

Abstract
Protein trafficking plays a central role in many aspects of neuronal function, from the release of neurotransmitters by exocytosis and the recycling of synaptic vesicle proteins to the regulation of receptor signalling. Synaptic function can be significantly modified on a short time scale by alterations in the levels of receptors, ion channels and transporters both pre- and postsynaptically. In many cases, these alterations appear to be mediated by acute changes in the rates at which the proteins are endocytosed from and exocytosed to the cell surface from intracellular pools. While our current understanding of the signalling mechanisms and the intracellular pathways responsible for these acute changes is still in its infancy, intriguing details are beginning to emerge from a number of systems.
Interest in regulatory mechanisms controlling the activity of synaptic proteins has directed renewed attention to protein trafficking as a potential mediator of synaptic plasticity. A major goal in neuroscience has been the elucidation of the molecular mechanisms that underlie the ability of higher organisms to learn and remember. At a cellular level, these processes are thought to be mediated by both long- and short-term changes in the signalling properties of neurons. Due to remarkable advances in molecular biology in the past 10 years, neuroscientists can now envision that such changes in synaptic function can be achieved by altering levels of key proteins involved in synaptic transmission, including synaptic vesicle proteins, neurotransmitter receptors and neurotransmitter transporters. While long-term changes in protein expression are frequently the result of alterations in mRNA levels and increased protein synthesis, acute changes in protein distribution may involve regulation of protein trafficking, in particular in the endosomal pathway. This review presents a number of different systems in which protein trafficking to and from the plasma membrane is directly implicated in the short-term modification of synaptic function. In addition, we will discuss a variety of molecular mechanisms that mediate redistribution of integral membrane proteins.
Accessory proteins and phosphorylation play a key role in regulated trafficking of membrane proteins
Trafficking of membrane proteins in the cell must always occur within the context of the lipid bilayer. Studies of protein trafficking from yeast to mammalian cells in the past decade have revealed the basic mechanisms by which proteins move between intracellular compartments and the plasma membrane (Jackson, 1998; Le Borgne & Hoflack, 1998; Gu & Gruenberg, 1999; Marsh & McMahon, 1999). Cytosolic coat proteins assemble on donor compartment membranes and select or are recruited by cargo proteins destined to be delivered to the target membrane. Simultaneously, they also serve to physically deform the donor membrane for vesicle formation. After fission, the transport vesicles uncoat and become competent for fusion with the target membrane.
Integral membrane proteins trafficking through the secretory and endocytic pathway achieve their steady state distributions by sequential and differential recognition of a variety of targeting signals encoded in their amino acid sequence. Recognition of these signals is thought to be mediated by vesicle coat proteins, including the adaptor proteins AP-1, AP-2 and AP-3, which operate predominantly in the late secretory and endocytic pathways, and the COPI and II complexes, which mediate vesicle formation in the ER and Golgi (Springer et al. 1999). Proteins can interact directly with the adaptor proteins, or indirectly through an adaptor binding protein. In addition, phosphorylation can regulate interaction with both adaptors and adaptor binding proteins. Rapid endocytosis from the plasma membrane is mediated by association with the clathrin adaptor AP-2. Plasma membrane proteins that lack specific signals that specify interaction with AP-2 undergo endocytosis at the rate of bulk membrane flow through the endocytic pathway, about 1 % min−1. This low but significant rate can be compared to the more rapid endocytic rate (∼10-12 % min−1) of proteins that interact in a constitutive or regulated fashion with the clathrin adaptor AP-2. Several different sequence motifs have been identified that can mediate interaction between the cytoplasmic domains of internalized proteins and AP-2 (Marsh & McMahon, 1999). A tyrosine-based consensus motif (YXXØ, where Ø is a large hydrophobic amino acid) interacts with the medium chain subunit μ2 of AP-2, while dileucine-containing endocytosis signals mediate rapid endocytosis by interacting with the β-chain of adaptors (Kirchhausen, 1999).
A variety of mechanisms can increase the rate of endocytosis of a plasma membrane protein by enhancing interaction with clathrin and its associated proteins. For many G-protein coupled receptors (GPCRs), ligand binding and phosphorylation of the receptor lead to the association of arrestin, a protein which binds both clathrin and the clathrin adaptor AP-2 (Krupnick & Benovic, 1998; Laporte et al. 1999). Rapid endocytosis of CD4, the primary receptor for HIV and SIV, is triggered following viral infection by the binding of the HIV-1 Nef protein to both the cytoplasmic tail of CD4 and to the clathrin adaptor protein AP-2 (Greenberg et al. 1998; Oldrige & Marsh, 1998).
Phosphorylation plays a key role in promoting internalization of plasma membrane receptors as well as intracellular targeting. Substrates for phosphorylation include both the internalized protein and the components of the clathrin-based endocytic machinery. Endocytosis mediated by dileucine motifs can be regulated by phosphorylation of adjacent serine residues (Pitcher et al. 1999). In the case of the receptor tyrosine kinase EGFR, ligand binding may lead to downregulation through conformational changes induced by tyrosine phosphorylation, causing exposure of cryptic adaptor interaction sites in the cytoplasmic domain of the receptor. Recruitment and activation of the tyrosine kinase Src is involved in the downregulation of both the EGFR and the β2-adrenergic receptor (Luttrell et al. 1999; Wilde et al. 1999). Elegant experiments analysing the targeting of furin, a secretory pathway endoprotease, demonstrate how phosphorylation and dephosphorylation can lead to differential targeting of a single protein (Molloy et al. 1999). Furin has two separate trafficking loops, one between the trans-Golgi network (TGN) and the endosome, and the other between the plasma membrane and endosomes. Casein kinase II (CKII)-mediated phosphorylation of furin leads to an association with phosphofurin acidic cluster sorting protein (PACS)-1, which then mediates furin association with clathrin-associated adaptors. Within the endosomal-to-plasma membrane loop, dephosphorylation of furin by protein phosphatases results in the redirection of the protein to the TGN.
These processes all involve functionally similar clathrin adaptor binding proteins, and/or a phosphorylation event necessary for inducible protein binding. They differ in the ultimate fate of the proteins following endocytosis: EGFR and CD4 are targeted to late endosomes and lysosomes for degradation, furin localization is shifted between two different recycling loops, and the β2-adrenergic receptor is recycled to the cell surface from the early endosome following dephosphorylation. These differences demonstrate that regulation of plasma membrane proteins by endocytosis must involve more than simple engagement with the clathrin-adaptor machinery. Additional information must be provided to specify the subsequent intracellular pathway, and this may be achieved by interactions with other adaptor or coat complexes, including AP-1, AP-3 and the COP proteins.
Biogenesis of synaptic vesicles
Synthesis of synaptic vesicle membrane proteins and subsequent post-translational modifications such as glycosylation take place in the ER and the Golgi of neurons and neuroendocrine cells. It was long assumed that synaptic vesicles were formed by budding from the trans-Golgi network, and that mature synaptic vesicles were subsequently transported to the nerve terminal by axonal transport. However, a number of different experiments have suggested that synaptic vesicles are most likely to be formed at the nerve terminal by endocytosis. In PC12 cells, a neuroendocrine cell line, newly synthesized synaptophysin is transported to the plasma membrane in constitutive secretory vesicles and then incorporated into mature synaptic vesicles by endocytosis (Regnier-Vigouroux et al. 1991). In neurons, the synaptic vesicle proteins SV2, synaptobrevin and synaptophysin are localized to different compartments in the cell body, while they co-localize at the nerve terminal (Mundigl et al. 1993). Axonal organelles containing different motor proteins also contain different complements of synaptic vesicle proteins (Okada et al. 1995). Finally, mutational studies of synaptic vesicle protein targeting in primary neurons supports the hypothesis that recognition of synaptic vesicle targeting signals occurs at the synapse (West et al. 1997). Taken together, these experiments suggest that newly synthesized synaptic vesicle proteins traffic to the nerve terminal in different transport organelles, and become incorporated into synaptic vesicles at the synapse. Once they are in the nerve terminal plasma membrane it is likely that newly synthesized synaptic vesicle proteins utilize the same pathways to reach synaptic vesicles as recycling synaptic vesicle proteins.
The role of the endocytic pathway in synaptic vesicle biogenesis
There is general consensus that synaptic vesicle formation relies predominantly on the endocytic pathway, but at least two different mechanisms appear to mediate synaptic vesicle formation in neurons and neuroendocrine cells (Fig. 1) (Hannah et al. 1999). These pathways have been termed the ‘direct pathway’, as synaptic vesicles appear to arise directly from the plasma membrane, and the ‘indirect pathway’, in which synaptic vesicle proteins are delivered first to an endosomal intermediate. Formation of the synaptic vesicles from each of the two pathways has different molecular requirements. Synaptic vesicle formation from the plasma membrane requires both AP-2, a clathrin-adaptor protein, and dynamin, a GTPase essential for clathrin-mediated endocytosis (Schmidt et al. 1997; Clift-O'Grady et al. 1998; Schmidt & Huttner, 1998; Shi et al. 1998). Targeting of the synaptic vesicle protein synaptobrevin to synaptic vesicles from endosomes is dependent on AP-3, a distinct adaptor protein, as well as ARF1, a small GTPase in the ADP-ribosylation factor family (Faundez et al. 1997, 1998; Salem et al. 1998; Shi et al. 1998). Morphological studies of synapses in Drosophila with a temperature-sensitive mutation in dynamin, shibirets, have indicated the direct pathway for synaptic vesicle formation is closely localized to the active zone, while the indirect pathway is located at the periphery of the synaptic contact (Koenig & Ikeda, 1996). Examination of vesicle recycling at hippocampal synapses in vitro using the membrane fluorescent dye FM1-43 also suggests that single vesicles can form from the plasma membrane and re-fuse without prior delivery to an endosomal intermediate (Murthy & Stevens, 1998).
Figure 1. The two mechanisms of endocytic formation of synaptic vesicles.
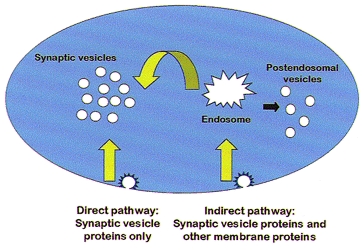
Synaptic vesicles are formed by endocytosis, directly from the plasma membrane and indirectly from an endosomal intermediate. Primary endocytic vesicles in the two pathways differ in their protein composition. Vesicles in the direct pathway are enriched for synaptic vesicle proteins only, while endocytic vesicles in the indirect pathway contain both synaptic vesicle proteins and other proteins that traffic through the endosomal pathway. At the nerve terminal, these proteins would presumably include plasma membrane proteins such as channels, neurotransmitter transporters and presynaptic receptors.
Two populations of primary endocytic vesicles have been described that are likely to correspond to the direct and indirect pathways for synaptic vesicle biogenesis. Coated vesicle preparations from brain have been shown to contain at least two distinct types of coated vesicles (Uriu et al. 1991; Thoidis et al. 1998, 1999). One population contains only synaptic vesicle proteins, while the second population contains synaptic vesicle proteins along with other potential nerve terminal proteins and can be labelled with antibodies to the extracellular domain of included proteins (Thoidis et al. 1998, 1999; Provoda et al. 2000). Finally, studies of synaptic targeting in hippocampal neurons have demonstrated the presence of a presynaptic endosome which may be important for synaptic vesicle formation and for sorting of other nerve terminal proteins (West et al. 1997).
Why should there be two different pathways for synaptic vesicle formation at the synapse? One obvious possibility is that high fidelity sorting of synaptic vesicle proteins cannot be maintained over time with only a direct pathway. The direct pathway may rely on a kiss-and-run mechanism (Ceccarelli et al. 1973; Fesce et al. 1994), interactions between synaptic vesicle proteins in a complex that ensures their simultaneous endocytosis (Bennett et al. 1992; Haucke & De Camilli, 1999), or endocytosis from a specialized area of the presynaptic membrane specifically enriched in synaptic vesicle proteins. In all cases, it is highly likely that some percentage of the synaptic vesicle proteins escape internalization by the direct pathway. These proteins may then be internalized by a general clathrin-mediated pathway, along with other nerve terminal proteins such as transporters, ion channels and presynaptic receptors, to the endosome, where they can be sorted into the appropriate vesicles, either synaptic or transport vesicles, depending on their eventual destinations. The presynaptic endosome also provides a mechanism to maintain the appropriate composition of synaptic vesicle proteins by adding newly synthesized synaptic vesicle proteins, removing aged proteins for degradation and sorting away non-synaptic vesicle proteins.
Regulation of neurotransmitter transporters by membrane trafficking
Neurotransmitter levels in the synaptic cleft are controlled by transporters that reside in the presynaptic plasma membrane. Neurotransmitter transporters depend on the transmembrane sodium gradient to transport extracellular neurotransmitter into the cytoplasm of the presynaptic terminal, where it can be repackaged into synaptic vesicles by vesicular neurotransmitter transporters (Masson et al. 1999). The plasma membrane transporters are critically important for regulation of the magnitude and time course of synaptic signalling. They have also been implicated in a number of neuropathologies, including depression, drug abuse and neurodegeneration. For this reason, the functional regulation of plasma membrane neurotransmitter transporters by a variety of mechanisms, including membrane trafficking, has recently been the focus of much attention, and there are excellent current reviews on this topic (Beckman & Quick, 1998; Blakely et al. 1998; Masson et al. 1999). In this section, I will discuss recent evidence for the contribution of protein trafficking to transporter downregulation.
Downregulation of most members of the monoamine/GABA family of neurotransmitter transporters can be achieved by activation of protein kinase C with phorbol esters or by agonists that activate GPCRs coupled to PKC (Beckman & Quick, 1998; Blakely et al. 1998; Beckman et al. 1999; Masson et al. 1999). Early studies of transporter regulation suggested that the observed changes in Vmax were due to a change in the levels of transporter in the plasma membrane, without a change in total cellular transporter. The amount of transporter in the plasma membrane at steady state is the sum of the transporter reaching the cell surface from the biosynthetic pathway, leaving the surface via the endocytic pathway, and returning to the plasma membrane from intracellular endosomal pools; consequently, a change in any of these parameters could be responsible for differences in transporter localization. To determine the role of intracellular compartments in the regulation of plasma membrane neurotransmitter levels, we analysed the distribution of the human dopamine transporter (DAT), stably expressed in PC12 cells, before and after stimulation with the phorbol ester β-phorobol 12-myristate 13-acetate (β-PMA) (Melikian & Buckley, 1999).
We discovered that at steady state a substantial proportion (∼60 %) of DAT is intracellular. We determined the identity of this intracellular compartment by separation of DAT-PC12 homogenates in sucrose equilibrium density gradients. Based on studies of other plasma membrane receptors and transporters, we suspected that at least some of the intracellular protein was localized to the endosomal pathway. The distribution of organelles in the gradient was examined by Western blotting gradient fractions with antibodies to specific markers characteristic for endosomal organelles. As illustrated in Fig. 2a, following endocytosis, plasma membrane proteins are endocytosed to the early endosome, which is characterized by the presence of EEA1, a protein that mediates early endosome fusion, and rab5a, an early endosome-specific small G-protein (Mu et al. 1995; Mills et al. 1998; Christoforidis et al. 1999; McBride et al. 1999). Recycling membrane proteins such as the transferrin receptor (TfR) traffic from the early endosome to the endosomal recycling compartment (ERC) before they return to the plasma membrane, while proteins destined to be degraded are targeted to late endosomes and lysosomes from the early endosome (Mellman, 1996). The TfR exits the early endosome rapidly (t½≈ 4 min) but leaves the recycling compartment more slowly (t½≈ 12 min), so at steady state the majority of the TfR is concentrated in the ERC.
Figure 2. Model for clathrin-mediated endocytosis of plasma membrane proteins (A) and suggested models for regulation of neurotransmitter receptors by endocytosis (B).

A, clathrin-mediated endocytosis (CME) of plasma membrane proteins such as the transferrin receptor (TfR) occurs at a rapid rate (≈10 % min−1) due to interactions between specific sequence motifs in the cytoplasmic domains of the internalized proteins and clathrin adaptor proteins such as AP-2. Following internalization into clathrin-coated vesicles, the TfR is delivered to the early endosome, located at the periphery of the cell and characterized by the presence of rab5a and EEA1. The TfR leaves the early endosome with a short half-time, and accumulates in the endosomal recycling compartment (ERC), located in the pericentriolar region of the cell, due to the longer time for exit from this compartment. Proteins destined for degradation in lysosomes are sorted away from recycling proteins in the early endosome. B, suggested models for regulation of neurotransmitter receptors by endocytosis. Plasma membrane receptors exist in two states, free receptor and immobilized receptor concentrated at synaptic sites. The relative proportions of free and immobilized receptor may vary for different receptor types. The amount of free receptor in the plasma membrane is regulated by the rate of delivery from the biosynthetic pathway, and the rate of recycling through the endosomal pathway. Downregulation of free receptor can be achieved by stimuli that lead to enhanced endocytosis (CME). Different stimuli may cause distinct modifications that determine whether the receptor can be rapidly recycled to the cell surface through the ERC (green diamond) or is destined to be degraded in the lysosome (red diamond). An acute increase in synaptic receptor may be achieved in the absence of new protein synthesis by varying combinations of lateral recruitment of free receptor to the synaptic pool and an increase in the level of free receptors by changes in kend or kexo.
When we examined the distribution of DAT at steady state, we found that virtually all of the intracellular DAT colocalizes with TfR, a marker of the ERC, rather than rab5a and EEA1, markers of the early endosome. Immunoisolation of organelles with antibodies to the cytoplasmic domain of the TfR followed by Western blotting with anti-DAT antibodies revealed that the majority of DAT was immunoprecipitated by antibodies to the TfR, indicating that the two proteins are colocalized in the same organelles. These results suggest that at steady state, DAT constitutively recycles between the plasma membrane and the endosomal recycling compartment.
Activation of PKC leads to sequestration of DAT and other neurotransmitter transporters, but the identity of the trafficking pathway had not been determined. Following endocytosis, DAT could be targeted to lysosomes for degradation, or it may instead recycle through the endosomal compartment back to the plasma membrane. The former pathway would potentially lead to long-term downregulation, while the recycling pathway is more likely to mediate short-term downregulation. To determine if DAT internalized in response to PKC activation was targeted to the endosomal recycling compartment or to the late endosome/lysosome pathway, we used biotinylation of cell surface proteins to monitor the trafficking of DAT from the plasma membrane to its intracellular destination during PKC activation.
To determine the subcellular localization of DAT internalized in response to PMA treatment, we first labelled cell surface DAT by biotinylation, then induced internalization with PMA, and removed the remaining cell surface biotin residues. The cells were homogenized and the distribution of biotinylated DAT in organelles in the sucrose equilibrium gradient was compared to the distribution of endosomal compartment markers as before. In untreated control cells, internalized DAT co-fractionated with the TfR peak. β-PMA treatment caused a significant increase in the amount of internalized DAT in the same compartment. Furthermore, precipitation of TfR-containing organelles after PMA treatment with antibodies to the TfR from these fractions revealed that immunoisolation of >85 % of the transferrin receptor in these fractions resulted in coprecipitation of >88 % of the DAT. Taken together, these results provide strong evidence that DAT internalized after PMA treatment is colocalized with the TfR in the endosomal recyling pathway.
These data and previous studies of regulated receptor trafficking suggest one possible model for DAT regulation (Fig. 3). Activation of PKC by stimulation of GPCRs leads to phosphorylation of DAT, enhancing the transporter's interaction with clathrin-associated proteins such as AP-2, and increasing the rate of DAT endocytosis. This effect could also be achieved indirectly through phosphorylation of an adaptor-binding protein analogous to arrestin, Nef, or PACS, which then binds with increased affinity to DAT. This event would increase the amount of DAT in the endosomal recycling compartment, by enhanced endocytosis from the plasma membrane. By analogy to GPCR downregulation, DAT may then be dephosphorylated in the endosome and recycled to the plasma membrane on a relatively short time scale. An alternative possibility is that phosphorylation of DAT changes its rate of exocytosis from the ERC. A decrease in the rate constant for exocytosis with the same rate of endocytosis would increase intracellular DAT and decrease plasma membrane transporter. A recent study of DAT trafficking in MDCK cells, a polarized epithelial cell line, provides evidence for targeting of DAT to lysosomes in response to activation of PKC (Daniels & Amara, 1999). MDCK cells are known to possess distinct trafficking pathways and may possess different adaptor or adaptor-binding proteins than neuroendocrine cells (Fölsch et al. 1999). Phorbol ester treatment could potentially cause enhanced flux through both the endosomal recycling pathway and the lysosomal pathway, while the relative balance of trafficking through each pathway may be determined by cellullar context.
Figure 3. Model for regulated endocytosis of the dopamine transporter.

Activation of protein kinase C, directly by application of phorbol ester or through GPCRs such as the muscarinic acetylcholine receptor, causes a change in the dopamine transporter that enhances its interaction with the clathrin-based endocytic machinery. This change may be phosphorylation of the transporter itself (represented by the small yellow circle), or an upstream protein, that increases kend, the endocytic rate constant for DAT. Following internalization, DAT is delivered to the early endosome, characterized by the presence of rab5a and EEA1. From the early endosome, DAT trafficks to the endosomal recycling compartment, rather than to lysosomes for degradation. By analogy to known mechanisms of GPCR downregulation, phosphorylation of DAT, if it occurs, may be reversed in the endosomal system, regenerating unmodified DAT for recycling to the cell surface. Alternatively, activation of PKC may cause a decrease in kexo, the rate constant for exocytosis from the ERC, leading to intracellular accumulation of DAT and a decrease in the cell surface pool of the transporter.
Recent experiments suggest that transporter trafficking can be influenced by the interaction of substrates and inhibitors with the transporter. Substrates of GABA transporter 1 (GAT1) upregulate transport and increase surface expression of the transporter, while inhibitors that are not transporter substrates downregulate transport (Bernstein & Quick, 1999). Similarly, substrates that can permeate the serotonin transporter (SERT), such as serotonin and amphetamines, can block PMA-mediated downregulation and phosphorylation of the transporter, while non-permeant inhibitors such as cocaine do not (Ramamoorthy & Blakely, 1999). Taken together, these studies suggest that neurons may be able to respond to changes in extracellular substrate concentrations and in the activity of individual transporters by regulating the amount of available transporter.
While trafficking appears coincident with transporter downregulation, changes in unitary transporter function may also occur, as described for GPCRs (Krupnick & Benovic, 1998; Pitcher et al. 1998). Co-expression of syntaxin with GAT1 increases GAT1 expression at the plasma membrane but decreases the permeation rates of GABA through the transporter, as suggested by a slowing in the time to activation of transport in the presence of syntaxin, and a slowing in the time to peak of the transporter-mediated currents (M. Quick, University of Alabama, personal communication). Experiments to determine the relative contributions of membrane trafficking and direct modification of unitary transporter function to downregulation are currently ongoing in a number of laboratories and should provide a more complete picture of the diversity in mechanisms of transporter regulation.
Regulation of neurotransmitter receptors by membrane trafficking
Interest in the role of membrane trafficking in the acute regulation of synaptic neurotransmitter receptors has intensified in the last few years. In CNS neurons, insulin treatment leads to a rapid increase in the expression of postsynaptic GABAA receptors, and this increase may be achieved by translocation from an intracellular compartment (Wan et al. 1997). N-ethylmaleimide-sensitive factor (NSF), a key protein in the process of vesicle fusion, interacts directly with glutamate receptor subunits (Jahn, 1998; Nishimune et al. 1998; Osten et al. 1998; Song et al. 1998; Lüthi et al. 1999; Noel et al. 1999). Recruitment of the AMPA receptor subunit GluR1 to dendritic spines following tetanic synaptic stimulation may occur via increased exocytosis from intracellular pools (Shi et al. 1999). Substantial intracellular pools in neurons have been described for the subunits of both the AMPA- and NMDA-type glutamate receptors (Hall & Soderling, 1997a, b). Reversible changes in the density of acetylcholine receptor at the neuromuscular junction involve redistribution of receptors from a synaptic localization to a peri-synaptic region of the plasma membrane, followed by internalization (Akaaboune et al. 1999). The perisynaptic pool of receptors can be also recruited into the synaptic pool, suggesting a dynamic regulation of the distribution of plasma membrane receptor between a freely diffusible and an immobilized state. Endocytosis of freely diffusible receptors and subsequent targeting to lysosomes contributes to the downregulation of acetylcholine receptor seen in the absence of synaptic transmission. The authors of this study hypothesize that at least four distinct compartments contribute to the steady state density of acetylcholine receptor at the neuromuscular junction: junctional receptors, extrajunctional receptors, an internal pool of newly synthesized receptors and an internalized pool destined for degradation. This model can be extended to include the potential constributions of the endosomal recycling compartment (Fig. 2b), which is well-suited to play a role in the steady state distribution of the receptors, both by providing a local supply of functional hetero-oligomeric receptor and by mediating regulated changes in receptor levels by changes in the rate constants of recycling through the endosomal compartment.
Conclusion
Many aspects of neuronal function are apt to be regulated by protein trafficking through the endocytic pathway, including the targeting of proteins to specific pre- and postsynaptic domains and the ability to vary levels of pre- and postsynaptic proteins on a short time scale in response to activation of signal transduction pathways by extracellular stimuli. Modulation of synaptic function through the endocytic pathway may be especially important presynaptically, due to the lack of biosynthetic pools of membrane proteins available in the neuronal cell body and dendrites. The combination of modern imaging techniques with current biochemical approaches to protein trafficking, applied to the study of synaptic function in neurons, will be key to revealing the pathways and compartments involved in the dynamic regulation of proteins and signalling at the synapse.
Acknowledgments
The work from K.M.B.‘s laboratory was supported by grants from the National Institutes of Health (NIH) (R01 NS27536) and the National Science Foundation (NSF) (IBN 9511017).
References
- Akaaboune M, Culican SM, Turney SG, Lichtman JW. Rapid and reversible effects of activity on acetylcholine receptor density at the neuromuscular junction in vivo. Science. 1999;286:503–507. doi: 10.1126/science.286.5439.503. [DOI] [PubMed] [Google Scholar]
- Beckman ML, Bernstein EM, Quick MW. Multiple G protein-coupled receptors initiate protein kinase C redistribution of GABA transporters in hippocampal neurons. Journal of Neuroscience. 1999;19:RC9. doi: 10.1523/JNEUROSCI.19-11-j0006.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman ML, Quick MW. Neurotransmitter transporters: regulators of function and functional regulation. Journal of Membrane Biology. 1998;164:1–10. doi: 10.1007/s002329900388. [DOI] [PubMed] [Google Scholar]
- Bennett MK, Calakos N, Kreiner T, Scheller RH. Synaptic vesicle membrane proteins interact to form a multimeric complex. Journal of Cell Biology. 1992;116:761–775. doi: 10.1083/jcb.116.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein EM, Quick MW. Regulation of gamma-aminobutyric acid (GABA) transporters by extracellular GABA. Journal of Biological Chemistry. 1999;274:889–895. doi: 10.1074/jbc.274.2.889. [DOI] [PubMed] [Google Scholar]
- Blakely RD, Ramamoorthy S, Schroeter S, Qian Y, Apparsundaram S, Galli A, DeFelice LJ. Regulated phosphorylation and trafficking of antidepressant-sensitive serotonin transporter proteins. Biological Psychiatry. 1998;44:169–178. doi: 10.1016/s0006-3223(98)00124-3. [DOI] [PubMed] [Google Scholar]
- Ceccarelli B, Hurlbut WP, Mauro A. Turnover of transmitter and synaptic vesicles at the frog neuromuscular junction. Journal of Cell Biology. 1973;57:499–524. doi: 10.1083/jcb.57.2.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoforidis S, McBride HM, Burgoyne RD, Zerial M. The Rab5 effector EEA1 is a core component of endosome docking. Nature. 1999;397:621–625. doi: 10.1038/17618. [DOI] [PubMed] [Google Scholar]
- Clift-O'Grady L, Desnos C, Lichtenstein Y, Faundez V, Horng JT, Kelly RB. Reconstitution of synaptic vesicle biogenesis from PC12 cell membranes. Methods. 1998;16:150–159. doi: 10.1006/meth.1998.0662. [DOI] [PubMed] [Google Scholar]
- Daniels GM, Amara SG. Regulated trafficking of the human dopamine transporter. Clathrin-mediated internalization and lysosomal degradation in response to phorbol esters. Journal of Biological Chemistry. 1999;274:35794–35801. doi: 10.1074/jbc.274.50.35794. [DOI] [PubMed] [Google Scholar]
- Faundez V, Horng JT, Kelly RB. ADP ribosylation factor 1 is required for synaptic vesicle budding in PC12 cells. Journal of Cell Biology. 1997;138:505–515. doi: 10.1083/jcb.138.3.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faundez V, Horng JT, Kelly RB. A function for the AP3 coat complex in synaptic vesicle formation from endosomes. Cell. 1998;93:423–432. doi: 10.1016/s0092-8674(00)81170-8. [DOI] [PubMed] [Google Scholar]
- Fesce R, Grohovaz F, Valtorta F, Meldolesi J. Neurotransmitter release: fusion or ‘kiss-and-run’? Trends in Cell Biology. 1994;4:1–4. doi: 10.1016/0962-8924(94)90025-6. [DOI] [PubMed] [Google Scholar]
- Fölsch H, Ohno H, Bonifacino JS, Mellman I. A novel clathrin adaptor complex mediates basolateral targeting in polarized epithelial cells. Cell. 1999;99:189–198. doi: 10.1016/s0092-8674(00)81650-5. [DOI] [PubMed] [Google Scholar]
- Greenberg M, Detulleo L, Rapoport I, Skowronski J, Kirchhausen T. A dileucine motif in HIV-1 Nef is essential for sorting into clathrin-coated pits and for downregulation of CD4. Current Biology. 1998;8:1239–1242. doi: 10.1016/s0960-9822(07)00518-0. [DOI] [PubMed] [Google Scholar]
- Gu F, Gruenberg J. Biogenesis of transport intermediates in the endocytic pathway. FEBS Letters. 1999;452:61–66. doi: 10.1016/s0014-5793(99)00561-x. [DOI] [PubMed] [Google Scholar]
- Hall RA, Soderling TR. Differential surface expression and phosphorylation of the N-methyl-D-aspartate receptor subunits NR1 and NR2 in cultured hippocampal neurons. Journal of Biological Chemistry. 1997a;272:4135–4140. doi: 10.1074/jbc.272.7.4135. [DOI] [PubMed] [Google Scholar]
- Hall RA, Soderling TR. Quantitation of AMPA receptor surface expression in cultured hippocampal neurons. Neuroscience. 1997b;78:361–371. doi: 10.1016/s0306-4522(96)00525-8. [DOI] [PubMed] [Google Scholar]
- Hannah MJ, Schmidt AA, Huttner WB. Synaptic vesicle biogenesis. Annual Review of Cell and Developmental Biology. 1999;15:733–798. doi: 10.1146/annurev.cellbio.15.1.733. [DOI] [PubMed] [Google Scholar]
- Haucke V, De Camilli P. AP-2 recruitment to synaptotagmin stimulated by tyrosine-based endocytic motifs. Science. 1999;285:1268–1271. doi: 10.1126/science.285.5431.1268. [DOI] [PubMed] [Google Scholar]
- Jackson T. Transport vesicles: coats of many colours. Current Biology. 1998;8:R609–612. doi: 10.1016/s0960-9822(98)70388-4. [DOI] [PubMed] [Google Scholar]
- Jahn R. Synaptic transmission: two players team up for a new tune. Current Biology. 1998;8:R856–858. doi: 10.1016/s0960-9822(07)00531-3. [DOI] [PubMed] [Google Scholar]
- Kirchhausen T. Adaptors for clathrin-mediated traffic. Annual Review of Cell and Developmental Biology. 1999;15:705–732. doi: 10.1146/annurev.cellbio.15.1.705. [DOI] [PubMed] [Google Scholar]
- Koenig JH, Ikeda K. Synaptic vesicles have two distinct recycling pathways. Journal of Cell Biology. 1996;135:797–808. doi: 10.1083/jcb.135.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupnick JG, Benovic JL. The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annual Review of Pharmacology and Toxicology. 1998;38:289–319. doi: 10.1146/annurev.pharmtox.38.1.289. [DOI] [PubMed] [Google Scholar]
- Laporte SA, Oakley RH, Zhang J, Holt JA, Ferguson SS, Caron MG, Barak LS. The β2-adrenergic receptor/βarrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proceedings of the National Academy of Sciences of the USA. 1999;96:3712–3717. doi: 10.1073/pnas.96.7.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- le Borgne R, Hoflack B. Mechanisms of protein sorting and coat assembly: insights from the clathrin-coated vesicle pathway. Current Opinion in Cell Biology. 1998;10:499–503. doi: 10.1016/s0955-0674(98)80065-3. [DOI] [PubMed] [Google Scholar]
- Lüthi A, Chittajallu R, Duprat F, Palmer MJ, Benke TA, Kidd FL, Henley JM, Isaac JTR, Collingridge GL. Hippocampal LTD expression involves a pool of AMPARs regulated by the NSF-GluR2 interaction. Neuron. 1999;24:389–399. doi: 10.1016/s0896-6273(00)80852-1. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, Caron MG, Lefkowitz RJ. β-Arrestin-dependent formation of β2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- McBride HM, Rybin V, Murphy C, Giner A, Teasdale R, Zerial M. Oligomeric complexes link Rab5 effectors with NSF and drive membrane fusion via interactions between EEA1 and syntaxin 13. Cell. 1999;98:377–386. doi: 10.1016/s0092-8674(00)81966-2. [DOI] [PubMed] [Google Scholar]
- Marsh M, McMahon HT. The structural era of endocytosis. Science. 1999;285:215–220. doi: 10.1126/science.285.5425.215. [DOI] [PubMed] [Google Scholar]
- Masson J, Sagne C, Hamon M, El Mistikawy S. Neurotransmitter transporters in the central nervous system. Pharmocological Reviews. 1999;51:439–464. [PubMed] [Google Scholar]
- Melikian HE, Buckley KM. Membrane trafficking regulates the activity of the human dopamine transporter. Journal of Neuroscience. 1999;19:7699–7710. doi: 10.1523/JNEUROSCI.19-18-07699.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellman I. Endocytosis and molecular sorting. Annual Review of Cell and Developmental Biology. 1996;12:575–625. doi: 10.1146/annurev.cellbio.12.1.575. [DOI] [PubMed] [Google Scholar]
- Mills IG, Jones AT, Clague MJ. Involvement of the endosomal autoantigen EEA1 in homotypic fusion of early endosomes. Current Biology. 1998;8:881–884. doi: 10.1016/s0960-9822(07)00351-x. [DOI] [PubMed] [Google Scholar]
- Molloy SS, Anderson ED, Jean F, Thomas G. Bi-cycling the furin pathway: from TGN localization to pathogen activation. Trends in Cell Biology. 1999;9:28–36. doi: 10.1016/s0962-8924(98)01382-8. [DOI] [PubMed] [Google Scholar]
- Mu FT, Callaghan JM, Steele-Mortimer O, Stenmark H, Parton RG, Campbell PL, McCluskey J, Yeo JP, Tock EP, Toh BH. EEA1, an early endosome-associated protein. EEA1 is a conserved α-helical peripheral membrane protein flanked by cysteine ‘fingers’ and contains a calmodulin-binding IQ motif. Journal of Biological Chemistry. 1995;270:13503–13511. doi: 10.1074/jbc.270.22.13503. [DOI] [PubMed] [Google Scholar]
- Mundigl O, Matteoli M, Daniell L, Thomas-Reetz A, Metcalf A, Jahn R, De Camilli P. Synaptic vesicle proteins and early endosomes in cultured hippocampal neurons: differential effects of Brefeldin A in axon and dendrites. Journal of Cell Biology. 1993;122:1207–1221. doi: 10.1083/jcb.122.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy VN, Stevens CF. Synaptic vesicles retain their identity through the endocytic cycle. Nature. 1998;392:497–501. doi: 10.1038/33152. [DOI] [PubMed] [Google Scholar]
- Nishimune A, Isaac JT, Molnar E, Noel J, Nash SR, Tagaya M, Collingridge GL, Nakanishi S, Henley JM. NSF binding to GluR2 regulates synaptic transmission. Neuron. 1998;21:87–97. doi: 10.1016/s0896-6273(00)80517-6. [DOI] [PubMed] [Google Scholar]
- Noel J, Ralph GS, Pickard L, Williams J, Molnar E, Uney JB, Collingridge GL, Henley JM. Surface expression of AMPA receptors in hippocampal neurons is regulated by an NSF-dependent mechanism. Neuron. 1999;23:365–376. doi: 10.1016/s0896-6273(00)80786-2. [DOI] [PubMed] [Google Scholar]
- Okada Y, Yamazaki H, Sekineaizawa Y, Hirokawa N. The neuron-specific kinesin superfamily protein kif1a is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell. 1995;81:769–780. doi: 10.1016/0092-8674(95)90538-3. [DOI] [PubMed] [Google Scholar]
- Oldrige J, Marsh M. Nef – an adaptor adaptor. Trends in Cell Biology. 1998;8:302–305. doi: 10.1016/s0962-8924(98)01318-x. [DOI] [PubMed] [Google Scholar]
- Osten P, Srivastava S, Inman GJ, Vilim FS, Khatri L, Lee LM, States BA, Einheber S, Milner TA, Hanson PI, Ziff EB. The AMPA receptor GluR2 C terminus can mediate a reversible, ATP-dependent interaction with NSF and α- and β-SNAPs. Neuron. 1998;21:99–110. doi: 10.1016/s0896-6273(00)80518-8. [DOI] [PubMed] [Google Scholar]
- Pitcher C, Honing S, Fingerhut A, Bowers K, Marsh M. Cluster of differentiation antigen 4 (CD4) endocytosis and adaptor complex binding require activation of the CD4 endocytosis signal by serine phosphorylation. Molecular Biology of the Cell. 1999;10:677–691. doi: 10.1091/mbc.10.3.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher JA, Freedman NJ, Lefkowitz RJ. G protein-coupled receptor kinases. Annual Review of Biochemistry. 1998;67:653–692. doi: 10.1146/annurev.biochem.67.1.653. [DOI] [PubMed] [Google Scholar]
- Provoda CJ, Waring MT, Buckley KM. Evidence for a primary endocytic vesicle involved in synaptic vesicle biogenesis. Journal of Biological Chemistry. 2000 doi: 10.1074/jbc.275.10.7004. in the Press. [DOI] [PubMed] [Google Scholar]
- Ramamoorthy S, Blakely RD. Phosphorylation and sequestration of serotonin transporters differentially modulated by psychostimulants. Science. 1999;285:763–766. doi: 10.1126/science.285.5428.763. [DOI] [PubMed] [Google Scholar]
- Regnier-Vigouroux A, Tooze SA, Huttner WB. Newly synthesized synaptophysin is transported to synaptic-like microvesicles via constitutive secretory vesicles and the plasma membrane. EMBO Journal. 1991;10:3589–3601. doi: 10.1002/j.1460-2075.1991.tb04925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salem N, Faundez V, Horng JT, Kelly RB. A v-SNARE participates in synaptic vesicle formation mediated by the AP-3 adaptor complex. Nature Neuroscience. 1998;1:551–556. doi: 10.1038/2787. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Hannah MJ, Huttner WB. Synaptic-like microvesicles of neuroendocrine cells originate from a novel compartment that is continuous with the plasma membrane and devoid of transferrin receptor. Journal of Cell Biology. 1997;137:445–458. doi: 10.1083/jcb.137.2.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Huttner WB. Biogenesis of synaptic-like microvesicles in perforated PC12 cells. Methods. 1998;16:160–169. doi: 10.1006/meth.1998.0663. [DOI] [PubMed] [Google Scholar]
- Shi G, Faundez V, Roos J, Dell'Angelica EC, Kelly RB. Neuroendocrine synaptic vesicles are formed in vitro by both clathrin-dependent and clathrin-independent pathways. Journal of Cell Biology. 1998;143:947–955. doi: 10.1083/jcb.143.4.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- Song I, Kamboj S, Xia J, Dong H, Liao D, Huganir RL. Interaction of the N-ethylmaleimide-sensitive factor with AMPA receptors. Neuron. 1998;21:393–400. doi: 10.1016/s0896-6273(00)80548-6. [DOI] [PubMed] [Google Scholar]
- Springer S, Spang A, Schekman R. A primer on vesicle budding. Cell. 1999;97:145–148. doi: 10.1016/s0092-8674(00)80722-9. [DOI] [PubMed] [Google Scholar]
- Thoidis G, Chen P, Pushkin AV, Vallega G, Leeman SE, Fine RE, Kandror KV. Two distinct populations of synaptic-like vesicles from rat brain. Proceedings of the National Academy of Sciences of the USA. 1998;95:183–188. doi: 10.1073/pnas.95.1.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoidis G, Kupriyanova T, Cunningham JM, Chen P, Cadel S, Foulon T, Cohen P, Fine RE, Kandror KV. Glucose transporter Glut3 is targeted to secretory vesicles in neurons and PC12 cells. Journal of Biological Chemistry. 1999;274:14062–14066. doi: 10.1074/jbc.274.20.14062. [DOI] [PubMed] [Google Scholar]
- Uriu T, Omori K, Yamamoto A, Inoue M, Inagaki C. Two types of clathrin-coated vesicles isolated from rat brain: analysis of biochemical properties and cellular origin. Journal of Neurochemistry. 1991;56:1548–1556. doi: 10.1111/j.1471-4159.1991.tb02050.x. [DOI] [PubMed] [Google Scholar]
- Wan Q, Xiong ZG, Man HY, Ackerley CA, Braunton J, Lu WY, Becker LE, MacDonald JF, Wang YT. Recruitment of functional GABA(A) receptors to postsynaptic domains by insulin. Nature. 1997;388:686–690. doi: 10.1038/41792. [DOI] [PubMed] [Google Scholar]
- West AE, Neve RL, Buckley KM. Targeting of the synaptic vesicle protein synaptobrevin in the axon of cultured hippocampal neurons: evidence for two distinct sorting steps. Journal of Cell Biology. 1997;139:917–927. doi: 10.1083/jcb.139.4.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilde A, Beattie EC, Lem L, Riethof DA, Liu SH, Mobley WC, Soriano P, Brodsky FM. EGF receptor signaling stimulates SRC kinase phosphorylation of clathrin, influencing clathrin redistribution and EGF uptake. Cell. 1999;96:677–687. doi: 10.1016/s0092-8674(00)80578-4. [DOI] [PubMed] [Google Scholar]