhabditis elegans where they are involved in mechanoperception. Recently, novel members that are required for co-ordinated movement have been identified in motor neurons (UNC-8, DEL-1; Tavernarakis et al. 1997) and muscle (UNC-105; Liu et al. 1996). However, the modes of activation of these channels remain uncertain since only an heterologous expression of gain-of-function mutants of UNC-105 has been described to date (Garcia-Añoveros et al. 1998). One interesting feature of these proteins is that gain-of-function mutations in their genes (deg-1, mec-4, mec-10, unc-8) cause the degeneration of some or all the neurons in which they are expressed, hence the name degenerin.
Remarkably, extrapolation in some mammalian members of this ion channel family (i.e. ASIC1, Bassilana et al. 1997, and ASIC2, previously named MDEG1, Waldmann et al. 1996) of a particular point mutation associated with gain-of-function and neuronal degeneration in Caenorhabditis elegans degenerins, also causes constitutive channel activation. Taken together, the implication of degenerin gain-of-function mutations in inherited neurodegeneration in the nematode Caenorhabditis elegans and the existence of gain-of-function mutations in mammalian neuronal NaC/DEG channels that can cause cell death (Waldmann et al. 1996) suggest that degenerin-like channels might also be involved in mammalian forms of neurodegeneration or muscle pathologies.
The number of mammalian NaC/DEG genes remains quite low compared with the more than 15 as yet uncharacterized degenerin homologues found in the Caenorhabditis elegans genome. In the present study, we report the cloning of a novel degenerin-like ion channel gene in rat and mouse and determine its tissue distribution and the biophysical properties of gain-of-function mutants.
METHODS
Cloning of rat and mouse BLINaC
Two degenerate oligonucleotides that correspond to sequences conserved between FaNaC, ASIC, MEC-4, MEC-10, DEG-1 and α, β and γ subunits of ENaC were used as primers for polymerase chain reaction (PCR) on rat brain cDNA obtained from poly A+ RNA reverse-transcribed with Superscript II reverse transcriptase (Gibco-BRL). The sense (5′-GGIAAYTGYTWYRYITTYAA-3′) and antisense (5′-ARICCIRDIDDICCICCIAIRTC-3′) primers correspond to amino acids 205-211 and 444-451 of rat BLINaC, respectively. After two rounds of amplification (30 s at 95°C, 1 min at 46°C, and 1 min at 72°C for 41 cycles with Taq DNA polymerase, Promega), amplified products of the expected size (∼740 bp) were subcloned into pBluescript SK− vector (Stratagene) and two clones containing a BLINaC fragment were obtained. The sequences upstream (5′) and downstream (3′) of the BLINaC fragment were obtained by rapid amplification of cDNA ends (RACE) as previously described (Waldmann et al. 1997a). The full-length BLINaC coding sequence was obtained from rat brain and small intestine cDNAs with Taq DNA polymerase (Promega) using a sense primer positioned just before the first ATG (nucleotide positions in rat 60-80: 5′-ACGCTAGCAGGACTTGGCATCACTGAGAG-3′) and an anti-sense primer positioned just after the stop codon (nucleotide positions in rat 1603-1626: 5′-CGCTCGAGTAGTATGAAAAGAAAACCAACACT-3′). The sense and anti-sense primers were supplied with a Nhe I site and a Xho I site, respectively. The PCR product was blunt-ended, ligated with EcoRI adaptor (Promega), cut by Xho I, and subcloned into EcoRI/Xho I-digested pBSK-SP6-globin vector (Waldmann et al. 1996). Eleven independent clones were sequenced on both strands. A443-mutated BLINaC clone was prepared as described (Vandeyar et al. 1988) and sequenced. For expression in mammalian cells, wild-type or mutated BLINaC cDNAs were excised from pBSK-SP6-globin vector with Nhe I/Xho I and subcloned into Nhe I/Xho I-digested pCI expression vector (Promega) or blunt-ended and subcloned into EcoRI-digested/blunt-ended pIRES-CD8 expression vector (Fink et al. 1998). The mouse BLINaC cDNA sequence was reconstructed from two overlapping PCR fragments located between a sense primer positioned just before the first rat ATG (nucleotide positions in rat 58-78: 5′-AGAGGACTTGGCATCACTGAG-3′) and an anti-sense primer starting at nucleotide 1603 in rat sequence (5′-CGCTCGAGTAGTATGAAAAGAAAACCAACACT-3′).
Hepatocyte preparation
Isolated rat (Wistar strain, male) and mouse (BALB/c strain, male) hepatocytes were prepared as described elsewhere (Sakai et al. 1996). Animals were anaesthetized by intraperitoneal administration of pentobarbital sodium (40 μg g−1), and killed by cervical dislocation. All experiments were carried out according to the guidelines of the Animal Care and Use Committee of the Centre National de la Recherche Scientifique. The liver was digested by portal infusion of modified Krebs-Henseleit solution supplemented with 100 units ml−1 of collagenase (Wako Pure Chemical Industries, Osaka, Japan) and 1 % BSA for 10 min at 37°C. The cell suspension was centrifuged at 45 g for 1 min, and the pellet was re-suspended with a fresh solution and centrifuged again (45 g for 1 min). After repeated washing (4 times) of the pellet, the purified hepatocyte suspension (> 99 % of hepatocytes) was obtained. Cell viability was determined to be > 95 % using the Trypan Blue exclusion test.
RNA isolation, Northern Blot and RT-PCR
Adult Wistar rats or BALB/c mice were anaesthetized and killed as described above. Rat total RNA was isolated by the acidic phenol method (Chomczynski & Sacchi, 1987). For RT-PCR, 1.5 μg rat total RNA were oligo(dT)-primed (500 ng) in a final volume of 20 μl in the presence of 200 units of Superscript II reverse transcriptase (Life Technologies, Inc.). The reaction was carried out at 37°C for 1 h and heated at 95°C for 4 min. One-twentieth of each sample was used with Taq DNA polymerase (Promega) and 100 ng of the sense primer (nucleotide positions 720-740: 5′-TATGGCGAGAATGTCCAAAGC-3′) and the anti-sense primer (nucleotide positions 1064-1084: 5′-AAGGGCAAACATCCACACAGC-3′) in a 15 μl reaction mixture. Forty cycles of PCR (95°C for 30 s, 60°C for 1 min, 72°C for 1 min) were performed, except for β-actin for which only 28 cycles were done. PCR products were gel electrophoresed and transferred onto a nylon membrane (Hybond N+, Amersham). The membrane was hybridized with a32P-labelled BLINaC cDNA probe, washed in 0.1 × SSC/0.1 % SDS at 65°C then exposed to Kodak X-Omat AR film at -70°C.
For the mouse RT-PCR described in Fig. 3A, total RNAs were extracted from mouse tissues with the SNAP total RNA isolation kit (Invitrogen) and treated with DNase. Fifteen micrograms of total RNA was reverse-transcribed according to the manufacturer's instructions (Gibco-BRL) and 1/40 of each sample was used as template for PCR amplification (Taq DNA polymerase, Promega) using BLINaC primers overlapping rat and mouse sequences (nucleotide positions in rat 182-201: 5′-TGACCGGAAGAAGTTTGATC-3′; and 898-918: 5′-GCATCCCCACAGGAGAAGACA-3′) or GAPDH primers (Clontech). BLINaC amplified fragments (95°C for 30 s, 55°C for 1 min, 72°C for 1 min; 38 cycles) were transferred onto a nylon membrane then probed with a32P-labelled cDNA probe corresponding to the whole rat BLINaC coding sequence.
Figure 3. Tissue distribution of BLINaC mRNA in different mouse and rat tissues.
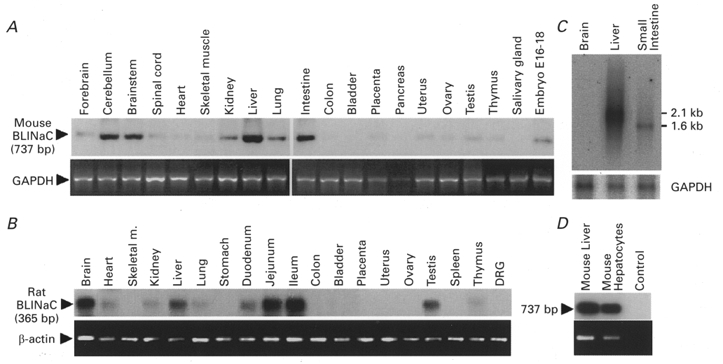
A, RT-PCR experiments performed with RNA isolated from different mouse tissues (indicated on top of each lane). PCR products were hybridized with an α-32P-labelled mouse BLINaC cDNA probe (upper panel). The expected PCR product size is given on the left. Amplification of GAPDH was used for control (lower panel, ethidium bromide staining). B, analysis of BLINaC expression on different rat tissues (indicated on top) by RT-PCR. Specificity was confirmed by hybridization of the expected 365 bp PCR product with an α-32P-labelled rat BLINaC cDNA probe (upper panel). A control amplification of β-actin was performed in parallel (lower panel, ethidium bromide staining). C, detection of BLINaC transcript on mouse brain, liver and small intestine poly A+ RNA (5 μg per lane) by Northern blot analysis. The probe used corresponds to an α-32P-labelled mouse BLINaC cDNA fragment. The filter was reprobed with a GAPDH probe as an RNA loading control (lower panel). A transcript of 2.1 kb was strongly detected in liver while a smaller transcript of 1.6 kb was found in small intestine. D, BLINaC mRNA expression in mouse liver and in freshly prepared hepatocytes was assessed by RT-PCR experiments. Specific amplification in liver and pure hepatocytes of a 737 bp product (upper panel, Southern blot) and β-actin control amplification (lower panel, ethidium bromide staining) are shown. The control corresponds to a PCR without cDNA.
For RT-PCR experiments on liver and isolated hepatocytes described in Fig. 3D, RNAs were prepared using the SV total RNA isolation system (Promega). Two micrograms of RNA was reverse-transcribed in the presence of random primers. One-twentieth of each RT was used for BLINaC amplification (95°C for 30 s, 55°C for 1 min, 72°C for 1 min; 26 cycles) using the previously described rat and mouse common primers. PCR products were gel electrophoresed, transferred onto a nylon membrane, then hybridized with a32P-labelled BLINaC cDNA probe. All the RT-PCR primers used span intron/exon junction(s) as confirmed by the partial sequencing of mouse BLINaC genomic DNA.
Northern blot analysis was carried out using standard techniques. Five micrograms of mouse poly A+ RNA prepared with the PolyATract mRNA isolation System (Promega) was separated on 1 % agarose/formaldehyde gels and transferred onto a nylon membrane (Zetaprobe GT, Biorad). The membranes were hybridized with a32P-labelled 737 bp mouse PCR fragment (nucleotide positions in rat: 182-918) overnight at 65°C in 0.25 M Na2HPO4 (pH 7.0)/7 % SDS, washed in 40 mM Na2HPO4 (pH 7.0)/1 % SDS at 65°C and subsequently analysed with a Fuji PhosphorImager according to the manufacturer's procedure.
Expression and electrophysiology in Xenopus oocytes and COS cells
Isolation, maintenance and injection of stage V and VI Xenopus oocytes with wild-type (WT) or mutated BLINaC was performed as described previously (Fink et al. 1998). Animals were anaesthetized by immersion in 1 g l−1 MS-222, according to the guidelines of the Animal Care and Use Committee of the CNRS. After extraction of oocytes, donors were sutured and allowed to recover in a separate holding tank where they were monitored for signs of infection. cRNA was synthesized from the Not I-digested pBSK-SP6-globin BLINaC vector using a kit from Stratagene. Xenopus oocytes were injected with 0.25-5 ng cRNA, and microelectrode voltage-clamp assays were performed 1-3 days after injection. The ND96 bathing solution contained (mM): 96 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, 5 Hepes, pH 7.4 (with NaOH). In some experiments, NaCl was replaced by N-methyl-D-glucamine chloride (NMDG-Cl), LiCl or KCl.
For expression in mammalian cells, COS-7 cells were transfected with the pIRES-CD8-BLINaC vector using DEAE-dextran. One to two days later, cells binding CD8 antibody-coated beads (Jurman et al. 1994) were used for experiments. Single channel currents were recorded using the patch-clamp technique as described previously (Champigny et al. 1998). For outside-out patches, the pipette solution contained (mM): 140 KCl, 2 MgCl2, 5 EGTA, 10 Hepes, pH 7.4 (with KOH). The bathing solution contained (mM): 140 NaCl, 2 MgCl2, 1.8 CaCl2, 10 Hepes, pH 7.4 (with NaOH). Data were low-pass filtered (1 kHz) and digitized every 100 μs for analysis. Open probabilities were calculated from the open-closed events of single channels during 20-30 s using Biopatch software (Biologic, Grenoble, France). Experiments were carried out at room temperature (22-24°C).
Fluorescence in situ chromosomal mapping
Metaphase spreads were prepared from a WMP male mouse, in which all the autosomes except chromosome 19 were in a form of metacentric Robertsonian translocations (Bonhomme & Guénet, 1989). The mouse was killed as described above and lymphocyte culture was performed immediately from freshly excised spleen. Concanavalin A-stimulated lymphocytes were cultured at 37°C for 72 h with 5-bromodeoxyuridine (60 μg ml−1) added for the final 6 h of culture to ensure a chromosomal R-banding of good quality. A BLINaC 5 kb Pst I genomic fragment was obtained from a mouse BAC clone (Genome Systems Inc.) and was biotinylated by nick translation with biotin-16-dUTP, as outlined by the Boerhinger Mannheim protocol. Hybridization to chromosome spreads was performed using standard protocols (Matsuda et al. 1992). The biotin-labelled DNA was mixed with hybridization solution at a final concentration of 10 μg ml−1 and 300 ng was used per slide. Before hybridization, the labelled probe was annealed with a 150-fold excess amount of murine Cot-1 DNA (Gibco-BRL) for 45 min at 37°C to compete with the aspecific repetitive sequences. The hybridized probe was detected by means of fluorescence isothiocyanate-conjugated avidin (Vector laboratories). Chromosomes were counterstained and R-banded with propidium iodide diluted in antifade solution (pH 11) as described in Lemieux et al. (1992).
RESULTS
Cloning, structure and chromosomal localization of BLINaC
Degenerate PCR using two primers directed against conserved sequences of the amiloride sensitive Na+ channel/ degenerin family allowed the isolation of a novel NaC/DEG homologue from rat brain. A full-length cDNA of 1887 base pairs was isolated from rat brain by 3′- and 5′-rapid amplification of cDNA ends (RACE/PCR) (accession number Y19034). It contains an open reading frame preceded by a stop codon, coding for a protein of 495 amino acids named BLINaC (Fig. 1A). A mouse cDNA clone was obtained by PCR (not shown, accession number Y19035) and the corresponding 495 amino acid protein was 94 % identical to the rat one. BLINaC displays significant identity with other members of the NaC/DEG family (up to 30 %; Fig. 2A and B) and had the same structural organization with two putative transmembrane domains flanking a large cysteine-rich region (Fig. 1A and B). The topological organization deduced from this channel structure and experimentally determined for the epithelial Na+ channel and the degenerin MEC-4 corresponds to intracellular N- and C-termini with a large extracellular loop (Fig. 1C). The phylogenetic analysis links BLINaC to the ligand-gated channel branch of the NaC/DEG family that includes the peptide-gated channel FaNaC and the mammalian proton-gated channel (ASIC) subunits (Fig. 2A). The closest relatives are the ASIC subunits (Fig. 2A) which share a similar overall structure with BLINaC (Fig. 1A), despite a rather low amino acid sequence identity (∼30 %; Fig. 2B). For comparison, the identity between the most distant ASIC subunits, i.e. ASIC1 and ASIC3 (previously named DRASIC), is about 53 % (Fig. 2B). The BLINaC murine gene was mapped by fluorescence in situ hybridization. A total of 30 metaphase cells were analysed, and about 80 % of the cells showed specific fluorescent spots on both chromosomes 3, in the 3E-3F1 region (Fig. 6). The surrounding region of mouse chromosome 3 includes an apoptosis susceptibility locus (Rapop3), a susceptibility locus to experimental allergic encephalomyelitis (Eae3), loci influencing body weight (late growth, Bglq3; abdominal fat weight, Afw1; abdominal fat percentage, Afpq1; and kidney weight, Kwq2) and a locus for familial combined hyperlipidaemia (FCHL), a common multifactorial disorder predisposing to premature coronary heart disease and associated with elevated levels of plasma triglyceride, cholesterol, or both (Mouse Genome Database, 1999). At present, the relation between BLINaC and any of these loci is not established and it is therefore not possible to associate BLINaC mutations with mouse genetic disorders.
Figure 1. Sequence analysis of the BLINaC protein.
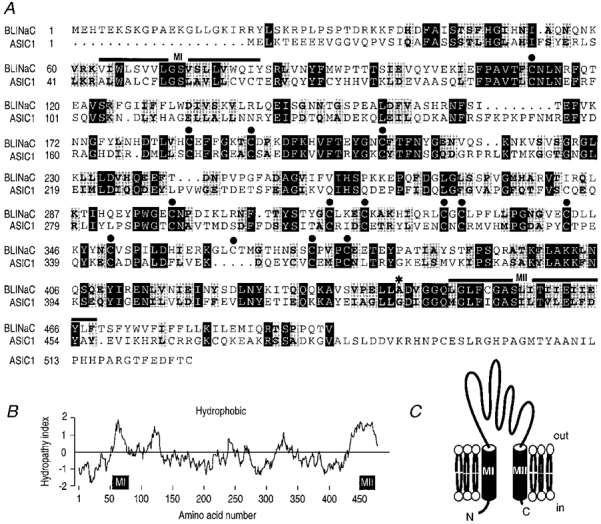
A, sequence comparison between rat BLINaC and the closest relative ASIC1 proteins. The two putative transmembrane regions MI and MII are indicated by bold lines above the sequence. External cysteines are marked with filled circles. The mutated alanine involved in the gain-of-function just before the TMII is indicated by an asterisk. Identical and similar residues are printed white on black and black on grey, respectively. The sequences were aligned using the Genetics Computer Group (GCG) Pileup program (Madison, WI, USA), with minor manual corrections when necessary. B, Kyte and Doolittle hydropathicity plot. Hydropathy values (positive is hydrophobic, negative is hydrophilic) represent the average over a 19 residues window. Amino acid numbering and putative transmembrane domains are given below the plot. C, BLINaC putative membrane topology deduced from its hydropathy profile and from the data obtained on other members of the NaC/DEG family. The extracellular loop represents most of the protein while the intracellular C-terminal part is extremely short.
Figure 2. Sequence comparison between BLINaC and the other NaC/DEG family members.
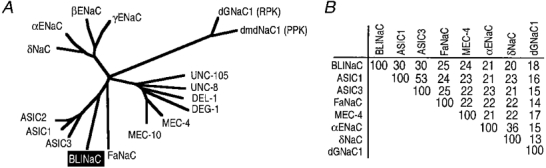
A, phylogenetic analysis. ASICs are acid-sensing ion channel subunits including ASIC1, ASIC2 (previously named MDEG1) and ASIC3 (previously named DRASIC). FaNaC is the FMRFamide-activated Na+ channel. MEC-4, MEC-10, DEG-1, DEL-1, UNC-8 and UNC-105 are Caenorhabditis elegans degenerins. α, β and γ ENaC are subunits of the epithelial Na+ channel and δNaC is an αENaC-like subunit. dGNaC1 (ripped pocket) and dmdNaC1 (pickpocket) are Drosophila members of the NaC/DEG family. The phylogenetic tree was established using the GCG Distances program with Kimura substitution followed by the GCG Growtree program with the UPGMA option, from an alignment obtained with the GCG Pileup program. Phylogenetic analysis clearly associates BLINaC with the subgroup of ligand-gated channels that comprise ASICs and FaNaC. B, percentage of amino acid sequence identity between BLINaC and other members of the amiloride-sensitive Na+ channel/degenerin family. Percentages were calculated using the GCG Distances program without correction for multiple substitutions. Accession numbers for ASIC1, ASIC3 (previously named DRASIC), FaNaC, MEC-4, rat αENaC, δNaC and dGNaC1 are U94403, AF013598, X92113, U53669, X70521, U38254 and Y16240, respectively. The overall identity with the other NaC/DEG channels remains low (below or equal to 30 %) while much higher identity may exist locally (e.g. in the second transmembrane domain region).
Figure 6. Localization of the BLINaC probe on WMP murine chromosomes.
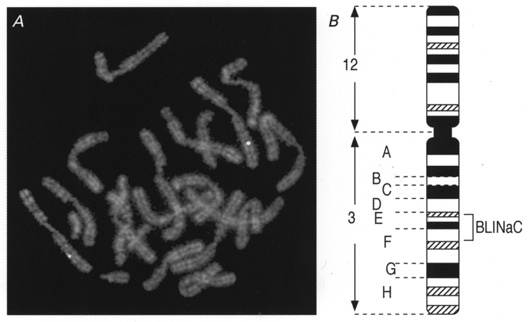
A, R-banded metaphase chromosomes show the fluorescent hybridization signals on chromosome 3 belonging to the (3;12) chromosomes. B, the corresponding localization (3E-3F1) is shown on the idiogram of the G-banded (3;12) Robertsonian translocation.
Distribution of BLINaC in rat and mouse
The tissue distribution of BLINaC in rat was assessed by the sensitive RT-PCR approach. A signal was detected after high sensitivity PCR (40 cycles) in whole brain, liver, small intestine (duodenum, jejunum and ileum) and testis (Fig. 3B). With the PCR conditions used, the transcript was also detected, although to a lower extent, in heart, kidney, lung and barely in thymus (Fig. 3B). No signal could be detected in dorsal root ganglia even after 40 cycles of PCR (Fig. 3B). A similar tissue distribution was obtained with RT-PCR on mouse tissues, i.e. in order of intensity, in liver, intestine, brain (mostly cerebellum), brainstem, kidney, lung and embryo (stage 16-18 postcoitus) with a difference for the testis signal which was almost absent (Fig. 3A). A significant expression of BLINaC in liver and intestine was confirmed by Northern blot analysis with mouse total RNA (not shown) or poly A+ RNA (Fig. 3C). A transcript of ∼2.1 kb was strongly detected in liver and a smaller transcript (∼1.6 kb) was detected in small intestine while no signal was found in brain (Fig. 3C). This could be explained by the apparently restricted expression in cerebellum leading to a signal dilution in total brain (Fig. 3A). The size difference between liver and intestine transcripts detected by Northern blot analysis might represent different transcriptional start sites, different mRNA polyadenylation or alternative splicing of the BLINaC gene. In the liver, BLINaC seems to be mostly expressed in hepatocytes since a strong signal was obtained by RT-PCR on total RNA prepared from isolated mouse hepatocytes (> 99 % pure) (Fig. 3D). The expression in hepatocytes was also confirmed in rat (not shown) but the overall level of BLINaC transcript was significantly lower than in mouse. It took at least six more rounds of amplification to achieve the same level of amplification in rat liver than in mouse liver under identical conditions and with the same primers (not shown).
BLINaC expression in Xenopus oocytes
When BLINaC cRNA was injected into Xenopus oocytes, a small constitutive current could be recorded in most experiments. Large variations in the amplitude of this current were observed with different oocyte batches. This constitutive current was abolished after replacement of Na+ by NMDG+ in the bathing solution (not shown) but it was not selective for Na+ over K+ (reversal potential, Er, -4.1 ± 1.3 mV with 96 mM NaCl bathing solution, n = 5; Fig. 4B) and it was only partially blocked by amiloride (IC50 > 1 mM; Fig. 4D). However, the relation of this current observed in oocytes with the physiological function of BLINaC remains unclear since such a current was never recorded after BLINaC expression in SF9 cells or mammalian COS cells (data not shown). Furthermore, the amplitude and properties of these currents were clearly different from those of the mutation-activated current (see below). A decrease in the external pH (to pH 4.0), an activation of protein kinases A and C, stretch, hypertonic stress (accomplished after addition of 100 mM sucrose) or addition of acetylcholine (100 μm), ATP (1 mM), ADP (1 mM), neuropeptide Y (0.5 μm), vasoactive intestinal peptide (VIP; 1 μm), calcitonin gene-related peptide (CGRP; 1 μm), N-formyl-methionyl-leucyl-phenylalanine (fMLP; 1 μm), angiotensin II (1 μm), thrombin (0.1 U ml−1), FMRFamide and related peptides (e.g. Met-enkephalin-Arg-Phe, F8Famide, A18Famide; 30 μm) failed to activate the channel or to alter the small constitutive current observed in Xenopus oocytes (data not shown). Co-expression of BLINaC with rat αENaC in Xenopus oocytes did not significantly alter the properties of the recorded current nor did it confer any sensitivity to hypertonic stress (not shown). On the other hand, a very large and consistent activation of BLINaC was obtained both in Xenopus oocyte and in COS cells after replacement of alanine 443, located just before the second hydrophobic domain (Fig. 1A, asterisk), by amino acids with larger lateral chains (Fig. 4A). The extent of BLINaC activation depends on the size of the side chain of the amino acid that replaces alanine 443. Replacement of alanine 443 by a glycine, i.e. an amino acid without a side chain, was without effect. Ser and Cys mutants were poorly active compared with the maximum activation found with bulkier amino acids like Lys, Val, Phe or Thr (Fig. 4A). The A443T gain-of-function mutant properties were analysed in more detail. The inversion of the amiloride-sensitive current at positive potential (Er, 36.8 ± 4.1 mV, n = 8) (Fig. 4B) and the ionic substitution experiments (Fig. 4C) suggested a much higher Na+ permeability over K+ and a slightly larger permeability for Li+ than for Na+. Amiloride and benzamil blocked the channel with half-inhibitory concentrations (IC50s) of 1.31 and 2.4 μm, respectively. Ethylisopropyl amiloride (EIPA) was less effective with an IC50 of 37 μm (Fig. 4D).
Figure 4. Properties of the rat wild-type (WT) and mutated BLINaC expressed in Xenopus oocytes.
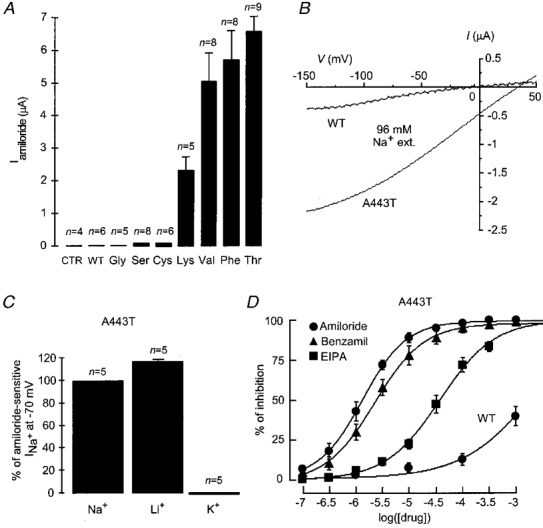
A, comparison of the amiloride-sensitive current (Iamiloride) recorded from oocytes injected with WT or A443 mutants (indicated below the columns) of BLINaC using the two-microelectrode voltage clamp. Currents were recorded at a holding potential of -70 mV in the presence or in the absence of 1 mM amiloride. CTR corresponds to water-injected oocytes. Columns and bars represent the means and s.e.m., the number of analysed oocytes is given above the columns. B, current-voltage relationship of the amiloride-sensitive current (1 mM amiloride) recorded from oocytes injected with WT BLINaC cRNA (3 ng oocyte−1; typical recording) and A443T mutant cRNA (0.25 ng oocyte−1; mean of 3 oocytes) obtained by voltage ramps from -150 to +50 mV in Na+-containing medium (ND96 solution). C, effect of Na+ replacement by Li+ or K+ in the external medium on the amiloride-sensitive current of the A443T mutant recorded at -70 mV. D, concentration-dependent inhibition of the A443T mutant currents by amiloride (•), benzamil (▴) and EIPA (▪) and inhibition of the WT BLINaC constitutive current by amiloride (•). The currents were measured at -70 mV in the ND96 bathing solution. Each point represents the mean value from four oocytes.
Single channel properties of gain-of-function mutants in COS cells
After expression in COS cells, gain-of-function mutants led to large currents that allowed single channel analysis of their properties. Unitary activities of A443S, A443C, A443F and A443T mutants were recorded in excised outside-out patches (Fig. 5A). Amiloride inhibits all these mutant channels at the single channel level by inducing a flickering behaviour as illustrated for the A443T mutant (Fig. 5B and D). The amiloride block is reversible, concentration dependent (Fig. 5B) and is seen at all clamped voltages (Fig. 5D). The four mutants have a high selectivity for Na+ over K+ (ratio of the permeabilities to the respective ions, PNa/PK≥ 10), as shown by the estimated reversal potential (> +58 mV) in bi-ionic conditions (Fig. 5C). They also have similar unitary conductances in the 140 mM NaCl bathing solution: 9.0 ± 0.5 pS (Vm between -50 mV and 0 mV; n = 3) for the A443S mutant, 10.6 ± 0.6 pS (n = 4) for the A443C mutant, 10.3 ± 0.5 pS (n = 3) for the A443F mutant, and 9.0 ± 0.1 pS (n = 2) for the A443T mutant (Fig. 5C). The open probability is very high for the A443F and A443T mutants (0.961 ± 0.020, n = 3 and 0.964 ± 0.022, n = 2, respectively) compared with the A443S and A443C mutants (≤ 0.07 ± 0.017, n = 3 and ≤0.196 ± 0.126, n = 3, respectively) (Fig. 5A). Because of their low open probability compared with the other gain-of-function mutants, the ability of stretch to modulate the activity of the A443S and A443C mutants recorded in inside-out or outside-out patches was examined. Stretch application was without effect on the open probability of the mutants (not shown).
Figure 5. Single-channel properties of BLINaC A443 gain-of-function mutants transiently expressed in COS cells.
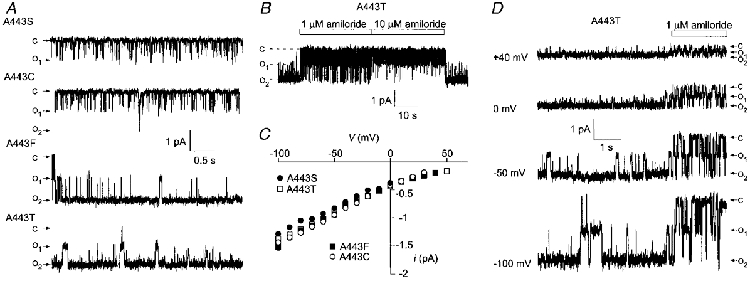
A, typical traces of single-channel activity in outside-out patches recorded at a membrane potential of -50 mV for four BLINaC gain-of-function mutants; c and o indicate the closed and opened states, respectively. B, effect of amiloride (1 and 10 μm) on the single-channel current recorded at -50 mV from an outside-out patch containing two A443T mutant channels. The amiloride block is reversible and concentration dependent. C, mean single-channel current-voltage relationships of the four BLINaC gain-of-function mutants recorded from outside-out patches in Na+-containing medium (140 mM Na+ in the medium). The estimated reversal potential was at least larger than 58 mV (PNa/PK≥ 10). Data were collected from two (A443T, □), three (A443S, •; and A443F, ▪) and four (A443C; ○) different patches. Unitary conductance corresponding to each mutant was estimated from the slope between -50 and 0 mV. D, single-channel current traces at different membrane potentials (indicated to the left) obtained from an outside-out patch containing two A443T mutant channels in the presence of 140 mM Na+ in the bathing solution. Amiloride application (1 μm) can block the channel at all potentials.
DISCUSSION
We have identified from rat and mouse a novel member of the NaC/DEG channel family named BLINaC. The closest relatives of this channel are the proton-gated channel subunits but BLINaC remains sufficiently different in terms of sequence to define a new subgroup in the family (Fig. 2A). In agreement with this observation, BLINaC is not activated by a decrease in the extracellular pH to 4.0 nor by the peptide FMRFamide.
BLINaC is activated by the same mutations that were previously described to be responsible for the dominant gain-of-function phenotypes associated with neurodegeneration in Caenorhabditis elegans (Chalfie & Wolinski, 1990; Driscoll & Chalfie, 1991). These mutations cause non-apoptotic, vacuolated degeneration of the expressing cells that shares features with the cell death of mammalian genetic disorders with alterations in channel subunits (Hall et al. 1997; Garcia-Añoveros et al. 1998). Equivalent mutations also increase the activity of the Drosophila ripped pocket channel (Adams et al. 1998a) and activate the Caenorhabditis elegans degenerin UNC-105 (Garcia-Añoveros et al. 1998). Therefore, activation of BLINaC by these gain-of-function mutations is not a novel property but is important since only two related channel proteins with such properties, ASIC1 (Bassilana et al. 1997) and ASIC2 previously named MDEG1 (Waldmann et al. 1996), are known in mammals. BLINaC is activated by very small structural changes at the level of the key amino acid responsible for the gain-of-function, some of them (e.g. the A443S mutation) being ineffective on MEC-4 and ASIC2. Therefore, any inherited mutation except glycine at this crucial position could lead to constitutive BLINaC activation and to cellular disorders which might cause cell death resembling that previously described for the Caenorhabditis elegans degenerin mutants.
Gain-of-function mutants of BLINaC were responsible for a large constitutive current with similar characteristics both in Xenopus oocytes and COS cells. This suggests that the mutated subunits are necessary and sufficient to form a functional channel. The activation is due to an increase in the open probability of the mutants in close relation with the side chain size of the amino acid at position 443 and without much affecting the selectivity and/or the conductance of the mutated channels. This confirms that the region surrounding the so-called ‘degeneration’ position just before the second transmembrane segment is important for the gating of NaC/DEG channels but is apparently not directly involved in the ion pore. All the mutants that provide a maximal activity (A443-T, -K, -V, -F) have nearly similar properties in terms of amiloride sensitivity and selectivity for Na+ over K+. They share, as determined for the A443T mutant, a lot of properties with the other members of the NaC/DEG family including a high selectivity for Na+versus K+ (PNa/PK≥ 10), an inhibition by the diuretic amiloride and its derivatives (with a novel pharmacological profile; amiloride ∼ benzamil > EIPA), no voltage-dependent gating and a low single-channel conductance (9.0 pS in 140 mM NaCl). Therefore, the characteristics of gain-of-function mutants conform to the properties commonly found in the NaC/DEG channel family. For the ligand-gated channel ASIC2, activation by the same gain-of-function mutations has been shown to be closely related to activation by proton, i.e. ASIC2 physiological activator (Adams et al. 1998b; Champigny et al. 1998). Both the activation of BLINaC by mutations and the properties of the mutated channels suggest the existence of an, as yet unknown, physiological activator for this new channel. BLINaC could be a ligand-gated channel (Barnard, 1996) although no extracellular ligand has yet been identified. The ligand could be a peptide, as for the peptide ionotropic receptor FaNaC.
The expression of wild-type BLINaC in Xenopus oocytes after cRNA injection allowed detection of a small constitutive channel activity. The recorded current is quite different from the classical NaC/DEG channel behaviour in its lack of significant amiloride sensitivity and Na+ over K+ selectivity. In addition, the current was of low amplitude (from several dozens to a few hundreds of nA) and saturates rapidly during injection of increased concentrations of cRNA (not shown), suggesting the presence of a limiting Xenopus oocyte-specific component that may be physiologically important to regulate the BLINaC channel activity. Consistent with this hypothesis, no basal activity was recorded after expression in other heterologous systems like SF9 cells or COS cells (not shown). At present, the physiological relevance of the basal current associated with wild-type BLINaC in the Xenopus expression system may be questioned. A similar situation was already described by Price et al. (1996) for the human BNC1 channel (corresponding to the ASIC2 channel). They described a very small basal Na+ current after expression in Xenopus oocytes while ASIC2 is a proton-gated channel that generates a very large transient current after a brief acidification of the extracellular medium (Champigny et al. 1998).
BLINaC is significantly expressed in the liver and the small intestine while a consistent but weaker expression can be detected in the central nervous system. Interestingly, differences in the level of BLINaC expression in resting conditions exist between rat and mouse, suggesting the existence of strong regulatory mechanisms for this channel. BLINaC has a structure close to that of ASIC channels and its expression in brain may be relevant to that of brain ASICs, i.e. the ASIC1, ASIC2 and ASIC2b subunits (Waldmann & Lazdunski, 1998). BLINaC could for example act as a regulatory subunit but it did not significantly modify the properties of the ASIC1 or ASIC2 channels expressed in Xenopus oocytes (not shown). In addition, BLINaC is present in tissues where no ASIC subunits have been described so far. These include the small intestine and the liver that appear as the two major sites of its expression. The epithelial Na+ channel is present in the intestine (Barbry & Hofman, 1997) but its expression is mostly restricted to the distal part of the colon. BLINaC is thus the first channel of this family which is substantially expressed in the small intestine. It is at present difficult to associate BLINaC to an already characterized ion channel and information on the native channel in vivo will probably be facilitated by its cell-specific localization along the small intestine. Of a special interest is the recent description in the Caenorhabditis elegans intestine of a novel ion channel belonging to the NaC/DEG superfamily and called FLR-1 (Take-Uchi et al. 1998). Genetic analysis suggests that FLR-1 may be a component of an intestinal regulatory system that controls the nematode defaecation rhythm. FLR-1 is clearly different from the already cloned degenerins and its possible relations with mammalian channels like BLINaC will have to be examined.
BLINaC is also strongly expressed in liver, and its expression in hepatocytes mainly accounts for its whole liver expression. The presence of NaC/DEG channels in liver may be related to the considerable increase in an hepatocyte membrane Na+ conductance associated with hypertonic stress. This conductance is inhibited by 10 μm amiloride, a concentration that did not affect Na+/H+ activity in this system (Wehner et al. 1995, 1997). The entry of Na+ plays a key role in the regulatory volume increase (RVI), i.e. in the cellular response that corresponds to the active readjustment of cell volume despite the continuous hypertonic challenge. However, wild-type BLINaC expressed in Xenopus oocytes was found to be insensitive to hypertonic stress (not shown). This does not completely disqualify BLINaC from being involved in such a physiological mechanism since additional subunits may be required for transforming BLINaC into a volume-activated channel, as additional subunits are necessary for the nematode degenerins to function as mechanosensitive channels (Gu et al. 1996). However, a small expression of the human epithelial Na+ channel α subunit has also been reported in the liver (McDonald et al. 1995) and rat αβγ-ENaC has been recently shown to enhance RVI in Xenopus oocytes (Ji et al. 1998). We did not find any interaction between BLINaC and rat α-or-βγ-ENaC subunits expressed in Xenopus oocytes (not shown). The exact participation of ENaC and the possible implication of BLINaC in hepatocyte RVI therefore remain to be clarified.
In conclusion, BLINaC is a member of the NaC/DEG family that has several interesting features. It seems to possess a novel, while unknown, mode of activation, it displays an original pattern of expression and it behaves as a new mammalian degenerin. At present, it is difficult to assign to this channel a clear physiological role in the different tissues where it is detected and no obvious association of BLINaC with genetic disorders has been found to date. However, similarly to Caenorhabditis elegans degenerins and to other channels involved in several inherited neurodegenerative diseases in mice (Zuo et al. 1997), the BLINaC degenerin-like behaviour resulting in an abnormal channel activation may be of pathophysiological relevance.
Acknowledgments
We are very grateful to Lionel Schaefer and François Maingret for help with some experiments and to Drs Rainer Waldmann, Pascal Barbry, Catherine Heurteaux, Pierre Pacaud, Anny Cupo, Sylvie Coscoy and Isabelle Darboux for fruitful discussions. We thank Dr Akira Ikari for help with hepatocyte preparation, Dr Florian Lesage for the generous gift of the mouse cDNA panel and Dr Amanda Patel for careful reading of the manuscript. Thanks are due to Franck Aguila for help with the artwork and to Valerie Friend, Martine Jodar, Gisèle Jarretou, Nathalie Leroudier, Valerie Briet and Dahvya Doume for technical assistance. H. S. thanks Professor Noriaki Takeguchi for great encouragement. This work was supported by the Centre National de la Recherche Scientifique (CNRS), the Institut National de la Santé et de la Recherche Médicale (INSERM), the Association Française contre les Myopathie (AFM) and the Association pour la Recherche sur le Cancer (ARC). H. S. was partly supported by the CNRS and by an Overseas Scholarship from the Ministry of Education, Science, Sport and Culture of Japan.
This paper is dedicated to the memory of our friend and colleague Dr Guy Champigny who has contributed in a major way to the current understanding of the properties of the NaC/DEG ion channel family.
References
- Adams CM, Anderson MG, Motto DG, Price MP, Johnson WA, Welsh MJ. Ripped pocket and pickpocket, novel Drosophila deg/ENaC subunits expressed in early development and in mechanosensory neurons. Journal of Cell Biology. 1998a;140:143–152. doi: 10.1083/jcb.140.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams CM, Snyder PM, Price MP, Welsh MJ. Protons activate brain Na+ channel 1 by inducing a conformational change that exposes a residue associated with neurodegeneration. Journal of Biological Chemistry. 1998b;273:30204–30207. doi: 10.1074/jbc.273.46.30204. [DOI] [PubMed] [Google Scholar]
- Barbry P, Hofman P. Molecular biology of Na+ absorption. American Journal of Physiology. 1997;273:G571–585. doi: 10.1152/ajpgi.1997.273.3.G571. [DOI] [PubMed] [Google Scholar]
- Barnard EA. The transmitter-gated channels: a range of receptor types and structures. Trends in Pharmacological Sciences. 1996;17:305–309. [PubMed] [Google Scholar]
- Bassilana F, Champigny G, Waldmann R, De Weille JR, Heurteaux C, Lazdunski M. The acid sensitive ionic channel subunit ASIC and the mammalian degenerin MDEG form a heteromultimeric H+-gated Na+ channel with novel properties. Journal of Biological Chemistry. 1997;272:28819–28822. doi: 10.1074/jbc.272.46.28819. [DOI] [PubMed] [Google Scholar]
- Bonhomme F, Guénet J-L. The wild house mouse and its relative. In: Lyon MF, Searle AG, editors. Genetic Variants and Strains of the Laboratory Mouse. Oxford: Oxford University Press; 1989. pp. 649–662. [Google Scholar]
- Chalfie M, Wolinski E. The identification and suppression of inherited neurodegeneration in Caenorhabditis elegans. Nature. 1990;345:410–416. doi: 10.1038/345410a0. [DOI] [PubMed] [Google Scholar]
- Champigny G, Voilley N, Waldmann R, Lazdunski M. Mutations causing neurodegeneration in Caenorhabditis elegans drastically alter the pH sensitivity and inactivation of the mammalian H+-gated Na+ channel MDEG1. Journal of Biological Chemistry. 1998;273:15418–15422. doi: 10.1074/jbc.273.25.15418. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Analytical Biochemistry. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Cottrell GA. The first peptide-gated ion channel. Journal of Experimental Biology. 1997;200:2377–2386. doi: 10.1242/jeb.200.18.2377. [DOI] [PubMed] [Google Scholar]
- Darboux I, Lingueglia E, Champigny G, Coscoy S, Barbry P, Lazdunski M. dGnaC1, a gonad-specific amiloride-sensitive Na+ channel. Journal of Biological Chemistry. 1998a;273:9424–9429. doi: 10.1074/jbc.273.16.9424. [DOI] [PubMed] [Google Scholar]
- Darboux I, Lingueglia E, Pauron D, Barbry P, Lazdunski M. A new member of the amiloride-sensitive sodium channel family in Drosophila melanogaster peripheral nervous system. Biochemical and Biophysical Research Communications. 1998b;246:210–216. doi: 10.1006/bbrc.1998.8183. [DOI] [PubMed] [Google Scholar]
- Driscoll M, Chalfie M. The Mec-4 gene is a member of a family of Caenorhabditis elegans genes that can mutate to induce neuronal degeneration. Nature. 1991;349:588–593. doi: 10.1038/349588a0. [DOI] [PubMed] [Google Scholar]
- Fink M, Lesage F, Duprat F, Heurteaux C, Reyes R, Fosset M, Lazdunski M. A neuronal two P domain K+ channel stimulated by arachidonic acid and polyunsaturated fatty acids. EMBO Journal. 1998;17:3297–3308. doi: 10.1093/emboj/17.12.3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Añoveros J, Garcia JA, Liu JD, Corey DP. The nematode degenerin UNC-105 forms ion channels that are activated by degeneration- or hypercontraction-causing mutations. Neuron. 1998;20:1231–1241. doi: 10.1016/s0896-6273(00)80503-6. [DOI] [PubMed] [Google Scholar]
- Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiological Reviews. 1997;77:359–396. doi: 10.1152/physrev.1997.77.2.359. [DOI] [PubMed] [Google Scholar]
- Gu G, Caldwell GA, Chalfie M. Genetic interactions affecting touch sensitivity in Caenorhabditis elegans. Proceedings of the National Academy of Sciences of the USA. 1996;93:6577–6582. doi: 10.1073/pnas.93.13.6577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DH, Gu G, García-Añoveros J, Gong L, Chalfie M, Driscoll M. Neuropathology of degenerative cell death in Caenorhabdistis elegans. Journal of Neuroscience. 1997;17:1033–1045. doi: 10.1523/JNEUROSCI.17-03-01033.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horisberger JD. Amiloride-sensitive Na channels. Current Opinion in Cell Biology. 1998;10:443–449. doi: 10.1016/s0955-0674(98)80056-2. [DOI] [PubMed] [Google Scholar]
- Huang M, Chalfie M. Gene interactions affecting mechanosensory transduction in Caenorhabditis elegans. Nature. 1994;367:467–470. doi: 10.1038/367467a0. [DOI] [PubMed] [Google Scholar]
- Ji HL, Fuller CM, Benos DJ. Osmotic pressure regulates alpha beta gamma-rENaC expressed in Xenopus oocytes. American Journal of Physiology. 1998;275:C1182–1190. doi: 10.1152/ajpcell.1998.275.5.C1182. [DOI] [PubMed] [Google Scholar]
- Jurman ME, Boland LM, Yellen G. Visual identification of individual transfected cells for electrophysiology using antibody-coated beads. BioTechniques. 1994;17:876–881. [PubMed] [Google Scholar]
- Lemieux N, Dutrillaux B, Viegas-Pequignot E. A simple method for simultaneous R- or G-banding and fluorescence in situ hybridization of small single-copy genes. Cytogenetics and Cell Genetics. 1992;59:311–312. doi: 10.1159/000133277. [DOI] [PubMed] [Google Scholar]
- Lingueglia E, Champigny G, Lazdunski M, Barbry P. Cloning of the amiloride-sensitive FMRFamide peptide-gated sodium channel. Nature. 1995;378:730–733. doi: 10.1038/378730a0. [DOI] [PubMed] [Google Scholar]
- Liu J, Schrank B, Waterston R. Interaction between a putative mechanosensory membrane channel and a collagen. Science. 1996;273:361–364. doi: 10.1126/science.273.5273.361. [DOI] [PubMed] [Google Scholar]
- McDonald FJ, Price MP, Snyder PM, Welsh MJ. Cloning and expression of the β- and γ-subunits of the human epithelial Na+ channel. American Journal of Physiology. 1995;268:C1157–1163. doi: 10.1152/ajpcell.1995.268.5.C1157. [DOI] [PubMed] [Google Scholar]
- Matsuda Y, Harada YN, Natsuume-Sakai S, Lee K, Shiomi T, Chapman VM. Location of the mouse complement factor H gene (cfh) by FISH analysis and replication R-banding. Cytogenetics and Cell Genetics. 1992;61:282–285. doi: 10.1159/000133423. [DOI] [PubMed] [Google Scholar]
- Mouse Genome Informatics. Bar Harbor, Maine: The Jackson Laboratory; 1999. Mouse Genome Database, (MGD) World Wide Web (URL: http://www.informatics.jax.org/) [Google Scholar]
- Price MP, Snyder PM, Welsh MJ. Cloning and expression of a novel brain Na+ channel. Journal of Biological Chemistry. 1996;271:7879–7882. doi: 10.1074/jbc.271.14.7879. [DOI] [PubMed] [Google Scholar]
- Sakai H, Kakinoki B, Diener M, Takeguchi N. Endogenous arachidonic acid inhibits hypotonically-activated Cl− channels in isolated rat hepatocytes. Japanese The Journal of Physiology. 1996;46:311–318. doi: 10.2170/jjphysiol.46.311. [DOI] [PubMed] [Google Scholar]
- Take-Uchi M, Kawakami M, Ishihara T, Amano T, Kondo K, Katsura I. An ion channel of the degenerin/epithelial sodium channel superfamily controls the defecation rhythm in Caenorhabditis elegans. Proceedings of the National Academy of Sciences of the USA. 1998;95:11775–11780. doi: 10.1073/pnas.95.20.11775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavernarakis N, Shreffler W, Wang S, Driscoll M. unc-8, a DEG/ENaC family member, encodes a subunit of a candidate mechanically gated channel that modulates C. elegans locomotion. Neuron. 1997;18:107–119. doi: 10.1016/s0896-6273(01)80050-7. [DOI] [PubMed] [Google Scholar]
- Vandeyar MA, Weiner MP, Hutton CJ, Batt CA. A simple and rapid method for the selection of oligodeoxynucleotide-directed mutants. Gene. 1988;65:129–133. doi: 10.1016/0378-1119(88)90425-8. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Bassilana F, De Weille JR, Champigny G, Heurteaux C, Lazdunski M. Molecular cloning of a non-inactivating proton-gated Na+ channel specific for sensory neurons. Journal of Biological Chemistry. 1997a;272:20975–20978. doi: 10.1074/jbc.272.34.20975. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M. A proton-gated cation channel involved in acid sensing. Nature. 1997b;386:173–177. doi: 10.1038/386173a0. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Champigny G, Voilley N, Lauritzen I, Lazdunski M. The mammalian degenerin MDEG, an amiloride-sensitive cation channel activated by mutations causing neurodegeneration in C. elegans. Journal of Biological Chemistry. 1996;271:10433–10436. doi: 10.1074/jbc.271.18.10433. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Lazdunski M. H+-gated cation channels: neuronal acid sensors in the NaC/DEG family of ion channels. Current Opinion in Neurobiology. 1998;8:418–424. doi: 10.1016/s0959-4388(98)80070-6. [DOI] [PubMed] [Google Scholar]
- Wehner F, Kinne RK, Tinel H. Hypertonicity-induced alkalinization of rat hepatocytes is not involved in activation of Na+ conductance or Na+,K+-ATPase. Biochimica et Biophysica Acta. 1997;1328:166–176. doi: 10.1016/s0005-2736(97)00085-0. [DOI] [PubMed] [Google Scholar]
- Wehner F, Sauer H, Kinne RK. Hypertonic stress increases the Na+ conductance of rat hepatocytes in primary culture. Journal of General Physiology. 1995;105:507–535. doi: 10.1085/jgp.105.4.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhainazarov AB, Cottrell GA. Single-channel currents of a peptide-gated sodium channel expressed in Xenopus oocytes. The Journal of Physiology. 1998;513:19–31. doi: 10.1111/j.1469-7793.1998.019by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo J, De Jager PL, Takahashi KA, Jiang W, Linden DJ, Heintz N. Neurodegeneration in Lurcher mice caused by mutation in delta2 glutamate receptor gene. Nature. 1997;388:769–773. doi: 10.1038/42009. [DOI] [PubMed] [Google Scholar]