

Abstract
We have investigated the roles of different voltage-dependent Ca2+ channels in the activation of the Cl− and K+ channels responsible for the afterdepolarization (ADP) and slow afterhyperpolarization (AHP) in sympathetic neurones of the isolated mouse superior cervical ganglion in vitro.
The ADP and its associated Ca2+-activated Cl− current were markedly decreased by ω-agatoxin IVA (40–200 nm) and nifedipine (1–10 μm), but not by ω-conotoxin GVIA (300 nm).
In contrast, the AHP and the apamin-sensitive Ca2+-activated K+ current that underlies this potential were blocked by ω-conotoxin GVIA, but were not affected by ω-agatoxin IVA and were only slightly reduced by nifedipine.
Ryanodine (20 μm) reduced the Ca2+-activated Cl− current following an action potential by 75 % but on average did not affect the Ca2+-activated K+ current.
Evidence that R-type channels provide a proportion of the Ca2+ activating both types of Ca2+-dependent channel was obtained.
We conclude that Ca2+ entering through L- and P-type Ca2+ channels preferentially activates the Cl− current responsible for the ADP in mouse sympathetic neurones, predominantly via Ca2+-induced Ca2+ release, whereas the Ca2+ that activates the K+ channels responsible for the AHP enters predominantly through N-type channels. The data can be explained by the selective association of each type of Ca2+ channel with particular intracellular mechanisms for activating other membrane channels, one indirect and the other direct, probably located at discrete sites on the soma and dendrites.
In mammalian sympathetic ganglion cells, Ca2+ entry during action potential firing activates K+ and Cl− conductances which contribute to slow afterpotentials (McAfee & Yarowsky, 1979; Sacchi et al. 1995; De Castro et al. 1997). In rat superior cervical ganglion (SCG) cells, which exhibit a long afterhyperpolarization (AHP), Ca2+ entry via N-type channels activates apamin-sensitive Ca2+-activated K+ channels (SK-type), which are responsible for most of the underlying conductance change (Davies et al. 1996). This conductance has been named gK,Ca1 (Cassell & McLachlan, 1987a), to distinguish it from the slower Ca2+-dependent conductance gK,Ca2, which underlies a prolonged AHP in one subclass of sympathetic neurone, and from IAHP, which differs in kinetic and pharmacological characteristics between several types of central neurone (Sah, 1996). On the other hand, in rat SCG cells, large conductance Ca2+-activated K+ channels (BK-type) are in part activated by Ca2+ entering through L-type channels, which has no effect on gK,Ca1 (Davies et al. 1996). These observations suggest that Ca2+ entering through a specific channel type during the action potential (AP) is directed predominantly to a particular type of K+ channel. Recently, N-type and BK-type channels in some cases, and L-type and SK-type channels in others, have been identified in single patches isolated from hippocampal somata, providing direct evidence for colocalization of pairs of functionally linked channels (Marrion & Tavalin, 1998). While the relative locations of Ca2+ and K+ channels must be crucial in determining the electrical behaviour of neurones, these data imply that particular associations between channels are not necessarily the same for all neurone types. Data from sympathetic neurones have confirmed that, even for this one type of neurone, there are species differences and differences between phenotypically distinct classes of neurone in the way Ca2+ entry is linked to activation of BK and SK channels (Ireland et al. 1998; Davies et al. 1999).
In mouse SCG cells, Ca2+ entry during the AP activates, in addition to gK,Ca1, a Cl− conductance (gCl,Ca) that produces an afterdepolarization (ADP; De Castro et al. 1997). When the cell is clamped at −55 mV, this is recorded as an inward current. About 45 % of the high-voltage-activated Ca2+ current evoked by a step depolarization in dissociated mouse SCG somata flows through P/Q-type channels, 40 % through N-type channels, 10 % through L-type channels and a small proportion through channels resistant to known selective organic antagonists (here called R-type channels; Namkung et al. 1998). The question then arises as to how Ca2+ entering through these diverse types of voltage-dependent Ca2+ channel during an AP activates the Cl− and K+ conductances.
In this study, we have used pharmacological antagonists to distinguish the origin of the Ca2+ responsible for activation of Ca2+-dependent afterpotentials in mouse sympathetic neurones. The experiments have utilized recordings from intact mouse SCG neurones, as the distribution of channels on the soma and dendrites is not uniform (e.g. Westenbroek et al. 1998) so that responses from dissociated somata will not necessarily reflect physiological events. Our data indicate that, as in rat SCG cells, the K+ channels responsible for the slow AHP are activated mainly by Ca2+ entering through N-type channels, whereas Cl− channel activation results from Ca2+ entering through L- and P-type Ca2+ channels. Ca2+-induced Ca2+ release from intracellular stores is a major contributor to Cl− channel activation but participates in K+ channel activation in only a subgroup of cells. Some of the results have been presented in abstract form (Martínez-Pinna et al. 1998).
Methods
The methods used for intracellular and voltage-clamp recording have been described previously (De Castro et al. 1997). Briefly, 5- to 7-week-old male mice (Swiss OF-1) were deeply anaesthetized by an i.p. injection of sodium pentobarbitone (40 mg kg−1) and perfused through the heart with cold oxygenated saline. The superior cervical ganglion was taken from the animal and pinned to the Sylgard (Dow-Corning, Midland, MI, USA) bottom of a chamber continuously perfused with saline (mM: NaCl, 128; KCl, 5; CaCl2, 2; NaH2PO4, 1; NaHCO3, 16; MgSO4, 1.2; and glucose, 5.5) equilibrated with 95 % O2-5 % CO2 (pH 7.4) at room temperature (22–25°C). The ganglion was illuminated from one side by a fine fibre optic light source, allowing the cells on the surface to be seen using a microscope (× 375–600). The experimental procedures were in accordance with the guidelines of the ethics committee of the Instituto de Neurociencias de la Universidad Miguel Hernández-CSIC.
Potentials were measured with respect to a Ag-AgCl pellet submerged in the outflow section of the bath. Cells were impaled with microelectrodes filled with 3 M KCl (50–90 MΩ) and data were collected only if the neurone generated APs of at least 70 mV in amplitude at the resting potential. The sampling frequency for discontinuous single-electrode current and voltage clamp was 2–6 kHz, with a duty cycle of 30/70 (Axoclamp 2B, Axon Instruments Inc.). Capacitance compensation was continuously monitored and adjusted to ensure head stage settling. Data were digitized and stored on a computer for subsequent analysis using commercial software (pCLAMP6, Axon Instruments Inc.). After impaling a cell, we waited for the membrane potential to stabilize and then measured the basic electrical properties at the resting membrane potential, including the amplitude and duration of the afterpotentials. Action potential half-width was measured as the duration at half the peak amplitude. Input resistance was calculated from the linear part of the I-V relationship determined near the end of 200 ms hyperpolarizing current pulses. APs were initiated after a 5 ms depolarizing current pulse or by a train, usually of six pulses at 40 Hz, and the ensuing afterpotentials were examined. To test the effect of the different drugs, we adjusted the membrane potential to −55 mV by passing current in discontinuous current-clamp mode before making measurements of APs and tail currents. Generally, we averaged five sweeps when a single AP was evoked and three when recording a train of discharges. To enable the degree of block to be quantified, cells were selected with relatively large outward tail currents (measures of gK,Ca1) or inward tail currents (measures of gCl,Ca) so that their responses are not necessarily representative of those of all SCG neurones.
Tail currents underlying the afterpotentials were measured using a hybrid clamp protocol in which APs were evoked with depolarizing current pulses and the amplifier was switched to voltage clamp mode, either after repolarization of a single AP, or 10 ms after the end of a brief train at 40 Hz. Tail currents were recorded under voltage clamp at −55 mV after filtering at 0.3 kHz and were analysed using commercial software (Origin, Microcal Software Inc., Northampton, MA, USA). After digital smoothing using a 9-point running average, the peak values were measured and the currents integrated from 20 ms after the peak of the AP in order to derive the amount of charge transferred. To test the effects of increasing Ca2+ loads on the current underlying the ADP, we could not use trains of > 10 APs because large ADPs were generated that reduced AP amplitude by inactivating Na+ currents. Therefore, for these experiments, the cells were voltage clamped in the presence of 1 μm TTX and 100 nm apamin and trains of 5 ms pulses (to 0 mV from a holding potential of −55 mV) of varying duration at 40 Hz were applied. All values are expressed as means ±s.e.m. Statistical comparisons were made using Student's paired t test (Sigmastat, Jandel Scientific Software, Erkrath, Germany).
All drugs were obtained from Sigma, except nifedipine (RBI, Natick, MA, USA) and ω-agatoxin IVA (Peptides International, KY, USA), and were applied dissolved in the perfusing solution. Cytochrome c (0.1 mg ml−1) was added to the toxin stock solutions to saturate non-specific peptide binding to the apparatus. Care was taken when using nifedipine to protect it from breakdown by light. The antagonists were used at concentrations that are relatively selective for different channel types. ω-Conotoxin GVIA blocks N-type channels quite selectively with an IC50 of 1–40 nm (Regan et al. 1991; Boland et al. 1994). ω-Agatoxin IVA blocks both P- and Q-type channels but with very different IC50 values of 1–20 nm and ∼100 nm, respectively (Mintz et al. 1992; Randall & Tsien, 1995). Dihydropyridines block neuronal L-type channels with an IC50 of 0.3–2 μm (Jones & Jacobs, 1990; Regan et al. 1991). We usually applied a relatively high concentration of 10 μm, which is commonly used in intact neurones to ensure full blockade. This concentration has no non-specific actions in rat SCG cells (Davies et al. 1996). It should be noted, however, that 1 and 10 μm nifedipine had similar effects in the present experiments. Application of the drugs or toxins had significant effects on the input resistance, time constant, the holding current required to maintain the resting potential at −55 mV or the amplitude and duration of the AP only in the cases that are noted specifically.
Results
Properties of SCG neurones of the mouse
The electrical properties of 71 mouse SCG neurones measured in control solution were: resting membrane potential, −51 ± 1.0 mV; AP amplitude, 99 ± 1.2 mV; AP half-width, 2.4 ± 0.1 ms; input resistance, 54 ± 3.1 MΩ; capacitance, 171 ± 10 pF; and time constant, 8.6 ± 0.6 ms.
Depolarizing and hyperpolarizing afterpotentials following AP firing varied greatly between neurones. In all cells, the fast AHP after repolarization of the AP was followed by a slow AHP (Fig. 1B). The slow AHP was separated from the fast AHP by a depolarizing inflection in 22 % of the cells (Fig. 1B, arrow) including those cells (6 %) in which the peak of this inflection was positive to the resting membrane potential, i.e. an ADP (Fig. 5Aa; see also De Castro et al. 1997). In all but six of the remaining cells, the transition from fast to slow AHP was marked by a change in the rate of depolarization (Fig. 2B a, arrow). A train of six APs at 40 Hz generated an ADP followed by a slow AHP in 46 % of cells (Fig. 1C; see also De Castro et al. 1997). In another 31 % of cells, the membrane potential at the end of the train was more positive than the initial fast AHP (Fig. 3Ab), indicating that a depolarizing conductance had been activated during the train. In the remaining cells, the train generated a slow AHP that built up from the fast AHP. Thus, the relative magnitude of the conductances underlying the AHP and ADP differed between cells.
Figure 1. Ca2+-dependent afterpotentials in mouse sympathetic ganglion cells.
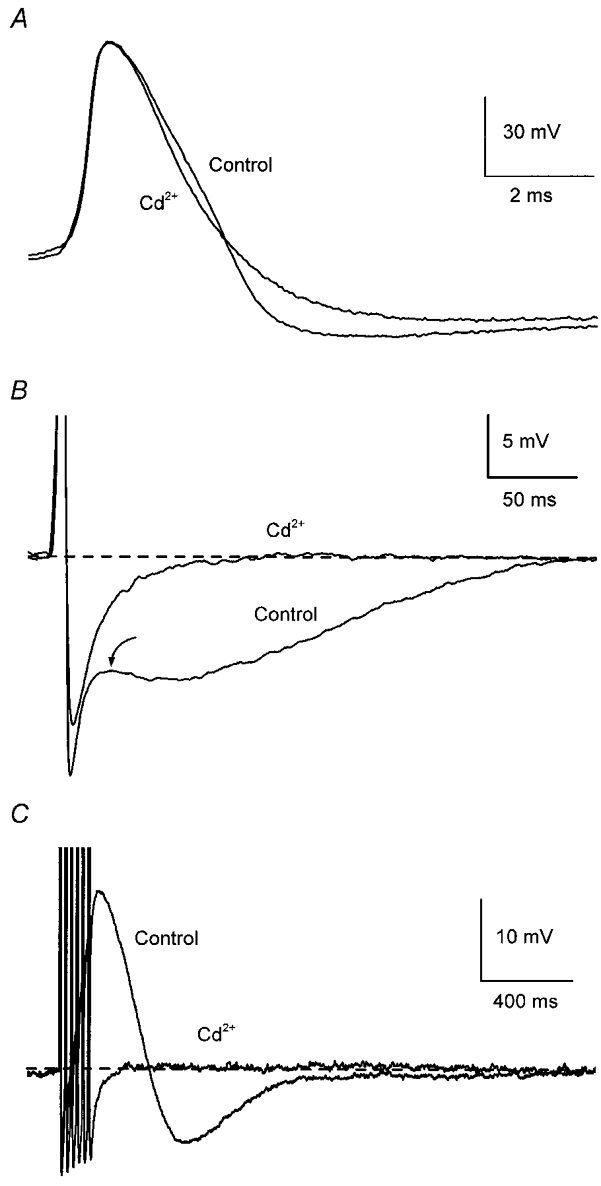
Records from a neurone in the superior cervical ganglion. A, addition of 500 μm Cd2+ reduced action potential (AP) half-width without affecting its amplitude. B, addition of Cd2+ abolished the depolarizing inflection following AP repolarization (arrow) and markedly reduced the afterhyperpolarization (AHP) following a single AP. C, addition of Cd2+ abolished the afterdepolarization (ADP) and reduced the AHP after a train of six APs at 40 Hz. APs in B and C are truncated (as in subsequent figures) and traces in control and test solutions have been superimposed with labels placed closest to the relevant trace. In these, and in the current-clamp records in subsequent figures, the dashed line indicates the baseline membrane potential, which was maintained close to −55 mV by passing current through the microelectrode.
Figure 5. L-type Ca2+ channels contribute to activation of the ADP but have little effect on the AHP.
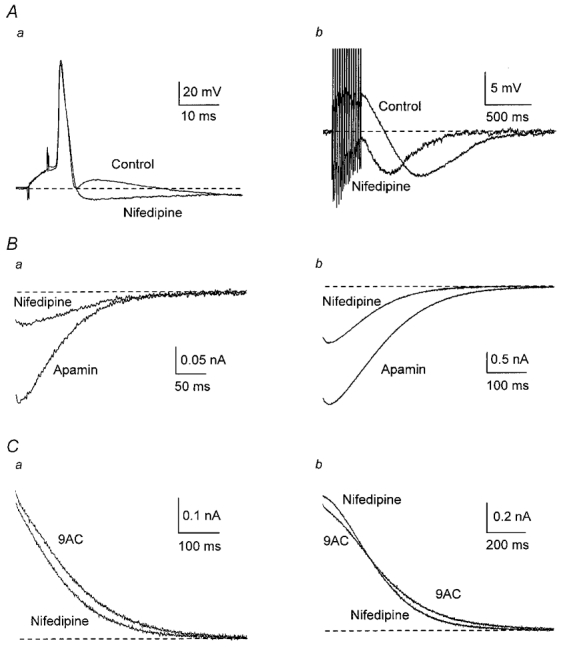
A, addition of 10 μm nifedipine abolished the ADP revealing an AHP after one AP (a) and reduced the ADP without affecting AHP amplitude after a train of 13 APs (b). B, averaged inward currents from nine cells in apamin were reduced when nifedipine was added. C, averaged outward currents from 11 cells in the presence of 9AC showed, when nifedipine was added, a small decrease after a single AP (a) and a change in the time course after a train of APs (b).
Figure 2. Apamin and 9AC reveal the Cl− and K+ currents underlying afterpotentials.
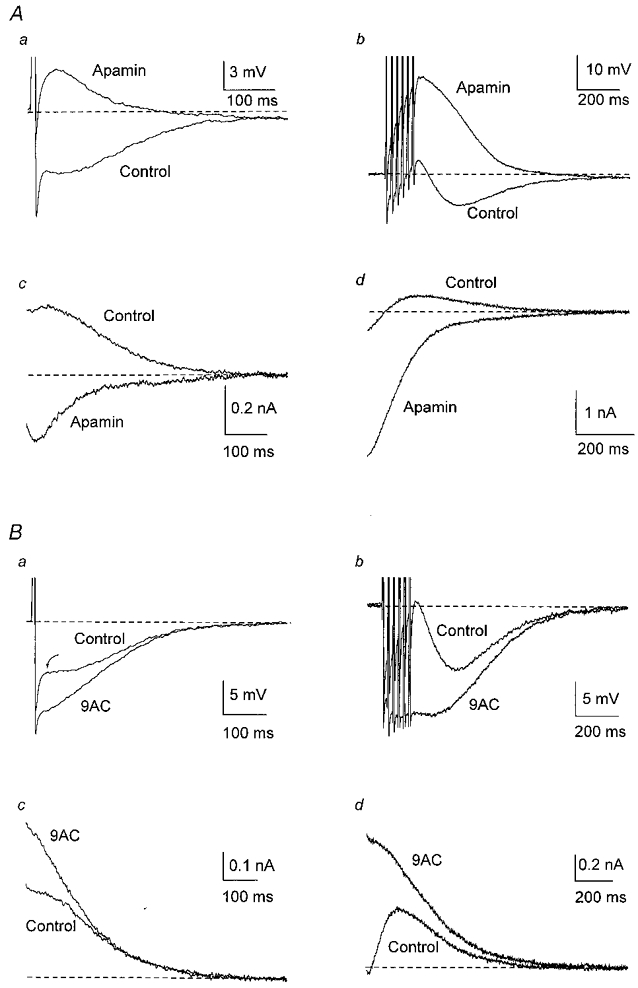
Responses following a single AP are shown on the left (a) and after a train of six APs at 40 Hz on the right (b) in this and subsequent figures. A, in one cell, addition of apamin (100 nm in this and subsequent figures) abolished the slow AHP, revealing an ADP after one AP (a) and enhancing the ADP after a train of six APs at 40 Hz (b). In the same neurone, outward tail currents following a single AP (c) or a train of APs (d) were blocked by apamin, revealing inward tail currents. B, in another cell, addition of 9AC (2 mm in this and subsequent figures) reduced the depolarizing inflexion in the AHP after a single AP (a) and the small ADP following a train of APs (b), enhancing the AHP. Arrow points to the transition from fast to slow AHP. In the same neurone, 9AC blocked the inward currents after a single AP (c) and after a train of APs (d), enhancing the outward tail currents. In these, and in voltage-clamp records in the following figures, the dashed line indicates the holding current needed to keep the membrane potential at −55 mV. In this, and in the following figures except Fig. 5Ab, the trains consisted of six APs at 40 Hz.
Figure 3. N-type Ca2+ channels contribute to activation of the AHP but not the ADP.
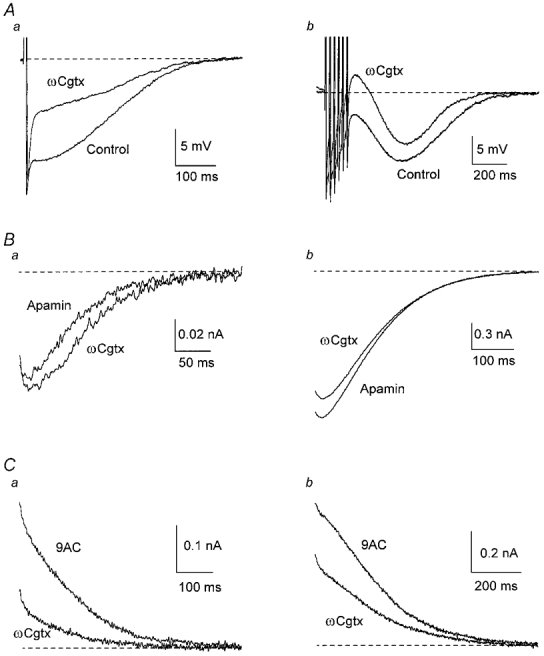
A, in one cell, addition of 300 nmωCgtx GVIA reduced the AHP, therefore increasing the ADP. B, averaged inward currents from six cells in the presence of apamin showed little variation when ωCgtx GVIA was added. C, averaged outward currents from five cells in the presence of 9AC were reduced when ωCgtx GVIA was added.
Afterpotentials depend on Ca2+ entry
As in other mammalian SCG cells (Adams & Harper, 1995), blockade of Ca2+ entry by addition of Cd2+ (100–500 μm) to the perfusate (n = 9) abolished both of the slower afterpotentials (when present) after a single AP (Fig. 1B) or a train of APs (Fig. 1C). Cd2+ also produced a 32 % decrease in AP half-width (Fig. 1A; control, 2.2 ± 0.20 ms; Cd2+, 1.5 ± 0.10 ms; P < 0.01).
Blockade of SK channels with apamin reveals gCl,Ca
As in the rat SCG (Kawai & Watanabe, 1986), apamin (100 nm) largely abolished the slow AHP (Fig. 2A). In 52 % of 37 cells, addition of apamin to the control solution revealed an ADP after a single AP (Fig. 2Aa) and the same occurred after a train of APs in all but one cell, including those neurones in which only an AHP was detected in control solution. Apamin increased the amplitude and duration of ADPs after a train (Fig. 2Ab), generating repetitive discharges in 11 cells. In contrast, very rarely one or two APs occurred during a large ADP in control solution. These data indicate that, in most cells that lacked an ADP in control solution, activation of the Cl− conductance after a single discharge was obscured by the K+ conductance underlying the AHP.
When the cells were voltage clamped in control solution, the currents recorded at −55 mV after an AP had different shapes due to the variable amounts of inward Cl− and outward K+ conductances. There was usually an early inward inflection or plateau in the outward current after a single AP (Fig. 2Ac and Bc) but, after a train, a small net inward current of variable amplitude was generated and was followed by an outward current (Fig. 2Ad and Bd). In the presence of apamin (100 nm), the outward tail currents were largely blocked, revealing a clear inward current after one AP (Fig. 2Ac) and increasing markedly the inward current that followed a train (Fig. 2Ad). The enhancement of the inward current after a train resulted in as much as 40 times the total charge transfer as that following a single AP (Table 1).
Table 1.
Effect of ωCgtx GVIA, ωAga IVA, nifedipine and ryanodine on gCl,Ca in the presence of apamin
| Single action potential | Action potential train | |||
|---|---|---|---|---|
| Current peak (nA) | Total charge (pC) | Current peak (nA) | Total charge (pC) | |
| Apamin (n = 6) | −0.08 ± 0.03 | −6.7 ± 3.7 | −1.7 ± 0.4 | −279 ± 69 |
| Apamin +ωCgtx GVIA | −0.09 ± 0.04 | −8.6 ± 5.1 | −1.5 ± 0.4 | −255 ± 80 |
| Apamin (n = 10) | −0.11 ± 0.01 | −8.4 ± 0.9 | −2.0 ± 0.3 | −334 ± 58 |
| ZApamin +ωAga IVA | −0.06 ± 0.01 ** | −3.5 ± 0.9 ** | −1.2 ± 0.2 ** | −165 ± 36 ** |
| Apamin (n = 9) | −0.22 ± 0.06 | −20.9 ± 7.6 | −3.0 ± 0.6 | −490 ± 119 |
| Apamin + nifedipine | −0.09 ± 0.03 ** | −6.6 ± 2.6 * | −1.5 ± 0.4 ** | −184 ± 53 ** |
| Apamin (n = 5) | −0.24 ± 0.05 | −30.9 ± 7.9 | −2.8 ± 0.9 | −481 ± 158 |
| Apamin + ryanodine | −0.09 ± 0.05 * | −7.6 ± 4.4 * | −2.2 ± 0.8 * | −319 ± 131 * |
Tail currents after a single action potential or after a train of six action potentials at 40 Hz were recorded in voltage clamp at a holding potential of −55 mV. Inward Ca2+activated Cl− currents were isolated by blocking gK,Ca1 with 100 nm apamin. Changes after adding nifedipine (10 μm), ωCgtx GVIA (300 nm),ωAga IVA (40 nm) or ryanodine (20 μm) were compared using Student's paired t test
P < 0.05
P < 0.01.
Thus application of 100 nm apamin reveals the inward Cl− current, providing a measure of the Ca2+-activated Cl− conductance, gCl,Ca (see Introduction). In all the voltage-clamp experiments on gCl,Ca described below, 100 nm apamin was present throughout.
In 14 cells in which electrical properties were measured in control solution, apamin decreased the holding current needed to keep the resting membrane potential at −55 mV (from −0.43 ± 0.10 to −0.29 ± 0.09 nA, P < 0.05) and also increased input resistance (from 43 ± 4 to 60 ± 8 MΩ, P < 0.05). Apamin also increased AP amplitude (from 94 ± 2 to 101 ± 2 mV, P < 0.01) but reduced AP duration (from 2.6 ± 0.16 to 2.3 ± 0.13 ms, P < 0.01). These results suggest that apamin blocks a K+ conductance that is active at −55 mV. Such a conductance was not detected in rat (Davies et al. 1996) or guinea-pig (Ireland et al. 1998) sympathetic neurones.
Blockade of Cl− channels with 9AC reveals gK,Ca1
As reported previously (De Castro et al. 1997), addition of the Cl− channel blocker anthracene-9-carboxylic acid (9AC; 2 mm, n = 21) substantially reduced the amplitude of the ADP, increasing the amplitude and duration of the AHP (Fig. 2Ba and b) and the outward current carried by gK,Ca1 (Fig. 2Bc and d). In general, the degree of block produced by 9AC was more marked after a single AP than after a train. The enhancement of the outward current after the train resulted in about 4 times as much total charge transfer as that following a single AP (Table 2).
Table 2.
Effects of ωCgtx GVIA, ωAga IVA, nifedipine and ryanodine on gK,Ca1 in the presence of 9AC
| Single action potential | Action potential train | |||
|---|---|---|---|---|
| Current peak (nA) | Total charge(pC) | Current peak(nA) | Total charge(pC) | |
| 9AC (n = 5) | 0.32 ± 0.06 | 41.9 ± 12.4 | 0.72 ± 0.08 | 190 ± 31 |
| 9AC +ωCgtx GVIA | 0.14 ± 0.03 ** | 12.1 ± 4.2 * | 0.46 ± 0.08 * | 111 ± 31 ** |
| 9AC (n = 5) | 0.47 ± 0.06 | 75.5 ± 16.7 | 0.80 ± 0.08 | 329 ± 48 |
| 9AC +ωAga IVA | 0.46 ± 0.05 | 74.0 ± 11.4 | 0.90 ± 0.08 | 371 ± 36 |
| 9AC (n = 11) | 0.44 ± 0.03 | 62.2 ± 4.8 | 0.87 ± 0.05 | 290 ± 24 |
| 9AC + nifedipine | 0.41 ± 0.03 | 50.0 ± 5.3 * | 0.94 ± 0.07 | 277 ± 21 |
| 9AC (n = 7) | 0.39 ± 0.03 | 82.7 ± 17.7 | 0.66 ± 0.07 | 352 ± 64 |
| 9AC + ryanodine | 0.33 ± 0.03 | 60.6 ± 8.0 | 0.63 ± 0.09 | 298 ± 47 |
Tail currents after a single action potential or after a train of six action potentils at 40 Hz were recorded in voltage clamp at a holding potential of −55 mV. Outward Ca2+activated K+ currents were isolated by blocking gCl,Ca with 2 nm. Changes after adding nifedipine (10 μm), ωCgtx GVIA (300 nm), ωAga IVA (40 nm) or ryanodine (20 μm) were compared using Student's paired t test
P < 0.05
P < 0.01.
Thus, application of 2 mm 9AC reveals the outward K+ current, providing a measure of the Ca2+-activated K+ conductance gK,Ca1 (see Introduction). In all the voltage-clamp experiments on gK,Ca1 described below, 2 mm 9AC was present throughout.
N-type Ca2+ channel block has no effect on gCl,Ca but reduces gK,Ca1
The N-type channel antagonist ω-conotoxin GVIA (ωCgtx GVIA, 300 nm) decreased the AHP in all cells tested (n = 9,Fig. 3Aa) and increased the ADP following a train of APs (Fig. 3Ab). On average, the membrane voltage measured at the end of the train shifted from −2.4 ± 6.1 mV to 4.2 ± 5.6 mV (P < 0.001). These results suggest that blockade of Ca2+ entry through N-type channels decreased activation of gK,Ca1, as occurs in rat SCG neurones (Davies et al. 1996), allowing for a greater effect of gCl,Ca in depolarizing the membrane following AP discharge.
N-type channels were not involved in the activation of gCl,Ca because 300 nmωCgtx GVIA had no significant effect on the mean inward current evoked after a single AP or a train of APs (n = 6; Fig. 3B and Table 1; see also Fig. 11A).
Figure 11. Percentage block of Ca2+-activated Cl− and K+ conductances following different treatments suggests the mechanisms of their activation by APs.
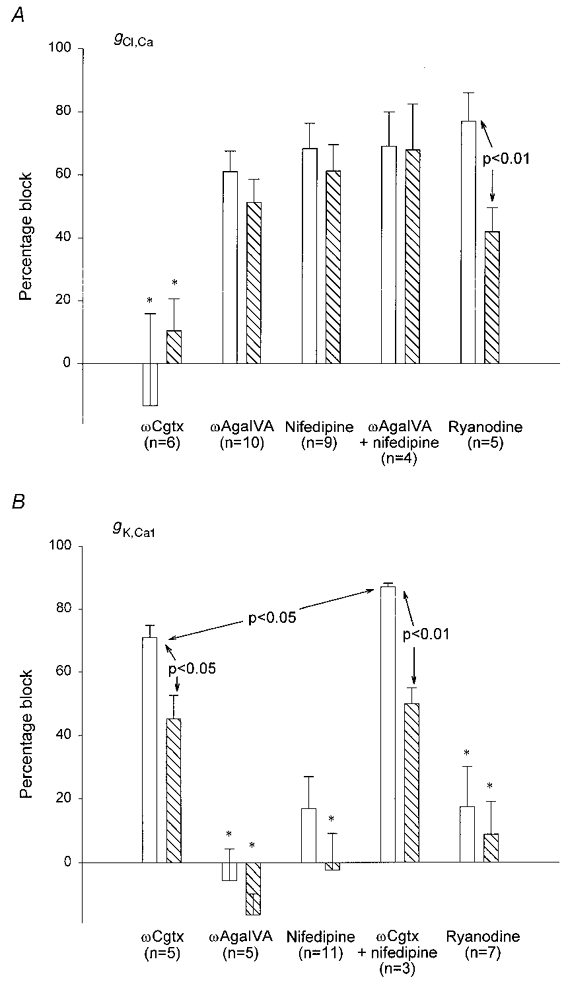
Changes in gCl,Ca (A) and changes in gK,Ca1 (B) (measured as total charge transfer) produced by ωCgtx GVIA, ωAga IVA, nifedipine and ryanodine (see also Tables 1 and 2). □, effect after a single AP;  , effect following a train. Data were obtained in the presence of 100 nm apamin (A) or 2 mm 9AC (B). Error bars indicate s.e.m. Statistically significant comparisons between different groups are indicated. All changes were significantly different from control (in the absence of any Ca2+ current blocker) except those marked * (see Tables 1 and 2). gCl,Ca was blocked to a similar extent by all treatments except ωCgtx GVIA. The data suggest that Ca2+ from P- and L-type channels must act together to release Ca2+ from intracellular stores that, in turn, activates Cl− channels underlying the ADP. In contrast, ωCgtx GVIA markedly reduced gK,Ca1, suggesting a direct link between Ca2+ entering through N-type channels and SK channels underlying the AHP. A small amount of Ca2+ entry through L-type channels also contributes to activation of SK channels in type II cells. See text for further discussion.
, effect following a train. Data were obtained in the presence of 100 nm apamin (A) or 2 mm 9AC (B). Error bars indicate s.e.m. Statistically significant comparisons between different groups are indicated. All changes were significantly different from control (in the absence of any Ca2+ current blocker) except those marked * (see Tables 1 and 2). gCl,Ca was blocked to a similar extent by all treatments except ωCgtx GVIA. The data suggest that Ca2+ from P- and L-type channels must act together to release Ca2+ from intracellular stores that, in turn, activates Cl− channels underlying the ADP. In contrast, ωCgtx GVIA markedly reduced gK,Ca1, suggesting a direct link between Ca2+ entering through N-type channels and SK channels underlying the AHP. A small amount of Ca2+ entry through L-type channels also contributes to activation of SK channels in type II cells. See text for further discussion.
In contrast, ω-Cgtx GVIA reduced gK,Ca1, the effect being larger on the outward tail current evoked by a single AP (Fig. 3Ca) than on that following a train (Fig. 3Cb), when measured as either charge transfer or mean peak outward current (n = 5; Table 2; see also Fig. 11B). Thus, Ca2+ entering through N-type channels activates gK,Ca1.
P-type Ca2+ channel block reduces gCl,Ca but has no effect on gK,Ca1
Relatively selective blockade of P-type channels can be achieved by using ω-agatoxin IVA (ωAga IVA) at concentrations < 50 nm, as the IC50 for Q-type channels is ∼100 nm (Randall & Tsien, 1995). Application of ωAga IVA (40 nm, n = 3) always enhanced the AHP, particularly after a train of APs (Fig. 4A), suggesting that P-type channel block reduces the activation of gCl,Ca without having much effect on gK,Ca1.
Figure 4. P-type Ca2+ channels contribute to activation of the ADP but not the AHP.
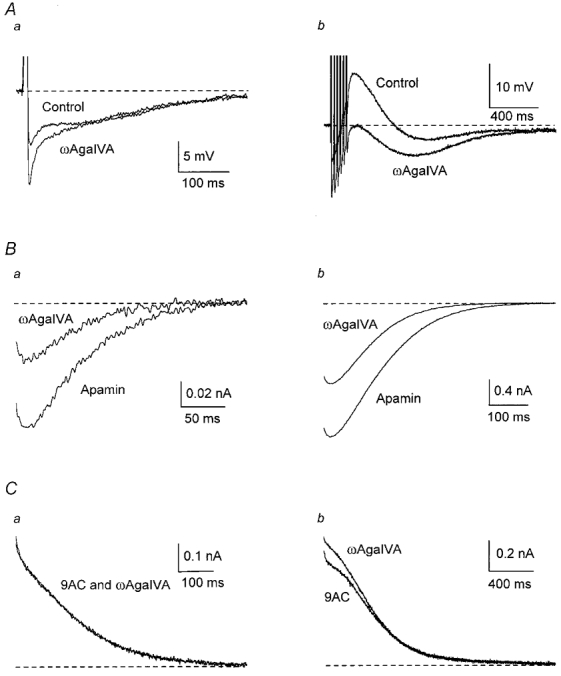
A, in one cell, addition of ωAga IVA (40 nm) decreased the ADP, therefore increasing the AHP. B, averaged inward currents from 10 cells in the presence of apamin were partially blocked by ωAga IVA. C, averaged outward currents from five cells in the presence of 9AC showed little variation when ωAga IVA was added.
In 10 cells, 40 nmωAga IVA decreased peak inward current by 45 % and charge transfer by about 60 % after a single AP, the effect being slightly smaller after a train (Fig. 4B and Table 1; see also Fig. 11A). The peak inward current after a single AP was not affected by 20 nmωAga IVA whereas peak current was reduced by 53 % and charge transfer by 61 % in 200 nmωAga IVA (n = 3). This latter change was not significantly different from the effect of 40 nmωAga IVA, suggesting that the toxin's action was more likely to have occurred by blockade of P-type than Q-type channels.
In contrast outward currents were not decreased by 40 nmωAga IVA in any of the five cells studied (Fig. 4C and Table 2; see also Fig. 11B). These data indicate that Ca2+ entering through P-type channels has no effect on gK,Ca1.
L-type Ca2+ channel block reduces gCl,Ca but has little effect on gK,Ca1
In 12 cells, the L-type Ca2+ channel antagonist nifedipine (10 μm) reduced or abolished the ADP that followed APs (Fig. 5A). In contrast, the peak amplitude of the slow AHP increased, consistent with the majority of the Ca2+ for activation of the slow AHP entering through channels other than L-type ones.
The amplitude and duration of gCl,Ca were substantially reduced by 10 μm nifedipine (n = 9), both after a single AP and after a train (Fig. 5B and Table 1; see also Fig. 11A). These results suggest that, during an AP, the great majority of Ca2+ entering through L-type channels is directed towards activation of Cl− channels with little effect on gK,Ca1.
In agreement with this idea, nifedipine (n = 11) had no significant effect on the peak amplitude of the outward current after a single AP, although it produced a small significant reduction in charge transfer (Fig. 5Ca and Table 2; see also Fig. 11B). There was no significant effect after a train of APs (Table 2). However, the responses to nifedipine differed between cells, which could be separated into two distinct groups on the basis of the time course of decay of gK,Ca1 (Fig. 6). In six of the 11 cells (type I), outward currents initially fell either above (Fig. 6Aa) or on the exponential fitted to the later decay of gK,Ca1. In the remaining five cells (type II), there was a plateau during the early part of the outward current, so that the data fell below the extrapolated exponential (Fig. 6Ba; see also Fig. 8Ca). In type I cells, nifedipine had no significant effect on gK,Ca1 after one AP and produced a small increase in the peak outward current after a train (from 0.87 ± 0.09 to 1.00 ± 0.08 nA, P < 0.05). This increase could be due to concurrent block of residual gCl,Ca (due to incomplete block by 9AC). In type II cells, nifedipine decreased gK,Ca1 after one AP (Fig. 6B), blocking 35 % of the charge transfer (from 69 ± 6 to 45 ± 10 pC, P < 0.05) and 17 % of the peak current (from 0.42 ± 0.04 to 0.35 ± 0.06 nA, P < 0.05), but had no significant effect on gK,Ca1 after a train. This suggests that, with low Ca2+ loads, such as those generated by a single AP, L-type channels are involved in the activation of gK,Ca1 only in type II SCG neurones but not in type I neurones.
Figure 6. L-type Ca2+ channels contribute to activation of gK,Ca1 in a subgroup of neurones.
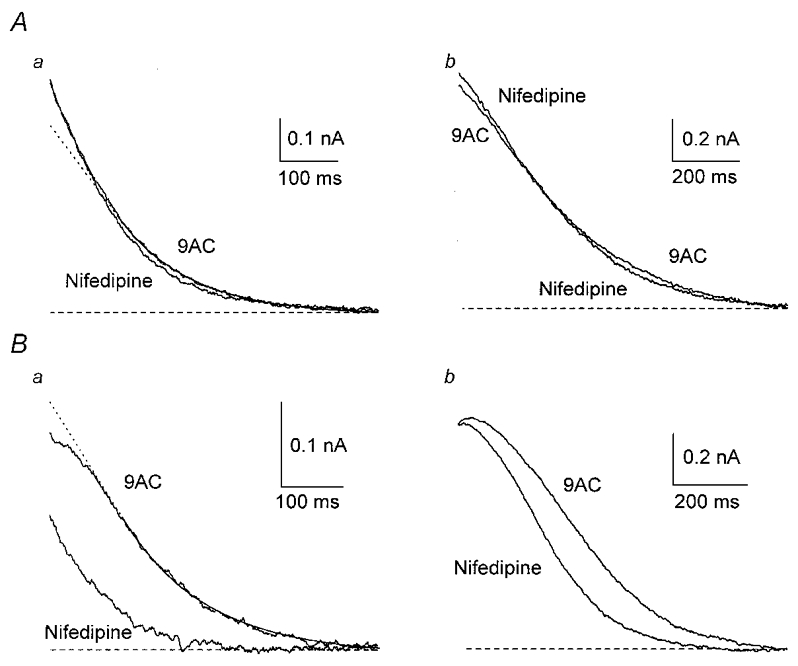
Two groups of cells were identified on the basis of the time course of the outward current recorded in the presence of 9AC. A, response of type I cells. When an exponential function was fitted to the outward current, starting 150 ms after the AP (continuous line), and extrapolated to the initial part (dotted line in this and subsequent figures), the outward current of type I cells either fitted the exponential well or initially fell above it (a). When 10 μm nifedipine was added, the outward current from a type I cell was not affected after one AP (a) and showed a small increase in peak current after a train (b). B, response of type II cells. The outward current of type II cells initially fell below the extrapolated exponential (a). In one type II cell, nifedipine markedly reduced gK,Ca1 after one AP (a) and had a smaller effect on the latter part of the current after a train (b).
Figure 8. Ca2+-induced Ca2+ release contributes to activation of gK,Ca1 only in type II cells.
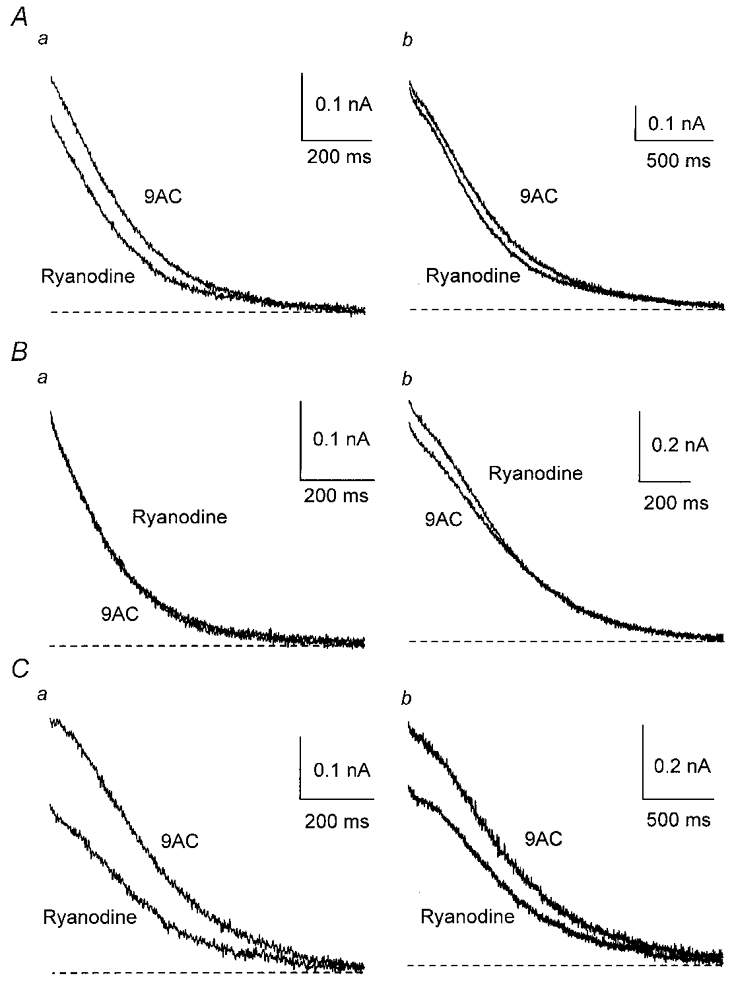
A, ryanodine (20 μm), applied after 9AC, had no significant effect on the averaged outward currents from seven cells. When the cells were separated into two groups as in Fig. 6, ryanodine did not significantly affect the averaged outward currents from four type I cells (B) but decreased gK,Ca1 in three type II cells (C) after a single AP (a) and after a train (b).
Ryanodine reduces gCl,Ca but has little effect on gK,Ca1
To investigate the contribution of Ca2+-induced Ca2+ release (CICR) in the activation of gCl,Ca and gK,Ca1, we applied ryanodine (20 μm), which blocks Ca2+ release from intracellular stores (Meissner, 1994).
Ryanodine (n = 5) markedly decreased the inward current following an AP and to a lesser extent that following a train (Figs 7A and 11A, Table 1). This suggests that, after a single AP, gCl,Ca is activated predominantly by CICR but, as intracellular Ca2+ concentration increases, the proportion of Cl− channels activated directly by Ca2+ entering the cell also increases. When the cells were challenged with pulse trains of increasing duration, the proportion of the peak inward current blocked by ryanodine decreased from about 75 % for a single pulse to about 35 % for the longer trains (n = 5; Fig. 7B). Thus, CICR plays an important role in activating gCl,Ca when intracellular Ca2+ concentration is low (i.e. in the physiological range).
Figure 7. Ca2+-induced Ca2+ release contributes to activation of gCl,Ca.
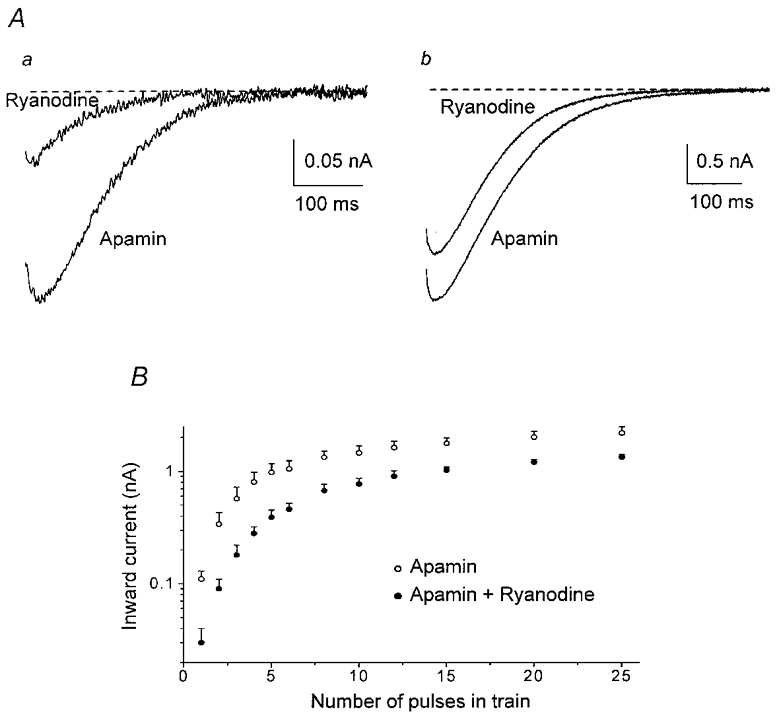
A, averaged inward currents from five cells in the presence of apamin were markedly reduced after one AP when 20 μm ryanodine was added (a) but less so after a train (b). B, mean inward currents from five cells following 5 ms depolarizing pulses (to 0 mV from −55 mV) applied at 40 Hz, plotted against the number of pulses in the train. The reduction in peak current was larger (75 %) when one or two pulses were applied than when the train was prolonged (35 %). Note the logarithmic ordinate. Error bars show s.e.m.
In contrast, ryanodine did not significantly affect the averaged outward currents (n = 7) recorded in 9AC (Fig. 8A and Table 2; see also Fig. 11B). However, if the above criteria were used to separate the cells on the basis of the decay of gK,Ca1, the responses to ryanodine differed between type I and type II cells. In four type I cells, ryanodine had no significant effect (Fig. 8B) but in three type II cells (Fig. 8C) ryanodine clearly decreased the outward current after one AP (charge transfer, 46 ± 1.1 % block, P < 0.01) and to a smaller degree after a train (32 ± 7.0 % block, P < 0.05). This suggests that CICR contributes to activation of gK,Ca1 only in type II cells, in which it delays the decline in the outward current after an AP, whereas gK,Ca1 is probably activated directly by Ca2+ entering through N-type channels in type I cells.
The effects of ryanodine on gCl,Ca and on gK,Ca1 in type II cells were almost identical to those of nifedipine. In other cell types, activation of CICR commonly occurs via the Ca2+ that enters through L-type channels (Shoshan-Barmatz & Ashley, 1998). We therefore tested the effects of adding ryanodine on the residual gCl,Ca and gK,Ca1 that remained after application of 10 μm nifedipine. In four cells, nifedipine blocked gCl,Ca as described above, but subsequent application of ryanodine produced no significant change in gCl,Ca (Fig. 9A). Similarly, nifedipine decreased gK,Ca1 in four type II cells, but ryanodine did not reduce the current further (Fig. 9B). Thus, Ca2+ entry through L-type channels triggers the ryanodine-sensitive mechanism that activates gCl,Ca and, in type II cells, gK,Ca1.
Figure 9. Ca2+-induced Ca2+ release is activated by Ca2+ entering through L-type channels.
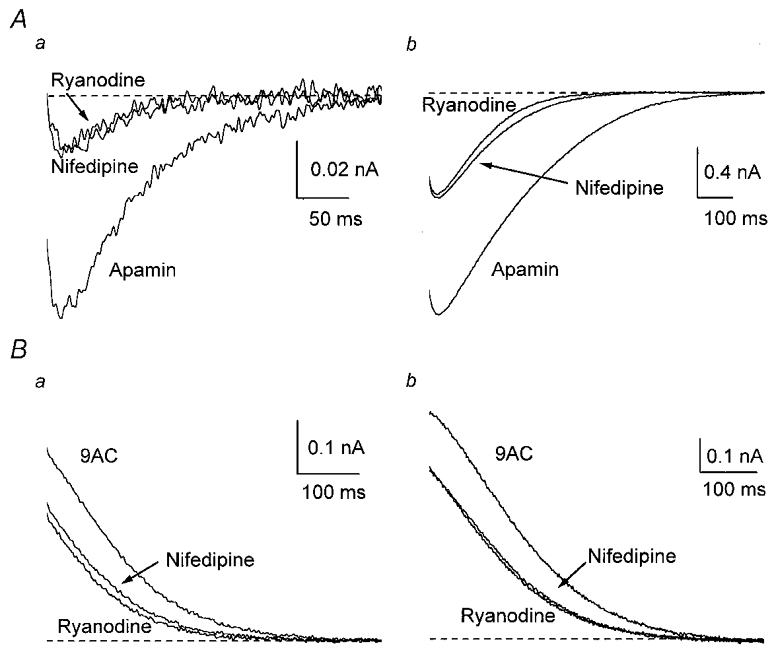
A, averaged inward tail currents from four cells in the presence of apamin were decreased by 10 μm nifedipine, but showed no significant reduction when 20 μm ryanodine was added. B, averaged outward tail currents from four type II cells were decreased by nifedipine, but not by subsequent addition of ryanodine.
Resistant Ca2+ channels contribute to both gCl,Ca and gK,Ca1
Because the degree of block of gCl,Ca was similar (about 65 %) when either P- or L-type channels were blocked, we examined whether R-type channels were involved in the activation of gCl,Ca by applying both ωAga IVA (40–200 nm) and nifedipine (1–10 μm) in the presence of 100 nm apamin (n = 4). After a single AP, charge transfer was blocked by about 70 % and peak current by about 65 %, whereas after a train, block was about 70 and 60 %, respectively (Fig 10A and Fig 11A). Given the lack of effect of N-type channel blockers on gCl,Ca, this result indicates that R-type channels contributed to activation of the inward current.
Figure 10. R-type channels contribute to activation of the currents underlying afterpotentials.
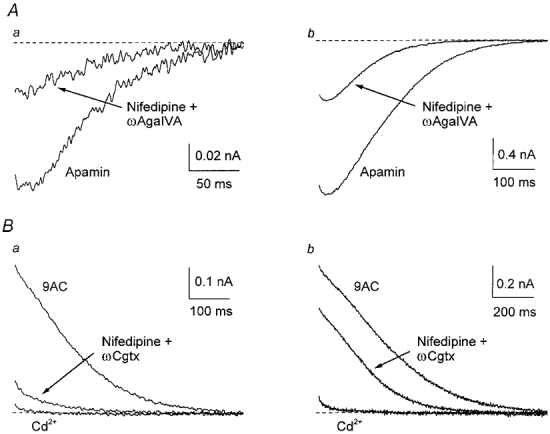
A, averaged inward tail currents from four cells in the presence of apamin were markedly reduced, but not completely abolished, in the presence of 40 nmωAga IVA and 10 μm nifedipine. B, averaged outward tail currents from three cells in the presence of 9AC were markedly reduced, but not completely blocked, in the presence of 10 μm nifedipine and 300 nmωCgtx GVIA. Subsequent addition of 300 μm Cd2+ blocked the remaining outward currents.
To test whether Ca2+ entering through R-type channels also activates gK,Ca1, we applied nifedipine (10 μm) and ωCgtx GVIA (300 nm) in the presence of 2 mm 9AC (n = 3). Overall block of the outward current was much greater after a single AP (charge transfer about 85 %, peak current about 75 %) than after a train (charge transfer about 50 %, peak current 30 %; Fig 10B and Fig 11B), but there was clearly a fraction of gK,Ca1 resistant to both drugs, even after a single AP, because 300 μm Cd2+ blocked the remaining current. Since P-type channel block did not affect gK,Ca1, this result suggests that Ca2+ entry through R-type channels contributes to activation of gK,Ca1.
Discussion
Our main conclusion is that activation of the Cl− conductance (gCl,Ca) responsible for the ADP following APs in mouse SCG neurones involves Ca2+ entering through P- and L-type channels and acting via Ca2+ release from ryanodine-sensitive intracellular stores. In contrast, the K+ conductance responsible for the slow AHP (gK,Ca1) is activated predominantly by Ca2+ entering through N-type channels. Assuming that our pharmacological tools provide selective and complete blockade, we can conclude that a smaller proportion of both currents is activated directly by Ca2+ entering through R-type channels.
These conclusions rest on the assumption that nifedipine, ωCgtx GVIA and ωAga IVA did not directly block Cl− or K+ channels. None of these drugs had effects on AP amplitude or passive properties, indicating that they do not interfere with Na+ currents or most K+ currents as previously shown in rat SCG neurones (Davies et al. 1996). This is in agreement with the known specificity of ωCgtx GVIA for N-type Ca2+ channels (Olivera et al. 1994) and of ωAga IVA for P/Q-type channels (Randall & Tsien, 1995). Also, ryanodine did not directly affect Ca2+, K+ or Cl− channels because AP shape and input resistance were not modified. Furthermore, the currents generated by gK,Ca1 in type I cells were not reduced in the presence of ryanodine.
Activation of gCl,Ca
Our results after blocking gK,Ca1 with apamin demonstrate that most, if not all, normal mouse SCG neurones express gCl,Ca, as suggested by De Castro et al. (1997). Also, when 9AC was applied to cells that apparently lacked an ADP, the AHP became larger, indicating that blockade of gCl,Ca allowed gK,Ca1 to hyperpolarize the membrane further.
The function of gCl,Ca in sympathetic neurones is not clear (e.g. Sánchez-Vives & Gallego, 1994). When SK channels were blocked with apamin, the ADP often generated APs, which is very rare in control solution (Sánchez-Vives & Gallego, 1994; De Castro et al. 1997). This suggests that decreases in the AHP, either indirectly following inhibition of N-type channels by e.g. catecholamines and neuropeptides (Toth & Miller, 1995), or directly following inhibition of SK-type channels by e.g. muscarinic agonists (Cassell & McLachlan, 1987b), might lead to repetitive discharge. Thus the Cl− conductance underlying the ADP is expressed to a greater or lesser extent in all mouse SCG neurones where it could play a role in generating bursts of activity. However, this mechanism to increase firing appears to be absent from normal sympathetic neurones of other species (Cassell & McLachlan, 1987a; Sánchez-Vives & Gallego, 1994).
Activation of gCl,Ca was dependent on Ca2+ entering through L-, P- and R-type channels (Fig. 11A). Functional coupling between L-type channels and Cl− channels in sensory neurones has been previously implied by the potentiation of Cl− tail currents by BayK8644, an L-type channel agonist (Scott et al. 1988). Although L- and N-type channels have been implicated in the activation of Ca2+-dependent Cl− currents in rabbit parasympathetic neurones (Akasu et al. 1990), ωCgtx GVIA did not affect gCl,Ca in mouse SCG neurones.
After one AP, gCl,Ca was blocked by about 70 % by ryanodine (Fig. 11A), indicating that CICR plays an important role in its activation. The fact that either L- or P-type channel blockade reduced gCl,Ca by approximately the same amount (Fig. 11A) suggests that Ca2+ entering through these channels activates CICR. The lack of effect of ryanodine on gCl,Ca and gK,Ca1 when applied after nifedipine confirms this for L-type channels, as has been shown in muscle cells (Shoshan-Barmatz & Ashley, 1998) and some neurones (Osmanovic & Shefner, 1993; Tanabe et al. 1998; Cordoba-Rodriguez et al. 1999). It appears, however, that both L- and P-type channels are needed simultaneously to trigger CICR in mouse SCG neurones, because a similar degree of block (70 %) was produced when the two drugs were applied together (Fig. 11A). As the amounts of Ca2+ entering through each type of channel during an AP are quite different (J. Martínez-Pinna & R. Gallego, unpublished observations), they presumably act co-operatively but non-linearly in activating CICR. As the amount of gCl,Ca (30 %) remaining after combined blockade of L- and P-type channels (Fig. 11A) was similar to the ryanodine-insensitive component, Ca2+ entry through R-type channels probably activates gCl,Ca directly.
The fact that the degree of block of gCl,Ca by nifedipine and ωAga IVA was similar after a single AP and after a train of six APs (when the peak inward current was about 16 times larger) implies that ‘spill over’ from channels other than L- and P-type was relatively ineffective in activating gCl,Ca. This suggests that CICR activation of gCl,Ca is spatially restricted (Berridge, 1998). This arrangement would allow CICR to deliver the concentration of Ca2+ necessary for activation of gCl,Ca because the Ca2+ sensitivity of the Cl− channels is relatively low (EC50∼25 μm; Gomez-Hernandez et al. 1997; Hallani et al. 1998). In contrast, after a train of six APs, ryanodine produced a smaller degree of block (40 %, Fig. 11A) than that produced by nifedipine and ωAga IVA (65 %), suggesting that Ca2+ entering through L- and P-type channels, as well as through R-type channels, can activate gCl,Ca directly when the neurone is overloaded with Ca2+ (see Fig. 7). Since sympathetic ganglion cells fire in vivo at low frequencies (McLachlan et al. 1997), the physiological activation of gCl,Ca probably depends primarily on CICR mechanisms. Recent evidence in dorsal root ganglion neurones confirms that Cl− channels can be activated by Ca2+ released from intracellular stores during one AP (Ayar & Scott, 1999).
Activation of gK,Ca1
Activation of gK,Ca1 after a single AP is clearly dependent on Ca2+ entry through N-type channels (Fig. 11B). This dependence is in agreement with findings in rat hypoglossal (Viana et al. 1993), vagal (Sah, 1995), sympathetic (Davies et al. 1996) and trigeminal (Kobayashi et al. 1997) neurones and guinea-pig sympathetic neurones (Ireland et al. 1998), as well as lamprey spinal neurones (Wikström & El Manira, 1998). Because gK,Ca1 primarily involves SK channels, the functional link between these and N-type Ca2+ channels is therefore common to many neurones (cf. Marrion & Tavalin, 1998). On the other hand, R-type channels also contribute to gK,Ca1 activation because the combined application of nifedipine and ωCgtx GVIA did not abolish the outward current (Fig. 11B).
Ryanodine had different effects in two subtypes of mouse SCG neurone that could be separated on the basis of the time course of gK,Ca1. In type II cells, in which gK,Ca1 showed a plateau before it declined, CICR activated about half of the current after one AP, whereas in type I cells in which gK,Ca1 declined exponentially, CICR played no significant role. A small contribution of CICR (30 %) to gK,Ca1 was previously reported in guinea-pig prevertebral sympathetic neurones (Jobling et al. 1993). Because nifedipine inhibited gK,Ca1 and blocked the effect of ryanodine only in type II cells, its action is most likely to have been to prevent activation of CICR. According to this interpretation, gK,Ca1 activation in type I neurones depends entirely on Ca2+ entering through N- and R-type channels, whereas in type II neurones, Ca2+ release induced by Ca2+ entering through L-type channels is also involved. L-type channels, CICR and Ca2+-activated K+ (SK) channels are functionally linked in vascular smooth muscle (Jaggar et al. 1998) and hippocampal neurones (Tanabe et al. 1998).
The contribution of R-type channels to gK,Ca1 activation was substantially larger after a train than after one AP (Fig. 11B), consistent with ‘spill over’ of accumulated Ca2+. Under these conditions, there was only slight inhibition of gK,Ca1 by nifedipine or ryanodine in type II cells (Figs 6Bb and 8Cb), consistent with the saturation of CICR by Ca2+ influx after a few APs.
Distinct intracellular mechanisms for activation of Cl− and K+ channels
The physical bases for the functional links between Ca2+ and K+/Cl− channels in intact neurones are not known. The present results could be explained if, in mouse SCG neurones, L- and P-type channels are linked to an intracellular pathway, perhaps the store in the subsurface cisternae (Berridge, 1998), from which Ca2+ release directly activates membrane Cl− channels, whereas N-type channels are closely juxtaposed to SK channels. R-type channels provide Ca2+ that activates both Cl− and SK channels, to a much greater extent following a train of APs, and so they may be more widely distributed. Colocalization of Ca2+ and K+ channels has been demonstrated directly at the neuromuscular junction, where BK channels are located adjacent to N-type channels in the presynaptic terminal (Robitaille et al. 1993), and in membrane patches from hippocampal somata (Marrion & Tavalin, 1998). In the latter case, N-type, SK and BK channels were present in the same somatic patch but SK channels were not activated by Ca2+ entering through the N-type channels, suggesting that some form of direct linkage between channels can exist at the molecular level.
Another possibility is that the different channels are located in separate regions of the cell (Davies et al. 1996). Although a dendritic location for high-threshold Ca2+ channels in sympathetic neurones has been proposed (Hirst & McLachlan, 1986), no data are available about the distribution of the different channel types in the somatodendritic membrane. In central neurones, L-type channels, although present in distal dendrites, are more common in the cell body and proximal dendrites, whereas N-type channels are more or less equally expressed in both cellular compartments (Westenbroek et al. 1998). Based on indirect evidence, it has been proposed that the Cl− channels responsible for the ADP are located in the dendrites (Sánchez-Vives & Gallego, 1994; De Castro et al. 1997) which, in mouse SCG neurones, are relatively few in number and short. If this were the case, L- and P-type Ca2+ channels may also be located on the dendrites. There might even be two distinct populations of Cl− channels in mouse SCG cells: a larger one that is activated via intracellular pathways dependent on Ca2+ entering via L- and P-type channels and another population that is activated by Ca2+ derived from R-type channels. While the second population of Cl− channels might be activated either directly or indirectly, it is likely to be spatially distinct from the Cl− channels dependent on Ca2+ entering via L- and P-type channels.
In conclusion, our results provide the first evidence for distinct mechanisms linking the physiological entry of Ca2+ through particular channel types during AP firing with the activation of Cl− and K+ membrane conductances in the same neurone. The data confirm that general intracellular Ca2+ levels are unlikely to reflect the localized consequences of Ca2+ influx through specific channels during physiological events.
Acknowledgments
This work was supported by grants PM95-0107 and PM98-0102-C02-01 from the DGICYT of Spain and by grant 970852 from the National Health and Medical Research Council of Australia. J.M.-P. holds a graduate fellowship from the DGES of Spain. We thank Eva Quintero and Alfonso Pérez-Vegara for technical assistance and Félix Viana and James A. Brock for helpful comments on the manuscript.
References
- Adams DJ, Harper AA. Electrophysiological properties of autonomic ganglion neurons. In: McLachlan EM, editor. Autonomic Ganglia. Luxembourg: Harwood Academic Publishers; 1995. pp. 153–212. [Google Scholar]
- Akasu T, Nishimura T, Tokimasa T. Calcium-dependent chloride current in neurones of the rabbit pelvic parasympathetic ganglia. The Journal of Physiology. 1990;422:303–320. doi: 10.1113/jphysiol.1990.sp017985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayar A, Scott RH. The actions of ryanodine on Ca2+-activated conductances in rat cultured DRG neurones; evidence for Ca2+-induced Ca2+ release. Naunyn-Schmiedeberg's Archives of Pharmacology. 1999;359:81–91. doi: 10.1007/pl00005335. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Neuronal calcium signalling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Boland LM, Morrill JA, Bean BP. ω-Conotoxin block of N-type calcium channels in frog and rat sympathetic neurons. Journal of Neuroscience. 1994;14:5011–5027. doi: 10.1523/JNEUROSCI.14-08-05011.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassell JF, McLachlan EM. Two calcium-activated potassium conductances in a subpopulation of coeliac neurones of guinea-pig and rabbit. The Journal of Physiology. 1987a;394:331–349. doi: 10.1113/jphysiol.1987.sp016873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassell JF, McLachlan EM. Muscarinic agonists block five different potassium conductances in guinea-pig sympathetic neurones. British Journal of Pharmacology. 1987b;91:259–261. doi: 10.1111/j.1476-5381.1987.tb10279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordoba-Rodriguez R, Moore KA, Kao JPY, Weinreich D. Calcium regulation of a slow post-spike hyperpolarization in vagal afferent neurons. Proceedings of the National Academy of Sciences of the USA. 1999;96:7650–7657. doi: 10.1073/pnas.96.14.7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies PJ, Ireland DR, McLachlan EM. Sources of Ca2+ for different Ca2+-activated K+ conductances in neurones of the rat superior cervical ganglion. The Journal of Physiology. 1996;495:353–366. doi: 10.1113/jphysiol.1996.sp021599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies PJ, Ireland DR, Martínez-Pinna J, McLachlan EM. Electrophysiological roles of L-type channels in different classes of guinea pig sympathetic neuron. Journal of Neurophysiology. 1999;82:818–828. doi: 10.1152/jn.1999.82.2.818. [DOI] [PubMed] [Google Scholar]
- De Castro F, Geijo-Barrientos E, Gallego R. Calcium-activated chloride current in normal mouse sympathetic ganglion cells. The Journal of Physiology. 1997;498:397–408. doi: 10.1113/jphysiol.1997.sp021866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Hernandez JM, Stühmer W, Parekh AB. Calcium dependence and distribution of calcium-activated chloride channels in Xenopus oocytes. The Journal of Physiology. 1997;502:569–574. doi: 10.1111/j.1469-7793.1997.569bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallani M, Lynch JW, Barry PH. Characterization of calcium-activated chloride channels in patches excised from the dendritic knob of mammalian olfactory receptor neurons. Journal of Membrane Biology. 1998;161:163–171. doi: 10.1007/s002329900323. [DOI] [PubMed] [Google Scholar]
- Hirst GDS, McLachlan E. Development of dendritic calcium currents in ganglion cells of the rat lower lumbar sympathetic chain. The Journal of Physiology. 1986;377:349–368. doi: 10.1113/jphysiol.1986.sp016191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireland DR, Davies PJ, McLachlan EM. The role of N-type Ca2+ channels in regulating excitability of guinea-pig sympathetic neurones. Journal of the Autonomic Nervous System. 1998;73:109–114. doi: 10.1016/s0165-1838(98)00127-1. [DOI] [PubMed] [Google Scholar]
- Jaggar JH, Wellman GC, Heppner TJ, Porter VA, Perez GJ, Gollasch M, Kleppisch T, Rubart M, Stevenson AS, Lederer WJ, Knot HJ, Bonev AD, Nelson MT. Ca2+ channels, ryanodine receptors and Ca2+-activated K+ channels: a functional unit for regulating arterial tone. Acta Physiologica Scandinavica. 1998;164:577–587. doi: 10.1046/j.1365-201X.1998.00462.x. [DOI] [PubMed] [Google Scholar]
- Jobling P, McLachlan EM, Sah P. Calcium induced calcium release is involved in the afterhyperpolarization in one class of guinea pig sympathetic neurone. Journal of the Autonomic Nervous System. 1993;42:251–258. doi: 10.1016/0165-1838(93)90370-a. [DOI] [PubMed] [Google Scholar]
- Jones SW, Jacobs LS. Dihydropyridine actions on calcium currents of frog sympathetic neurons. Journal of Neuroscience. 1990;10:2261–2267. doi: 10.1523/JNEUROSCI.10-07-02261.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Watanabe M. Blockade of Ca-activated K conductance by apamin in rat sympathetic neurones. British Journal of Pharmacology. 1986;87:225–232. doi: 10.1111/j.1476-5381.1986.tb10175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Inoue T, Matsuo R, Masuda Y, Hidaka O, Kang YN, Morimoto T. Role of calcium conductances on spike afterpotentials in rat trigeminal motoneurons. Journal of Neurophysiology. 1997;77:3273–3283. doi: 10.1152/jn.1997.77.6.3273. [DOI] [PubMed] [Google Scholar]
- McAfee DA, Yarowsky PJ. Calcium-dependent potentials in the mammalian sympathetic neurone. The Journal of Physiology. 1979;290:507–523. doi: 10.1113/jphysiol.1979.sp012787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan EM, Davies PJ, Häbler HJ, Jamieson J. On-going and reflex synaptic events in rat superior cervical ganglion cells. The Journal of Physiology. 1997;501:165–181. doi: 10.1111/j.1469-7793.1997.165bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrion NV, Tavalin SJ. Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature. 1998;395:900–905. doi: 10.1038/27674. [DOI] [PubMed] [Google Scholar]
- Martínez-Pinna J, McLachlan EM, Gallego R. Different calcium sources for activation of chloride or potassium channels in sympathetic ganglion cells. European Journal of Neuroscience. 1998;10(suppl. 10):72. [Google Scholar]
- Meissner G. Ryanodine receptor/Ca2+ release channels and their regulation by endogenous effectors. Annual Review of Physiology. 1994;56:485–508. doi: 10.1146/annurev.ph.56.030194.002413. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Adams ME, Bean BP. P-type calcium channels in rat central and peripheral neurons. Neuron. 1992;9:85–95. doi: 10.1016/0896-6273(92)90223-z. [DOI] [PubMed] [Google Scholar]
- Namkung Y, Smith SM, Lee SB, Skrypnyk NV, Kim H-L, Chin H, Scheller RH, Tsien RW, Shin H-S. Targeted disruption of the Ca2+ channel b3 subunit reduces N- and L-type Ca2+ channel activity and alters the voltage-dependent activation of P/Q-type Ca2+ channels in neurons. Proceedings of the National Academy of Sciences of the USA. 1998;95:12010–12015. doi: 10.1073/pnas.95.20.12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivera BM, Miljanich GP, Ramachandran J, Adams ME. Calcium channel diversity and neurotransmitter release: the ω-conotoxins and ω-agatoxins. Annual Review of Biochemistry. 1994;63:823–867. doi: 10.1146/annurev.bi.63.070194.004135. [DOI] [PubMed] [Google Scholar]
- Osmanovic SS, Shefner SA. Calcium-activated hyperpolarizations in rat locus coeruleus neurons in vitro. The Journal of Physiology. 1993;469:89–109. doi: 10.1113/jphysiol.1993.sp019806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall A, Tsien RW. Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. Journal of Neuroscience. 1995;15:2995–3012. doi: 10.1523/JNEUROSCI.15-04-02995.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan LJ, Sah DWY, Bean BP. Ca2+ channels in rat central and peripheral neurons: High-threshold current resistant to dihydropyridine blockers and omega-conotoxin. Neuron. 1991;6:269–280. doi: 10.1016/0896-6273(91)90362-4. [DOI] [PubMed] [Google Scholar]
- Robitaille R, Garcia ML, Kaczorowski GJ, Charlton MP. Functional colocalization of calcium and calcium-gated potassium channels in control of transmitter release. Neuron. 1993;11:645–655. doi: 10.1016/0896-6273(93)90076-4. [DOI] [PubMed] [Google Scholar]
- Sacchi O, Rossi ML, Canella R. The slow Ca2+-activated K+ current, IAHP, in the rat sympathetic neurone. The Journal of Physiology. 1995;483:15–27. doi: 10.1113/jphysiol.1995.sp020564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P. Different calcium channels are coupled to potassium channels with distinct electrophysiological roles in vagal neurons. Proceedings of the Royal Society B. 1995;260:105–111. doi: 10.1098/rspb.1995.0066. [DOI] [PubMed] [Google Scholar]
- Sah P. Ca2+-activated K+ currents in neurones: types, physiological roles and modulation. Trends in Neurosciences. 1996;19:150–154. doi: 10.1016/s0166-2236(96)80026-9. [DOI] [PubMed] [Google Scholar]
- Sánchez-Vives MV, Gallego R. Calcium-dependent chloride current induced by axotomy in rat sympathetic neurons. The Journal of Physiology. 1994;475:391–400. doi: 10.1113/jphysiol.1994.sp020080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott RH, McGuirk SM, Dolphin AC. Modulation of divalent cation-activated chloride ion currents. British Journal of Pharmacology. 1988;94:653–662. doi: 10.1111/j.1476-5381.1988.tb11572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoshan-Barmatz V, Ashley RH. The structure, function and cellular regulation of ryanodine-sensitive Ca2+ release channels. International Review of Cytology. 1998;183:185. doi: 10.1016/s0074-7696(08)60145-x. [DOI] [PubMed] [Google Scholar]
- Tanabe M, Gähwiler BH, Gerber U. L-type Ca2+ channels mediate the slow Ca2+-dependent afterhyperpolarization current in rat CA3 pyramidal cells in vitro. Journal of Neurophysiology. 1998;80:2268–2273. doi: 10.1152/jn.1998.80.5.2268. [DOI] [PubMed] [Google Scholar]
- Toth PT, Miller RJ. Calcium and sodium currents evoked by action potential waveforms in rat sympathetic neurones. The Journal of Physiology. 1995;485:43–57. doi: 10.1113/jphysiol.1995.sp020711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viana F, Bayliss DA, Berger AJ. Multiple potassium conductances and their role in action potential repolarization and repetitive firing behavior of neonatal rat hypoglossal motoneurons. Journal of Neurophysiology. 1993;69:2150–2163. doi: 10.1152/jn.1993.69.6.2150. [DOI] [PubMed] [Google Scholar]
- Westenbroek RE, Hoskins L, Catterall WA. Localization of Ca2+ channel subtypes on rat spinal motor neurons, interneurons, and nerve terminals. Journal of Neuroscience. 1998;18:6319–6330. doi: 10.1523/JNEUROSCI.18-16-06319.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikström MA, El Manira A. Calcium influx through N- and P/Q-type channels activate apamin-sensitive calcium-dependent potassium channels generating the late hyperpolarization in lamprey spinal neurons. European Journal of Neuroscience. 1998;10:1528–1532. doi: 10.1046/j.1460-9568.1998.00194.x. [DOI] [PubMed] [Google Scholar]