

Abstract
The large conductance, calcium-sensitive K+ channel (BKCa channel) is a voltage-activated ion channel in which direct calcium binding shifts gating to more negative cellular membrane potentials. We hypothesized that the calcium-binding domain of BKCa channels may mimic the role played by calmodulin (CaM) in the activation of calcium-CaM-dependent enzymes, in which a tonic inhibitory constraint is removed on CaM binding.
To examine such a hypothesis, we used peptides from the autoregulatory domains of CaM kinase II (CK291–317) and cNOS (the constitutive nitric oxide synthase; cNOS725–747) as probes for the calcium-dependent activation of murine BKCa channels transiently expressed in HEK 293 cells. We found that these CaM-binding peptides produced potent, time-dependent inhibition of mammalian BKCa channel current following voltage-dependent activation. Inhibition was observed in both the presence and the absence of cytosolic free calcium.
Similar application of CK291–31 had no effect on either the amplitude or kinetics of voltage-dependent, macroscopic currents recorded from rabbit smooth muscle Kv1.5 potassium channels transiently expressed in HEK 293 cells.
Cytosolic application of both CK291–317 and tetraethylammonium (TEA) produced an additive and non-competitive block of BKCa current. This finding suggests that the peptide-binding site is distinct (e.g. outside the pore region of the channel) from that of TEA.
Our results are thus consistent with a model in which the BKCa channel's voltage-dependent gating process is under an intramolecular constraint that is relieved upon calcium binding. The intrinsic calcium sensor of the channel may thus interact with an inhibitory domain present in the BKCa channel, and by doing so, remove an inhibitory ‘constraint’ that permits voltage-dependent gating to occur at more negative potentials.
Potassium channels form a large family of ion-selective pores that are found in both excitable and non-excitable cells. The large conductance, calcium-sensitive K+ channel (maxi-K or BKCa) is a unique member of this family, in that channel opening probability is increased by both membrane depolarization and intracellular free calcium (Rudy, 1988; Latorre et al. 1989; McManus, 1991). As a result of this behaviour, BKCa channels can ‘couple’ rises in intracellular calcium with changes in membrane potential, and thereby provide rapid ‘feedback’ regulation of cellular events initiated by depolarization-induced elevations in cytosolic calcium. Such a physiological role for BKCa channels is suggested by experiments demonstrating that blocking these channels increases myogenic tone in cerebral, femoral and mesenteric arteries (Brayden & Nelson, 1992; Asano et al. 1993, 1995), and enhances the presynaptic calcium-dependent release of neurotransmitter at neuromuscular junctions (Robitaille & Charlton, 1992; Robitaille et al. 1993).
Recent experiments taking advantage of heterologous expression of cloned mammalian BKCa channels have shown that these channels can be gated by voltage alone (Meera et al. 1996; Cui et al. 1997), much like the purely voltage-dependent ‘Kv’ classes of K+ channels. However, calcium binding by the BKCa channel is critical since it strongly influences this voltage-dependent process, thus allowing channel activation to occur over a more negative range of cellular membrane potentials (McManus, 1991; Meera et al. 1996; Cui et al. 1997). Recent observations demonstrating that ‘calcium sparks’ occur in intact cerebral arteries (Jaggar et al. 1998) and can activate normally quiescent BKCa channels in isolated vascular smooth muscle myocytes (Nelson et al. 1995) are thus consistent with these properties.
Given the importance of calcium binding and its subsequent effects on channel activation, we examined whether intracellular calcium shifts BKCa channel gating by a molecular process that may be mechanistically similar to that by which the ubiquitous calcium sensor calmodulin (CaM) activates calcium-dependent enzymes, such as protein kinases. In many of these molecules, calcium-bound CaM interacts with a regulatory domain in the target enzyme, thereby relieving an inhibitory steric constraint and exposing the active site of the enzyme (Kemp & Pearson, 1991; Ito et al. 1991; Knighton et al. 1992; Nairn & Picciotto, 1994). By analogy with these CaM-dependent enzymes, we hypothesized that in BKCa channels, calcium binding to the intrinsic calcium sensor may lead to interaction of this site with a putative ‘inhibitory’ domain of the channel, thereby relieving a molecular constraint on the voltage-dependent gating mechanism.
To explore whether the calcium-dependent activation of BKCa channels is mechanistically similar to that of calcium-sensitive enzymes, we took advantage of reagents that are known to specifically interfere with the calcium-CaM-dependent activation of calcium-CaM-dependent protein kinase II (CaM kinase II; Braun & Schulman, 1995a, b). These reagents were then used as probes to examine the voltage- and calcium-dependent gating of mammalian BKCa channels transiently expressed in HEK 293 cells.
METHODS
Construction and transfection of cDNA plasmids
The cDNA encoding the mouse brain mSlo α subunit (Pallanck & Ganetzky, 1994) was obtained from Dr Leo Pallanck (University of Wisconsin) and a ∼3.7 kb fragment was subcloned into the SV40 promoter-based mammalian expression plasmid SRα (Takebe et al. 1988) as follows. The EcoR I site of SRα and the BamH I site at the 5′ end of the mSlo cDNA were blunted with Klenow fragment and a Not I linker was ligated to these ends. The Xba I site at the 3′ end of the mSlo cDNA was then blunted with Klenow fragment and the insert cDNA was ligated between the Not I and EcoR V restriction sites in the plasmid's polylinker region. The cDNA encoding green fluorescent protein (GFP; Chalfie et al. 1994) was obtained from Dr Martin Chalfie (Columbia University) and was subcloned into the SRα plasmid between the Pst I and EcoR I sites. A cDNA construct encoding the rabbit vascular smooth muscle Kv1.5 channel (Clément-Chomienne et al. 1999) in the pcDNA3.1 plasmid was kindly provided by Dr W. C. Cole (University of Calgary).
Transient transfection of HEK 293 cells (50–80 % confluency) was carried out in 35 mm tissue culture dishes using the lipofection technique. Briefly, 6–8 μl of Lipofectamine (Gibco/BRL) was mixed together with ∼1.5 μg of plasmid cDNA in 1 ml of serum-free culture medium (Dulbecco's modified Eagle's medium supplemented with L-glutamine and 4.5 g l−1 D-glucose) and placed on cells for 4–6 h in a humidified incubator containing 5 % CO2 at 37°C. DNA-containing medium was then aspirated and replaced with serum-containing medium. The following day, cells were detached from the dish by treatment with 0.05 % (v/v) trypsin-EDTA and replated onto sterile glass coverslips. Electrophysiological recordings were typically performed on days 3–5 following transfection.
Electrophysiology
Macroscopic and single channel currents were recorded at 35 ± 0.5°C from excised inside-out membrane patches of HEK 293 cells using an Axopatch 200 patch clamp amplifier and pCLAMP 6.03 software (Axon Instruments). BKCa channel currents were activated by voltage clamp pulses delivered from a holding potential of 0 mV to a membrane potential ranging from −180 to 240 mV; tail currents were recorded at +50, −80 or −120 mV. Current traces were filtered at 2–5 kHz (4-pole Bessel filter) and acquired on a Gateway 486 or Dell Pentium II-based computer at a sampling frequency of 8–10 kHz using a TL-1 analog/digital interface. Kv1.5 channel currents were recorded at 35 ± 0.5°C from a holding potential of −70 mV, with voltage clamp steps delivered at a frequency of 0.1 Hz. Recording micropipettes were pulled from thin-walled borosilicate glass capillaries (1.2 mm i.d., 1.5 mm o.d., WPI, Sarasota, FL, USA) using a Sutter P-87 horizontal electrode puller. Micropipettes were filled with a solution containing (mm): 5 KCl, 140 KOH, 1 MgCl2, 1 CaCl2 and 10 Hepes (pH adjusted to 7.3 with methanesulfonic acid), and had tip resistances of 2–5 MΩ. The bath solution contained (mm): 5 KCl, 140 KOH, 1 MgCl2, 5 EGTA and 10 Hepes (pH adjusted to 7.2 with methanesulfonic acid); variable amounts of a 0.1 M CaCl2 solution were added to give the desired free calcium concentration. The level of free calcium in solution was confirmed using a calcium electrode (Orion) with calibration standards (WPI) ranging from pCa 8 to 2. The recording chamber (∼0.3 ml volume) was perfused at a constant rate of 1–1.5 ml min−1, using a set of manually controlled solenoid valves to switch between various solutions. Reagents were added directly to the solution reservoir tubes at the indicated final concentrations.
Transfected HEK 293 cells seeded on coverslips were placed in a temperature-controlled recording chamber on the stage of a Nikon Diaphot inverted microscope. Individual cells expressing BKCa channels were then identified visually by co-expression of the marker protein GFP under epifluorescence using 480 nm excitation and 510 nm emission filters.
Peptides and reagents
Calmidazolium chloride was purchased from Calbiochem (San Diego, CA, USA). The CaM kinase-derived peptides CK291–317 and CK281–302 were prepared using a solid-phase peptide synthesizer (Applied Biosystems) and purified by reverse-phase high-performance liquid chromatography. Peptide sequences were confirmed by automated sequencing. The CaM-binding peptide derived from nitric oxide synthase (cNOS) was kindly provided by Dr M. P. Walsh (Department of Biochemistry and Molecular Biology, University of Calgary). All other chemicals were of reagent grade or higher and obtained from Sigma/Aldrich Co.
RESULTS
Expression of mSlocDNA gives rise to voltage- and calcium-dependent currents
In HEK 293 cells transfected with GFP alone, excised inside-out membrane patches displayed no significant endogenous macroscopic current in response to voltage clamp steps from −90 to +180 mV in the presence of 31 μm cytoplasmic free Ca2+ (Fig. 1A). Co-expression of the mouse brain mSlo α subunit, which is the pore-forming subunit of a large conductance, calcium-sensitive K+ channel, gave rise to macroscopic currents that appeared quite noisy and displayed strong outward rectification under the same recording conditions (Fig. 1B). Increasing cytoplasmic free Ca2+ from 0.8 to 31 μm had several effects on the macroscopic currents: (1) a shift in the voltage for half-maximal activation of the current to more negative membrane potentials, (2) an increase in the maximal current amplitude, and (3) an increase in the speed of current activation (Fig. 1B–E). Normalized conductance-voltage (G–V) relations derived from tail current measurements (Fig. 1F) document the leftward shifts in the G-V relations along the voltage axis with increasing free intracellular calcium. These observations are thus similar to those recently reported by others (Meera et al. 1996; Cui et al. 1997) for heterologously expressed mammalian BKCa channels.
Figure 1. Transient expression of mSlo α subunit in HEK 293 cells.
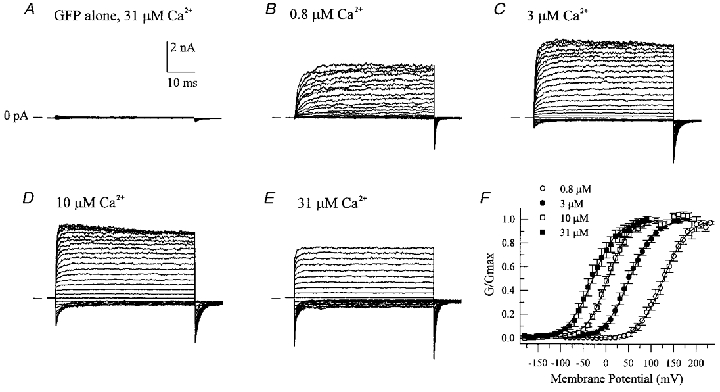
Macroscopic currents were recorded from inside-out patches excised from cells expressing either GFP alone (A) or GFP + mSlo α subunit (B-E). Membrane voltage was stepped from −90 to +180 mV (A-D) or −180 to +90 mV (E) in the presence of the indicated amount of cytoplasmic free calcium. Tail currents recorded at either −80 mV (A-D) or −120 mV (E) were then used to calculate normalized conductance-voltage (G-V) relations. F shows the average G-V relations (±s.e.m.) calculated from 4–8 individual membrane patches; smooth curves represent fits to single Boltzmann functions, i.e. G/Gmax = 1/(1 + exp((V½–Vm)/slope)) where 1/slope =zF/RT (Vm is membrane potential, V½ is the membrane potential at which 50% of the channels are open, and z, F, R and T have their usual meanings). Derived V½ and slope parameters are as follows: 0.8 μm Ca2+, 127.4 mV and 26.5 mV per e-fold change in G; 3 μm Ca2+, 51.7 mV and 26.9 mV per e-fold change in G; 10.5 μm Ca2+, 3.9 mV and 23.2 mV per e-fold change in G; 31 μm Ca2+, −29.1 mV and 27.0 mV per e-fold change in G.
Further characterization of these expressed channels demonstrated that (1) ∼80 % of macroscopic currents could be blocked by 2 mm external TEA, (2) single channel events recorded in the inside-out mode from mSlo-expressing cells displayed a large conductance (∼280 pS) under conditions of symmetrical 145 mm K+, and (3) a tenfold decrease in cytoplasmic K+ (replaced with Na+) shifted the reversal potential ∼+50 mV, indicating a strong K+ selectivity (data not shown). Taken together, these observed properties for heterologously expressed mSlo channels in HEK 293 cells are consistent with those of native large conductance, calcium-sensitive K+ channels from nerve or muscle reported in the literature (Rudy, 1988; Latorre et al. 1989; McManus, 1991).
Mechanistic model of BKCa channel gating by calcium
In a strict sense, BKCa channels can be considered as voltage-gated K+ channels, in which the role of intracellular calcium is to shift the voltage dependence of gating to a more negative range of membrane potentials. Therefore, calcium binding and the molecular mechanism by which it influences channel gating are critically important for the biological activity of BKCa channels. We asked whether the calcium-dependent ‘activation’ of BKCa channels may be mechanistically similar to the activation of other calcium-dependent, but functionally distinct, cellular molecules, such as calcium-CaM-dependent protein kinase II (CaM kinase II), myosin light chain kinase (MLCK) or calcium-dependent protein kinase (CDPK) from plants. The mechanism of calcium-dependent activation of these enzymes has been more thoroughly studied and may thus serve as a model to examine a potentially similar process in BKCa channels.
In the case of CaM kinase II and MLCK, it is known that activation first involves calcium binding to CaM, a ubiquitous, high-affinity intracellular calcium-binding protein (Klee & Vanaman, 1982), followed by the interaction of calcium-CaM with an autoregulatory domain in the enzyme (Kemp & Pearson, 1991; Nairn & Picciotto, 1994). This interaction appears to relieve an inhibitory constraint imposed by the autoregulatory domain, allowing the enzymes's catalytic domain to function. In the case of CDPK, however, a CaM function is built into the kinase as an intrinsic CaM-like, calcium-binding domain that similarly interacts with an autoregulatory region to produce activation (Roberts & Harmon, 1992; see Fig. 2B). We hypothesized that in BKCa channels, calcium binding may also act to relieve some form of intramolecular constraint, thereby positively influencing channel gating (Fig. 2A). In a kinetic model, such an effect could resemble a stabilization of the channel's open state, for example, by changes in the appropriate rate constants. This would be consistent with earlier observations that cytoplasmic calcium increases the duration of open time events at the single channel level in native BKCa channels (Blatz & Magleby, 1987; McManus, 1991).
Figure 2. Hypothetical mechanism of calcium-dependent gating in BKCa channels.
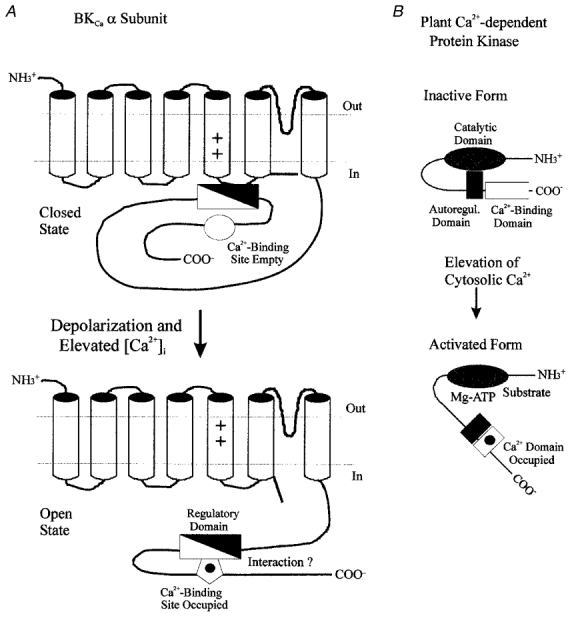
The calcium-induced leftward shift of voltage-dependent gating in BKCa channels (A) may occur by a mechanism similar to that described for the activation of Ca2+-dependent protein kinases by calcium-CaM (Kemp & Pearson, 1991; Ito et al. 1991; Knighton et al. 1992; Nairn & Picciotto, 1994). In the latter situation, an intrinsic autoregulatory domain interacts with the enzyme's catalytic site, keeping it inactive. In the plant Ca2+-dependent protein kinase (B), this inactivation is removed by calcium binding to a CaM-like domain contained within the enzyme's primary structure, thereby allowing substrate and ATP to access the catalytic site. In BKCa channels, an analogous regulatory segment (filled triangle) may also exist, that could, for example, constrain movement of the channel's voltage sensor. Binding of calcium to its intrinsic site may then allow interaction of this site with a region of the regulatory segment (open triangle), and thus remove the constraint on the channel's gating mechanism. The intrinsic calcium-binding site of the BKCa channel may thus act in a similar fashion to Ca2+-CaM in the activation of Ca2+-dependent protein kinases.
For calcium-CaM-dependent enzymes, synthetic peptides derived from the CaM-binding site within their autoregulatory domains act as inhibitors by competing with the enzyme for the binding of free calcium-bound CaM. By analogy, we hypothesized that if the intrinsic calcium-binding site of the BKCa channel interacted with an intrinsic autoregulatory domain to affect channel gating, then cytoplasmic application of synthetic peptides also capable of interacting with an intrinsic calcium sensor may competitively inhibit channel activation. It should be emphasized that in our hypothesis, we have not assumed that CaM is the true calcium sensor of the BKCa channel. Rather, our scheme explores the possibility that the channel's intrinsic calcium sensor may have some characteristics of CaM, most notably (1) the generation of a peptide-binding site via a calcium-induced conformational change and (2) an EF-hand-like calcium-binding motif (Marsden et al. 1990; Falke et al. 1994). Given that the EF-hand motif appears to be the most widely used calcium-binding structure in nature (Moncrief et al. 1990), some similarities as presented in the above scheme would not be unexpected.
CaM-binding peptides inhibit BKCa channels
Application of a CaM-binding peptide (CK291–317) derived from the autoregulatory domain of CaM kinase II (Hanley et al. 1987; Payne et al. 1988) to the cytosolic surface of BKCa channels in excised inside-out membrane patches was found to produce a dose-dependent inhibition of macroscopic currents (Fig. 3A–C). This inhibition involved a decrease of both the outward current component at positive potentials and the inward component, as judged by the decreased tail current magnitude. Therefore, peptide-induced inactivation of ion flux through the pore is bidirectional. There did not appear to be a significant effect of the peptide on the kinetics of activation at the start of the voltage clamp step or on deactivation of the tail currents. This inhibitory effect appeared to be selective for CaM-binding peptides as two other synthetic peptide inhibitors of CaM kinase II (CK281–302, AC3-I), also derived from its autoregulatory domain, but with little or no affinity for CaM, were without effect when applied under identical conditions (data not shown). Conductance-voltage relations of BKCa macroscopic currents in the absence and presence of CK291–317 demonstrated that the peptide did not shift the voltage dependence of BKCa channel gating, but rather decreased the current magnitude (Fig. 3D). If the CK291–317 peptide were inhibitory due to its ability to mimic a CaM-like target sequence in the BKCa channel, then another CaM-binding peptide with a different primary sequence would be predicted to also act as an inhibitor. To examine this prediction, we tested a CaM-binding peptide derived from the calcium-CaM-dependent form of nitric oxide synthase (cNOS; Bredt et al. 1991; Zhang & Vogel, 1994). We observed that this cNOS-derived peptide produced a similar inhibition of BKCa current, but with a somewhat lower potency (Fig. 4A and B). In the case of cNOS725–747, a rapid, time-dependent inhibition of current was observed during the positive voltage clamp steps. As discussed later, this effect may represent a state-dependent interaction of the peptide with the channel. The inhibitory effects of the two CaM-binding peptides are summarized in a plot of peptide concentration versus fraction of remaining current (Fig. 5); CK291–317 was found to inhibit BKCa current with a calculated IC50 of ∼240 nm and a Hill coefficient approximating unity, whereas the cNOS peptide appeared to be ∼5-fold less effective. It is noteworthy that the inactive peptide CK281–302 has a similar length and net overall positive charge to CK291–317 and cNOS725–747, yet does not produce block of the channel. This suggests that additional properties of CK291–317 and cNOS725–747 contribute to their inhibitory actions.
Figure 3. Inhibition of BKCa channels by a CaM-binding peptide.
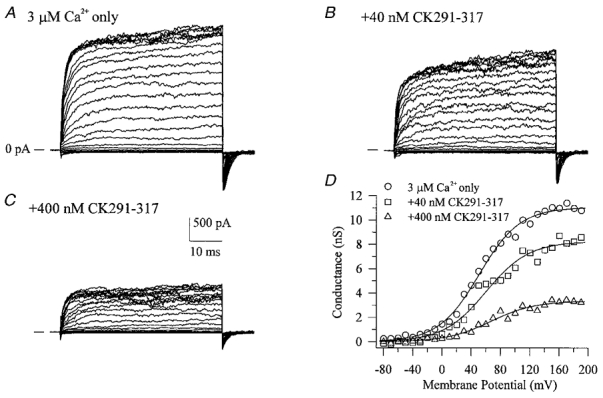
A shows macroscopic currents recorded during voltage clamp steps from −90 to +180 mV in the presence of 3 μm cytoplasmic free calcium. Addition of a CaM-binding peptide (CK219–317) derived from the autoregulatory domain of CaM kinase II (Payne et al. 1988) to the cytoplasmic face of the excised patch produced a dose-dependent decrease in both outward and inward current amplitude (B and C). Conductance-voltage relations derived from tail current data in the absence and presence of CK291–317 are shown in D.
Figure 4. A CaM-binding peptide from cNOS inhibits BKCa channels.
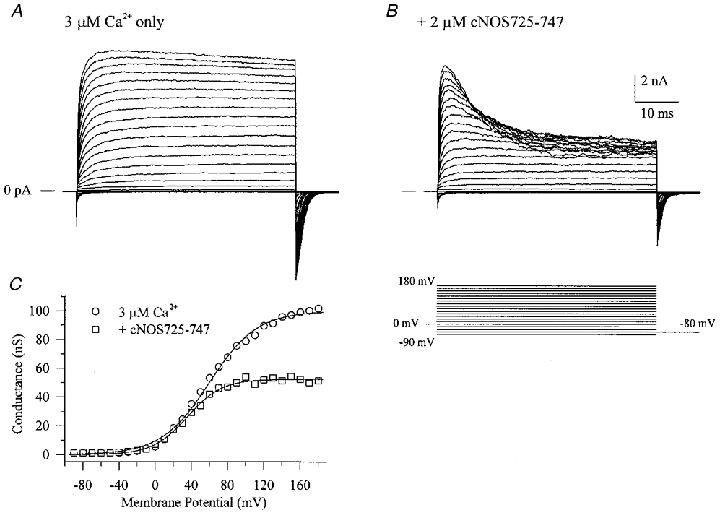
Macroscopic currents were recorded in the absence (A) or presence (B) of a CaM-binding peptide (cNOS725–747) applied to the cytoplasmic surface of the patch. cNOS725–747 was derived from the CaM-binding domain of constitutive nitric oxide synthase (Bredt et al. 1991; Zhang & Vogel, 1994). C shows conductance-voltage relations calculated from tail current data that have been fitted with single Boltzmann functions.
Figure 5. Plot of dose-dependent inhibition of BKCa channels by the CaM-binding peptides CK291–317 and cNOS725–747.
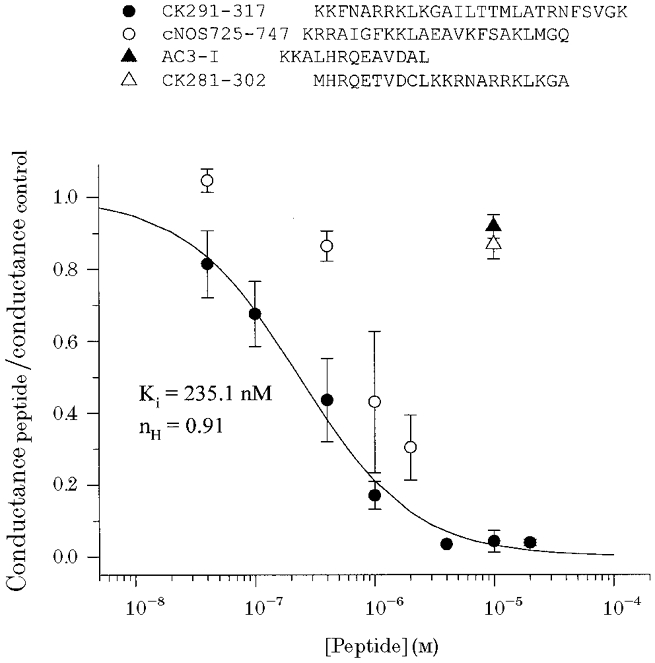
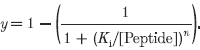 |
To explore the mechanism by which inhibition occurred, single BKCa channel activity was examined in the absence and presence of 2 μm CK291–317 (Fig. 6). The peptide did not decrease single channel amplitude, but rather it appeared to decrease the overall channel open probability (NPo). No obvious change in the channel open time distribution was noted; however, a small increase in the time constant of closed durations was observed (data not shown).
Figure 6. The CaM-binding peptide CK291–317 inhibits BKCa single channel events.
In the presence of ∼200 μm cytoplasmic free calcium, single channel activity was recorded at a membrane voltage of −60 mV. A shows an excised inside-out patch containing at least 3 BKCa channels under control conditions; C denotes the closed level, O1 and O2 denote the amplitude levels of a single open channel and 2 simultaneously open channels, respectively. B shows the same patch following addition of 2 μm CK291–317 to the cytoplasmic surface. The all-points amplitude histograms below the current records were constructed using bin widths of 1 pA. Binned data in A and B were then fitted with Gaussian distributions, from which the peak amplitudes of each level were derived.
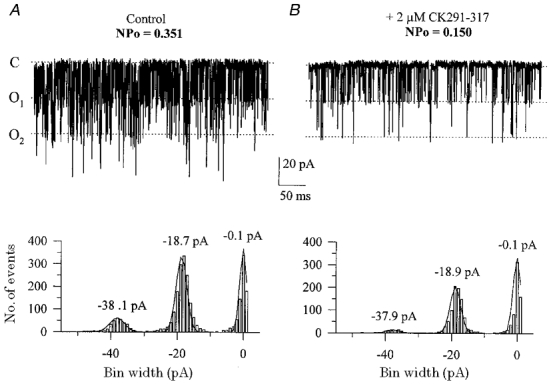
The observation that CaM-binding peptides, but not other kinase inhibitory peptides with different sites of action (i.e. CK281–302, AC3-I), produced strong inhibition of BKCa channels is consistent with the hypothesis presented in Fig. 2 and prompted us to examine whether a different, non-peptide inhibitor of CaM may also inhibit BKCa channels. Figure 7 shows that the potent CaM antagonist calmidazolium (van Belle, 1981; 1 μm), caused strong inhibition of BKCa current similar to that observed with CK291–317. A stronger inhibition of BKCa channels was further observed with 5 μm calmidazolium (data not shown). Although calmidalozium has also been observed to block other ion channels, such as L-type Ca2+ channels (Klockner & Isenberg, 1987; Nakazawa et al. 1993), our finding is in agreement with our hypothesis (see Fig. 2) that agents which interfere with the actions of CaM may be inhibitors of BKCa channels due to the calcium-dependent mechanism of activation of these channels and its potential similarity to the activation of calcium-CaM-dependent enzymes.
Figure 7. The CaM antagonist calmidazolium inhibits BKCa channels.
Cytoplasmic application of 1 μm calmidazolium produced inhibition of both inward and outward macroscopic currents (B) compared to control currents in the presence of 3 μm cytoplasmic free calcium only (A). C shows conductance-voltage relations of the data in A and B; data points have been fitted with single Boltzmann functions.
If the CaM-like functional domain of the BKCa channel strictly required calcium binding in order to interact with a potential target, then one would predict that channel inhibition by CK291–317 and cNOS725–747 should only occur in the presence of internal free calcium. This situation holds true for the binding of CaM to these same peptides (Kelly et al. 1988; Zhang & Vogel, 1994). We tested this prediction by examining the effect of CK219–317 on BKCa currents activated in the absence of intracellular calcium. Other investigators have already shown that voltage alone can activate both BKCa ionic and BKCa gating currents (Cui et al. 1997; Stefani et al. 1997; Horrigan et al. 1999a,b). Using a Ca2+-free intracellular solution (zero added Ca2+, b 5 mm EGTA), BKCa currents could be activated by strongly positive voltage clamp steps, up to +240 mV (Fig. 8A). Addition of 2 μm CK291–317 to the cytoplasmic surface of the channels led to a rapid, time-dependent inhibition of macroscopic current following initial activation of the current (Fig. 8B). This observation indicates that the BKCa channel need not have calcium bound in order for CK291–317 to produce a block of the current.
Figure 8. The CaM-binding peptide CK291–317 inhibits BKCa channels in the absence of cytoplasmic free calcium.
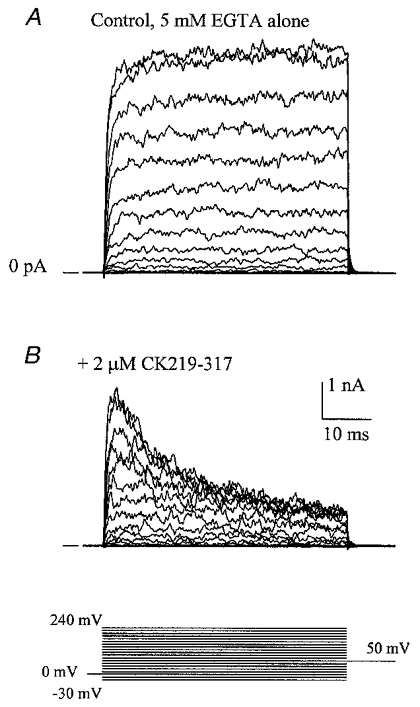
In the presence of 5 mm EGTA and no added calcium, macroscopic BKCa currents were recorded in response to voltage clamp steps from −30 to +240 mV (A). Following cytoplasmic application of 2 μm CK291–317 for 1 min, a time-dependent inhibition of current was observed during the 50 ms voltage clamp step (B). The voltage clamp protocol is shown at the bottom of the figure.
CK291–317 does not block Kv1.5 channels
To examine whether the CaM-binding peptide CK291–317 could also inhibit other voltage-dependent K+ channels, we expressed voltage-gated Kv1.5 channels cloned from rabbit vascular smooth muscle (Clément-Chomienne et al. 1999) in HEK 293 cells. Figure 9 shows macroscopic currents recorded from excised inside-out membrane patches in the absence (Fig. 9A) or presence (Fig. 9B) of internal 2 μm CK291–317. We observed that this peptide had no effect on either the voltage dependence of activation, or the current magnitude at either the start or end of the voltage clamp steps (90 ± 13 % of control values, n = 4). Following washout of CK291–317, application of 0.5 mm 4-aminopyridine (4-AP) to the cytoplasmic face of the patch produced a significant blockade of outward current (36 ± 4 % of control, n = 3; Fig. 9C), consistent with its reported IC50 value for this channel (0.18 mm; Clément-Chomienne et al. 1999). Washout of 4-AP from the bath (Fig. 9D) led to almost complete recovery of current magnitude.
Figure 9. CK291–317 does not inhibit Kv1.5 voltage-dependent potassium channels.
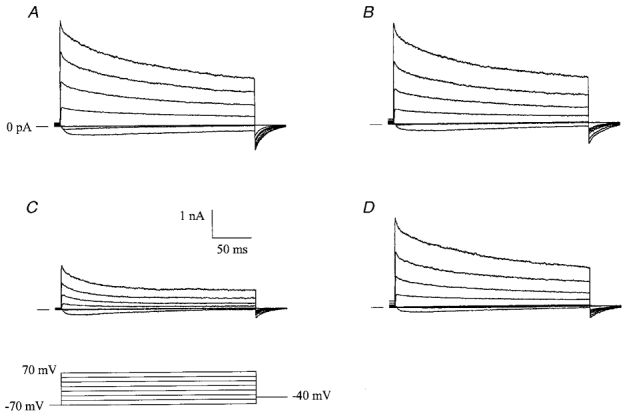
Macroscopic currents were recorded from recombinant rabbit vascular smooth muscle Kv1.5 channels in excised, inside-out membrane patches at 35 °C from a holding potential of −70 mV. Positive-going voltage clamp steps were delivered at a rate of 0.1 Hz, in 20 mV increments, followed by repolarization to a tail potential of −40 mV (refer to protocol in C). Following a series of voltage clamp steps under control conditions (A), 2 μm CK219–317 was applied to the cytoplasmic face of the patch and the same voltage clamp protocol was repeated (B). After washout of CK219–317, voltage clamp pulses were delivered in the presence of internal 0.5 mm 4-AP (C). Recovery from inhibition of the macroscopic currents was observed following washout of 4-AP from the bath (D). The horizontal and vertical scale bars shown apply to all four panels.
Effects of tetraethylammonium on blockade by CK291–317
To further address the question of whether CK291–317 may be acting as an internal blocker of the channel pore itself, we examined the effect of CK291–317 in the presence of internal tetraethylammonium (TEA), which is known to block BKCa channels by acting at the cytoplasmic mouth of the pore (Vergara et al. 1984; Yellen, 1984; Villarroel et al. 1988). This strategy is thus similar to that reported for blockade of BKCa channels by a Shaker inactivating ‘ball’ peptide (Toro et al. 1992; Foster et al. 1992), in which internal TEA was observed to slow the rate of block of these channels by internal application of the ball peptide. These data were interpreted to indicate that TEA and the ball peptide compete for a common binding site in the BKCa channel (i.e. the internal mouth of the pore) and that TEA acts by preventing access of the ball peptide to the pore. In our own experiments (see Fig. 10A), we observed that internal 40 mm TEA-Cl produced a reversible decrease of outward current amplitude to 63.8 ± 13.0 % (mean ±s.e.m., n = 8) of control, along with a slight time-dependent inactivation of current (Fig. 10A, trace 2). Further addition of 2 μm CK291–317 on top of TEA led to a greater reduction in peak current amplitude and a pronounced time-dependent inactivation of current with a single exponential time constant of 5.3 ± 0.9 ms (mean ±s.e.m., n = 6; Fig. 10A, trace 3). Subsequent washout of TEA-Cl, with CK291–317 remaining in the bath, led to a significant recovery of current amplitude, but no change in the time course of current inactivation (τ = 7.2 ± 4.4 ms, mean ±s.e.m.; Fig. 10A, trace 4). This lack of effect of TEA on the CK291–317-dependent inactivation of current is shown more clearly by normalization of traces to the peak control current (Fig. 10B). Our data thus indicate that, unlike the reported effects of TEA on the Shaker ball peptide, internal TEA does not interfere with the blocking action of CK291–317 on BKCa channels.
Figure 10. Internal TEA does not affect the time course of current inactivation by CK291–317.
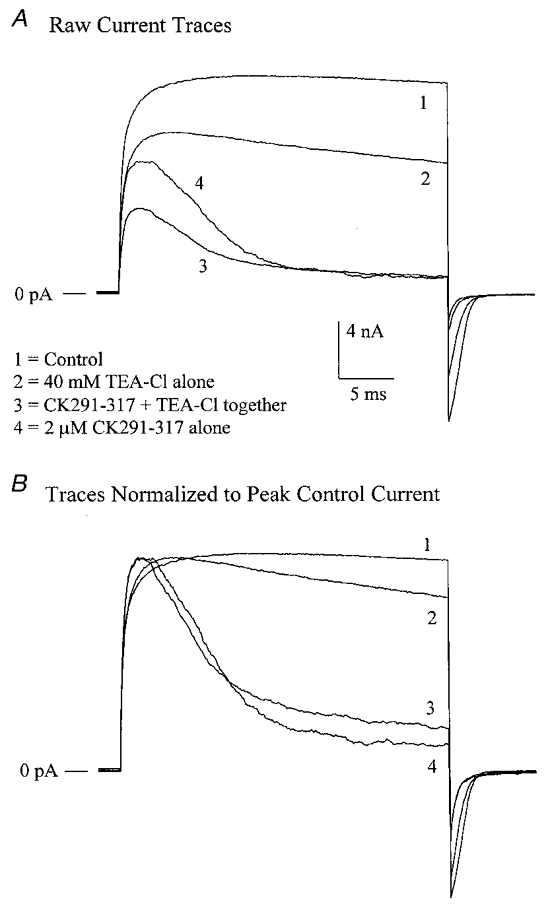
A, in the presence of 2 μm cytoplasmic free Ca2+, macroscopic BKCa channel currents were recorded from an inside-out patch in response to voltage clamps steps from 0 to +140 mV once every 4 s. Following control recordings (trace 1), 40 mm TEA-Cl was added to the bath solution, producing a significant decrease in amplitude (trace 2). The bath solution was then switched to one containing 40 mm TEA-Cl + 2 μm CK219–317, leading to a time-dependent inactivation of current (trace 3). TEA-Cl was then washed out, leaving only 2 μm CK291–317 (trace 4). B shows the relative time courses of current decay for traces 1–4, following normalization of each current to the peak current amplitude of trace 1. CK291–317 produces a similar rate of decay in the absence and presence of internal TEA. Note that traces 1–4 represent the averages of 5–8 individual sweeps recorded under each condition.
DISCUSSION
Calcium binding by BKCa channels is essential for their physiological activity as calcium shifts the voltage-dependent gating of these channels, thus allowing activation to occur within the physiological range of membrane potentials. To date, neither the calcium-binding site in the BKCaα subunit, nor the molecular mechanism by which calcium binding influences channel gating has been clearly defined. In the present study, we have hypothesized that BKCa channel gating may be controlled in a manner similar to the activation of calcium-CaM-dependent enzymes, such as protein kinases (Kemp & Pearson, 1991; Nairn & Picciotto, 1994). In these molecules, free CaM first binds calcium, then interacts with its target site in the autoregulatory domain of the enzyme (Klee & Vanaman, 1982). This interaction with calcium-bound CaM relieves a tonic inhibition imposed by the autoregulatory domain of the protein kinase, thus allowing the catalytic site to become exposed and active (Crivici & Ikura, 1995). The plant calcium-CaM-dependent protein kinase CDPK utilizes a very similar mechanism, except that a CaM-like sequence is built into the primary structure of the molecule, thereby eliminating the need for exogenous CaM. This situation therefore more closely resembles that of the BKCa channel (see Fig. 2).
In the present study, we have examined the possibility that the intrinsic calcium sensor of the BKCa channel functions in a fashion that is mechanistically similar to CaM. In this scheme, calcium binding to the intrinsic sensor of the channel would allow it to interact with an intramolecular target domain, thereby relieving some type of ‘tonic constraint’ within the channel. This ‘relief’ would translate into a shift of channel activation to more negative membrane potentials (Fig. 2A). It is known for Ca2+-CaM-dependent enzymes that a synthetic peptide derived from the autoregulatory region of the enzyme can prevent activation by acting as a ‘pseudo-target’ and compete for the binding of available calcium-bound CaM. If we imagine the BKCa channel to contain a similar target for its intrinsic calcium sensor, then CaM-binding peptides may also act as pseudo-targets for the calcium-bound sensor of the channel and inhibit channel activity by blocking interaction of the intrinsic calcium sensor with its endogenous target.
The presence of such a hypothetical target domain acting as an ‘inhibitory constraint’ is consistent with recent observations by Salkoff and co-workers (Schreiber et al. 1999). In these elegant studies, chimeric BKCa channels were constructed containing the transmembrane segments and pore of the BKCa channel (‘core’ domain) and the entire C-terminus (‘tail’ domain) from the calcium-insensitive mSlo3 channel. As predicted, this chimeric construct displayed calcium-insensitive gating, but importantly, the half-maximal voltage of activation, in the absence of calcium, was shifted ∼100 mV more negative compared to wild-type BKCa channels. Such observations led to the interpretation that the BKCa channel C-terminus contains a region that acts as an inhibitory constraint, which when removed, allows the channel to gate at significantly more negative membrane voltages. This interpretation would thus be consistent with the model presented in Fig. 2A.
Identification of the CaM-binding domains within a number of CaM-dependent enzymes has led to the synthesis of peptides which antagonize CaM-dependent events by competitive binding of calcium-bound CaM (O'Neil & DeGrado, 1990). In our experiments, cytoplasmic application of such CaM-binding peptides derived from the calcium-CaM-dependent enzymes CaM kinase II (Payne et al. 1988) and cNOS (Bredt et al. 1991; Zhang & Vogel, 1994) was observed to potently block BKCa channel activity in a dose-dependent manner (Figs 3–5). Similarly, a non-peptide CaM antagonist, calmidazolium, was also found to inhibit macroscopic currents (see Fig. 7) at concentrations (1–5 μm) similar to those required for inhibition of the CaM-containing enzyme phosphorylase kinase (van Belle, 1981). Taken together, these observations demonstrate that certain classes of CaM antagonists are potent inhibitors of BKCa channels when applied to the intracellular face.
An interesting finding is that inhibition of BKCa channel activity by CK291–317 or cNOS725–747 could be seen as a time-dependent decay of macroscopic current during a depolarizing step (4B and 8B). However, in the presence of somewhat higher free calcium (3 Figs 4B and 8B). However, in the presence of somewhat higher free calcium (3–4 μm), inhibition was not time dependent (Fig. 3B and C 4 μm), inhibition was not time dependent (Fig. 3B and C). This could occur if BKCa channel inhibition by the peptide occurred very rapidly during the start of the voltage clamp step and could not be resolved at the sampling rate used, or was complete prior to the start of the voltage clamp steps. At a holding potential of 0 mV and with 3–4 μm free cytosolic calcium, BKCa channel open probability (Popen) is ∼0.1, based on conductance-voltage relations (see Fig. 1F and Cui et al. 1997). It is thus likely that under these steady-state conditions, Popen is sufficient to allow the high affinity binding of CK291–317 to equilibrate with its target site in the channel.
Collectively, our observations indicate that the CaM antagonists used in this study inactivate BKCa channels, rather than simply preventing the action of calcium to shift voltage-dependent gating. In the latter situation, we would expect to see two populations of active channels, unaffected channels with calcium bound in which gating is shifted to more negative potentials, and those with bound calcium, but which behave like purely voltage-gated channels due to interference by CaM inhibitors of the leftward shifts of gating by calcium. However, in the presence of either CK291–317 or cNOS725–747, conductance-voltage curves were well fitted by single Boltzmann functions and displayed voltage- and calcium-dependent properties similar to those of control curves. This observation suggests that CaM-binding peptides reduce the number of functional channels, rather than allow gating of affected channels in a strictly voltage-dependent fashion.
One result that differs from the mechanistic model for the inhibition of calcium-dependent enzymes by CaM-binding peptides is the observed inhibition of BKCa channel activity by CK291–317 in the absence of intracellular free calcium (Fig. 8B). In the case of CaM, it is known that calcium binding induces a conformational change, allowing the molecule to fold around its target site (Ikura et al. 1992; Ikura, 1996). In the absence of calcium, CaM is unable to bind such a target and competitive peptides such as CK291–317 are ineffective inhibitors of enzyme activity (Hanley et al. 1987; Kelly et al. 1988). Similarly, it may be expected that the calcium sensor of BKCa channels should be unoccupied in the absence of cytosolic calcium, and by strict analogy with CaM, this site would be unable to interact with a potential target such as the CK291–317 peptide. Can such an observation still fit within the model of BKCa channel activation by calcium as presented in Fig. 2? One possibility is that the calcium sensor of the channel may be able to bind these peptides in either the absence or the presence of free calcium. This situation would be analogous to the observation that proteins with ‘IQ’ motifs, such as neuromodulin, neurogranin, myosin (Rhoads & Friedberg, 1997) and PEP-19 (Slemmon et al. 1996), bind CaM in both the absence and the presence of calcium. More recently, small conductance, calcium-activated K+ channels (SK channels) have been shown to use CaM as their intrinsic calcium sensor, which may remain constitutively bound to the channel protein even in the absence of calcium (Xia et al. 1998; Keen et al. 1999). In BKCa channels, it is thus possible that the intrinsic calcium sensor also interacts with a target domain in a ‘quasi-permanent’ fashion, and calcium binding induces a conformational change that is transferred via the target to another domain within the molecule.
A more simple interpretation of our results could be that inhibition of BKCa channel current by CaM-binding peptides reflects a direct blockade of the channel pore itself. In this case, peptide binding would occur at or near the inner mouth of the pore and inhibit the channel by directly plugging the conduction pathway. For example, the observed time dependence of inhibition (B and 8B), the micromolar affinity of peptide inhibition and a Hill coefficient near unity would be consistent with such a mechanism. To address this question, we utilized a strategy previously developed to examine the mechanism of block of BKCa channels by an inactivating ‘ball’ peptide derived from the N-terminus of the Shaker K+ channel (Toro et al. 1992; Foster et al. 1992). In these experiments, the actions of the ball peptide were compared in the absence and presence of internal TEA, a compound known to block the BKCa channel pore directly (Vergara et al. 1984; Yellen, 1984; Villarroel et al. 1988), and expected to compete with another agent acting at a similar site. As shown in Fig. 10, application of internal TEA did not interfere with the blocking action of CK291 Figs 4B and 8B), the micromolar affinity of peptide inhibition and a Hill coefficient near unity would be consistent with such a mechanism. To address this question, we utilized a strategy previously developed to examine the mechanism of block of BKCa channels by an inactivating ‘ball’ peptide derived from the N-terminus of the Shaker K+ channel (Toro et al. 1992; Foster et al. 1992). In these experiments, the actions of the ball peptide were compared in the absence and presence of internal TEA, a compound known to block the BKCa channel pore directly (Vergara et al. 1984; Yellen, 1984; Villarroel et al. 1988), and expected to compete with another agent acting at a similar site. As shown in Fig. 10, application of internal TEA did not interfere with the blocking action of CK291–317, in contrast with the reported effects of TEA on blockade of BKCa channels by the Shaker ball peptide. This observation suggests that CK291–317 acts at a site distinct from that of TEA, most probably outside the pore region of the channel. Our conclusion is similar to that recently reported for the simultaneous blockade of single BKCa channels by the two positively charged peptides bovine pancreatic trypsin inhibitor (BPTI) and dendrotoxin-I (Favre & Moczydlowski, 1999) 317, in contrast with the reported effects of TEA on blockade of BKCa channels by the Shaker ball peptide. This observation suggests that CK291–317 acts at a site distinct from that of TEA, most probably outside the pore region of the channel. Our conclusion is similar to that recently reported for the simultaneous blockade of single BKCa channels by the two positively charged peptides bovine pancreatic trypsin inhibitor (BPTI) and dendrotoxin-I (Favre & Moczydlowski, 1999). CK291–317, also a basic peptide, may thus act like BPTI and/or dendrotoxin and cause allosteric inhibition of the channel, in line with the model presented in Fig. 2.
In conclusion, our data are consistent with the hypothesis that the calcium-dependent alteration of BKCa channel gating may occur by a process in which the gating mechanism of the channel is under a ‘tonic constraint’ that is somehow relieved following calcium binding to the intrinsic calcium sensor. By analogy with Ca2+-CaM-dependent enzymes, this relief may involve a physical interaction of the calcium sensor with a putative autoregulatory domain within the channel.
Acknowledgments
The authors thank Dr R. W. Aldrich and members of his laboratory for their helpful discussions during the early phases of these studies and Dr W. C. Cole for providing the cDNA for the rabbit Kv1.5 α subunit. This work was supported in part by an AHFMR Establishment grant and MRCC grant MT-14066 to A.P.B.
References
- Asano M, Masuzawa-Ito K, Matsuda T. Charybdotoxin-sensitive K+ channels regulate the myogenic tone in the resting state of arteries from spontaneously hypertensive rats. British Journal of Pharmacology. 1993;108:214–222. doi: 10.1111/j.1476-5381.1993.tb13465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano M, Nomura Y, Ito K, Uyama Y, Imaizumi Y, Watanabe M. Increased function of voltage-dependent Ca2+ channels and Ca2+-activated K+ channels in resting state of femoral arteries from spontaneously hypertensive rats at prehypertensive stage. Journal of Pharmacology and Experimental Therapeutics. 1995;275:775–783. [PubMed] [Google Scholar]
- Blatz AL, Magleby KL. Calcium-activated potassium channels. Trends in Neurosciences. 1987;10:463–467. [Google Scholar]
- Braun AP, Schulman H. A non-selective cation current activated via the multifunctional Ca2+-calmodulin-dependent protein kinase in human epithelial cells. The Journal of Physiology. 1995a;488:37–55. doi: 10.1113/jphysiol.1995.sp020944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun AP, Schulman H. The multifunctional calcium/calmodulin-dependent protein kinase: from form to function. Annual Review of Physiology. 1995b;57:417–445. doi: 10.1146/annurev.ph.57.030195.002221. [DOI] [PubMed] [Google Scholar]
- Brayden JE, Nelson MT. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science. 1992;256:532–535. doi: 10.1126/science.1373909. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Hwang PM, Glatt CE, Lowenstein C, Reed RR, Snyder SH. Cloned and expressed nitric oxide synthase structurally resembles cytochrome P-450 reductase. Nature. 1991;351:714–718. doi: 10.1038/351714a0. [DOI] [PubMed] [Google Scholar]
- Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- Clément-Chomienne O, Ishii K, Walsh MP, Cole WC. Identification, cloning and expression of rabbit vascular smooth muscle Kv1.5 and comparison with native delayed rectifier K+ currents. The Journal of Physiology. 1999;515:653–667. doi: 10.1111/j.1469-7793.1999.653ab.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crivici A, Ikura M. Molecular and structural basis of target recognition by calmodulin. Annual Review of Biophysics and Biomolecular Structure. 1995;24:85–116. doi: 10.1146/annurev.bb.24.060195.000505. [DOI] [PubMed] [Google Scholar]
- Cui J, Cox DH, Aldrich RW. Intrinsic voltage dependence and Ca2+ regulation of mSlo large conductance Ca-activated K+ channels. Journal of General Physiology. 1997;109:647–673. doi: 10.1085/jgp.109.5.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falke JJ, Drake SK, Hazard AL, Peersen OB. Molecular tuning of ion binding to calcium signaling proteins. Quarterly Review of Biophysics. 1994;27:219–290. doi: 10.1017/s0033583500003012. [DOI] [PubMed] [Google Scholar]
- Favre I, Moczydlowski E. Simultaneous binding of basic peptides at intracellular sites on a large conductance, Ca2+-activated K+ channel. Journal of General Physiology. 1999;113:295–320. doi: 10.1085/jgp.113.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster CD, Chung S, Zagotta WN, Aldrich RW, Levitan IB. A peptide derived from the Shaker B K+ channel produces short and long blocks of reconstituted Ca2+-dependent K+ channels. Neuron. 1992;9:229–236. doi: 10.1016/0896-6273(92)90162-7. [DOI] [PubMed] [Google Scholar]
- Hanley RM, Means AR, Ono T, Kemp BE, Burgin KE, Waxham N, Kelly PT. Functional analysis of a complementary DNA for the 50-kilodalton subunit of calmodulin kinase II. Science. 1987;237:293–297. doi: 10.1126/science.3037704. [DOI] [PubMed] [Google Scholar]
- Horrigan FT, Cui J, Aldrich RW. Allosteric voltage gating of potassium channels I. mSlo ionic currrents in the absence of calcium. Journal of General Physiology. 1999a;114:277–304. doi: 10.1085/jgp.114.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horrigan FT, Cui J, Aldrich RW. Allosteric voltage gating of potassium channels II. mSlo channel gating charge movement in the absence of calcium. Journal of General Physiology. 1999b;114:305–336. doi: 10.1085/jgp.114.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikura M. Calcium binding and conformational response in EF-hand proteins. Trends in Biochemical Sciences. 1996;21:14–17. [PubMed] [Google Scholar]
- Ikura M, Clore GM, Gronenborn AM, Zhu G, Klee CB, Bax A. Solution structure of a calmodulin-target peptide complex by multidimensional NMR. Science. 1992;256:632–638. doi: 10.1126/science.1585175. [DOI] [PubMed] [Google Scholar]
- Ito M, Guerriero V, Hartshorne DJ. Structure-function relationships in smooth muscle myosin light chain kinase. Advances in Experimental Medicine and Biology. 1991;304:3–10. doi: 10.1007/978-1-4684-6003-2_2. [DOI] [PubMed] [Google Scholar]
- Jaggar JH, Stevenson AS, Nelson MT. Voltage dependence of Ca2+ sparks in intact cerebral arteries. American Journal of Physiology. 1998;274:C1755–1761. doi: 10.1152/ajpcell.1998.274.6.C1755. [DOI] [PubMed] [Google Scholar]
- Keen JE, Khawaled R, Farrens DL, Neelands T, Rivard A, Bond CT, Janowsky A, Fakler B, Adelman JP, Maylie J. Domains responsible for constitutive and Ca2+-dependent interactions between calmodulin and small conductance Ca2+-activated potassium channels. Journal of Neuroscience. 1999;19:8830–8838. doi: 10.1523/JNEUROSCI.19-20-08830.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly PT, Weinberger RP, Waxham MN. Active site-directed inhibition of Ca2+/calmodulin-dependent protein kinase type II by a bifunctional calmodulin-binding peptide. Proceedings of the National Academy of Sciences of the USA. 1988;85:4991–4995. doi: 10.1073/pnas.85.14.4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp BE, Pearson RB. Intrasteric regulation of protein kinases and phosphatases. Biochimica et Biophysica Acta. 1991;1094:67–76. doi: 10.1016/0167-4889(91)90027-u. [DOI] [PubMed] [Google Scholar]
- Klee CB, Vanaman TC. Calmodulin. Advances in Protein Chemistry. 1982;35:213–321. doi: 10.1016/s0065-3233(08)60470-2. [DOI] [PubMed] [Google Scholar]
- Klockner U, Isenberg G. Calmodulin antagonists depress calcium and potassium currents in ventricular and vascular myocytes. American Journal of Physiology. 1987;253:H1601–1611. doi: 10.1152/ajpheart.1987.253.6.H1601. [DOI] [PubMed] [Google Scholar]
- Knighton DR, Pearson RB, Sowadski JM, Means AR, Ten Eyck LF, Taylor SS, Kemp BE. Structural basis of the intrasteric regulation of myosin light chain kinases. Science. 1992;258:130–135. doi: 10.1126/science.1439761. [DOI] [PubMed] [Google Scholar]
- Latorre R, Oberhauser A, Labarca P, Alvarez O. Varieties of calcium-activated potassium channels. Annual Review of Physiology. 1989;51:385–399. doi: 10.1146/annurev.ph.51.030189.002125. [DOI] [PubMed] [Google Scholar]
- McManus OB. Calcium-activated potassium channels: Regulation by calcium. Journal of Bioenergetics and Biomembranes. 1991;23:537–560. doi: 10.1007/BF00785810. [DOI] [PubMed] [Google Scholar]
- Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- Marsden BJ, Shaw GS, Sykes BD. Calcium binding proteins. Elucidating the contributions to calcium affinity from an analysis of species variants and peptide fragments. Biochemistry and Cell Biology. 1990;68:587–601. doi: 10.1139/o90-084. [DOI] [PubMed] [Google Scholar]
- Meera P, Wallner M, Jiang Z, Toro L. A calcium switch for the functional coupling between α (hslo) and β subunits (Kv,Caβ) of maxi K channels. FEBS Letters. 1996;382:84–88. doi: 10.1016/0014-5793(96)00151-2. [DOI] [PubMed] [Google Scholar]
- Moncrief ND, Kretsinger RH, Goodman M. Evolution of EF-hand calcium-modulated proteins. I. Relationships based on amino acid sequences. Journal of Molecular Evolution. 1990;30:522–562. doi: 10.1007/BF02101108. [DOI] [PubMed] [Google Scholar]
- Nairn AC, Picciotto MR. Calcium/calmodulin-dependent protein kinases. Seminars in Cancer Biology. 1994;5:295–303. [PubMed] [Google Scholar]
- Nakazawa K, Higo K, Abe K, Tanaka Y, Saito H, Norio M. Blockade by calmodulin inhibitors of Ca2+ channels in smooth muscle from rat vas deferens. British Journal of Pharmacology. 1993;109:137–141. doi: 10.1111/j.1476-5381.1993.tb13543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- O'Neil KT, DeGrado WF. How calmodulin binds its targets: sequence independent recognition of amphiphilic α-helices. Trends in Biochemical Sciences. 1990;15:59–64. doi: 10.1016/0968-0004(90)90177-d. [DOI] [PubMed] [Google Scholar]
- Pallanck L, Ganetzky B. Cloning and characterization of human and mouse homologs of the Drosophilia calcium-activated potassium channel gene, slowpoke. Human Molecular Genetics. 1994;3:1239–1243. doi: 10.1093/hmg/3.8.1239. [DOI] [PubMed] [Google Scholar]
- Payne ME, Fong Y-L, Ono T, Colbran RJ, Kemp BE, Soderling TR, Means AR. Calcium/calmodulin-dependent protein kinase II. Characterization of distinct calmodulin binding and inhibitory domains. Journal of Biological Chemistry. 1988;263:7190–7195. [PubMed] [Google Scholar]
- Rhoads AR, Friedberg F. Sequence motifs for calmodulin recognition. FASEB Journal. 1997;11:331–340. doi: 10.1096/fasebj.11.5.9141499. [DOI] [PubMed] [Google Scholar]
- Roberts DM, Harmon AC. Calcium-modulated proteins: Targets of intracellular calcium signals in higher plants. Annual Review of Plant Physiology and Plant Molecular Biology. 1992;43:375–414. [Google Scholar]
- Robitaille R, Charlton MP. Presynaptic calcium signals and transmitter release are modulated by calcium-activated potassium channels. Journal of Neuroscience. 1992;12:297–305. doi: 10.1523/JNEUROSCI.12-01-00297.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille R, Garcia ML, Kaczorowski GJ, Charlton MP. Functional colocalization of calcium and calcium-gated potassium channels in control of transmitter release. Neuron. 1993;11:645–655. doi: 10.1016/0896-6273(93)90076-4. [DOI] [PubMed] [Google Scholar]
- Rudy B. Diversity and ubiquity of K channels. Neuroscience. 1988;25:729–749. doi: 10.1016/0306-4522(88)90033-4. [DOI] [PubMed] [Google Scholar]
- Schreiber M, Yuan A, Salkoff L. Transplantable sites confer calcium sensitivity to BK channels. Nature Neuroscience. 1999;2:416–421. doi: 10.1038/8077. [DOI] [PubMed] [Google Scholar]
- Slemmon JR, Morgan JI, Fullerton SM, Danho W, Hilbush BS, Wengenack TM. Camstatins are peptide antagonists of calmodulin based upon a conserved structural motif in PEP-19, neurogranin and neuromodulin. Journal of Biological Chemistry. 1996;271:15911–15917. doi: 10.1074/jbc.271.27.15911. [DOI] [PubMed] [Google Scholar]
- Stefani E, Ottolia M, Noceti F, Olcese R, Wallner M, Latorre R, Toro L. Voltage-controlled gating in a large conductance Ca2+-sensitive K+ channel (hslo) Proceedings of the National Academy of Sciences of the USA. 1997;94:5427–5431. doi: 10.1073/pnas.94.10.5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takebe Y, Seiki M, Fujisawa J-I, Hoy P, Yokota K, Arai K-I, Yoshida M, Arai N. SRα promoter: An efficient and versatile mammalian cDNA expression system composed of the Simian virus 40 early promoter and the R-U5 segment of human T-cell leukemia virus type 1 long terminal repeat. Molecular and Cellular Biology. 1988;8:466–472. doi: 10.1128/mcb.8.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toro L, Stefani E, Latorre R. Internal blockade of a Ca2+-activated K+ channel by Shaker B inactivating ‘ball’ peptide. Neuron. 1992;9:237–245. doi: 10.1016/0896-6273(92)90163-8. [DOI] [PubMed] [Google Scholar]
- van Belle H. R 24571: A potent inhibitor of calmodulin-activated enzymes. Cell Calcium. 1981;2:483–494. [Google Scholar]
- Vergara C, Moczydlowski E, Latorre R. Conduction, blockade and gating in a Ca2+-activated K+ channel incorporated into planar lipid bilayer. Biophysical Journal. 1984;45:73–76. doi: 10.1016/S0006-3495(84)84114-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarroel A, Alvarez O, Oberhauser A, Latorre R. Probing a Ca2+-activated K+ channel with quaternary ammonium ions. Pflügers Archiv. 1988;413:118–126. doi: 10.1007/BF00582521. [DOI] [PubMed] [Google Scholar]
- Xia X-M, Fakler B, Rivard A, Wayman G, Johnson-Pais T, Keen JE, Ishii T, Hirshberg B, Bond CT, Lutsenko S, Maylie J, Adelman JP. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature. 1998;395:503–507. doi: 10.1038/26758. [DOI] [PubMed] [Google Scholar]
- Yellen G. Ionic permeation and blockade in Ca2+-activated K+ channels of bovine chromaffin cells. Journal of General Physiology. 1984;84:157–186. doi: 10.1085/jgp.84.2.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Vogel HJ. Characterization of the calmodulin-binding domain of rat cerebellar nitric oxide synthase. Journal of Biological Chemistry. 1994;269:981–985. [PubMed] [Google Scholar]