

Abstract
The quaternary ammonium (QA) lidocaine derivative QX-314 (2-(triethylamino)-N--(2,6-dimethylphenyl)-acetamide) induces internal pore blockade of single cardiac Na+ channels enzymatically modified (papain) to eliminate fast inactivation. The mechanism involves dual, interacting blocking modes (rapid and discrete) with binding domains deep in the pore from the cytoplasmic mouth, and where the rapid blocked configuration serves as a transition-intermediate for the development of discrete block. The primary goals of this study were to test for this mechanism in a recombinant Na+ channel genetically engineered to selectively lack fast inactivation, and if present, to explore the underlying structural features.
Fast inactivation was removed in rat skeletal muscle μ1 Na+ channels (RSkM1) with an IFM-QQQ mutation in the cytoplasmic III-IV interdomain (QQQ). QQQ was expressed in Xenopus oocytes and single-channel activity was studied in cell-free, inside-out membrane patches. Application of QX-314 (QX, 0-4 mM) to the cytoplasmic membrane surface caused two distinct modalities of single-channel blockade: reduction of unitary current and interruptions of current lasting tens of milliseconds. These are consistent with rapid and discrete pore block, respectively. The voltage and concentration dependence of block indicates that the modes interact and have binding sites that share a deep location in the pore, at ≈65 % of the membrane electric field in from the cytoplasmic mouth.
Mutation of phenylalanine (F1579) in domain IV-S6, critical in local anaesthetic block, to alanine in QQQ (QQQ-F1579A) disabled discrete block but notably failed to alter rapid block, single-channel gating and slope conductance.
Amplitude distribution analysis was applied to long bursts (> 50 ms) of QQQ-F1579A activity to investigate the kinetics of rapid block. Computed rapid blocking and unblocking rate constants are 42 000 ± 18 000 M−1 ms−1 and 82 ± 22 ms−1, respectively (n = 3, -20 mV).
The results support a general transition-intermediate mechanism that governs internal QX and local anaesthetic pore block of voltage-gated Na+ channels and provide insight into underlying structural features.
Quaternary ammonium (QA) ions have been utilized as probes of the conduction pathway of voltage-gated K+ (Armstrong, 1971; Miller, 1982; Kirsch et al. 1991) and Na+ channels (Moczydlowski, 1986; O'Leary & Horn, 1994). Inhibition of Na+ channels by the QA QX-314 (QX) has been particularly well studied (Frazier et al. 1970; Strichartz, 1973; Hille, 1977; Cahalan, 1978; Cahalan & Almers, 1979; Wang et al. 1987; Zamponi et al. 1993; Gingrich et al. 1993; Balser et al. 1996). QX is a permanently charged analogue of the local anaesthetic lidocaine. As a result, investigating QX interactions with the pore of Na+ channels provides important biophysical information and offers insight into the molecular pharmacology of local anaesthetics.
We have previously studied the effects of internal QX on single native cardiac Na+ channels enzymatically (papain) treated to remove fast inactivation, thereby enabling unobscured observation of QX block of open channels (Gingrich et al. 1993). Block occurred on two distinct time scales: one so rapid that it was manifested as a reduction of apparent unitary current and the other produced interruptions of current lasting tens of milliseconds, consistent with rapid and discrete pore blockade, respectively. Associated binding sites shared an apparently deep physical location in the pore, as reported by electrical distances that traversed ∼70 % of the electric field in from the cytoplasmic channel mouth. In addition, blocking modes interacted such that the rapid quaternary ammonium complex modulated the formation of the longer-lived, discrete blocked state. The results led us to propose a mechanism of QX pore block involving two distinct binding domains and where the rapid-block configuration serves as a ‘transition-intermediate’ state that catalyses formation of discrete block.
In this study, we wished to test for this mechanism in a recombinant Na+ channel genetically engineered to selectively lack fast inactivation. We studied internal QX block of single rat skeletal muscle μ1 Na+ channels (RSkM1) containing an IFM-QQQ mutation in the III-IV interdomain (QQQ) that eliminates fast inactivation (West et al. 1992; Hartmann et al. 1994). The results reproduce key features of our previous findings, thereby supporting a transition-intermediate mechanism, and discounting the possibility of previous confounding non-specific enzyme effects. Alanine (A) substitution for phenyalanine (F) at location 1579 (QQQ-F1579A) in domain IV-S6, a residue important in local anaesthetic block, selectively disabled discrete block. This indicates that F1579 plays a critical role in discrete block which probably arises from its contribution to a distinct binding domain or through conformational influences. Overall, the results support a general mechanism involving a transition intermediate that governs internal QX and local anaesthetic pore block of voltage-gated Na+ channels, and provides insight into the underlying structural features. A preliminary report of this work has appeared in abstract form (Kimbrough & Gingrich, 1998).
METHODS
Molecular biology
Rat skeletal muscle Na+ channel μ1 α-subunit (RSkM1; graciously provided by Dr David Yue, Department of Biomedical Engineering, The Johns Hopkins University) was inserted in pBluescript (KS-) at the Eco RI site. Mutations were performed using sequential PCR-based mutagenesis (Sambrook et al. 1989). RSkM1 was mutated from wild type (αWT: [I1303, F1304, M1305]) to the QQQ genotype (αQQQ: [Q1303, Q1304, Q1305]). This mutation eliminates fast inactivation (West et al. 1992; Hartmann et al. 1994). The additional point mutation F1579A in domain IV-S6 was performed in QQQ and is referred to as QQQ-F1579A. Oligonucleotides were synthesized on Applied Biosystems DNA synthesizers. All mutations were confirmed using dideoxynucleotide sequencing. The Xenopusβ-globin untranslated region flanked the channel-coding sequence in pBluescript (KS-) to enhance expression. Rat brain Na+ channel accessory β1 subunit was kindly provided by Dr William Catterall (Department of Pharmacology, University of Washington), in pBluescript (KS+) inserted at the EcoRI site. cRNAs were synthesized using T3 RNA polymerase after a linearizing cut at Sal I, and followed by 5′ capping.
Channel expression in oocytes
Oocytes were obtained from Xenopus laevis purchased from Nasco (Fort Atkinson, WI, USA) or Xenopus Express (Homosassa, FL, USA). Oocytes were collected under anaesthesia (3.7 g tricaine l−1) from frogs that were humanely killed after the final collection. This protocol (UCAR 97-9-1) was approved by the University Committee on Animal Resources, University of Rochester.
Oocytes were defolliculated by agitating in frog OR2 solution ((mm): 100 NaCl, 2 KCl, 1 MgCl2, 5 Hepes, pH 7.6 with NaOH) containing collagenase (0.2 mg ml−1 collagenase type 1A, Sigma) for 1-3 h at 20°C. Oocytes were injected with 30-50 nl (5-20 ng) of 5′ capped cRNA of αWT with β1 (1:1 ratio by weight which provides saturating β1 effects on whole-cell current kinetics (Cannon & Strittmatter, 1993)), referred to as WT-β1 or QQQ alone. The oocytes were incubated in media containing (mm): 100 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, 5 Hepes and 2.5 sodium pyruvate; plus 1 % penicillin-streptomycin, 2 % horse serum, pH 7.6, for 1-7 days at 16°C prior to experimentation.
Single channel recordings
Currents of single Na+ channels were studied using inside-out, cell-free, membrane patches. Oocytes were immersed in a hypertonic stripping solution ((mm): 200 aspartate, 20 CsCl, 2 MgCl2, 2 EGTA, 10 Hepes, pH 7.35 with TEA-OH); devitellinized and transferred to a bath solution ((mm): 120 CsF, 20 CsCl, 2 MgCl2, 2 EGTA, 10 Hepes, pH 7.35 with CsOH). Pipettes were pulled from borosilicate glass to a resistance of 2-20 MΩ, coated with Sylgard (Dow Corning, NY, USA), fire polished and filled with (mm): 145 NaCl, 2 MgCl2, 10 Hepes, pH 7.35 with TEA-OH. The cytoplasmic surface was exposed to control (bath) and test (QX: 0-4 mm) solutions via a local solution changer. Single channel currents were measured with an Axopatch 200A amplifier (Axon Instruments, Foster City, CA, USA), lowpass filtered at 1-2 kHz (-3 db, 4-pole, Bessel), and digitally sampled at ≤ 100 μs intervals. The rise time of the patch-clamp recording system is < 0.25 ms. Na+ channel activity was elicited every 1-5 s by voltage steps lasting ∼200 ms.
Data analysis
These methods have been previously described (Gingrich et al. 1993). Briefly, single channel records were compensated for leak and capacity currents by subtracting smooth functions, and were idealized by half-height criteria. Adjustments were made for two-channel patches: in some patches, two channels rather than one were observed, as determined by the stacking of unitary events. In such cases, for the analysis of the discrete block of open channels, only open durations from non-stacked openings were considered. As well, only the intervals between openings with both channels closed were compiled for closed time analysis. Corrections were made for missed events: open and closed durations, as detected by a half-height criterion, suffer from artifactual lengthening intrinsic to this detection method (see Colquhoun & Sigworth, 1995). Dwell time histograms were binned logarithmically (Sigworth & Sine, 1987). Artifactual lengthening was addressed by fitting transformed multi-exponential probability density functions to histograms only considering durations greater than the system dead time (td≡ 0.2 ms). Parameters derived from such function fits still, however, suffer from distortions owing to missed events. To account for these, we took advantage of the equations of Blatz & Magelby (1986) that predict what the observed exponential parameters, distorted by missed events, should be, given an assumed set of true rate constants for the four transitions of the gating equation C ⇌ O ⇌ D, where C and O are native closed and open states, respectively, and D is the open state after open channel blockade in the discrete configuration. Using a Levenberg-Marquart method (Matlab, Natick, MA, USA), we could then iteratively solve the set of rate constants (including discrete block and unblock rates) that best predicts the observed exponential parameters as distorted by missed events. Although the corrected transition rates for the equivalent one-channel model can be used to predict the closed time histograms for two-channels, Pcth,2(t), this will differ significantly from the analogous distribution for one of the two channels, Pcth,1(t). However, binomial statistics allows Pcth,1(t) to be calculated from Pcth,2(t) according to the following quadratic equation:
where Fo,1 is the fraction of sweeps with activity for a single channel, derived from the measured overall fraction of active sweeps in the two-channel patch. Parameters for single- and bi-exponential fits to Poth,1(t) and Pcth,1(t) (for open and closed channels, respectively), can then be used to calculate the discrete block and unblock rates that would be anticipated for either one of the two channels in the patch.
Rapid block with kinetics beyond the response time of our recording system results in brief interruptions in open channel current that are low-pass filtered to appear as a uniform reduction in unitary current (Yellen, 1984b). We employed the technique detailed by Yellen (1984b) to deduce the kinetics of rapid block. Briefly, blocking and unblocking rates can be inferred by assuming a simple two-state (open and blocked) Poisson process filtered through a single time constant filter. The effect of this filter closely approximates that of the 4-pole Bessel filter used in this study (Yellen, 1984b). The steady-state solution to the related set of equations gives the amplitude distribution of the filter output, which is a β distribution. Sections of single channel records with one active channel, which included some sections of baseline, were included in amplitude histograms. At least two histograms were compiled for each experimental condition. A Gaussian relation was fitted to the peak corresponding to the baseline, which was then subtracted from the histogram. In order to compensate for the widening of the experimental distribution by unrelated noise, the theoretical distribution was also broadened by convolving it with the Gaussian curve used to fit the amplitude distribution of the baseline noise. Blocking and unblocking rate constants were then adjusted to give a good fit by eye of the empirical distribution by the theoretical.
Statistical tests were performed as described in the figure legends. Maximum stacking of unitary events in control at high open probability was taken as channel number. Grouped data are presented as means ±s.e.m. One-tailed P values ≤ 0.05 were considered to have statistical significance.
RESULTS
Elimination of fast inactivation slows native gating and allows clear observation of QX interactions with the channel. We previously reported that QX induced dual (rapid and discrete) interacting modes of pore blockade suggesting a transition-intermediate mechanism in cardiac Na+ channels enzymatically modified with papain to remove fast inactivation (Gingrich et al. 1993). We wished to confirm these findings in a channel modified by other means since enzymatic modification may alter local anaesthetic pharmacology (Yeh, 1978; Cahalan, 1978; Cahalan & Almers, 1979; Wang et al. 1987; Yeh & TenEick, 1987). We decided against channel modification using toxins since they may interfere with local anaesthetic binding (Postma & Catterall, 1984; McHugh et al. 1995; Linford et al. 1998; Wang et al. 1998b). A genetically engineered channel is a powerful platform that provides for targeted changes in channel structure and the investigation of underlying structure-function relationships. Therefore, we studied single channel currents of rat skeletal muscle μ1 Na+ channels (RSkM1) containing an IFM-QQQ mutation in the III-IV interdomain that selectively eliminates fast inactivation (West et al. 1992; Hartmann et al. 1994), referred to as QQQ.
QX induces rapid and discrete blockade of QQQ currents
Figure 1A shows the activity of an inside-out, cell-free, membrane patch containing a single QQQ channel. The channel remained primarily open and the ensemble current decayed little over the 200 ms depolarization demonstrating the absence of fast inactivation consistent with previous reports (Hirschberg et al. 1995). The permeation pathway appeared unaffected by this mutation since slope conductance was similar to wild type (Fig. 4F). Application of 2 mm QX produced two distinct effects (Fig. 1B): (1) a uniform reduction in unitary current, and (2) discrete interruptions in current lasting tens of milliseconds. These observations accord with rapid and discrete pore block, respectively. The kinetics of rapid block are probably more than 10-fold faster than the rise time of our recording system, such that the interruptions in open channel current would be low-pass filtered to appear as a uniform reduction in unitary current (Yellen, 1984b). These observations agree with our previous results and support a transition-intermediate mechanism of pore blockade (Gingrich et al. 1993). This mechanism is summarized in eqn (10) of (Gingrich et al. 1993)) and reproduced here in modified form as: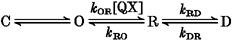 where C and O signify closed (non-conductive) and open (conductive) states that are intrinsic to native channel gating. A single QX molecule may rapidly enter the cytoplasmic mouth led by the charged QA head, bind to a deep intrapore site, and reach the rapid blocked configuration (R). This is a bimolecular binding reaction in which the forward rate constant is proportional to QX concentration [QX]. In order for the QX molecule to proceed to the higher-affinity discrete blocked state (D), we proposed that the hydrophobic aromatic tail finds its way into a more avid configuration, perhaps by entry of the hydrophobic aromatic tail into a specific hydrophobic pocket. In this way the R state serves as a transition intermediate that catalyses the formation of D. Rapid block involves movement of the charged QA head from the bulk solution to its deepest position in the pore (see Fig. 7). In contrast, discrete block is independent of voltage because it reflects stabilization of the rapid block configuration that lacks translation of the QX head within the membrane electric field. Since rapid block is likely to be orders of magnitude faster than discrete block then, practically speaking, rapid block can be considered to be in equilibrium independent of discrete block (Gingrich et al. 1993). Thus, reasoning from Scheme 1, the effective discrete block rate constant (kRD,effective) becomes equal to the product of kRD and the probability (Prob) that a channel is in the rapid blocked state (R) dependent on whether it is open (O) or rapid blocked (Prob{R|O or R}). Prob{R|O or R} =kOR[QX]/(kOR[QX]+kRO) which can be simplified to {}[QX]/([QX]+KD(V)} by substituting KD(V) =kRO/kOR. KD(V) is the rapid block dissociation constant which is voltage dependent and can be determined experimentally. As a result:
where C and O signify closed (non-conductive) and open (conductive) states that are intrinsic to native channel gating. A single QX molecule may rapidly enter the cytoplasmic mouth led by the charged QA head, bind to a deep intrapore site, and reach the rapid blocked configuration (R). This is a bimolecular binding reaction in which the forward rate constant is proportional to QX concentration [QX]. In order for the QX molecule to proceed to the higher-affinity discrete blocked state (D), we proposed that the hydrophobic aromatic tail finds its way into a more avid configuration, perhaps by entry of the hydrophobic aromatic tail into a specific hydrophobic pocket. In this way the R state serves as a transition intermediate that catalyses the formation of D. Rapid block involves movement of the charged QA head from the bulk solution to its deepest position in the pore (see Fig. 7). In contrast, discrete block is independent of voltage because it reflects stabilization of the rapid block configuration that lacks translation of the QX head within the membrane electric field. Since rapid block is likely to be orders of magnitude faster than discrete block then, practically speaking, rapid block can be considered to be in equilibrium independent of discrete block (Gingrich et al. 1993). Thus, reasoning from Scheme 1, the effective discrete block rate constant (kRD,effective) becomes equal to the product of kRD and the probability (Prob) that a channel is in the rapid blocked state (R) dependent on whether it is open (O) or rapid blocked (Prob{R|O or R}). Prob{R|O or R} =kOR[QX]/(kOR[QX]+kRO) which can be simplified to {}[QX]/([QX]+KD(V)} by substituting KD(V) =kRO/kOR. KD(V) is the rapid block dissociation constant which is voltage dependent and can be determined experimentally. As a result:
| (1) |
Figure 1. QX-314 induces discrete and rapid block of single QQQ channels.
A, individual current records from a single QQQ channel triggered by the indicated voltage protocol (top, holding potential = -100 mV). Openings are downward to a unitary current level marked by the dashed line. The ensemble average current (bottom) illustrates the absence of fast inactivation. B, response of the same channel to application of 2 mm QX-314. Individual records now show long non-conducting periods interposed by brief openings, and the unitary current amplitude is reduced. The ensemble average current (bottom) shows a marked overall inhibition of current by QX-314. Currents filtered at 1 kHz.
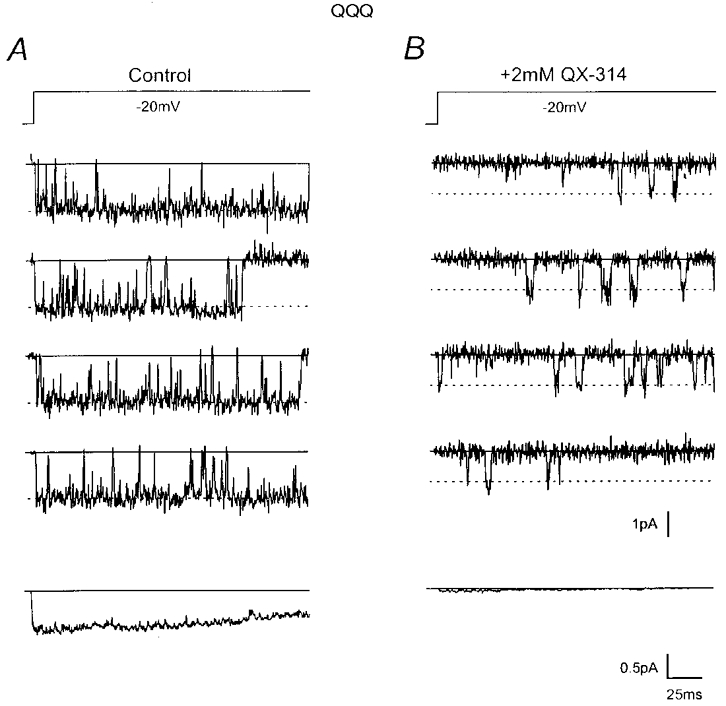
Figure 4. F1579A selectively disables discrete block.
A, individual current records of a single QQQ-F1579A Na+ channel triggered by the indicated voltage protocol (top, holding potential = -100 mV). QQQ-F1579A is predominantly open during the depolarization, similar to QQQ (cf. Fig. 1A). The ensemble average current (bottom) illustrates slow inactivation. B, response of the same channel in the presence of 1 mm QX-314. Records show reduction of unitary current, but lack discrete current interruptions as seen with QQQ (cf. Fig. 1B). Currents filtered at 1 kHz. C, open and closed time histograms in control conditions. D, open and closed time histograms in 1 mm QX-314. In both C and D, thick continuous lines are fits by transformed mono- and bi-exponential functions with the parameters as shown. Histograms were binned logarithmically. A1 and A2 are the individual coefficients of bi-exponential functions. Dashed lines represent component mono-exponential functions. The thin line is the fit of the control open time histogram replotted in D for comparison. E, bar graph of mean open time (Open), fast(ClosedF) and slow (ClosedS) closed times, and the fraction of fast closures (FractionF) for QQQ, and QQQ-F1579A in control and plus 1 mm QX-314. Parameters were derived directly from histograms and are displayed as means with s.e.m. bars (n = 3-5 patches). F, unitary current amplitude plotted versus membrane voltage for WT-β1, QQQ and QQQ-F1579A, as indicated (means ±s.e.m., n = 3-6 patches). G, plot of fractional reduction of QQQ-F1579A unitary current (1 – (iQX/ictrl), •) versus[QX-314] during voltage steps to -20 mV. H, normalized unitary current for QQQ-F1579A (•) versus membrane voltage, with 1 mm QX-314 present throughout. Data for QQQ (○) and functions fits (continuous lines) replotted from Fig. 2 for comparison in G and H. Standard error bars are shown around means derived from 3-5 patches.
This expression indicates that kRD,effective is indeed concentration and voltage dependent in contrast to kRD. Qualitatively kRD,effective saturates with increasing [QX]. Also, since KD(V) is reduced by depolarization (see eqn (2) below), kRD,effective is enhanced by the same change. In contrast, lifetimes in D are unaffected by rapid block, hence kDR is independent of both concentration and voltage. We next tested whether our empirical observations of rapid and discrete block in QQQ channels accord with Scheme 1 and eqn (1) to determine the appropriateness of the transition-intermediate mechanism.
Rapid block of open channels
Figure 2A clearly demonstrates in both raw data and amplitude histograms the reduction in unitary current induced by QX, which is consistent with rapid pore block. The degree of rapid block can be gauged by the fractional reduction in unitary current. We examined the concentration dependence of rapid block by plotting the fractional reduction in unitary current versus QX concentration (Fig. 2B). The relationship shows saturation and is well fitted by a Langmuir isotherm relationship (continuous line) consistent with involvement of a bimolecular binding reaction. The location of the binding site within the membrane electric field can be inferred by analysing the voltage dependence of rapid block. Normalized unitary current, which reports the rapid unblocked fraction, declines steeply with voltage (Fig. 2C). A Boltzmann function fit (continuous line) indicates that a QX molecule traverses 65 % of the transmembrane voltage from the cytoplasmic mouth to reach the rapid binding site deep in the pore. This deep location within the membrane electric field in conjunction with antagonism of rapid block by increased Na+ influx (Gingrich et al. 1993) supports the argument for a binding site deep in the channel pore.
Figure 2. Concentration- and voltage dependence of QQQ rapid block.
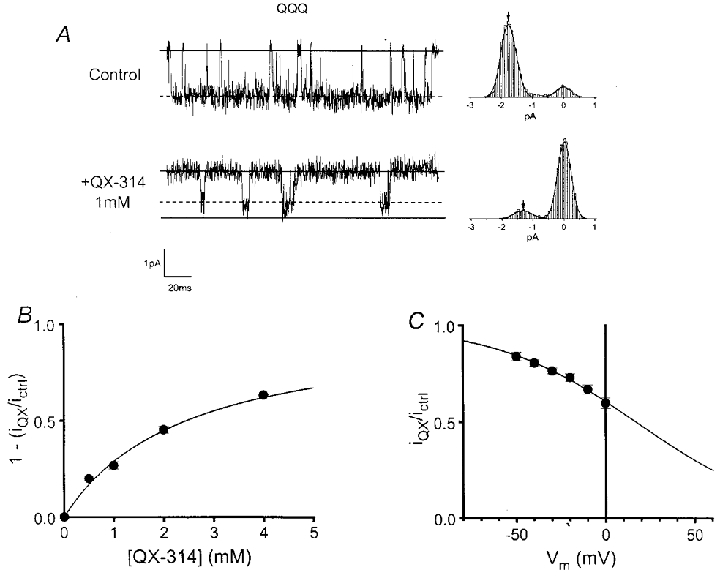
A, left, the upper trace shows a current record of a single QQQ channel during a depolarizing pulse to -20 mV in control conditions (holding potential = -100 mV). The lower trace shows activity of the same channel in the presence of 1 mm QX-314. The reduction in unitary current (lowest continuous line – control) is consistent with a rapid open channel blocking process. Upper continuous lines mark baselines and dashed lines signify open channel current. Records filtered at 2 kHz. Right, corresponding amplitude histograms fitted with Gaussian functions (smooth continuous lines) to allow clear observation of peaks. Arrows mark peaks corresponding to open channel current. B, plot of fractional reduction of unitary current (1 – (iQX/ictrl)) versus[QX-314] during voltage steps to -20 mV. The data are well fitted by a Langmuir isotherm relationship with a maximum of 1 (continuous curve), KD = 2.45 mm, as gauged by regression of a linearized version of the data. C, plot of normalized unitary current (i) versus membrane voltage, with 1 mm QX-314 present throughout. The reduction of unitary current is steeply voltage dependent; the Boltzmann function (continuous curve), fitted by regression of a linearized version of the data, shows an e-fold change in 39.1 mV and half-blocking voltage = 17.1 mV. Combining this information with that froB provides an expression for the voltage dependence of the dissociation constant: KD (V) = 1.45 mm (exp(-0.65FV/RT)). Standard error bars are shown around means derived from 3-5 patches.
From the analysis of the voltage dependence of rapid block (Fig. 2C) and the determination of the rapid block, dissociation constant (KD) at -20 mV (Fig. 2B), the expression for KD(V) is:
| (2) |
where F, R and T have their usual meaning. The location of the binding site within the membrane electric field is similar to our previous results in native cardiac channels, but the affinity of this binding reaction is appreciably reduced as measured by a nearly 50 % increase in KD(0).
Discrete block of open channels supports a transition-intermediate mechanism
Figure 3A (top) shows an open time histogram plotted in log-binned fashion (Sigworth & Sine, 1987) under control conditions. The distribution is well fitted by a transformed mono-exponential function, indicating a single, kinetically distinct, open state. The closed time histogram is dominated by a faster component that represents nearly 90 % of all closures (Fig. 3A, bottom). These findings are well approximated by a simple closed-open gating model similar to native cardiac channels, and described by Scheme 1 in the absence of QX. QX moves the open time histogram towards briefer openings while preserving its mono-exponential character (Fig. 3B, top). This supports the argument that QX introduces a new closing rate constant operating on the same open state as in control. Closed time histograms are now dominated by a new, slow component (Fig. 3B, bottom). The fast component has a time constant nearly that of control probably representing native closures; the new, slow component probably reflects long-lived, drug-bound conformations. The simplest explanation for this is discrete open channel blockade, which accords with Scheme 1 when rapid blocked state is considered invisible. Alternatively, drug binding may promote the pre-existing slow closed state through an allosteric mechanism. Such a mechanism is less compelling because of the six-fold difference in mean closed times of the slow component in control versus drug and the greater complexity.
Figure 3. Kinetics of discrete open channel block in QQQ.
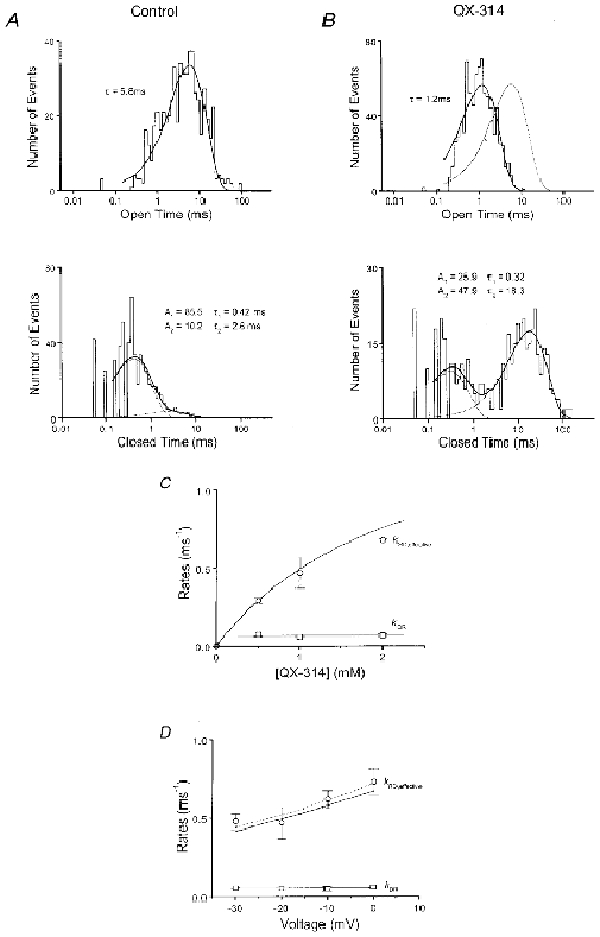
A, open and closed time histograms binned logarithmically (Sigworth & Sine, 1987) in control conditions from the patch of Fig. 1 (depolarization -20 mV). B, open and closed time histograms in 2 mm QX-314. In both A and B, thick continuous lines are fits by transformed mono- and bi-exponential functions with the parameters as shown. A1 and A2 are the individual coefficients of bi-exponential functions. Dotted lines represent component mono-exponential functions. QX-314 shortens open times and induces a new slow component in the closed time histogram. The thin line is the fit of the control open time histogram replotted in B for comparison. C, dependence of effective discrete block (kRD,effective, ○) and unblock (kDR, □) rate constants on [QX-314], all at -20 mV. D, effective discrete (un)block rates as a function of voltage, with 1 mm QX-314 present. kRD,effective changes e-fold per 62 mV, as shown by the dashed exponential curve (determined from linear regression analysis of a logarithmic version of the data). In C and D, standard errors are shown bracketing the means of 3-5 patches. kRD,effective responses were simultaneously well fitted by eqn (3) (curvilinear continuous lines) using a least squares technique (see text). kDR response showed no statistically significant change by linear regression analysis (continuous straight lines).
Open and closed time histograms in the presence of QX (e.g. Fig. 3B) were used to specify effective discrete blocking and unblocking rate constants (kRD,effective and kDR, respectively) in the following manner. We fitted transformed mono- and bi-exponential probability density functions to all open and closed intervals exceeding the dead time of our system (∼0.2 ms). The fits yield time constants and amplitudes that differ somewhat from those measured in the absence of missed opening and closing transitions inherent to the half-height criteria for identifying events. To derive precise values for kRD,effective and kDR, this distortion was accounted for using the iterative procedure of Blatz & Magelby (1986) and using a simplified version of Scheme 1 that, for the time being, explicitly ignores the rapid blocked state (see Methods for details):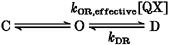
In this simplified gating scheme, the effects of rapid block are manifested in the dependence of kRD,effective on concentration and voltage (see eqn (1)). Figure 3C shows the concentration dependence of effective discrete (un)block rate constants resulting from missed events compensation. kDR (□) is independent of QX concentration. In contrast, kRD,effective (○) saturates with increasing QX concentration. These results roughly accord with the kinetics discerned from lidocaine block of macroscopic currents in hH1-QQQ channels (Bennett et al. 1995). We next examined voltage dependence. kDR (□, Fig. 3D) is also independent of voltage whereas kRD,effective (○, Fig. 3D) increases moderately with depolarization, changing e-fold per 62 mV (dashed line). These observations accord with qualitative predictions of Scheme 1 and eqn (1), and support a transition-intermediate mechanism.
We next determined the rate constant governing transitions from R to D in Scheme 1, kRD. Equation (1) specifies a relationship between kRD, kRD,effective and KD(V). Combining eqns (1) and (2) yields:
| (3) |
which describes kRD,effective in terms of kRD, QX concentration and voltage. Equation (3) permits straightforward determination of kRD when combined with our empirically derived kRD,effective responses (Fig. 3C and D). At the same time, a single value of kRD that allows eqn (3) to reproduce the concentration and voltage dependence of empirical kRD,effective, supports the argument for the appropriateness of a transition-intermediate mechanism. We estimated the value of kRD to provide an optimal fit of concentration and voltage dependence of empirical kRD,effective (Fig. 3C and D). kRD = 1.61 ms−1 provides a good fit of eqn (3) (continuous curved lines, Fig. 3C and D) to both empirical relationships, indicating that the data are quantitatively consistent with a transition-intermediate mechanism. Overall, these results strongly support the idea that a transition-intermediate relationship governs pore block of QQQ channels by QX. We next explored the structural underpinnings of discrete and rapid block.
F1579A selectively disables discrete block
The mutation F1764A in domain IV-S6 in rat brain IIA Na+ channels profoundly impairs local anaesthetic block consistent with F1764 contributing to an intrapore receptor (Ragsdale et al. 1994). The analogous mutation (F1579A) produces similar effects in RSkM1 (Wang et al. 1998a; Wagner et al. 1999). To examine the role of F1579 in pore blockade we performed the additional substitution F1579A in QQQ, referred to as QQQ-F1579A. We studied the effects of QX on this channel to gain insight into the possible involvement of a putative intrapore, local anaesthetic receptor.
QQQ-F1579A activity also showed long openings punctuated by brief closures (Fig. 4A) much like QQQ (cf. Fig. 1A). Comparisons of control gating (Fig. 4E) and single channel slope conduction (Fig. 4F) indicate that F1579A introduced little change in the underlying primary functions of QQQ. We also compared slow inactivation between these channels since slow inactivated states have been suggested as targets of local anaesthetic action (Kambouris et al. 1998). Slow inactivation manifested in ensemble current averages was also unchanged as gauged by single exponential function fits (time constants: QQQ, 246 ± 57 ms, n = 4; QQQ-F1579A, 323 ± 93 ms, n = 4; P = 0.50) and the percentage magnitude of current reduction (QQQ, 54 ± 7.6 %, n = 4; QQQ-F1579A, 58 ± 6.2 %, n = 4; P = 0.73). QX application reduced unitary current but remarkably failed to induce discrete current interruptions (Fig. 4B) pointing to selective disabling of discrete block. QX caused little change in open and closed time histograms (Fig. 4C–E) further supporting disabling of discrete block. In contrast, the voltage and concentration dependence of rapid block was unchanged (Fig. 4G and H) confirming that it was unaffected by the mutation. The results indicate that F1579A selectively disables discrete block.
Amplitude distributions analysis of rapid block in QQQ-F1579A
The transition-intermediate mechanism assumes that rapid block is more than an order of magnitude faster than discrete block. Therefore characterization of rapid block kinetics is important to validate this assumption as well as to provide insight into possible underlying physical mechanisms. QX application to QQQ-F1579A resulted in channel activity with long bursts (> 50 ms), which appeared noisy or flickery, and exhibited reduced apparent unitary currents (Fig. 4B). Amplitude distribution analysis can be applied to this activity allowing deduction of rapid block kinetics (Yellen, 1984a). However, the presence of infrequent, short duration (< 10 ms) bursts seen with QX-triggered block of QQQ channels (see Fig. 1B) precludes analysis of rapid block kinetics using this technique. Therefore, QQQ-F1579A provides a unique opportunity to characterize the kinetics of rapid block. We analysed amplitude distributions as described by Yellen (1984a) (see Methods). Figure 5 shows records of a single QQQ-F1579A channel in control and 1 mm QX (panel A), and the corresponding amplitude histograms (panel B) with baseline noise removed. Rapid (un)block rate constants were adjusted to give the best fit of the theoretical to the empirical distribution. The theoretical fit (smooth continuous line) is superimposed on the empirical distribution (Fig. 5B, bottom). The results of the analysis of this patch are consistent with eqn (1), and reveal very rapid kinetics: the blocking rate (kOR) = 30 000 m−1 ms−1 and the unblocking rate (kRO) = 87 ms−1. In grouped data (n = 3, -20 mV) the results are kOR = 42 000 ± 18 000 m−1 ms−1 and kRO = 82 ± 22 ms−1. These rate constants are nearly two orders of magnitude greater than those of discrete block (Fig. 3C and D) thereby validating an essential assumption of the transition-intermediate mechanism.
Figure 5. Amplitude distribution analysis of rapid block of QQQ-F1579A.
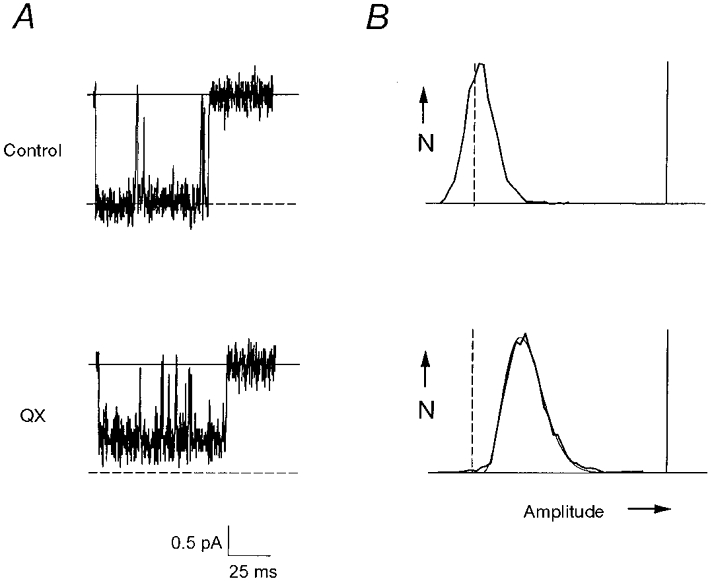
A, single QQQ-F1579A channel records (-20 mV) without (control, top) and with 1 mm QX-314 (bottom), filtered at 2 kHz and sampled at 20 kHz. Continuous lines denote baseline and dashed lines indicate open channel current. B, amplitude histograms for the corresponding individual records, plotted as the number of occurrences (N) versus current amplitude (abscissa). The continuous vertical line on the right corresponds to the baseline amplitude and the dashed line on the left to the open channel current amplitude (1.6 pA). Ordinate scaling is arbitrary. The continuous curve imposed on the histogram is a theoretical distribution fit of empirical data (see text).
DISCUSSION
We investigated the effects of the quaternary ammonium QX on single-channel currents of RSkM1 Na+ channels genetically engineered to selectively lack fast inactivation. Internal QX appeared to traverse ∼65 % of the membrane electric field to induce deep discrete and rapid pore blockade. Blocking modes interacted in a fashion consistent with a transition-intermediate mechanism of pore block. The results reproduce our previous key findings of QX block of single cardiac Na+ channels in which fast inactivation was abolished by papain exposure (Gingrich et al. 1993) and thereby discount the possibility of earlier confounding, non-specific enzyme effects. F1579 in domain IV-S6, implicated in local anaesthetic block, when mutated to alanine (A) selectively disabled discrete block suggesting that F1579 critically contributes to a distinct binding domain mediating discrete block, and provides further support for a transition-intermediate mechanism. Overall, the results support a general mechanism involving a transition intermediate that governs internal QX and local anaesthetic pore blockade in voltage-gated Na+ channels, and provides insight into underlying structural features. The results advance our understanding of the binding of quaternary ammonium and drug molecules within the inner pore of voltage-gated Na+ channels.
Transition-intermediate mechanism of pore block
Figure 6A summarizes our earlier proposals regarding the physical processes involved in a transition-intermediate mechanism of QX block of voltage-gated Na+ channels (Gingrich et al. 1993). In this arrangement, rapid and discrete block are coordinated by distinct binding domains that are physically separated. The intramolecular distance from the QA head to the hydrophobic tail of ∼9 Å (assuming a similar distance to that of the tertiary amine and the distal edge of the aromatic ring of lidocaine (Courtney, 1988) may then bridge the corresponding physical separation between the binding domains. This allows for simultaneous occupation of both binding domains which represents an essential element of a transition-intermediate mechanism.
Figure 6. Transition-intermediate mechanism of pore block.
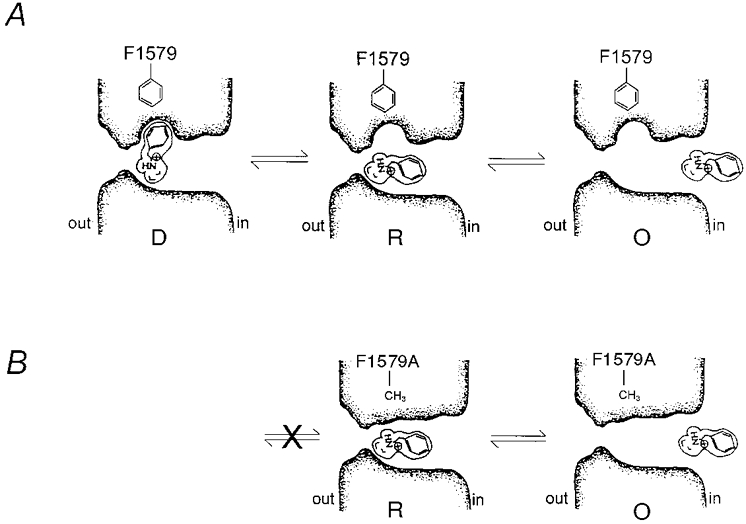
A, right, initially a QX-314 molecule rapidly enters the cytoplasmic mouth of the open channel (O) led by the charged quaternary ammonium head from the cytoplasmic solution. Middle, the quaternary ammonium head binds to a deep site located ≈70 % of the transmembrane electric field from the cytoplasmic entrance to form the rapid blocked state (R). The aromatic portion of QX-314 is not, however, coordinated by the high-affinity, hydrophobic pocket, thereby rendering the complex relatively unstable. The aromatic hydrophobic side group of F1579 is shown in close proximity to the hydrophobic pocket which represents its involvement with the pocket. From the R state the hydrophobic aromatic tail must find its way into a more avid configuration, perhaps by entry of the hydrophobic aromatic tail into a specific hydrophobic pocket (left). In this way the R state serves as a transition intermediate that catalyses the formation of D. B, mutation F1579A introduces a relatively hydrophilic methyl side group that may disrupt the hydrophobic binding pocket. Therefore, from R, QX-314 can only rapid unblock leading to O since state D is unlikely. This is represented by the cross in the transition pathway and the absence of D.
Several findings provide strong support for the proposed mechanism of block. These include reproduction of our earlier key findings of discrete and rapid block (Fig. 1), and a quantitative accounting of both by a transition-intermediate mechanism (Figs 2 and 3). Our observation that F1579A selectively disables discrete block provides additional support for the proposed mechanism in three ways. First, this mutation fails to alter appreciably single-channel gating and slope conductance (Fig. 4E and F). The simplest interpretation is a disruption of a binding domain mediating discrete pore block. This conclusion, in conjunction with unaltered rapid block, indicates that F1579 contributes exclusively to a binding domain mediating discrete block, and thereby supports the idea of two distinct binding domains. Second, the phenylalanine side group is relatively more aromatic and hydrophobic than alanine. Thus, alanine substitution could alter a hydrophobic binding pocket either by simply reducing its affinity or by other structural changes arising from conformational effects. We favour discrete block arising from hydrophobic-hydrophobic interactions between the hydrophobic tail of QX and such a pocket. However, the involvement of cation-π interactions involving the QA head cannot be excluded. Third, Scheme 1 predicts that the elimination of the discrete block state will leave rapid block intact. Furthermore, since rapid block is more than two orders of magnitude faster than discrete block (Fig. 5), then the kinetics of rapid block should appear, practically speaking, unmodified. This reasoning leads to the prediction that disrupting the discrete-block binding domain will exclusively disable discrete block while leaving rapid block unchanged. This is exactly our experimental result.
We extended our proposed model in Fig. 6A to include the contribution of F1579 to a hydrophobic binding pocket involved in discrete block. Figure 6B shows the proposed disruption of the binding pocket by F1579A precluding hydrophobic tail docking and the discrete block configuration. Overall, these findings strongly support a transition-intermediate mechanism of pore blockade and provide new mechanistic insight.
Possible nature and structural underpinnings of block
The transition-intermediate mechanism proposes that rapid and discrete block are individually subserved by the charged QA head and the hydrophobic tail of the QX molecule, respectively. What other evidence lends support to this proposal? Internal application of the QA tetraethylammonium (TEA) reduces unitary current in accord with rapid pore block in wild type hH1 Na+ channels (O'Leary & Horn, 1994) and in a mutant form, similar to QQQ in this study, that lacks fast inactivation (O'Leary et al. 1993). The structure of TEA is strikingly similar to the QA head of QX, permitting the proposition that rapid block arises from the interaction of the QA head with the channel pore. This proposal also accords with the likely deeper location of the rapid site (Fig. 2C) since the membrane electric field would cause the charged QA head to lead QX into the pore and bind more deeply. Our data indicate a specific binding reaction underlying rapid block (Fig. 2B). We speculate that the charged QA head participates in an interaction that is simple coulombic or cation-π (Heginbotham & MacKinnon, 1992) in nature.
What evidence grants support to the involvement of the aromatic tail in discrete block?O'Leary & Horn (1994) also reported that the QA tetrabutylammonium (TBA) reduced mean open times in addition to reducing unitary current, as with TEA. These dual effects are consistent with rapid and discrete pore block and are remarkably similar to those of QX in this study. Elongating aliphatic side groups from ethyl in TEA to butyl in TBA introduces hydrophobicity at ∼8 Å from the QA head (O'Leary et al. 1994) similar to the ∼9 Å separating the QA head and hydrophobic tail of QX. As a result, TBA can be seen to crudely resemble the QX molecule, thus supporting the proposal that the hydrophobic aromatic tail participates in discrete blockade.
Overall, we favour a transition-intermediate mechanism of block in which rapid block arises from binding of the QA head with structures deep in the pore near the selectivity filter. Electrostatic or cation-π reactions may stabilize the rapid block configuration. In contrast, discrete block develops from hydrophobic-hydrophobic interactions involving the tail of QX and a binding domain in IV-S6 that includes F1579.
Comparison with cardiac Na+ channels and structural insights
These results broadly confirm our previous results in cardiac channels but at the same time point to quantitative differences in rapid block between these similar Na+ channel isoforms. The following discussion is valid to the extent that the observed differences arise from isoform differences and not methods used to eliminate fast inactivation or obtain functional channels (native versus recombinant). The location of the rapid block binding domain within the membrane electric field is similar to our previous results in native cardiac channels, but the affinity of binding is appreciably reduced as indicated by nearly a 50 % increase in KD. This suggests possible structural divergence of the inner vestibule between skeletal muscle and cardiac isoforms. The inner vestibule of Na+ channels is composed of S6 α helices and S5-S6 loops from domains I-IV. S6 comprises the walls of the inner vestibule reaching from the cytoplasmic mouth to the selectivity filter deep in the pore. S5-S6 loop is a hairpin infolding of hydrophobic regions and contains a region critically involved in selectivity referred to as the permeation or P-loop (for reviews see Catterall, 1995; Fozzard & Hanck, 1996). It seems reasonable that structural differences in these regions may underlie differences in rapid block. The amino acid residues of P-loop and S6 transmembrane segments are nearly 95 % conserved between rat skeletal muscle and cardiac isoforms (Gellens et al. 1992) whereas the adjoining stretches of S5-S6 show significant divergence: S5-P-loop (∼60 %) and P-loop-S6 (∼77 %). Thus, it is plausible that S5-S6 loops may contain structural differences that give rise to the different affinities of rapid block.
Local anaesthetic receptor
We investigated the role of F1579 because this residue contributes to a putative local anaesthetic receptor, and QX is a QA derivative of the local anaesthetic lidocaine. F1764A in domain IV-S6 of rat brain IIA Na+ channels markedly reduces tonic and phasic inhibition of macroscopic currents by the local anaesthetic etidocaine (Ragsdale et al. 1994). The analogous mutation in RSkM1 (F1579A) causes similar effects (Wang et al. 1998a; Wagner et al. 1999). Study of pore block using single-channel QQQ currents provides a more direct examination of underlying intrapore binding than is possible with macroscopic currents. Remarkably, F1579A disables discrete block in QQQ without affecting rapid block, gating and slope conductance. The results suggest that F1579 contributes heavily to a discrete binding domain and is pore lining, as proposed for the analgous F1764 in rat brain IIA (Ragsdale et al. 1994). Our conclusions regarding F1579 overlap with those of Li et al. (1999). They investigated the role of the homologous residue (F1710) in local anaesthetic inhibition of the type αIII brain Na+ channel by substituting a series of amino acids that varied in hydrophobicity and aromaticity while examining the block of macroscopic currents by tetracaine. The authors concluded that F1710 contributes to an intrapore, local anaesthetic receptor and that drug binding to open channels involves either cation-π or aromatic-aromatic interactions.
Other structure-function studies involving Na+ channels and local anaesthetics may provide further insight into structural features underlying QX block. Sunami et al. (1997) reported that increased net negative P-loop charge enhanced block by tertiary amine but not neutral local anaesthetics and alcohols. The authors concluded that the P-loop influences binding of the charged moiety of local anaesthetic molecules consistent with our proposal regarding binding of the charged QA head of QX binding site. In contrast, Wang et al. (1998b) have demonstrated that mutations that add net charge to the middle of I-S6 of RSkM1 influence block by QX and etidocaine. The authors suggested that I-S6 contributes to a local anaesthetic receptor and a specific binding domain for the charged head of local anaesthetics.
Comparison with QA block of other channels
Could internal QA block of other ion channels share features with QX block of Na+ channels?Choi et al. (1993) examined macroscopic inhibition of inactivation-deficient mutant Shaker K+ channels by series of alkyl TEA molecules (Cn-TEA, where n is the number of carbons in the alkyl backbone) applied internally. The molecules are coarsely similar to QX, with a charged QA head and hydrophobic tail with varying lengths and hydrophobicities. Smaller QAs (TEA, C3-TEA and C4-TEA) simply attenuated whole-cell current consistent with rapid, open channel block. In fact, internal TEA induces rapid, open channel block in single-channel studies with the structurally similar RCK2 K+ channel (Kirsch et al. 1991). Those with longer alkyl tails (C6-TEA, C8-TEA and C10-TEA) induced time-dependent current reduction as in discrete open channel blockade in squid giant axon studied by Armstrong (1971). Therefore, longer molecules may induce both rapid and discrete open channel block similar to QX in Na+ channels. Interestingly, the M440I mutation in the P-loop preferentially reduced the affinity of TEA block by nearly 50-fold, implicating this region in what is perhaps rapid, open channel block. Furthermore, the mutation T469I in the middle of S6, preferentially increased the affinity of C8-TEA by 27-fold. The authors proposed that the P-loop contributes to a binding determinant for the QA head and T469 of S6 to a hydrophobic binding pocket that stabilizes the alkyl tail. The similarity to QX block of Na+ channels is notable. This suggests, by analogy, that F1579 contributes to a hydrophobic binding pocket which stabilizes the aromatic tail and that P-loop structures influence binding of the QA head. These similarities point to the possibility of a general mechanism which governs the interaction of QAs with common, fundamental, structural features within the inner pore of voltage-gated ion channels.
Acknowledgments
We thank Larry Wagner II for his excellent technical assistance. We also thank Ted Begenisich, Robert Dirksen, Paul Burkat and Larry Wagner II for the critical reading of preliminary manuscripts. This study was supported by University of Rochester Anesthesiology Group Research Fund and the National Institute of General Medical Sciences Grant RO1-GM56958.
References
- Armstrong CM. Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axons. Journal of General Physiology. 1971;58:413–437. doi: 10.1085/jgp.58.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balser JR, Nuss HB, Orias DW, Johns DC, Marban E, Tomaselli GF, Lawrence JH. Local anesthetics as effectors of allosteric gating. Lidocaine effects on inactivation-deficient rat skeletal muscle Na channels. Journal of Clinical Investigation. 1996;98:2874–2886. doi: 10.1172/JCI119116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett PB, Valenzuela C, Chen LQ, Kallen RG. On the molecular nature of the lidocaine receptor of cardiac Na+ channels. Modification of block by alterations in the alpha- subunit III-IV interdomain. Circulation Research. 1995;77:584–592. doi: 10.1161/01.res.77.3.584. [DOI] [PubMed] [Google Scholar]
- Blatz AL, Magelby KL. Correcting single channel data for missed events. Biophysical Journal. 1986;49:968–980. doi: 10.1016/S0006-3495(86)83725-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahalan MD. Local anesthetic block of sodium channels in normal and pronase-treated squid giant axons. Biophysical Journal. 1978;23:285–311. doi: 10.1016/S0006-3495(78)85449-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahalan MD, Almers W. Interactions between quaternary lidocaine, the sodium channel gates, and tetrodotoxin. Biophysical Journal. 1979;27:39–55. doi: 10.1016/S0006-3495(79)85201-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon SC, Strittmatter SM. Functional expression of sodium channel mutations identified in families with periodic paralysis. Neuron. 1993;10:317–326. doi: 10.1016/0896-6273(93)90321-h. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Structure and function of voltage-gated ion channels. Annual Review of Biochemistry. 1995;64:493–531. doi: 10.1146/annurev.bi.64.070195.002425. [DOI] [PubMed] [Google Scholar]
- Choi KL, Mossman C, Aube J, Yellen G. The internal quaternary ammonium receptor site of Shaker potassium channels. Neuron. 1993;10:533–541. doi: 10.1016/0896-6273(93)90340-w. [DOI] [PubMed] [Google Scholar]
- Colquhoun D, Sigworth FJ. Fitting and statistical analysis of single-channel records. In: Sakmann B, Neher E, editors. Single-Channel Recording. New York: Plenum Press; 1995. pp. 483–585. [Google Scholar]
- Courtney KR. Why do some drugs preferentially block open sodium channels? Journal of Molecular and Cellular Cardiology. 1988;20:461–464. doi: 10.1016/s0022-2828(88)80073-7. [DOI] [PubMed] [Google Scholar]
- Fozzard HA, Hanck DA. Structure and function of voltage-dependent sodium channels: comparison of brain II and cardiac isoforms. Physiological Reviews. 1996;76:887–926. doi: 10.1152/physrev.1996.76.3.887. [DOI] [PubMed] [Google Scholar]
- Frazier DT, Narahashi T, Yamada M. The site of action and active form of local anesthetics. II. Experiments with quaternary compounds. Journal of Pharmacology and Experimental Therapeutics. 1970;171:45–51. [PubMed] [Google Scholar]
- Gellens ME, George AL, Chen LQ, Chahine M, Horn R, Barchi RL, Kallen RG. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proceedings of the National Academy of Sciences of the USA. 1992;89:554–558. doi: 10.1073/pnas.89.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingrich KJ, Beardsley D, Yue DT. Ultra-deep blockade of Na+ channels by a quaternary ammonium ion: catalysis by a transition-intermediate state? The Journal of Physiology. 1993;471:319–341. doi: 10.1113/jphysiol.1993.sp019903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann HA, Tiedeman AA, Chen SF, Brown AM, Kirsch GE. Effects of III-IV linker mutations on human heart Na+ channel inactivation gating. Circulation Research. 1994;75:114–122. doi: 10.1161/01.res.75.1.114. [DOI] [PubMed] [Google Scholar]
- Heginbotham L, MacKinnon R. The aromatic binding site for tetraethylammonium ion on potassium channels. Neuron. 1992;8:483–491. doi: 10.1016/0896-6273(92)90276-j. [DOI] [PubMed] [Google Scholar]
- Hille B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. Journal of General Physiology. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschberg B, Rovner A, Lieberman M, Patlak J. Transfer of twelve charges is needed to open skeletal muscle Na+ channels. Journal of General Physiology. 1995;106:1053–1068. doi: 10.1085/jgp.106.6.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambouris NG, Hastings LA, Stepanovic S, Marban E, Tomaselli GF, Balser JR. Mechanistic link between lidocaine block and inactivation probed by outer pore mutations in the rat μ1 skeletal muscle sodium channel. The Journal of Physiology. 1998;512:693–705. doi: 10.1111/j.1469-7793.1998.693bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimbrough JT, Gingrich KJ. Mutation in the putative local anesthetic receptor in IVS6 of rat skeletal muscle mu1 Na+ channel alters single channel interactions. Biophysical Journal. 1998;74:A452. [Google Scholar]
- Kirsch GE, Taglialatela M, Brown AM. Internal and external TEA block in single cloned K+ channels. American Journal of Physiology. 1991;261:C583–590. doi: 10.1152/ajpcell.1991.261.4.C583. [DOI] [PubMed] [Google Scholar]
- Li HL, Galue A, Meadows L, Ragsdale DS. A molecular basis for the different local anesthetic affinities of resting versus open and inactivated states of the sodium channel. Molecular Pharmacology. 1999;55:134–141. doi: 10.1124/mol.55.1.134. [DOI] [PubMed] [Google Scholar]
- Linford NJ, Cantrell AR, Qu Y, Scheuer T, Catterall WA. Interaction of batrachotoxin with the local anesthetic receptor site in transmembrane segment IVS6 of the voltage-gated sodium channel. Proceedings of the National Academy of Sciences of the USA. 1998;95:13947–13952. doi: 10.1073/pnas.95.23.13947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh J, Mok WM, Wang GK, Strichartz G. Irreversible inhibition of sodium current and batrachotoxin binding by a photoaffinity-derivatized local anesthetic. Journal of General Physiology. 1995;105:267–287. doi: 10.1085/jgp.105.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C. Bis-quaternary ammonium blockers as structural probes of the sarcoplasmic reticulum K+ channel. Journal of General Physiology. 1982;79:869–891. doi: 10.1085/jgp.79.5.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moczydlowski E. Blocking pharmacology of batrachotocin-activated sodium channels. In: Miller C, editor. Ion Channel Reconstitution. New York: Plenum Press; 1986. pp. 405–428. [Google Scholar]
- O'Leary ME, Chahine M, Chen LQ, Kallen RG, Horn R. External and internal blockers of the human heart sodium channel. Biophysical Journal. 1993;64:A91. abstract. [Google Scholar]
- O'Leary ME, Horn R. Internal block of human heart sodium channels by symmetrical tetra-alkylammoniums. Journal of General Physiology. 1994;104:507–522. doi: 10.1085/jgp.104.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary ME, Kallen RG, Horn R. Evidence for a direct interaction between internal tetra-alkylammonium cations and the inactivation gate of cardiac sodium channels. Journal of General Physiology. 1994;104:523–539. doi: 10.1085/jgp.104.3.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postma SW, Catterall WA. Inhibition of binding of [3H]batrachotoxinin A 20-alpha-benzoate to sodium channels by local anesthetics. Molecular Pharmacology. 1984;25:219–227. [PubMed] [Google Scholar]
- Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science. 1994;265:1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning. Plainview, New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Sigworth FJ, Sine SM. Data transformations for improved display and fitting of single-channel dwell time histograms. Biophysical Journal. 1987;52:1047–1054. doi: 10.1016/S0006-3495(87)83298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strichartz GR. The inhibition of sodium currents in myelinated nerve by quaternary derivatives of lidocaine. Journal of General Physiology. 1973;62:37–57. doi: 10.1085/jgp.62.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunami A, Dudley SCJ, Fozzard HA. Sodium channel selectivity filter regulates antiarrhythmic drug binding. Proceedings of the National Academy of Sciences of the USA. 1997;94:14126–14131. doi: 10.1073/pnas.94.25.14126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner LE, II, Eaton M, Sabnis SS, Gingrich KJ. Meperidine and lidocaine block of recombinant voltage-dependent Na+ channels: evidence that meperidine is a local anesthetic. Anesthesiology. 1999;91:1481–1490. doi: 10.1097/00000542-199911000-00042. [DOI] [PubMed] [Google Scholar]
- Wang GK, Brodwick MS, Eaton DC, Strichartz GR. Inhibition of sodium currents by local anesthetics in chloramine-T-treated squid axons. The role of channel activation. Journal of General Physiology. 1987;89:645–667. doi: 10.1085/jgp.89.4.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GK, Quan C, Wang S. A common local anesthetic receptor for benzocaine and etidocaine in voltage-gated mu1 Na+ channels. Pflügers Archiv. 1998a;435:293–302. doi: 10.1007/s004240050515. [DOI] [PubMed] [Google Scholar]
- Wang GK, Quan C, Wang SY. Local anesthetic block of batrachotoxin-resistant muscle Na+ channels. Molecular Pharmacology. 1998b;54:389–396. doi: 10.1124/mol.54.2.389. [DOI] [PubMed] [Google Scholar]
- West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na(+)-channel inactivation. Proceedings of the National Academy of Sciences of the USA. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh JZ. Sodium inactivation mechanism modulates QX-314 block of sodium channels in squid axons. Biophysical Journal. 1978;24:569–574. doi: 10.1016/S0006-3495(78)85403-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh JZ, TenEick RE. Molecular and structural basis of resting and use-dependent block of sodium current defined using disopyramide analogues. Biophysical Journal. 1987;51:123–135. doi: 10.1016/S0006-3495(87)83317-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G. Ionic permeation and blockade in Ca2+-activated K+ channels of bovine chromaffin cells. Journal of General Physiology. 1984a;84:157–186. doi: 10.1085/jgp.84.2.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G. Relief of Na+ block of Ca2+-activated K+ channels by external cations. Journal of General Physiology. 1984b;84:187–199. doi: 10.1085/jgp.84.2.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi GW, Doyle DD, French RJ. Fast lidocaine block of cardiac and skeletal muscle sodium channels: one site with two routes of access. Biophysical Journal. 1993;65:80–90. doi: 10.1016/S0006-3495(93)81042-7. [DOI] [PMC free article] [PubMed] [Google Scholar]