

Abstract
Genetic programs promoting cell cycle progression, DNA repair, and survival are coordinately induced in developing T cells and require rapid turnover of effector molecules. As the COP9 signalosome (CSN) has been placed at the crossroads of these programs in lower organisms, we addressed its role by conditionally deleting CSN5/JAB1, its catalytic subunit, in developing thymocytes. CSN5/JAB1del/del thymocytes show defective S phase progression and massive apoptosis at the double-negative (DN) 4–double-positive (DP) transition stage, which is paralleled by altered turnover of selected CSN-controlled substrates, including p53, IκB-α, and β-catenin. Combined dysregulation of the p53 and NF-κB pathways affects thymocyte survival by altering the mRNA and protein levels of selected Bcl-2 family members. Genetic complementation analysis performed on p53−/−, Bcl-xL/Bcl-2A1, or T cell receptor transgenic backgrounds indicates that CSN5/JAB1 acts at distinct developmental stages to coordinate proliferation, survival, and positive selection of thymocytes by controlling the induction of defined genetic programs acting downstream of CSN-regulated transcription factors.
Timely coordination of cell cycle progression, DNA rearrangement and repair, and adaptation to proapoptotic stimuli are hallmark features of developing thymocytes (1). After successfully rearranging the TCR β subunit genes, double-negative (DN) thymocytes undergo a wave of proliferation driven by ligand-independent activation signals originating from the preTCR at the DN3 stage of development. Such signals involve the down-regulation of p53-dependent cell cycle arrest and apoptotic programs as part of the β selection process (2, 3). At a later stage of development, double-positive (DP) thymocytes no longer proliferate, but are instead exposed to a proapoptotic environment that is at least partially dependent on circulating steroid hormones, as well as on other factors inducing oxidative stress (4). These factors promote massive apoptosis of DP thymocytes (“death by neglect”) through p53-dependent and -independent mechanisms, which can be rescued by TCR-driven signals, to allow positive selection and further thymocyte maturation (5). Thus, coordination of DNA rearrangement and repair with proliferative and survival signals appears to be critical for thymocyte progression through various developmental stages. The mechanisms underlying this finely regulated process are incompletely understood, as are the relative roles played by major pathways controlling survival. As progression from the DN to the DP stage is a swift process being completed in ∼24 h (6), all of these steps have to rely on an extremely rapid course of action, possibly involving posttranscriptional, as well as posttranslational, control mechanisms.
The COP9 signalosome (CSN) is an evolutionarily conserved, ubiquitously expressed (7) multimolecular complex controlling the function of cullin-based ubiquitin ligase complexes (8, 9). CSN5/JAB1 is the only subunit of the complex endowed with catalytic activity (10), and it was shown by both our group (11) and another group (12) to be an adhesion-regulated molecule required for adhesion- and TCR-dependent gene expression in T lymphocytes. Constitutive knockouts of various subunits of the CSN (13, 14), including CSN5/JAB1 (15), lead to an early embryonic phenotype featuring accumulation of potential substrates of CSN-regulated E3 ligases, such as p27kip1, cyclin E, and p53, suggesting that in vivo the CSN is a positive regulator of such ubiquitin ligase complexes. Several studies have implicated the CSN in the regulation of proteins critical for the control of cellular proliferation. Among the E3 ligase substrates that could become altered via altered CSN function are the cyclin-dependent kinase inhibitors p27 kip1 (16, 17), p57, p21cip1 (18), and their transcriptional regulators (i.e., SMAD4 and SMAD7) (19, 20), as well as several cyclins (cyclin D1, E, and B1) (21). Multiple subunits in the CSN have also been implicated in DNA damage sensitivity and repair (22). A factor potentially affected by CSN misregulation is Cdt1 (23, 24), which is a replication initiator protein that is regulated by the CSN after irradiation damage. Interestingly, CSN interaction with p53 has been shown to target this tumor suppressor for degradation, greatly affecting the ability of the cells to respond to DNA damage and proceed through the cell cycle or signal for apoptosis (25, 26).
We hypothesize that the CSN could play a central role in the coordinate regulation of cell proliferation, DNA repair, and apoptosis in developing thymocytes. To test this hypothesis, as well as to gain insight into the function of the CSN in vivo, we conditionally deleted CSN5/JAB1, which is the CSN's catalytic subunit, in developing thymocytes, based on prior evidence (8) showing that ablation of CSN5/JAB1 would lead to functional inactivation of the entire CSN.
RESULTS
CSN5/JAB1 is essential for T cell lineage development
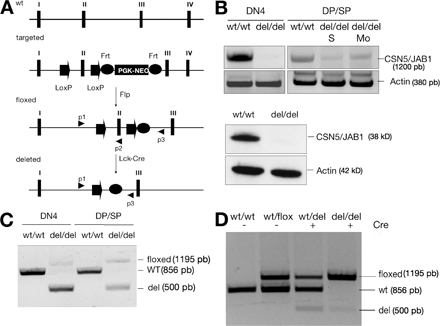
To address the function of CSN5/JAB1 in thymic development, we generated mice targeted with loxP sites at the CSN5/JAB1 locus for conditional deletion by Cre recombinase (Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20070725/DC1). We used a strategy based on both Cre- and Flp-mediated recombination to generate a selection marker–free locus flanked by recombinase-specific sites. Cre-mediated removal of exon 2 resulted in an early frameshift and translation termination. Heterozygous CSN5/JAB1+/Neoflox mice were viable and fertile. In vivo Flp-mediated excision of the PGK-Neo cassette was obtained by mating the CSN5/JAB1+/Neoflox mice to the FLPe deleter strain. After germ line transmission of the CSN5/JAB1flox allele to the progeny, correct excision of the selection marker cassette was confirmed by PCR and Southern blot analysis. Mice homozygous for the floxed CSN5/JAB1 allele (CSN5/JAB1flox/flox mice), which were developed normally with unperturbed CSN5/JAB1 expression (unpublished data), were generated at the expected Mendelian ratio and appeared phenotypically indistinguishable from their age-matched WT. We crossed CSN5/JAB1flox/flox mice with the LckCre deleter strain to generate thymocyte-specific CSN5/JAB1-deficient mice (CSN5/JAB1del/del). The lack of CSN5/JAB1 mRNA and protein in DN and DP thymocytes was demonstrated by semiquantitative RT-PCR and Western blot analysis (Fig. S1 B). In addition, genomic DNA prepared from sorted DN and DP/single-positive (SP) cells was analyzed by PCR using primers detecting WT, floxed, or deleted alleles (Fig. S1 C). Most T cells in CSN5/JAB1del/del mice showed a complete CSN5/JAB1 deletion, as indicated by the presence of the deleted allele in thymocytes and peripheral T cells. At the DN stage, CSN5/JAB1del/del thymocytes yielded exclusively the PCR product corresponding to deleted allele. In peripheral T cells, however, we detected considerable levels of the CSN5/JAB1flox allele, suggesting that the residual mature T cells had escaped Cre-mediated deletion (Fig. S1 D). In support of this interpretation, peripheral T cells, although severely reduced in number, displayed a predominant memory-effector phenotype (CD44highCD62L−) in CSN5/JAB1-deficient animals, suggesting homeostatic expansion of a small pool of phenotypically normal, mature T lymphocytes (Fig. S2).
Thymic morphology, thymocyte counts, and subset distribution were comparatively assessed 5–6 wk after birth in CSN5/JAB1flox/flox mice expressing the LckCre transgene (herein defined as CSN5/JAB1del/del) and in littermates lacking LckCre. Thymi from CSN5/JAB1del/del animals showed a severely hypoplastic medulla (Fig. 1 A), a marked decrease in the proportion of DP and SP thymocytes (Fig. 1 B), and an 80–90% reduction in thymocyte number (Fig. 1 C), whereas the various subsets of DN thymocytes, as determined by the expression of the CD44 and CD25 stage-specific markers, were not significantly altered compared with the CSN5/JAB1flox/flox littermate controls, both percentage-wise (Fig. 1 B) and in absolute numbers (Fig. 1 D). These findings suggest that without a functional CSN, thymic development is blocked at a transition stage between DN4 and DP thymocytes or that emerging DP thymocytes are selectively lost as a consequence of CSN5/JAB1 deletion.
Figure 1.

Impaired thymocyte development in CSN5/JAB1del/del mice. (A) Low magnification, hematoxylin and eosin–stained sections of thymi from WT (top) or CSN5/JAB1del/del (bottom) 5-wk-old mice show a greatly reduced medullary area, which is indicative of defective maturation of thymocytes in the absence of a functional CSN. Bar, 100 μm. M, medulla; C, cortex. (B) Thymocyte suspensions were stained with anti-CD4 plus anti-CD8 antibody to identify the major thymocyte subsets, and with anti-CD44 plus anti-CD25 antibody to detect the various subsets of DN thymocytes in WT (left) versus CSN5/JAB1del/del (right) mice. Percentage values for the various subsets are shown in the respective quadrants. (C) Thymic cellularity, as estimated by calculation of the number of thymocytes from individual thymi after electronic gating of the various subsets. Numbers are the mean ± the SD of values obtained from 25 mice per genotype at 5–6 wk of age. *, P < 0.001 (Student's two-tailed t test with equal variance). (D) DN subset distribution, calculated as in C, but after assessing the various DN subsets with anti-CD44 and -CD25 antibodies to identify DN1 (CD44+; CD25−), DN2 (CD44+; CD25+), DN3 (CD44−; CD25+), and DN4 (CD44−; CD25−) subsets.
Defective cell cycle progression in CSN5/JAB1-deficient thymocyte subsets
The aforementioned defects in thymocyte maturation, as well as the reduced thymic cellularity in CSN5/JAB1del/del animals, could be explained by a decreased proliferative rate and/or by an increased apoptotic death. To discriminate between these possibilities, we assessed thymocytes' competence to progress in the cell cycle by giving a short pulse (40 min) of BrdU in vivo, followed by simultaneous flow cytometric analysis of BrdU incorporation and DNA content with 7-amino-actinomycin staining, to quantitatively assess the various stages of the cell cycle. Overall, CSN5/JAB1del/del thymocytes did not show quantitative defects in BrdU incorporation (Fig. 2 A), suggesting that G1-to-S progression is not altered in the absence of CSN5/JAB1. However, a variable fraction of DN thymocytes, ranging from 12 to 28% in several independent experiments, appeared to be arrested in the S/G2/M phase in CSN5/JAB1del/del mice and failed to incorporate BrdU. By electronically gating onto the DN versus DP subsets, we found that S/G2/M phase–arrested thymocytes were also present in the DP fraction (Fig. 2 A). To follow developmental progression of actively proliferating cells, we chased BrdU+ thymocytes at various time points after the initial pulse label in separate groups of mice. As shown in Fig. 3 B, BrdU+ DN thymocytes accumulate throughout the chase period in CSN5/JAB1del/del mice, with minimal progression toward the DP stage. In contrast, DN thymocytes appear to rapidly progress into DP subsets in WT littermates (Fig. 2 B), with a fraction of them turning into SP cells that migrate to the periphery at later time points (not depicted). This, again, suggests that DN thymocytes show delayed/arrested S phase progression in CSN5/JAB1-deficient animals.
Figure 2.

Altered cell cycle progression in developing CSN5/JAB1del/del thymocytes. (A) Cell cycle progression was assessed by a short (40-min) pulse of i.p. injected BrdU, followed by total thymocyte isolation and staining with FITC-conjugated anti-BrdU antibody, to detect DNA-replicating cells, combined with 7-amino-actinomycin staining as a measure of DNA content. Flow cytometry was performed on electronically gated DN (top) or DP (bottom) thymocytes from WT (left) or CSN5/JAB1del/del thymocytes. Percentage values for G0/G1 (2N DNA content), S (intermediate DNA content), and G2/M (4N DNA content) cells are shown in the respective regions. Shown is a representative experiment out of eight independent ones performed. (B) BrdU pulse-chase analysis. 4 mice per time point for each genotype (WT, filled squares; CSN5/JAB1del/del, empty squares) were given a 40-min pulse with i.p. injected BrdU, after which one group of mice per genotype was killed immediately (time 0) or at the indicated time points. Thymocytes were isolated, counted, and stained with FITC-conjugated anti-BrdU combined with surface staining with anti-CD4 and -CD8 antibodies to electronically gate DN and DP cells by flow cytometry. Shown is the number of BrdU+ thymocytes (mean ± the SD for each group of animals) gated as DN or DP by flow cytometry. *, P < 0.005; **, P < 0.001 (Student's two-tailed t test with equal variance).
Figure 3.

Increased apoptosis in developing CSN5/JAB1del/del thymocytes. (A) Immunohistochemical sections of 5-wk-old thymi from WT (left) or CSN5/JAB1del/del (right) mice stained with anti-cleaved caspase 3 antibody to detect apoptotic cells (as revealed by HRP immunoenzymatic reaction). Bar, 50 μm. (B) Isolated thymocytes from WT (filled squares) or CSN5/JAB1del/del (empty squares) mice were cultured in complete tissue culture medium for the indicated time points, after which the rate of apoptosis was assessed by flow cytometry as indicated in the Materials and methods section. Electronic gating was performed to quantify apoptosis in DN and DP thymocyte subsets. Data are the mean ± the SD of values obtained from eight independent experiments. *, P < 0.05; **, P < 0.001 (Student's two-tailed t test with equal variance).
Increased apoptosis in DN and DP thymocytes in CSN5/JAB1del/del mice
The aforementioned findings suggest that defective cell cycle progression by DN thymocytes, although clearly detectable in a fraction of proliferating cells, is unlikely to account for the severe reduction in DP thymocyte numbers observed in CSN5/JAB1del/del mice. Therefore, we comparatively assessed the rate of apoptotic death in developing CSN5/JAB1del/delversus WT thymocytes. Immunohistochemical detection of apoptotic cells by cleaved effector caspase 3 (ClC3) staining revealed that the apoptotic rate was increased by more than threefold in CSN5/JAB1del/del thymi (Fig. 3 A). ClC3+ cells ranged from 1.5 ± 0.4% in WT to 5.6 ± 0.7% in CSN5/JAB1del/del thymi in three separate experiments. Increased propensity to undergo apoptosis by CSN5/JAB1del/del thymocytes was confirmed by assessing apoptotic death in cells cultured ex vivo for various time points (Fig. 3 B). As previously reported, normal thymocytes undergo spontaneous apoptosis after 24 h in culture, with 38 ± 12% apoptotic cells at the end of the culture period, particularly affecting the DP subset. Thymocytes from CSN5/JAB1del/del mice were more prone to undergo spontaneous apoptosis than CSN5/JAB1flox/flox littermates in both DN and DP subsets, with values being already higher at time 0 after isolation and approaching 100% after 24 h in culture (Fig. 3 B). Thus, the greatly reduced thymic cellularity and the developmental block in CSN5/JAB1del/del mice are compatible with a combined defect in cell cycle progression and survival of developing thymocytes at the DN–DP transition.
TCR gene rearrangement proceeds normally in CSN5/JAB1-deficient thymocytes
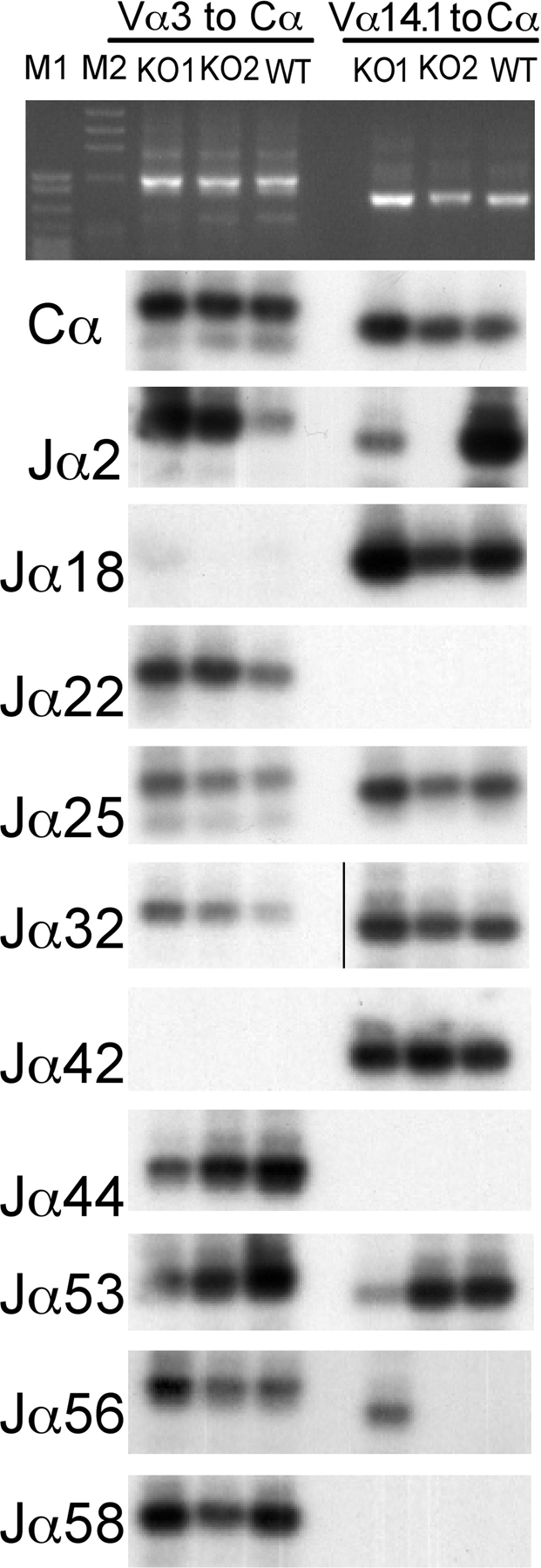
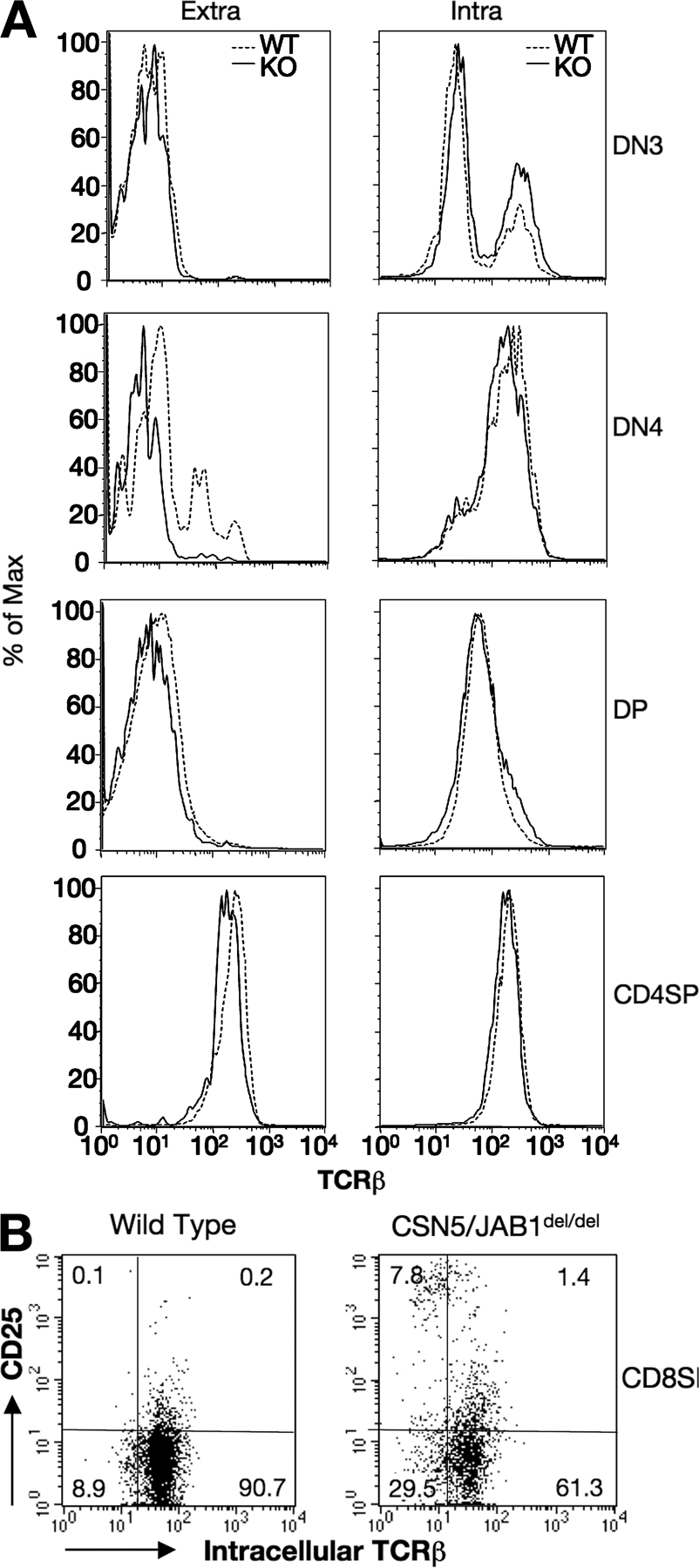
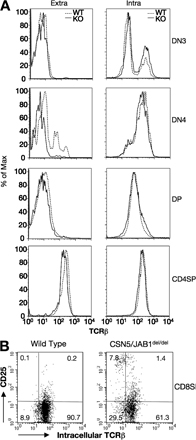
Although CSN5/JAB1del/del thymocytes had no apparent defects in DN3–DN4 stage progression, suggesting that a preTCR was correctly formed and was competent to transduce proliferative signals inherent to the β selection process, we assessed the expression and rearrangement of TCR β subunits in CSN5/JAB1del/del versus WT littermates. Expression of a productively rearranged TCR β subunit was detected intracellularly in DN and DP thymocytes. To characterize the efficiency of the β selection process and the subsequent steps in TCR rearrangement at the DP stage, we assessed the extent of V(D)J recombination at the TCRα and β loci. To this aim, we extracted total RNA from isolated DP/SP populations and amplified the cDNA by PCR using a panel of Vα oligos in combination with a Cα-specific primer, followed by hybridization of Vα-Cα PCR products with a panel of Jα oligoprobes selected from the 5′-most to the 3′-most Jα segments. Results showed no skewing in Jα segment usage in CSN5/JAB1del/del thymocytes (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20070725/DC1). Furthermore, we performed heteroduplex analysis to evaluate the heterogeneity of the Vα and Vβ repertoire on three representative Vα segments, and within each Vβ family. We observed that both TCR α and β subunit rearrangement is qualitatively not affected in CSN5/JAB1del/del mice (unpublished data). Collectively, these findings indicate that rearrangement of the TCR is qualitatively not affected by the lack of a functional CSN in developing thymocytes. However, we found that a measurable fraction of CSN5/JAB1del/del thymocytes underwent progression from DN to DP/SP stages (the latter is more evident in SP CD8+ cells) without expressing a productively rearranged, intracellular TCR β subunit, suggesting a partial impairment of the β selection process (Fig. S4).
CSN5/JAB1 is required for TCR-driven signals involvedin positive selection
The greatly enhanced apoptotic rate of CSN5/JAB1del/del DP cells suggested that most developing thymocytes in CSN5/JAB1del/del animals die “by neglect” before undergoing positive selection, as this process is believed to rescue just a small fraction of DP cells from apoptotic death (1). To further address this issue, we crossed the OT-II transgenic TCRs, which are restricted by MHC class II molecules and undergo positive selection in C57BL6 mice not expressing the antigenic peptide (27), onto the CSN5/JAB1del/del background. As in CSN5/JAB1del/del mice, thymic cellularity was reduced ∼90% in compound CSN5/JAB1del/del OT-II mice compared with the CSN5/JAB1flox/flox OT-II controls (Table S1, available at http://www.jem.org/cgi/content/full/jem.20070725/DC1). As expected, thymi from 5–6-wk-old OT-II mice contained mainly CD4+ SP cells expressing the transgenic TCR, as detected by antibodies to Va2/Vb5. In contrast, thymi from CSN5/JAB1del/del OT-II mice had only 2–8% the number of CD4+ SP cells found in control animals (Table S1), and such impairment in transgenic TCR differentiation and survival was even more evident in peripheral blood (Table S1). Thus, providing a productively rearranged TCR that is positively selected on a MHC class II molecule is not sufficient to rescue survival and/or differentiation of CD4+ SP thymocytes in the absence of a functional CSN5/JAB1 locus.
Selected E3 ligase substrates accumulatein CSN5/JAB1del/del thymocytes
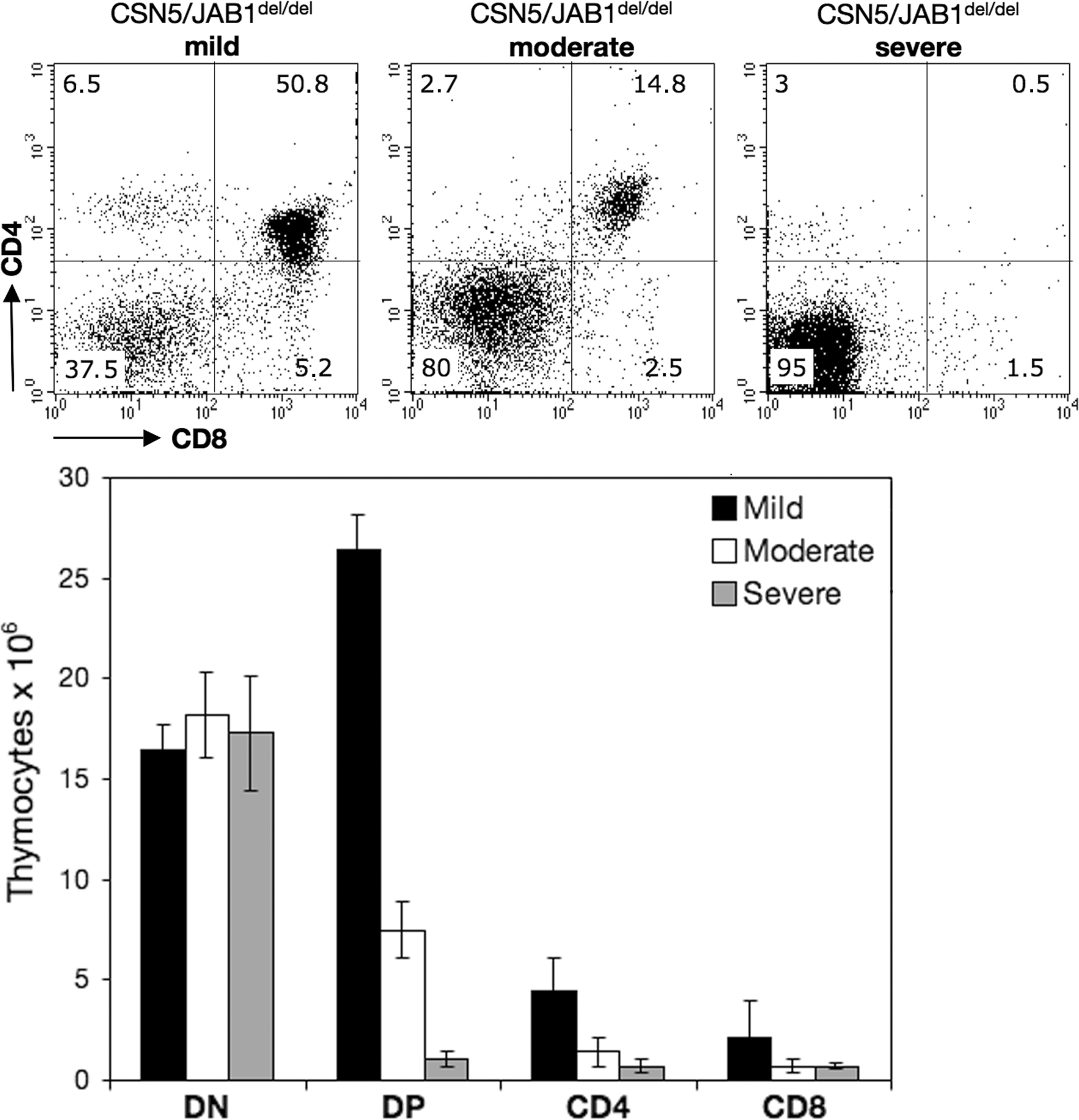
Previous studies have shown that inactivation of the CSN in vivo impairs the function of cullin-based ubiquitin ligase complexes, resulting in the accumulation of substrates that largely account for the observed phenotypes (9). Given the large number of substrates that are potentially controlled by the CSN, we sought to establish a correlation between the intracellular levels of such substrates and the phenotypes observed in CSN5/JAB1del/del mice. To this aim, purified DN and DP thymocytes from multiple animals were pooled (4–5 animals/pool on average) based on the severity of the thymic phenotype, as determined by assessing the ratio between DN and DP thymocyte subpopulations. Such ratio ranges from <0.05 in WT mice, to >4 in CSN5/JAB1del/del animals displaying a severe phenotype. Intermediate phenotypes may be explained by the incomplete penetrance of the Lck promoter-driven expression of Cre recombinase, resulting in inefficient deletion of the CSN5/JAB1 locus in a variable fraction of developing T cells (Fig. S5, available at http://www.jem.org/cgi/content/full/jem.20070725/DC1) (28). We assessed candidate substrates of CSN-controlled ubiquitin ligases whose dysregulated steady-state levels, which were caused by defective ubiquitin-proteasome system (UPS)–dependent degradation, might account for the observed cell cycle and survival defects in CSN5/JAB1del/del thymocytes. Within the former category, we included the cyclin-dependent kinase inhibitors p27kip1 and p21cip1, cyclin D3, cyclin E, and the DNA replication licensing factor Cdt1, whereas c-Myc, p53, β-catenin, the glucocorticoid receptor, and IκB-α were investigated as UPS-controlled proteins involved in the regulation of survival/apoptotic programs. Finally, to assess the efficiency of CSN5/JAB1del/del locus deletion in the pooled DN and DP/SP populations and to gain insight into the reduced function of the CSN, we biochemically analyzed the steady-state levels of CSN5/JAB1, cullin1, and the Cre recombinase (Fig. 4 A). Some substrates were poorly detectable in purified thymocytes (e.g., c-Myc and p21cip1; not depicted). Conversely, as shown in Fig. 4 B, β-catenin and p53 were the only substrates whose steady-state levels paralleled the severity of the thymic phenotype in CSN5/JAB1del/del mice. β-catenin, in particular, showed a mean 20-fold accumulation in DP thymocytes from the most severely affected animals, whereas p53, which is barely detectable in WT thymocytes, accumulated to clearly detectable levels in CSN5/JAB1del/del developing DP cells. Likewise, cyclin D3 and Cdt1 levels were moderately increased in DN thymocytes from CSN5/JAB1del/del mice, whereas in DP thymocytes the levels of Cdt1 were directly proportional to the severity of the phenotype (Fig. 4 B). At variance with the aforementioned findings, the levels of IκB-α showed an inverse correlation with the severity of the phenotype in both DN and DP cells from CSN5/JAB1del/del mice (Fig. 4 B). Unexpectedly, the levels of p27kip1 were also consistently reduced in CSN5/JAB1del/del DN cells, whereas they did not differ from controls in DP thymocytes (Fig. 4 B). Finally, cyclin E (unpublished data), did not show significant variations in developing thymocytes from CSN5/JAB1del/del animals when compared with WT littermates. Quantitative PCR analysis for β-catenin and p53 expression confirmed that the observed increase in their protein levels could not be accounted for by an increase in steady-state transcript levels (Table S2).
Figure 4.

Substrates of CSN-regulated E3 ligases are dysregulated CSN5/JAB1del/del thymocytes. Purified DN and DP/SP thymocytes from pools of 4 to 5 animals belonging to either WT or to each of the indicated CSN5/JAB1 knockout phenotypes (S, severe; Mo, moderate; Mi, mild; Fig. S5 shows phenotypic clustering of CSN5/JAB1del/del thymocytes based on the DN:DP/SP ratio), likely resulting from incomplete penetrance of Lck-Cre-mediated recombination, were lysed and subjected to SDS-PAGE followed by immunoblotting with the indicated antibodies. (A) Immunoblot analysis for CSN5/JAB1, Cre, and Cul1 in purified DP/SP thymocytes. Cul1 is visualized as a doublet with the upper band representing the hyperneddylated isoform. (B) Immunoblot analysis for candidate substrates of CSN-regulated E3 ligases. Films were processed by densitometric scanning and normalized to values for β-actin for each lane to assess the relative values in each lane. Fig. S5 is available at http://www.jem.org/cgi/content/full/jem.20070725/DC1.
Altered levels of Bcl-2 family proteins and defectiveNF-κB activation in developing thymocytesof CSN5/JAB1del/del mice
We were intrigued by the finding that the levels of β-catenin and IκB-α, both of which are known substrates of the CSN-controlled SCFβTrcP1 E3 ligase, showed opposite variations in CSN5/JAB1del/del DP thymocytes when compared with WT littermates. We reasoned that because IκB-α is a direct transcriptional target of NF-κB as part of a well-characterized regulatory feedback loop (29), its reduced levels could be explained by defective induction of NF-κB, possibly caused by delayed or inefficient degradation of its cytoplasmic inhibitor. To test this possibility, we used several approaches. First, we quantitatively assessed the steady-state levels of the IκB-α mRNA, which showed a marked reduction in both CSN5/JAB1del/del DN and DP cells compared with WT controls and paralleled the severity of the phenotype (Table S2). Second, we performed short-term activation assays in freshly purified DN4 thymocytes by exposing them in vitro to combined stimulation via PMA plus ionomycin (P + I), which is a known NF-κB–activating condition that promotes rapid phosphorylation, polyubiquitination, and UPS-dependent degradation of IκB-α, followed by nuclear translocation of transcription-competent NF-κB dimers. As shown in Fig. 5, steady levels of IκB-α decreased rapidly in WT thymocytes upon P + I treatment. Nuclear levels of p65RelA were clearly detectable in unstimulated cells, which is consistent with a constitutive induction of NF-κB in DN4 cells (30), and accumulated further upon P + I stimulation. In contrast, both steady-state IκB-α and nuclear p65 levels were greatly reduced in CSN5/JAB1del/del, unstimulated DN4 thymocytes. However, a sizeable fraction of p65RelA did accumulate in the nucleus of P + I–stimulated CSN5/JAB1del/del cells.
Figure 5.

Deregulated NF-κB activation in developing thymocytes from CSN5/JAB1del/del mice. Purified DN4 thymocytes were pooled from 4–5 WT or CSN5/JAB1del/del mice and either left untreated or stimulated with a combination of 50 nM PMA plus 250 μM ionomycin for 45 min, after which cells were lysed and cytosolic and nuclear extracts were prepared as described in the Materials and methods and run on a SDS-PAGE. Immunoblots were performed with the indicated antibodies.
The apparent dysregulation of NF-κB observed in CSN5/JAB1del/del thymocytes prompted us to assess the levels of selected members of the Bcl-2 protein family, some of which are known direct NF-κB targets and are prominent effectors of the survival programs controlled by this transcription factor in developing thymocytes (31, 32). To this aim, we used a combination of biochemical, immunohistochemical, and genetic approaches. Although Bcl-2 levels, which are up-regulated in DP thymocytes, did not appear to be affected in CSN5/JAB1del/del animals (not depicted), the levels of Bcl-xL were markedly down-regulated in both DN and DP cells from knockout animals. Such down-regulation was evident both at the mRNA and at the protein level (Fig. 6, A and B). Likewise, the antiapoptotic Bcl-2A1 protein, which is also a target of NF-κB, was down-regulated in CSN5/JAB1del/del thymocytes (Fig. 6 A). Conversely, the proapoptotic protein Bax, which is a target of p53, was up-regulated in CSN5/JAB1del/del DN and DP cells (Fig. 6 A), which is consistent with the increased levels of p53 in these cells. As a result, the ratio between Bax and Bcl-xL or Bax and Bcl-2A1 was completely subverted in both DN and DP cells from CSN5/JAB1del/del animals compared with WT littermates (Fig. 6 A), thus potentially accounting for the increased apoptotic rate observed in developing thymocytes from the knockout mice.
Figure 6.

Altered mRNA and protein levels of Bcl-2 family members in CSN5/JAB1del/del thymocytes. (A) Semiquantitative RT-PCR for the indicated Bcl-2 family members was performed on retrotranscribed total RNA extracted from purified DN and DP thymocytes obtained from pools of 4–5 animals, belonging to either WT or to each of the indicated CSN5/JAB1 knockout phenotypes (KOm, mild; KOs, severe; Fig. S5 shows phenotypic clustering of CSN5/JAB1del/del thymocytes based on the DN:DP/SP ratio). Values were normalized on β-actin for each sample and expressed as fold induction over the expression levels detected in WT, DN thymocytes for each Bcl-2 family member. (right) The normalized densitometric ratios of Bax:Bcl-xL and Bax:Bcl-2A1 values. Error bars indicate the mean ± the SD of three separate experiments. (B) Immunohistochemical sections of 5-wk-old thymi from WT (left) or CSN5/JAB1del/del (right) mice stained with anti-Bcl-xL antibody (as revealed by HRP immunoenzymatic reaction). M, medulla; C, cortex. Bcl-xL shows a typical dotted distribution in WT thymocytes, which is particularly evident in the thymic cortex. (right) Arrows indicate isolated, strongly positive thymic epithelial cells in CSN5/JAB1del/del thymi, which otherwise show barely detectable expression of Bcl-xL compared with WT controls (left). Fig. S5 is available at http://www.jem.org/cgi/content/full/jem.20070725/DC1. Bar, 20 μm.
Impaired survival in CSN5/JAB1-deficient thymocytesis independent of p53, but can be rescued by transgenic Bcl-xL expression
To genetically assess the pathways involved in CSN5/JAB1del/del-defective T cell development and enhanced apoptosis, and to gain insight into the role of the CSN in p53 functional regulation, the CSN5/JAB1del/del mutation was introduced into the p53−/− background. Phenotypic characterization of developing thymocytes in CSN5/JAB1del/del p53−/− animals showed that lack of p53 does not rescue the reduced thymic cellularity resulting from CSN5/JAB1 deficiency. However, a defined subset of thymocytes (intermediate SP [ISP]) reflecting a transitional state between DN and DP cells, was greatly increased in the compound CSN5/JAB1del/del p53−/− knockout mice compared with the individual knockouts (Fig. 7, A and B). Thus, inactivation of CSN5/JAB1 unveils a critical role of p53-dependent apoptosis in this particular stage of thymocyte development.
Figure 7.

Genetic complementation by a p53-deficient background only partially rescues thymic development in CSN5/JAB1del/del mice. (A) Thymocyte suspensions from the indicated genotypes were stained with anti-CD4 plus anti-CD8 antibody to identify the major thymocyte subsets. Dashed vertical lines were set to separate DN from ISP cells on the horizontal axis (showing CD8 expression levels). (B) Thymic cellularity of the indicated genotypes, as estimated by calculation of the number of thymocytes from individual thymi after electronic gating of the various subsets as shown in (A). Numbers are the mean ± the SD of values obtained from 3 mice per genotype at 5–6 wk of age.
To directly assess if reduced expression of selected Bcl-2 family members was responsible for the increased apoptotic rate of CSN5/JAB1del/del thymocytes, we transduced CSN5/JAB1del/del hematopoietic progenitor cells with self-inactivating lentiviral vectors expressing Bcl-xL, Bcl-2A1, or GFP (as a control) and transplanted transduced cells into sublethally irradiated RAG1−/− recipients. At 6 wk after transplant, we monitored the phenotypes and absolute counts of developing thymocytes and performed ex vivo cultures to assess the survival ability of transduced cells. As shown in Fig. 8 A, forced expression of Bcl-xL, but not Bcl-2A1, favored the accumulation of both DN4 and SP CSN5/JAB KO thymocytes to levels comparable to those found in floxed, Cre−-irradiated controls and RAG1−/− recipients reconstituted with WT hematopoietic progenitor cells (Fig. 8 A and not depicted). The ability of Bcl-xL to rescue survival of CSN5/JAB1del/del thymocytes was confirmed by assessing the apoptotic rate of ex vivo–cultured, purified thymocytes (Fig. 8 B). The apparent inability of Bcl-2A1 to affect survival of CSN5/JAB1del/del thymocytes was not caused by inefficient integration of the lentiviral vector carrying the Bcl-2A1 transgene (not depicted) or to reduced expression of the Bcl-2A1 protein, as shown by the increase in protein levels (three- to fivefold over the endogenous proteins for both Bcl-2A1 and Bcl-xL) in long-term cultures of virally transduced bone marrow precursors (Fig. 8 C).
Figure 8.

Transgenic expression of Bcl-xL rescues the number and late developmental stages of CSN5/JAB1del/del thymocytes. (A) DN, DP, and SP thymocyte counts from individual Rag1−/− animals 6 wk after reconstitution with bone marrow progenitors transduced with the indicated transgenes (CSN5/JAB1 flox/flox animals were used as controls). (B) Viability of DN and DP thymocytes from chimeric animals as described above, after ex vivo culture for the indicated time points and flow cytometric analysis with FITC-Annexin V staining and electronic gating on the indicated subsets; (C) Immunoblot analysis for Bcl-2A1 and Bcl-xL expression in bone marrow precursors 2 wk after transduction with lentiviral vectors carrying the indicated transgenes. β-actin was used for normalization. *, P < 0.05; **, P < 0.005; ***, P < 0.001 (Student's two-tailed t test with equal variance).
DISCUSSION
In this study, we addressed the function of the CSN in mammalian T cell development, as prior evidence in lower organisms suggested that it plays a pivotal role in the coordination of developmental programs entailing proliferation, differentiation, and survival in response to microenvironmental cues. The current model states that the hierarchical role of the CSN hinges on the functional regulation of cullin-based ubiquitin ligases, whose substrates are specifically devoted to directing the onset and maintenance of the aforementioned developmental programs (8, 9). Thus far, genetic evidence supporting this assumption is lacking in mammals, as the constitutive deletion of various CSN subunits leads to early embryonic lethality.
In view of our findings, it appears that timely control of the half-life of protein substrates of a defined subset of CSN-regulated E3 ligases is essential for the rapid and coordinate progression of maturing T cells through the various checkpoints imposed by TCR gene rearrangement, expression, and signaling. Overall, preTCR expression and proximal signaling appear to be preserved in CSN5/JAB1-deficient mice, as CSN5/JAB1del/del DN3 thymocytes progress normally toward the DN4 stage and undergo qualitatively normal rearrangements of TCR β and α subunit genes. In contrast, both DN4 and DP developmental stages are substantially altered in CSN5/JAB1del/del thymocytes. CSN5/JAB1del/del DN4 cells do not show gross numerical abnormalities compared with WT littermates. However, a sizeable fraction of DN4 cells displays defective progression in the cell cycle, as indicated by a delayed/arrested S phase progression. This suggests that CSN5/JAB1-deficient DN4 thymocytes fail to override a critical S phase checkpoint, resulting in cell cycle delay/arrest. We interpret the reduced steady-state levels of p27kip1 observed in CSN5/JAB1del/del DN cells as a further indication that they are arrested in S phase, as Skp2-mediated degradation of p27kip1 is maximal in cells undergoing DNA replication (33). Altered cell cycle progression in CSN5/JAB1del/del DN thymocytes does not appear to trigger a generalized apoptotic response, at least during the experimental time window (48 h) we examined. However, CSN5/JAB1del/del DN4 thymocytes fail to accumulate, suggesting that they do undergo apoptotic death (see the following paragraphs in Discussion) and that a variable fraction of these cells does progress toward further stages of maturation. The finding that forced expression of a rearranged transgenic TCR (OT-II) fails to rescue DP and SP development further supports the possibility that the TCR directly regulates the activity of the CSN while signaling selection/survival of DP thymocytes.
Among the candidate substrates we assessed as potentially accounting for the above cell cycle abnormalities in DN cells, we found no accumulation of p27kip1 and cyclin E, which have been proposed to alter cell cycle progression in CSN-deficient, transformed cell lines (17, 21). Instead, we observed marked accumulation of β-catenin and p53, which cannot be accounted for by their increased steady-state mRNA levels. Further, we observed a sizeable increase in the levels of the DNA replication initiation factor Cdt1, which paralleled the severity of the phenotype.
As a key effector of the Wnt signaling pathway, β-catenin is polyubiquitinated and routed to proteasomal degradation by the SCFβTrCP1 ubiquitin ligase complex, whose function is under direct positive regulation by the CSN (34). Thus, it is possible that a dysregulated turnover of β-catenin underlies selected phenotypic traits in CSN5/JAB1del/del T cells. The existing literature concerning the role of β-catenin throughout thymocyte development is somewhat controversial. Conditional deletion of a floxed β-catenin locus by LckCre-mediated excision results in defective G1-to-S transition of DN4 cells and in moderately decreased thymocyte numbers (35). Conversely, expression of a stabilized β-catenin mutant in developing thymocytes by in-frame deletion of a floxed exon 3 yields a striking phenotype, featuring forced maturation of DN cells into DP subsets before productive TCR gene rearrangement, as well as increased apoptosis of DP cells and reduced cellularity (36). In apparent contrast, transgenic overexpression of stabilized β-catenin under a CD4 promoter results in increased DP cell survival, correlating with increased steady-state levels of antiapoptotic Bcl-2 family members (37). In agreement with the study by Gounari et al. (36), and with evidence obtained in SIP-1 (38) and βTrCP1-null cells (34), we observed that in CSN5/JAB1del/del thymocytes β-catenin accumulation correlates with defective cell cycle progression and a propensity to undergo apoptotic cell death. We also noticed that a measurable fraction of CSN5/JAB1del/del DP/SP cells fails to express a rearranged intracellular TCR compared with WT controls, which is compatible with an abnormal gain of function phenotype caused by dysregulated turnover of β-catenin (36, 39). Notably, prior evidence suggested that overexpression of β-catenin leads to enhanced stability of p53 in selected cell types (40). However, notable phenotypic differences exist between CSN5/JAB1del/del mice and the β-catenin–overexpressing thymocytes reported by Gounari et al. (36). This, along with the incomplete rescue of the CSN5/JAB1del/del phenotype by the p53−/− background, suggests that elevation of p53 is not a direct a consequence of β-catenin accumulation in CSN5/JAB1del/del T cells and is unlikely to account for the observed phenotype. Rather, direct control of p53 protein stability by CSN-regulated E3 ligase complexes, such as Mdm2 itself or COP1 (26), might be impaired in the absence of a functional CSN. Notably, breeding the CSN5/JAB1 deficiency onto a constitutive p53null background does promote a relevant shift in thymocyte differentiation toward an ISP phenotype. This indicates that a critical p53-dependent checkpoint lies between DN4 cells and the immediate next step in thymocyte maturation, which involves exit from the cell cycle. A similar role for p53 has been suggested by a prior study investigating the role of the transcriptional regulator Bcl11b in thymic development (41). Further dissection of the aforementioned processes awaits the comprehensive identification of cell cycle checkpoint regulators that are under direct control by the CSN.
Increased propensity to undergo apoptotic death is a prominent feature of CSN5/JAB1del/del thymocytes at multiple developmental stages, and it is likely to account for the marked decrease in thymic cellularity observed in these mice. CSN5/JAB1del/del DN and DP cells display increased apoptosis both in vivo and after in vitro culture, suggesting an imbalance in critical effectors of death and survival programs. We uncovered a major decrease of both Bcl-xL and Bcl-2A1 transcript and protein levels in CSN5/JAB1del/del DN, as well as in DP cells, whereas the relative levels of Bcl-2 seemed unchanged compared with WT littermates. Coupled to the increased levels of the p53 transcriptional target Bax, which we also observed in both DN and DP cells of CSN5/JAB1-deficient mice, this yielded a complete reversion of the ratio between anti- and proapoptotic members of the Bcl-2 family of apoptosis regulators. We propose that defective activation of NF-κB–induced survival programs may partially explain the prominent decrease in antiapoptotic Bcl-2 family members observed in CSN5/JAB1del/del T cells. This interpretation is supported by several findings in this study, as follows: first, steady-state p65RelA nuclear localization is markedly reduced in CSN5/JAB1del/del DN4 thymocytes compared with WT controls, although further investigation is needed to address the underlying mechanisms, as well as the signals leading to induction of NF-κB that require a functional CSN; second, similar to β-catenin, IκB-α is a substrate of the SCFβTrcP complex, which is a CSN-regulated ubiquitin ligase that appears to be dysfunctional in CSN5/JAB1del/del mice (34); third, steady-state levels of IκB-α are markedly decreased in CSN5/JAB1del/del thymocytes, both at the mRNA and protein level. This finding might be a consequence of defective NF-κB activation, as IκB-α is a direct NF-κB transcriptional target as part of a well-characterized feed-back regulatory loop (29) and is consistent with recent reports linking CSN function to IκB-α regulation, both in Drosophila melanogaster (42) and in mammalian cell lines (43). Finally, both Bcl-xL and Bcl-2A1, whose levels are strongly reduced in CSN5/JAB1del/del mice, have been shown to be directly induced by NF-κB in developing thymocytes and to play essential roles in overall thymocyte survival and TCR-driven positive selection (31, 32). Our finding that transgenic expression of Bcl-xL in bone marrow reconstitution experiments rescues the number and late developmental stages of CSN5/JAB1del/del thymocytes provides further experimental support to our interpretation. A large body of evidence supports the notion that NF-κB–induced survival programs are essential in thymic development, although it is unclear if thymocytes rely on NF-κB prosurvival functions at a particular stage of development (44). A prior study based on genetic manipulation of NF-κB activation and on the use of a NF-κB activity reporter transgene (30) proposes that transcriptional activity is maximal in the final stage of DN4 cell development, before which high Bcl-2 levels replace the antiapoptotic function of NF-κB–induced effectors. Consistent with these findings, the antiapoptotic function of Bcl-2A1 has been shown to be both dependent on NF-κB and involved in DN thymocyte survival (45), whereas Bcl-xL overexpression allows selective survival and expansion of DP thymocytes (46). Our findings suggest that defective NF-κB activation may underlie increased apoptosis of both DN and DP cells in CSN5/JAB1del/del mice, as both subsets display a major reversion of the stoichiometric ratio of anti- vs. proapoptotic Bcl-2 family members. Possibly, pre-TCR–induced activation of NF-κB exerts relatively long-lasting effects, which contribute to limit death by neglect in DP cells undertaking editing of the TCR. Accordingly, defective activation of NF-κB would result in a large fraction of DP cells dying by apoptosis before productive engagement of the TCR. However, our findings also provide an indication that the CSN is required to allow survival of positively selected DP thymocytes, as shown by the failure of the OT-II transgenic TCR to rescue survival and maturation of DP cells in a CSN5/JAB1-deficient genetic background.
Proliferation and survival of developing thymocytes depend on appropriate signals delivered by the preTCR, the successful completion of a poorly defined process of DNA recombination and repair, and the appropriate coordination of processes affecting progression through and exit from the cell cycle. All of the aforementioned processes occur extremely rapidly, as the transition between DN4 and DP stages takes on average <24 h to be completed (6). Collectively, our findings suggest that the CSN exerts a general coordinating role on key signaling pathways in thymic development by controlling rapid posttranslational turnover of critical substrates. Although the CSN may extend its role throughout thymic development, the transition stage between actively proliferating DN4 cells and emerging DP thymocytes, which must survive a hostile microenvironment while editing a functional TCR, seem most critically dependent on a functional CSN. By conditionally deleting CSN5/JAB1 in other cell types, it will be important to address whether such a role of the CSN is context-dependent or can be extended to other developmental processes entailing a fine balance between proliferation and survival.
MATERIALS AND METHODS
Generation of mice carrying floxed CSN5/JAB1 alleles and Lck-Cre transgenes.
A targeting vector was constructed by subcloning the desired region of a BAC clone containing the entire CSN5/JAB1 genomic sequence into the pFlrt-Neo vector. The targeting construct includes a neomycin resistance cassette flanked by two Frt sites and the CSN5/JAB1 exon 2 flanked by LoxP sites. The mouse 129 embryonic stem (ES) cell line (R1) (SV129) was electroporated with the construct and subject to G418 selection. Homologous recombinants were identified by Southern blot analysis (SacI digest, 5′ probe). Two CSN5/JAB1flox ES clones were used to derive two independent lines of CSN5/JAB1+/Flox-Neo mice. The Neo cassette was removed by crossing mice with FLPe deleter mice. The CSN5/JAB1Flox/Flox mice were further crossed with Lck-Cre transgenic mice to promote deletion of the CSN5/JAB1 locus in T cells (CSN5/JAB1del/del mice). The mutant phenotype was analyzed in 129/C57BL/6 and C57BL/6 genetic backgrounds; no significant differences were observed. All mice were backcrossed at least eight times onto the C57/BL/6J background. FLPe deleter mice, Lck transgenic mice (provided by D. Wallach, Rehovot, Israel on behalf of Bristol-Myers Squibb), p53−/− mice, Ink4a−/− mice harboring deletion of exon 2 of the INK4a locus, which inactivates both p16INK4a and p19Arf (purchased from the NCI Mouse Models of Human Cancer Consortium) and OTII transgenic mice (imported from The Jackson Laboratory) were used in this study for crossbreeding. Mice were maintained under specific pathogen–free conditions and routinely analyzed at 5–8 wk after birth. All procedures were approved by the local Institutional Animal Care and Use Committee and conform to the local regulations and the European Union dispositions no. 86 and 609.
Genotyping by PCR.
Conventional PCR was performed on a GeneAmp 2400 machine (PerkinElmer). Primer sequences were as follows: InII1for(p1) 5′-GGTCAGAAAGCTAGGCCTAAGAAGG-3′, ExII1rev(p2) 5′-GGCATGCATCACCATTTTCAGTAG-3′, InIIIrev(p3) 5′-GGGCTTAGGAATGCCAAGC-3′. Sequences for Cre locus detection were 5′-GCCTGCATTACCGGTCGATGCAACGA-3′ and rev 5′-GTGGCAGATGGCGCGGCA-3′. Primer p1 was used in combination with primer p2 to identify floxed and wt alleles and the primer p1 with the primer p3 to assess Cre-mediated deletion.
Flow cytometry.
Complete medium consisted of RPMI 1640 with Glutamax (Invitrogen) supplemented with 10% FCS (Euroclone), 5.5 × 10−5 M 2-ME (Invitrogen), 100 U/ml penicillin, and 100 mg/ml streptomycin (Invitrogen). Thymi, spleens, LNs (inguinal, cervical, axillary, and brachial), and peripheral blood samples were obtained from littermate mice that were 5–6 wk old in most experiments. 5 × 105–106 dissociated thymocytes were suspended in staining buffer (PBS containing 3% FBS and 0.1% NaN3) for 15 min on ice for two, three, or four colors and analyzed by a FACScan or FACSCalibur flow cytometer (BD Biosciences). For intracellular stainings, cells were fixed in 2% paraformaldehyde in PBS for 10 min at room temperature and permeabilized with 0.5% saponin in PBS containing 3% FBS. The following directly conjugated antibodies (BD Biosciences) were used: CD4, CD8, CD3, CD25, CD44, CD69, TCRb, B220, and Bcl2. Data analysis was performed with CellQuest software (BD Biosciences).
BrdU incorporation assay.
1 mg BrdU was injected i.p., and thymi were explanted 1 h after injection. Cell cycle analysis was performed with BrdU-flow kit (BD Biosciences) according to the manufacturer's recommendations. PerCP-labeled CD4 and CD8 were also included with the BrdU-flow kit to examine thymocyte subpopulations.
Quantitation of apoptosis by Annexin V staining.
Thymocytes were incubated in RPMI complete medium at a concentration of 2.5 × 106 cells/ml in 12-well plates. Cells were analyzed either directly after isolation (time 0) or after incubation for the indicated time points. Cells were washed and stained using the Annexin V-FITC Apoptosis Detection kit (Bender MedSystems) with propidium iodide (PI) to stain dead cells, according to the manufacturer's recommendations. PerCP-labeled CD4 and CD8 were also included with the AnnexinV-FITC kit to examine thymocyte subpopulations. Apoptotic rate was assessed on cells gated as “viable” based on physical parameters (forward and side scatter), by scoring events consisting of early (AnnexinV-FITC+, PI−) and late (AnnexinV-FITC+, PI+) cells.
Immunoblotting.
Cells were washed in PBS and lysed in RIPA buffer. Protein extracts (30 mg each) were resolved by 8–10% sodium dodecyl sulfate-PAGE and transferred to nitrocellulose membranes (Hybond C Extra; GE Healthcare) in a blotting apparatus. Antibodies to p53 (Cell Signaling Technology), p27 (BD Biosciences), CSN5/JAB1 (Sigma-Aldrich), Cre (Covance), β-catenin (BD Biosciences), actin (Sigma-Aldrich), glycocorticoid receptor (Santa Cruz Biotechnology), Cdt1 (Santa Cruz Biotechnology), monocyte-derived macrophage 2 (Santa Cruz Biotechnology), p65RelA (Santa Cruz Biotechnology), IκBα (Cell Signaling Technology), Cul1 (Zymed Laboratories), and CycD3 (Santa Cruz Biotechnology) were used for immunoblot analysis. To obtain purified thymocyte subsets, cell suspensions were treated with anti-CD4 and -CD8 antibodies, followed by incubation with anti–rat IgG-conjugated magnetic beads, to recover DP/SP thymocytes (positive isolation). Nonadherent cells were subject to a second round of negative isolation by treatment with anti-CD4, -CD8, -B220, and -CD25 antibodies to recover DN1/DN4 thymocytes. Purity of the obtained DN1/DN4 and DP/SP subsets was assessed by flow cytometry and typically exceeded 90% (unpublished data). NF-κB activation assays and subcellular fractionations were performed as previously described (47). NF-κB immunoblotting was performed according to the manufacturer's instructions.
RT-PCR analysis.
Total RNA was extracted from sorted thymocytes by RNeasy Micro kit (QIAGEN) according to the manufacturer's recommendations, and the RNA was reverse transcribed with M-MLV (Promega) using random hexamers following the instructions of the manufacturer. Each sample was normalized by PCR for β-actin mRNA content. For semiquantitative RT-PCR analysis, serially diluted cDNA was amplified as follows: 94°C for 1 min, followed by 28–30 cycles of 94°C for 1 min, 60°C for 1 min, and 72°C for 2 min. Quantitative RT-PCR by Taqman (Applied Biosystems) was performed for p53, IκB-α, and β-catenin mRNAs using manufacturer-provided, predesigned primers, probes, and conditions. The following primer sets were used for the other transcripts: CSN5/Jab1, 5′-GAAGAAGCTTGCATCTTGATTGTGGAGCGAC-3′ and 5′-CTCAGGATCCCACACAGCACCTTAGTCTTC-3′; Bcl-xL, 5′-AGCAACCGGGAGCTGGTGGTCGAC-3′ and 5′-GACTGAAGAGTGAGCCCAGCAGA-3′; Bax, 5′-TCAGCCCATCTTCTTCCAGATGGT-3′ and 5′-GATTGCTGACGTGGACACGGACT-3′; β-actin, 5′-AGGTCCAGACGCAGGATGGC-3′ and 5′-ACTCCTATGTGGGTGACGAG-3′; Bcl2-A1, 5′-GCATCATTAACTGGGGAAGG-3′ and 5′-TCTTCCCAACCTCCATTCTG-3′; IκB-α, 5′-CTTGGCTGTGATCACCAACCAG-3′ and 5′-CGAAACCAGGTCAGGATTCTGC-3′; p53, 5′-CTTCCCAGCAGGGTGTCACGC-3′ and 5′-GTGCTGTGACTTCTTGTAGATGGC-3′.
Jα hybridization analysis.
RNA was extracted with TRIzol (Invitrogen) from DP/SP cells and reverse transcribed into cDNA. TCR Vαs transcripts were amplified for 35 cycles, followed by a nested PCR for 30 cycles using the same TCR V-specific oligo and internal Cα primers. The following primers were used: Up mVα3, 5′-ACCCAGACAGAAGGCCTGGTCA-3′; Up mVα14.1, 5′-GTGTCCCTGACAGTCCTGGTTGAC-3′; and Up mVα19, 5′-GCTTCTGACAGAGCTGATCAA-3′; also, respectively, dw mCα-ext, 5′-GCTTTTCTCGGTCAACGTGGCACT-3′; and for the nested PCR Dw mCα-int, 5′-ACTGGGGTAGGTGGCGTTGGTCTCT-3′. Amplification efficacy and uniformity of yield were evaluated by agarose gel electrophoresis. For Jα hybridization analysis, TCR Vα-Cα PCR products were electrophoresed on 1.5% agarose gel and transferred onto Hybond N+ nylon membranes (GE Healthcare) using alkaline solution. Hybridizations were performed with γ-32P end–labeled, Jα-specific oligonucleotide probes; Jα oligo sequences and hybridization conditions were performed as previously described (48).
Immunohistochemical analysis.
In brief, the thymus was rapidly removed and stored in 4% formaldehyde/saline solution. 2-mm-thick serial sections were obtained and then embedded in paraffin. These sections stained with hematoxylin and eosin. 4-mm-thick sections were prepared from formalin-fixed, paraffin-embedded mouse thymus, deparaffinized in xylene, and rehydrated in graded alcohol. Immunolocalization for CSN5/JAB1, Bcl-XL, CCl3, and Ki67 were performed with quenching of endogenous peroxidase in 3% H2O2, followed by blocking in 5% normal horse serum for 30 min at room temperature. The immunoreaction was revealed by horseradish peroxidase (HRP), using 3,3′-diaminobenzidine as chromogen (BioGenex), and the slides were slightly counterstained with Harris' hematoxylin.
LV production and titration.
Vesicular stomatitis virus–pseudotyped LV stocks were produced by transient cotransfection of the transfer constructs pCCLsin.cPPT.hPGK.eGFP.Wpre or pCCLsin.cPPT.hPGK.BclXL.Wpre and pCCLsin.cPPT.hPGK.Bcl-2A1.Wpre, the third-generation packaging constructs pMD2.Lg/p and pRSV.Rev, and the pMD2.G envelope construct in 293T cells followed by ultracentrifugation of conditioned medium, as previously described (49). Stocks were titered by endpoint expression titer in HeLa cells and quantified for particle content by HIV-1 Gag p24 immunocapture assay. The following primer sets were used to amplify the coding sequences of Bcl-2A1 and BclxL: Bcl-2A1 forward 5′-TTTTGGATCCATGTCTGAGTACGAGTTCATG-3′ and reverse 5′-TTTTCTCGAGTTACTTGAGGAGAAAGAGCATTTCC-3′; BclxL forward 5′-TTTTAGATCTATGTCTCAGAGCAACCGGGAGC-3′ and reverse 5′-TTTTCTCGAGTCACTTCCGACTGAAGAGTGCC-3′.
Isolation and transduction of hematopoietic progenitors.
6-wk-old CSN5/JAB1del/del mice were killed by CO2 inhalation, and bone marrow was harvested by flushing femurs and tibias with PBS-2% FBS (Invitrogen). Lin− cells were purified and transduced as previously described (49).
Transplantation procedures.
6-wk-old WT female Rag1−/− mice were lethally irradiated with 11.5 Gy administered in 2 doses 1 h apart. 2 h after irradiation, vector- and mock-transduced cells (7.5 × 105 cells/mouse) were injected via the tail vein. The bone marrow chimeras are used 6 wk after the transplant to allow complete reconstitution of immune system.
Online supplemental material.
Fig. S1 illustrates the strategy to conditionally delete the CSN5/JAB1 locus. Fig. S2 shows T cell subset distribution in peripheral lymphoid organs. Fig. S3 show normal patterns of TCR gene rearrangements in CSN5/JAB1del/del thymocytes. Fig. S4 shows intracellular and surface TCR expression in CSN5/JAB1del/del thymocytes; Fig. S5 shows incomplete penetrance of thymic phenotype in CSN5/JAB1del/del mice. Table S1 shows T cell counts in CSN5/JAB1del/del mice crossed onto the OT-II transgenic background. Table S2 shows the relative amounts of mRNAs coding for p53, IκBα, and β-catenin in CSN5/JAB1del/del thymocytes. The online version of this article is available at http://www.jem.org/cgi/content/full/jem.20070725/DC1.
Supplemental Material
Acknowledgments
We thank Barbara Clissi for technical help and Fabio Grassi for critical discussion. We thank the Telethon Core Facility for Conditional Mutagenesis for help in the generation of CSN5/JAB1flox ES clones and for blastocyst injection.
This work was supported in part by grants from the EU MAIN Network of Excellence (LSGH-CT-2003-502935), MIUR (PRIN projects) to R. Pardi, AIRC to R. Pardi, A. Mondino and G. Casorati, Compagnia San Paolo IMI to A. Mondino, and from the National Institutes of Health grant R01 HL43331 and the Raymond and Beverly Sackler Foundation to J.R. Bender.
The authors have no conflicting financial interests.
Abbreviations used: ClC, cleaved effector caspase; CSN, COP9 signalosome; DN, double-negative; DP, double-positive; HRP, horseradish peroxidase; ISP, intermediate single-positive; PI, propidium iodide; SP, single-positive; UPS, ubiquitin-proteasome system.
References
- 1.Spits, H. 2002. Development of alphabeta T cells in the human thymus. Nat. Rev. Immunol. 2:760–772. [DOI] [PubMed] [Google Scholar]
- 2.Haks, M.C., P. Krimpenfort, J.H. van den Brakel, and A.M. Kruisbeek. 1999. Pre-TCR signaling and inactivation of p53 induces crucial cell survival pathways in pre-T cells. Immunity. 11:91–101. [DOI] [PubMed] [Google Scholar]
- 3.Haines, B.B., C.J. Ryu, S. Chang, A. Protopopov, A. Luch, Y.H. Kang, D.D. Draganov, M.F. Fragoso, S.G. Paik, H.J. Hong, et al. 2006. Block of T cell development in P53-deficient mice accelerates development of lymphomas with characteristic RAG-dependent cytogenetic alterations. Cancer Cell. 9:109–120. [DOI] [PubMed] [Google Scholar]
- 4.Jondal, M., A. Pazirandeh, and S. Okret. 2004. Different roles for glucocorticoids in thymocyte homeostasis? Trends Immunol. 25:595–600. [DOI] [PubMed] [Google Scholar]
- 5.Chung, H., Y.I. Choi, M.G. Ko, and R.H. Seong. 2002. Rescuing developing thymocytes from death by neglect. J. Biochem. Mol. Biol. 35:7–18. [DOI] [PubMed] [Google Scholar]
- 6.Vasseur, F., A. Le Campion, and C. Penit. 2001. Scheduled kinetics of cell proliferation and phenotypic changes during immature thymocyte generation. Eur. J. Immunol. 31:3038–3047. [DOI] [PubMed] [Google Scholar]
- 7.Carrabino, S., E. Carminati, D. Talarico, R. Pardi, and E. Bianchi. 2004. Expression pattern of the JAB1/CSN5 gene during murine embryogenesis: colocalization with NEDD8. Gene Expr. Patterns. 4:423–431. [DOI] [PubMed] [Google Scholar]
- 8.Wei, N., and X.W. Deng. 2003. The COP9 signalosome. Annu. Rev. Cell Dev. Biol. 19:261–286. [DOI] [PubMed] [Google Scholar]
- 9.Cope, G.A., and R.J. Deshaies. 2003. COP9 signalosome: a multifunctional regulator of SCF and other cullin-based ubiquitin ligases. Cell. 114:663–671. [DOI] [PubMed] [Google Scholar]
- 10.Cope, G.A., G.S. Suh, L. Aravind, S.E. Schwarz, S.L. Zipursky, E.V. Koonin, and R.J. Deshaies. 2002. Role of predicted metalloprotease motif of Jab1/Csn5 in cleavage of Nedd8 from Cul1. Science. 298:608–611. [DOI] [PubMed] [Google Scholar]
- 11.Bianchi, E., S. Denti, A. Granata, G. Bossi, J. Geginat, A. Villa, L. Rogge, and R. Pardi. 2000. Integrin LFA-1 interacts with the transcriptional co-activator JAB1 to modulate AP-1 activity. Nature. 404:617–621. [DOI] [PubMed] [Google Scholar]
- 12.Perez, O.D., D. Mitchell, G.C. Jager, S. South, C. Murriel, J. McBride, L.A. Herzenberg, S. Kinoshita, and G.P. Nolan. 2003. Leukocyte functional antigen 1 lowers T cell activation thresholds and signaling through cytohesin-1 and Jun-activating binding protein 1. Nat. Immunol. 4:1083–1092. [DOI] [PubMed] [Google Scholar]
- 13.Lykke-Andersen, K., L. Schaefer, S. Menon, X.W. Deng, J.B. Miller, and N. Wei. 2003. Disruption of the COP9 signalosome Csn2 subunit in mice causes deficient cell proliferation, accumulation of p53 and cyclin E, and early embryonic death. Mol. Cell. Biol. 23:6790–6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan, J., K. Walz, H. Nakamura, S. Carattini-Rivera, Q. Zhao, H. Vogel, N. Wei, M.J. Justice, A. Bradley, and J.R. Lupski. 2003. COP9 signalosome subunit 3 is essential for maintenance of cell proliferation in the mouse embryonic epiblast. Mol. Cell. Biol. 23:6798–6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tomoda, K., N. Yoneda-Kato, A. Fukumoto, S. Yamanaka, and J.Y. Kato. 2004. Multiple functions of Jab1 are required for early embryonic development and growth potential in mice. J. Biol. Chem. 279:43013–43018. [DOI] [PubMed] [Google Scholar]
- 16.Yang, X., S. Menon, K. Lykke-Andersen, T. Tsuge, X. Di, X. Wang, R.J. Rodriguez-Suarez, H. Zhang, and N. Wei. 2002. The COP9 signalosome inhibits p27(kip1) degradation and impedes G1-S phase progression via deneddylation of SCF Cul1. Curr. Biol. 12:667–672. [DOI] [PubMed] [Google Scholar]
- 17.Denti, S., M.E. Fernandez-Sanchez, L. Rogge, and E. Bianchi. 2006. The COP9 signalosome regulates Skp2 levels and proliferation of human cells. J. Biol. Chem. 281:32188–32196. [DOI] [PubMed] [Google Scholar]
- 18.Peng, Z., Y. Shen, S. Feng, X. Wang, B.N. Chitteti, R.D. Vierstra, and X.W. Deng. 2003. Evidence for a physical association of the COP9 signalosome, the proteasome, and specific SCF E3 ligases in vivo. Curr. Biol. 13:R504–R505. [DOI] [PubMed] [Google Scholar]
- 19.Wan, M., Y. Tang, E.M. Tytler, C. Lu, B. Jin, S.M. Vickers, L. Yang, X. Shi, and X. Cao. 2004. Smad4 protein stability is regulated by ubiquitin ligase SCF beta-TrCP1. J. Biol. Chem. 279:14484–14487. [DOI] [PubMed] [Google Scholar]
- 20.Kim, B.C., H.J. Lee, S.H. Park, S.R. Lee, T.S. Karpova, J.G. McNally, A. Felici, D.K. Lee, and S.J. Kim. 2004. Jab1/CSN5, a component of the COP9 signalosome, regulates transforming growth factor beta signaling by binding to Smad7 and promoting its degradation. Mol. Cell. Biol. 24:2251–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cope, G.A., and R.J. Deshaies. 2006. Targeted silencing of Jab1/Csn5 in human cells downregulates SCF activity through reduction of F-box protein levels. BMC Biochem. 7:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Groisman, R., J. Polanowska, I. Kuraoka, J. Sawada, M. Saijo, R. Drapkin, A.F. Kisselev, K. Tanaka, and Y. Nakatani. 2003. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell. 113:357–367. [DOI] [PubMed] [Google Scholar]
- 23.Hu, J., C.M. McCall, T. Ohta, and Y. Xiong. 2004. Targeted ubiquitination of CDT1 by the DDB1-CUL4A-ROC1 ligase in response to DNA damage. Nat. Cell Biol. 6:1003–1009. [DOI] [PubMed] [Google Scholar]
- 24.Zhong, W., H. Feng, F.E. Santiago, and E.T. Kipreos. 2003. CUL-4 ubiquitin ligase maintains genome stability by restraining DNA-replication licensing. Nature. 423:885–889. [DOI] [PubMed] [Google Scholar]
- 25.Bech-Otschir, D., R. Kraft, X. Huang, P. Henklein, B. Kapelari, C. Pollmann, and W. Dubiel. 2001. COP9 signalosome-specific phosphorylation targets p53 to degradation by the ubiquitin system. EMBO J. 20:1630–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dornan, D., I. Wertz, H. Shimizu, D. Arnott, G.D. Frantz, P. Dowd, K. O'Rourke, H. Koeppen, and V.M. Dixit. 2004. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature. 429:86–92. [DOI] [PubMed] [Google Scholar]
- 27.Li, M., G.M. Davey, R.M. Sutherland, C. Kurts, A.M. Lew, C. Hirst, F.R. Carbone, and W.R. Heath. 2001. Cell-associated ovalbumin is cross-presented much more efficiently than soluble ovalbumin in vivo. J. Immunol. 166:6099–6103. [DOI] [PubMed] [Google Scholar]
- 28.Mao, X., Y. Fujiwara, and S.H. Orkin. 1999. Improved reporter strain for monitoring Cre recombinase-mediated DNA excisions in mice. Proc. Natl. Acad. Sci. USA. 96:5037–5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun, S.C., P.A. Ganchi, D.W. Ballard, and W.C. Greene. 1993. NF-kappa B controls expression of inhibitor I kappa B alpha: evidence for an inducible autoregulatory pathway. Science. 259:1912–1915. [DOI] [PubMed] [Google Scholar]
- 30.Voll, R.E., E. Jimi, R.J. Phillips, D.F. Barber, M. Rincon, A.C. Hayday, R.A. Flavell, and S. Ghosh. 2000. NF-kappa B activation by the pre-T cell receptor serves as a selective survival signal in T lymphocyte development. Immunity. 13:677–689. [DOI] [PubMed] [Google Scholar]
- 31.Khoshnan, A., C. Tindell, I. Laux, D. Bae, B. Bennett, and A.E. Nel. 2000. The NF-kappa B cascade is important in Bcl-xL expression and for the anti-apoptotic effects of the CD28 receptor in primary human CD4+ lymphocytes. J. Immunol. 165:1743–1754. [DOI] [PubMed] [Google Scholar]
- 32.Zong, W.X., L.C. Edelstein, C. Chen, J. Bash, and C. Gelinas. 1999. The prosurvival Bcl-2 homolog Bfl-1/A1 is a direct transcriptional target of NF-kappaB that blocks TNFalpha-induced apoptosis. Genes Dev. 13:382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kamura, T., T. Hara, M. Matsumoto, N. Ishida, F. Okumura, S. Hatakeyama, M. Yoshida, K. Nakayama, and K.I. Nakayama. 2004. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat. Cell Biol. 6:1229–1235. [DOI] [PubMed] [Google Scholar]
- 34.Nakayama, K., S. Hatakeyama, S. Maruyama, A. Kikuchi, K. Onoe, R.A. Good, and K.I. Nakayama. 2003. Impaired degradation of inhibitory subunit of NF-kappa B (I kappa B) and beta-catenin as a result of targeted disruption of the beta-TrCP1 gene. Proc. Natl. Acad. Sci. USA. 100:8752–8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu, Y., D. Banerjee, J. Huelsken, W. Birchmeier, and J.M. Sen. 2003. Deletion of beta-catenin impairs T cell development. Nat. Immunol. 4:1177–1182. [DOI] [PubMed] [Google Scholar]
- 36.Gounari, F., I. Aifantis, K. Khazaie, S. Hoeflinger, N. Harada, M.M. Taketo, and H. von Boehmer. 2001. Somatic activation of beta-catenin bypasses pre-TCR signaling and TCR selection in thymocyte development. Nat. Immunol. 2:863–869. [DOI] [PubMed] [Google Scholar]
- 37.Xie, H., Z. Huang, M.S. Sadim, and Z. Sun. 2005. Stabilized beta-catenin extends thymocyte survival by up-regulating Bcl-xL. J. Immunol. 175:7981–7988. [DOI] [PubMed] [Google Scholar]
- 38.Fukushima, T., J.M. Zapata, N.C. Singha, M. Thomas, C.L. Kress, M. Krajewska, S. Krajewski, Z. Ronai, J.C. Reed, and S. Matsuzawa. 2006. Critical function for SIP, a ubiquitin E3 ligase component of the beta-catenin degradation pathway, for thymocyte development and G1 checkpoint. Immunity. 24:29–39. [DOI] [PubMed] [Google Scholar]
- 39.Gounari, F., R. Chang, J. Cowan, Z. Guo, M. Dose, E. Gounaris, and K. Khazaie. 2005. Loss of adenomatous polyposis coli gene function disrupts thymic development. Nat. Immunol. 6:800–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Damalas, A., A. Ben-Ze'ev, I. Simcha, M. Shtutman, J.F. Leal, J. Zhurinsky, B. Geiger, and M. Oren. 1999. Excess beta-catenin promotes accumulation of transcriptionally active p53. EMBO J. 18:3054–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okazuka, K., Y. Wakabayashi, M. Kashihara, J. Inoue, T. Sato, M. Yokoyama, S. Aizawa, Y. Aizawa, Y. Mishima, and R. Kominami. 2005. p53 prevents maturation of T cell development to the immature CD4-CD8+ stage in Bcl11b−/− mice. Biochem. Biophys. Res. Commun. 328:545–549. [DOI] [PubMed] [Google Scholar]
- 42.Harari-Steinberg, O., R. Cantera, S. Denti, E. Bianchi, E. Oron, D. Segal, and D.A. Chamovitz. 2007. COP9 signalosome subunit 5 (CSN5/Jab1) regulates the development of the Drosophila immune system: effects on Cactus, Dorsal and hematopoiesis. Genes Cells. 12:183–195. [DOI] [PubMed] [Google Scholar]
- 43.Schweitzer, K., P.M. Bozko, W. Dubiel, and M. Naumann. 2007. CSN controls NF-kappaB by deubiquitinylation of IkappaBalpha. EMBO J. 26:1532–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Siebenlist, U., K. Brown, and E. Claudio. 2005. Control of lymphocyte development by nuclear factor-kappaB. Nat. Rev. Immunol. 5:435–445. [DOI] [PubMed] [Google Scholar]
- 45.Mandal, M., C. Borowski, T. Palomero, A.A. Ferrando, P. Oberdoerffer, F. Meng, A. Ruiz-Vela, M. Ciofani, J.C. Zuniga-Pflucker, I. Screpanti, et al. 2005. The BCL2A1 gene as a pre-T cell receptor-induced regulator of thymocyte survival. J. Exp. Med. 201:603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grillot, D.A., R. Merino, and G. Nunez. 1995. Bcl-xL displays restricted distribution during T cell development and inhibits multiple forms of apoptosis but not clonal deletion in transgenic mice. J. Exp. Med. 182:1973–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Geginat, J., B. Clissi, M. Moro, P. Dellabona, J.R. Bender, and R. Pardi. 2000. CD28 and LFA-1 contribute to cyclosporin A-resistant T cell growth by stabilizing the IL-2 mRNA through distinct signaling pathways. Eur. J. Immunol. 30:1136–1144. [DOI] [PubMed] [Google Scholar]
- 48.Villey, I., D. Caillol, F. Selz, P. Ferrier, and J.P. de Villartay. 1996. Defect in rearrangement of the most 5′ TCR-J alpha following targeted deletion of T early alpha (TEA): implications for TCR alpha locus accessibility. Immunity. 5:331–342. [DOI] [PubMed] [Google Scholar]
- 49.Biffi, A., M. De Palma, A. Quattrini, U. Del Carro, S. Amadio, I. Visigalli, M. Sessa, S. Fasano, R. Brambilla, S. Marchesini, et al. 2004. Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J. Clin. Invest. 113:1118–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}