

Abstract
Originally thought of as a stomach-derived endocrine peptide acting via its receptors in the central nervous system to stimulate food intake and growth hormone expression, ghrelin and its receptor (growth hormone secretagogue receptor (GHS-R)) are widely expressed in a number of organ systems, including cancer cells. However, the direct functional role of ghrelin and its receptor in tumors of central nervous system origin remains to be defined. Here, we demonstrate that the human astrocytoma cell lines U-118, U-87, CCF-STTG1, and SW1088 express 6-, 11-, 15-, and 29-fold higher levels of GHS-R compared with primary normal human astrocytes. The ligation of GHS-R by ghrelin on these cells resulted in an increase in intracellular calcium mobilization, protein kinase C activation, actin polymerization, matrix metalloproteinase-2 activity, and astrocytoma motility. In addition, ghrelin led to actin polymerization and membrane ruffling on cells, with the specific co-localization of the small GTPase Rac1 with GHS-R on the leading edge of the astrocytoma cells and imparting the tumor cells with a motile phenotype. Disruption of the endogenous ghrelin/GHS-R pathway by RNA interference resulted in diminished motility, matrix metalloproteinase activity, and Rac expression, whereas tumor cells stably overexpressing GHS-R exhibited increased cell motility. The relevance of ghrelin and GHS-R expression was verified in clinically relevant tissues from 20 patients with oligodendrogliomas and grade II–IV astrocytomas. Analysis of a central nervous system tumor tissue microarray revealed that strong GHS-R and ghrelin expression was significantly more common in high grade tumors compared with low grade ones. Together, these findings suggest a novel role for the ghrelin/GHS-R axis in astrocytoma cell migration and invasiveness of cancers of central nervous system origin.
Ghrelin, a 28-amino acid acylated orexigenic peptide, is believed to be largely secreted from X/A-like cells of the stomach (1, 2). The ghrelin gene encodes a 117-amino acid precursor peptide (preproghrelin) that shares 82% homology between rat and human (1). A mutation in the preproghrelin sequence (R51Q) has been shown to be prevalent in obese subjects (3). Ghrelin binds to the third transmembrane domain of the G protein-coupled receptor (GPCR)2 known as the growth hormone secretagogue receptor (GHS-R) (4). Ghrelin is currently known to be the most potent endogenous inducer of the growth hormone (GH)/insulin-like growth factor-1 (IGF-1) axis (5) and of food intake in mammals (6). The post-translational acyl modification of ghrelin is believed to be critical for its ability to bind to GHS-R and to mediate its biological activities (1, 7). Given that this octanoic group confers hydrophobicity to the N terminus of ghrelin, it may be that this group is responsible for specific targeting of ghrelin to lipid rafts (8) and also for its efficient transport across the blood-brain barrier (9). The deacylated (desacyl) form of ghrelin does not appear to mediate effects attributed to GHS-R; however, it has been reported recently that transgenic mice over expressing the desacyl form of ghrelin are small and have a blunted GH/IGF-1 axis, supporting a biologically relevant regulatory role for this isoform (10). The mRNAs for ghrelin and GHS-R are expressed in a wide variety of tissues and cell types (11). More recent studies have described a number of additional functions for ghrelin, including its ability to inhibit inflammatory cytokine expression (8, 12), to promote neurogenesis (13), to enhance memory retention (14), to induce adiposity (6, 15), and to exert prosurvival effects on cardiomyocytes (16). Repeated administration of ghrelin improves left ventricular function, exercise capacity, and muscle wasting in patients with chronic heart failure (17). In addition, a recent report has demonstrated an association of several single nucleotide polymorphisms and haplotypes within the GHS-R gene region with human obesity (18). The GHS-R gene is located within the quantitative trait locus on chromosome 3q26-q29, which is involved in the traits of metabolic syndrome and obesity (19). Given the association between obesity and the susceptibility to develop various cancers (20), it seems feasible that ghrelin/GHS-R interactions may play a pathophysiological role in both these complex conditions.
Among the metabolic hormones, the growth hormone-releasing hormone/GH/IGF axis has been most widely studied in the development and progression of various cancers, including tumors of the central nervous system (21-23). Most cancer cells express components of the IGF system, and it has been demonstrated that elevated IGF-1 levels are associated with brain tumor development (24, 25). Moreover, inhibition of IGF-1 (21, 26, 27) and growth hormone-releasing hormone (28) production blocks glioblastoma growth. Somatostatin, a negative regulator of the GH/IGF axis, has also been shown to exert potent anti-migratory and anti-invasive effects in neuroblastoma cells (29). A recent report has hypothesized that the ghrelin/GHS-R axis may operate through a similar autocrine/paracrine role in cancer biology (30). Ghrelin and GHS-R have been reported to be expressed in variety of cancers, including endocrine tumors (31), breast carcinomas (32), prostate cancer cells (30, 33), lung carcinomas (34), hepatomas (35), thyroid carcinomas (36), ovarian cancer (37), and gastrointestinal cancer (38). However, little is known about the expression of ghrelin or GHS-R in central nervous system cancers.
Astrocytomas are the most common central nervous system tumors, and the malignant forms include anaplastic astrocytoma (grade III) and glioblastoma (grade IV). These brain tumors are a major cause of cancer-related mortality, with a life expectancy of 1–3 years despite a multimodal therapy (39). These tumors are especially difficult to treat due to their high proliferation rate, increased angiogenesis, and invasion into surrounding brain tissues (40). Given that ghrelin has been identified recently as a positive regulator of the somatotropic axis, which has been shown to influence central nervous system tumor growth, we sought to determine whether this protein or its receptor plays any direct role in astrocytoma cell growth and function. Our findings provide the first evidence for the existence of ghrelin/GHS-R signaling pathways in astrocytomas and demonstrate that an endogenous hormonal loop may play a critical role in astrocytoma motility and invasiveness.
MATERIALS AND METHODS
Cells and Reagents
Four human glial cell lines (CCF-STTG1, U-87, U-118, and SW1088 cells) were obtained from the American Type Culture Collection and cultured as recommended by the supplier, and primary human astrocytes were purchased from Cambrex Corp. (Baltimore, MD). The octanoylated ghrelin-(1-18) fragment (Peptides International, Inc., Louisville, KY) was used to treat the cells in culture.
Intracellular Calcium Mobilization and Actin Polymerization
Measurement of intracellular calcium release in response to ghrelin was performed as described previously (8). Briefly, cells were incubated in phosphate-buffered saline containing Ca2+ and Mg2+ along with 5 μm Fura-2 acetoxymethyl ester (Molecular Probes, Eugene OR) for 30 min at room temperature. The cells were subsequently washed and then resuspended at 1 × 106/ml in phosphate-buffered saline. A total of 2 ml of the cell suspension was placed in a continuously stirred cuvette at room temperature in a PerkinElmer Life Sciences LS50B spectrophotometer. Fluorescence was monitored at λex1 = 340 nm, λex2 = 380 nm, and λem = 510 nm. The data are presented as the relative ratio of fluorescence excited at 340 and 380 nm.
For the actin polymerization studies, astrocytoma cells were incubated with ghrelin (100 ng/ml) for 20 min, after which the cells were fixed and permeabilized in 2% paraformaldehyde solution containing 0.1% Triton X-100. The fixed and permeabilized cells were subsequently labeled for F-actin using Alexa Fluor 594-labeled phalloidin, and the nucleus was counterstained with 4′,6-diamidino-2-phenylindole (Molecular Probes).
Immunofluorescence Staining
Immunofluorescence staining and microscopy were performed as described previously (8). Briefly, cells were fixed and permeabilized using 2% paraformaldehyde plus 0.1% Triton X-100, and nonspecific binding sites were blocked using an Fc block (Pharmingen) and 5% bovine serum albumin for 30 min. Cells were incubated overnight at 4 °C with different combinations of human anti-GHS-R goat IgG (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), anti-ghrelin rabbit IgG and anti-preproghrelin rabbit IgG (Phoenix Pharmaceuticals, Inc., Belmont, CA), anti-Rac mouse IgG (Sigma), and anti-golgin-97 mouse IgG (Molecular Probes). Cells were examined by high resolution microscopy, and images were acquired using SPOT Advanced software (Diagnostic Instruments, Sterling Heights, MI) on a Zeiss Axiovert S100 microscope under an ×100 objective lens.
Small Interfering RNA Knockdown Experiments
For all transfection studies, each of the astrocytoma lines was seeded in 6-well plates until 80% confluent. Control scrambled and GHS-R1a small interfering RNAs (siRNAs) were designed, synthesized, and labeled with rhodamine (Qiagen Inc., Valencia, CA). In addition, two separate ghrelin siRNA sequences (Sequence 1 from Qiagen Inc. and Sequence 2 from Dharmacon, Inc. (Lafayette, CO)) were also utilized to specifically knockdown ghrelin expression by these cell lines. For each of the siRNA constructs, the astrocytoma cells were transfected using Lipofectamine-based RNAifect reagent (Qiagen Inc.), after which these cells were utilized for the functional studies 24–48 h later.
Generation of Cell Lines Stably Expressing GHS-R1a
The full-length human GHS-R 1a cDNA (GenBank™ accession number AY429112) cloned into the pcDNA3.1+ vector was purchased from University of Missouri cDNA Resource Center (Rolla, MO). The open reading frame was amplified by PCR from human brain cDNA (Clontech), and the insert was sequenced-verified at 1101 bp. U-87 cells were cultured to 80% confluency and transfected with 5 μg of pcDNA3.1 expression plasmid (Invitrogen) containing cDNA for GHS-R1a or with empty vector alone using LyoVec reagent (InvivoGen, San Diego, CA) following the manufacturer’s instructions. Selection was carried out by adding Geneticin (Invitrogen) at a final concentration of 500 μg/ml over a period of 30 days, and colonies were picked, expanded, and screened for expression by PCR.
Real-time Reverse Transcription-PCR Analysis
Total RNA was isolated from cells using QIAshredder and RNeasy kits (Qiagen Inc.) and from different grades of tumor tissues using standard procedures (8). The isolated RNA (2 μg) and oligo(dT) primers were utilized to synthesize single-stranded cDNA using a reverse transcription kit (Invitrogen) according to the manufacturer’s instructions. The PCR was set up using SYBR Green Master Mix (Applied Biosystems, Foster City, CA) along with 1 μl of cDNA and the gene-specific primers at a final concentration of 0.3 μm. Thermal cycling was carried out on an Applied Biosystems GeneAmp 7700 sequence detector, and SYBR green dye intensity was analyzed using GeneAmp 7700 SDS software. The human GHS-R1a and ghrelin primers utilized in these studies were described previously (11). No PCR products were generated from genomic versus cDNA template.
Ghrelin Assay
Immunoreactive ghrelin was measured in duplicate by an enzyme-linked immunosorbent assay using a rabbit polyclonal antibody against the full-length ghrelin peptide (Phoenix Pharmaceuticals, Inc.) according to the manufacturer’s instructions.
Motility and Invasion Assays
Scratch and Transwell invasion assays were performed as described previously (41). For the scratch assays, astrocytoma cells were placed in 12-well plates coated with fibronectin (Pharmingen). After the cells were allowed to attach and reach confluency, a scratch was made through the fibronectin, and photographs of cells invading the scratch were taken at the indicated time points. Alternatively, cells were also fixed and stained with hematoxylin and Alexa Fluor 488-labeled phalloidin. For the Transwell invasion assays, cells were labeled with 5 mm calcein acetoxymethyl ester at 37 °C for 1 h and then placed on Matrigel-coated Transwell filters as described (41). A total of 800 μl of media containing various concentrations of ghrelin was placed in the lower chambers to generate a chemoattractant gradient. Each experiment was repeated a minimum of three times, and readings were performed on a Cytofluor 4000 fluorescent plate reader at excitation and emission wavelengths of 480 and 530 nm, respectively. Data are represented as the means ± S.E., and analysis of variance with Tukey’s test was used to determine the statistical significance (p < 0.05).
Gelatin Zymography
Cells were cultured in the presence or absence of ghrelin for 24–48 h, after which the conditioned medium was collected, diluted in sample buffer (50 mmol/liter Tris (pH 6.8), 0.5% SDS, 10% glycerol, and 0.2% bromphenol blue), and separated by electrophoresis on a 7.5% polyacrylamide gel containing 1 mg/ml gelatin as a substrate. SDS was removed by washing the gel with 2.5% Triton X-100 for 1 h at room temperature, followed by overnight incubation in reaction buffer (50 mm Tris (pH 7.6), 150 mm NaCl, 2.5 mm CaCl2, and 0.02% sodium azide). Gel staining was performed for 1 h at room temperature with 0.25% Coomassie Blue R-250, 50% methanol, and 7.5% acetic acid, after which the gel was destained with 10% acetic acid until clear bands representing proteinase activity appeared over a dark background.
Western Blot Analysis
Control and treated cells were lysed in radioimmune precipitation assay buffer supplemented with protease and phosphatase inhibitor mixture (Sigma), and protein concentrations of cell lysates were determined. These lysates (30 μg) were diluted with sample buffer, separated on 4–20% Tris-HCl/SDS-polyacrylamide gels (Bio-Rad), and electrophoretically transferred to nitrocellulose membranes (Schleicher & Schuüll). The blots were then incubated with rabbit antibody raised against phosphorylated protein kinase C (PKC) or total PKC (Cell Signaling Technology, Beverly, MA) or with an actin-specific mouse monoclonal antibody (Sigma). Immune complexes were visualized by incubation with horseradish peroxidase-conjugated anti-rabbit or anti-mouse secondary antibody (Amersham Biosciences). Immunoreactive bands were visualized by enhanced chemiluminescence (PerkinElmer Life Sciences).
Tissue Microarrays and Immunohistochemistry
The tissue microarrays utilized in these studies contained cores from 20 glioblastomas, 20 anaplastic astrocytomas, 20 infiltrating astrocytomas, and 20 oligodendroglial lesions (42). Microscopic examination of these arrays by a certified neuropathologist confirmed that the appearance of the tumor tissue cores corresponded to that in the donor blocks. To assess the expression of ghrelin and GHS-R on these tissue arrays, paraffin-embedded tumors were sectioned and deparaffinized using xylene, 100% EtOH, 95% EtOH, 75% EtOH, and H2O series, and antigens were retrieved by steaming samples in Dako target retrieval buffer for 20 min as described previously (41). After blocking the nonspecific binding sites, sections were incubated overnight at 4 °C with the appropriate antibodies. The sections were stained with 3,3′-diaminobenzidine substrate and counterstained with Meyer’s hematoxylin. Negative controls (obtained by occulting the primary antibody or by using an unrelated IgG) demonstrated no specific labeling. An investigator was blinded to the tissue array, which was then graded for immunopositivity of GHS-R and ghrelin based on the strength of the specific cytoplasmic labeling on a scale of 0 (no staining), 1+ (weak staining), 2+ (moderate staining), and 3+ (strong staining). The significance of the differences in GHS-R and ghrelin expression was examined by performing a Fisher’s exact test analysis on a four-way contingency table containing low grade versus high grade tumors and weak/no versus moderate/strong staining.
RESULTS
Functional Ghrelin Receptor Is Overexpressed in Human Astrocytoma Cells
The presence of GHS-R mRNA has been detected in many forms of cancer (30); however, GHS-R gene and protein expression in astrocytoma cells is currently unknown. Using immunofluorescence labeling, we observed that the GHS-R protein is widely expressed in the CCF-STTG1 cell line (Fig. 1A) and in the astrocytoma cell lines U-87, U-118, and SW1088 (Fig. 1B). It is interesting that normal human astrocytes (NHAs) demonstrated only very weak immunopositivity for GHS-R (Fig. 1B) compared with the astrocytoma cell lines. Moreover, ghrelin treatment resulted in the redistribution of GHS-R to the membrane ruffles on the leading edges of these cells (Fig. 1A, lower left panel), suggesting a role for GHS-R in interactions with adjacent cells or in cell migration. Quantitative real-time PCR analysis also revealed overexpression of GHS-R1a mRNA in all astrocytoma cell lines tested, with the highest expression (30-fold) in SW1088 cells compared with NHAs (Fig. 1C).
FIGURE 1. GHS-R expression in human astrocytoma cells.
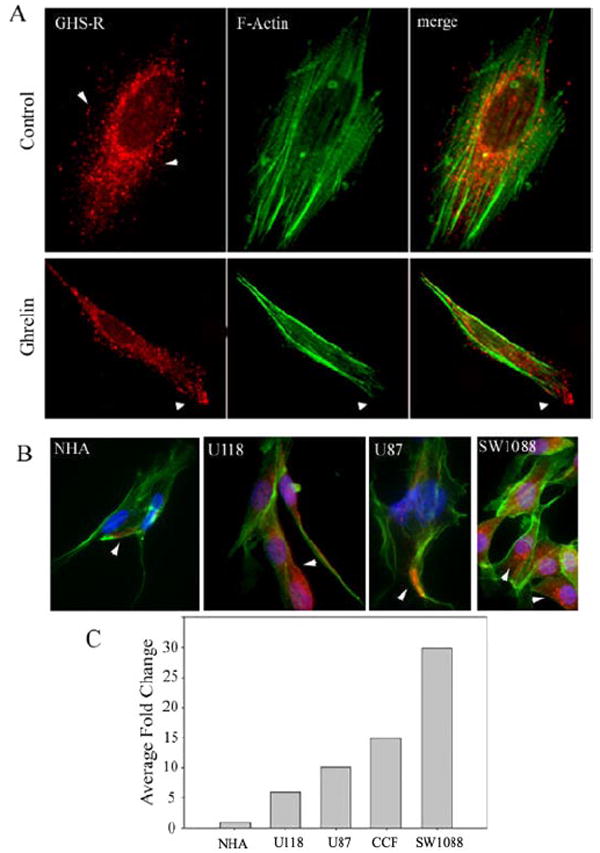
A, CCF-STTG1 (CCFS) cells were labeled for GHS-R (red), and F-actin was visualized using Alexa Fluor 488-labeled phalloidin (green). Control cells (upper panels) displayed a diffuse distribution of GHS-R. Upon ghrelin treatment (100 ng/ml), GHS-R was redistributed to the leading edge of the cell (lower panels). B, GHS-R protein (red) was widely expressed in U-118, U-87, and CCF-STTG1 cells compared with NHAs. C, shown is the -fold change in GHS-R 1a mRNA expression after normalization with glyceraldehyde-3-phosphate dehydrogenase as measured by real-time reverse transcription-PCR.
We next investigated the functionality and responsiveness of GHS-R-positive astrocytoma cells to acylated ghrelin. Ligation of GPCRs is often accompanied by the dramatic remodeling of the actin cytoskeleton and cell-surface molecules and the formation of membrane ruffles (43). Moreover, ligand binding to many functional GPCRs also results in mobilization of calcium from the intracellular stores via the generation of inositol triphosphate (44). This intracellular calcium results in the activation of PKC, which plays a role in actin polymerization, cell motility, and glioma progression (45). Similar to other GPCRs, ghrelin treatment of astrocytoma cells induced a marked increase in actin polymerization along with membrane ruffling and development of structures resembling lamellipodia compared with the vehicle control (Fig. 2A). In addition, we also observed a significant increase in intracellular calcium release in the U-87 astrocytoma cell line upon ghrelin treatment (Fig. 2C). Because activation of PKC downstream of intracellular calcium release is associated with cytoskeletal changes as well as with increases in motility and invasion in several cancers, including melanoma (41), colon carcinoma (46), breast carcinoma (47), and glioma (45), we next examined whether ghrelin activates PKC in astrocytoma cells. We observed a time-dependent increase in PKC phosphorylation in U-87 cells (Fig. 2B) that was sustained for 60 min after ghrelin treatment.
FIGURE 2. GHS-R is a functional receptor on astrocytoma cells.
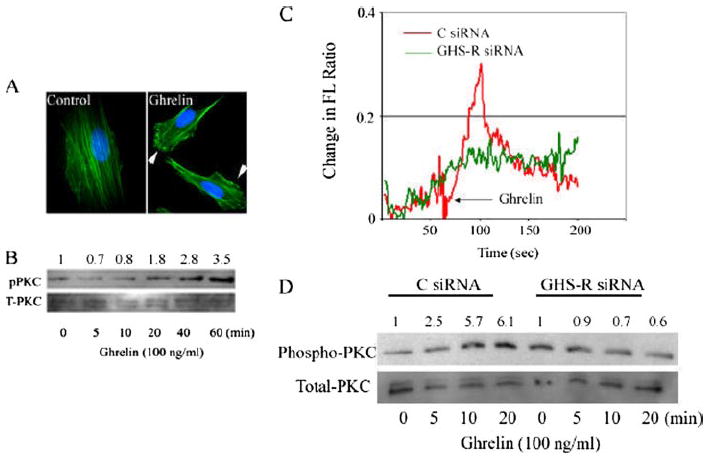
A, ghrelin induces membrane ruffling and actin polymerization in U-87 cells. Cells were fixed, permeabilized, and stained with Alexa Fluor 488-labeled phalloidin 20 min after ghrelin treatment (100 ng/ml). B, ghrelin treatment induces a time-dependent increase in PKC phosphorylation in U-87 cells. pPKC, phospho-PKC; T-PKC, total PKC. C, ghrelin elicits intracellular calcium release in control (C) scrambled siRNA-transfected U-87 cells, but not in cells subjected to GHS-R silencing by RNA interference. FL, fluorescence. D, ghrelin activates PKC via a GHS-R-dependent pathway. There was an increase in PKC phosphorylation 10 min after ghrelin treatment in cells transfected with control siRNA, but no induction in GHS-R siRNA-transfected U-87 cells.
Several recent studies have suggested that ghrelin may also bind and function through an unknown receptor distinct from GHS-R1a in cardiomyocytes (16), human erythroleukemic cells (48), and prostate cancer cells (33). In an effort to determine that the effects of ghrelin on astrocytoma cells are mediated through GHS-R, we down-regulated GHS-R1a in U-87 cells using specific siRNA constructs. Control (scrambled) and GHS-R siRNA sequences were labeled with rhodamine to track their entry and transfection efficiency (supplemental Fig. 1). In support of the studies described above, we also found that ghrelin treatment induced a rapid spike in intracellular calcium release in control scrambled siRNA-transfected cells, whereas GHS-R1a siRNA-transfected cells demonstrated a significantly diminished response upon ghrelin exposure (Fig. 2C). Similarly, we also found that ghrelin treatment resulted in a time-dependent increase in PKC activation in control siRNA-treated U-87 cells (Fig. 2D) and SW1088 cells (data not shown). However, cells in which GHS-R expression was inhibited by RNA interference failed to induce a time-dependent increase in PKC activity (Fig. 2D). Together, these results demonstrate that ghrelin is capable of stimulating astrocytoma cell lines via functional GHS-R1a.
GHS-R Co-localizes with Rac in Membrane Ruffles on the Leading Edge of Astrocytoma Cells, and Ghrelin Treatment Promotes Astrocytoma Cell Motility and Invasion
Given that ghrelin increases PKC activity, we next studied the spatial localization of GHS-R with Rac. Rac belongs to the Rho family of small GTPase proteins and plays a key role in the reorganization of the actin cytoskeletal network and the formation of lamellipodia and thereby regulates cell migration and adhesion (48). Localization of Rac on the leading edge of the cell is believed to increase motility and also to impart directionality to cellular movement (49). Given the actin polymerization and PKC activation upon ghrelin treatment of astrocytoma cells lines, we examined the localization of GHS-R, Rac, and F-actin at various times after ghrelin treatment of CCF-STTG1 cells. Ghrelin treatment led to the redistribution of GHS-R on the polar ends of the cells after 15 min (Fig. 3A) and 30 min (data not shown) of ghrelin treatment. In addition, GHS-R was found to be tightly co-localized with F-actin and Rac on the leading edge of astrocytoma cells in structures resembling lamellipodia. Such a pattern of receptor redistribution and location suggests a potential role for GHS-R in astrocytoma cell motility.
FIGURE 3. Ghrelin treatment causes aggregation of GHS-R and Rac in membrane ruffles and promotes astrocytoma cell motility and invasion.
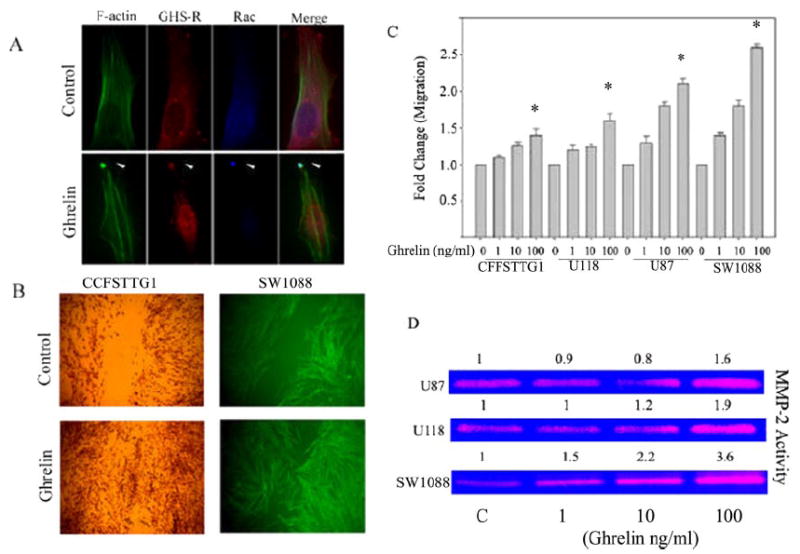
A, cells were triple-labeled for F-actin using Alexa Fluor 488-conjugated phalloidin (green), anti-goat GHS-R antibody, and anti-mouse Rac antibody, followed by specific secondary antibodies conjugated to Alexa Fluor 594 (red) and Alexa Fluor 355 (blue). Untreated cells (upper panels) displayed a broad distribution phenotype of GHS-R and Rac upon ghrelin treatment (15 min), and GHS-R was tightly co-localized with Rac on the leading edge of cells in membrane ruffles. B, ghrelin increased CCF-STTG1 and SW1088 cell motility in a scratch assay. Note the almost complete closure of the scratch in cells treated with ghrelin on a fibronectin-coated plate. C, Matrigel-coated Transwell chamber assay revealed a dose-dependent increase in cell invasion after ghrelin treatment. D, ghrelin increased gelatinolytic MMP2 activity in U-87, U-118, and SW1088 cells. Note that SW1088 cells expressing maximum GHS-R displayed the greatest increase in MMP activity after ghrelin exposure.
On the basis of above findings that astrocytoma cell lines express functional GHS-Rs and appear to induce changes in actin organization and PKC and Rac activation, we next examined the ability of ghrelin to induce astrocytoma cell motility and invasion. Astrocytoma cells (CCF-STTG1 and SW1088) were grown to confluency on fibronectin-coated plates, and then a scratch was inflicted with a sterile micropipette. These scratched monolayers were subsequently treated with ghrelin or vehicle control for various time periods. The results revealed a significant ghrelin-induced increase in the motility of other astrocytoma cell lines, including U-87 and U-118 (data not shown), as assessed by cells migrating into the scratch (Fig. 3B), whereas desacyl ghrelin failed to show any significant increase in cell motility (data not shown). This increased motility was also supported through the use of Matrigel-coated Transwell chamber invasion assays that mimic the three-step process of invasion, viz. adhesion, proteolysis of the extracellular matrix, and cell migration (50). As shown in Fig. 3C, ghrelin led to a dose-dependent increase (p < 0.05) in the migration and invasion of CCF-STTG1, U-118, U-87, and SW1088 cells. The SW1088 and U-87 cell lines demonstrated the greatest degree of migration in response to ghrelin, most likely due to increased GHS-R expression by these cells.
Matrix metalloproteinases (MMPs) are a family of zinc-dependent proteinases that degrade the extracellular matrix, promoting tumor invasion and metastasis (51). To determine whether ghrelin-induced invasion is associated with an increase in MMP activity, we performed gelatin and casein zymography. Ghrelin led to a dose-dependent increase in gelatinolytic activity corresponding to MMP2 in U-87, U-118, and SW1088 cells (Fig. 3D), with no change in MMP3, MMP9, MMP10, and MMP7 activity after zymography (data not shown). It is interesting that the SW1088 cells, which expressed the highest levels of GHS-R, displayed maximum invasion and increased MMP2 activity after ghrelin treatment, suggesting an essential role for the ghrelin/GHS-R pathway in the invasion of these cells.
Ghrelin Expression in Astrocytoma Cells Is Critical for Their Motility
Although the majority of ghrelin in the circulation is produced by the stomach, recent studies have demonstrated that ghrelin is also expressed and secreted from human immune cells (8) and in a variety of cancers (30). We therefore investigated the presence of ghrelin mRNA and protein in astrocytoma cells. As demonstrated by real-time reverse transcription-PCR analysis, ghrelin mRNA was constitutively expressed in all astrocytoma cell lines tested (Fig. 4A). The SW1088 cells expressed ~8-fold higher levels of ghrelin compared with NHAs (similar to the levels observed in human T cells and monocytes), whereas the U-118 and CCF-STTG1 lines demonstrated no significant differences in ghrelin mRNA expression compared with primary human astrocytes. Ghrelin was also secreted into the supernatants in substantial amounts (200 pg/ml) in these cells (Fig. 4B). Similar to the mRNA levels, SW1088 cells produced the highest levels of ghrelin protein. Treatment of CCF-STTG1 cells with interleukin-1β, a pro-inflammatory cytokine known to be a potent activator of astrocytes, did not significantly affect ghrelin message or protein expression (supplemental Fig. 2C). We further studied the subcellular distribution and expression of ghrelin (supplemental Fig. 2A) as well as the 117-amino acid prepro form of ghrelin (supplemental Fig. 2B). Our data suggest that ghrelin and preproghrelin are expressed in astrocytoma cells, demonstrating a diffuse cytoplasmic labeling. Neither preproghrelin nor ghrelin demonstrated any exclusive localization in Golgi bodies. However, as with many other proteins involved in tumorigenesis, the processing and localization of this protein may be distinct from that in primary cells, as this is in contrast to human T lymphocytes, in which preproghrelin is specifically localized to the Golgi apparatus (8).
FIGURE 4. Ghrelin isexpressedinandsecretedby astrocytoma cells, and specific ghrelin knockdown reveals an autocrine role in cell motility.
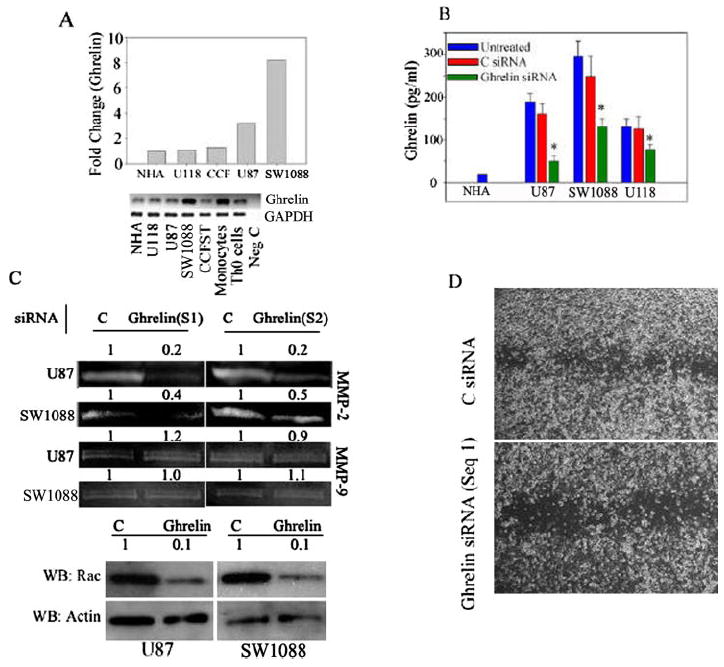
A, shown are the results from real-time PCR analysis of ghrelin mRNA expression in astrocytoma cell lines normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). All primers spanned the exon boundaries to prevent amplification of genomic DNA. Compared with NHAs, human U-87 and SW1088 astrocytoma cells expressed the highest levels of ghrelin mRNA. The PCR product was visualized on 2% agarose gel (lower panel); no product was generated from cDNA samples prepared by omitting the reverse transcriptase. CCT and CCFST, CCF-STTG1; Neg C, negative control. B, ghrelin secretion by astrocytoma cells is shown. SW1088 cells secreted the highest levels of ghrelin compared with NHAs, and ghrelin siRNA resulted in significant reduction of secreted ghrelin protein. C, control. C, ghrelin siRNA (Sequences 1 (S1) and 2 (S2)) inhibited MMP2 activity and Rac expression (lower panels) in U-87 and SW1088 cells without affecting MMP9 activity. WB, Western blot. D, specific ghrelin down-regulation by siRNA resulted in significant inhibition of U-87 cell motility in a scratch assay. Seq 1, Sequence 1.
Most cancer cells express the components of the somatotropic GH axis. Our data now suggest that ghrelin, a novel inducer of the GH/IGF axis, is also present in astrocytoma cells. However, the functional significance of hormone expression in cancer cells remains to be elucidated. To understand the functional role of ghrelin expression in astrocytoma cells, we down-regulated the ghrelin using two separate siRNA constructs and examined the effects of the loss of endogenous ghrelin on MMP activity, Rac activation, and cell motility. Compared with control scrambled siRNA (supplemental Fig. 1), ghrelin siRNA resulted in significant inhibition of ghrelin secretion into supernatants in all of the cell lines (Fig. 4B). Ghrelin knockdown by both siRNA sequences (Sequences 1 and 2) resulted specifically in marked inhibition of MMP2 activity in U-87 and SW1088 cells (Fig. 4C) without significantly affecting the MMP9 gelatinolytic activity (Fig. 4C). Moreover, down-regulation of endogenously produced ghrelin in both U-87 and SW1088 cells also led to strong inhibition of Rac expression (Fig. 4C, lower panels) and reduced motility and invasion (Fig. 4D) in a scratch assay. These data strongly support a role for tumor cell-derived ghrelin in astrocytoma motility and invasion.
GHS-R Knockdown and Stable Overexpression Reveal an Autocrine/Paracrine Role for Ghrelin in Astrocytoma Motility
A recent study indicates that, in certain cell types and tumors, ghrelin might mediate its effects independently of GHS-R (16). Therefore, we next investigated whether endogenously produced ghrelin mediates its effects on astrocytoma cell motility via a GHS-R-specific interaction. The transfection of U-87 cells with GHS-R siRNA led to significant reduction of GHS-R mRNA (Fig. 5A), and immunolabeling revealed reduced GHS-R protein levels (Fig. 5B). To examine the GHS-R-specific mechanism of ghrelin action on astrocytoma motility, U-87 cells transfected with control and GHS-R siRNAs were treated with ghrelin (100 ng/ml), and a scratch test was performed. As shown in Fig. 5C, ghrelin treatment led to an increase in motility and efficient closure of the scratch in the control siRNA-treated cells, but failed to demonstrate any effect on the GHS-R-down-regulated U-87 cells. It is interesting that the GHS-R siRNA-transfected cells without any ghrelin treatment also displayed reduced motility compared with control cells, suggesting the inability of endogenously produced ghrelin to promote cell migration. In addition, we observed a significant reduction of constitutive MMP2 activity in GHS-R siRNA-transfected U-87 and SW1088 cells, but not U-118 cells (Fig. 5D). To further confirm the role of GHS-R in cell motility, we stably overexpressed human GHS-R1a in U-87 cells. Real-time PCR analysis demonstrated a >200-fold increase in GHS-R expression (Fig. 5, E and F) in transfected cells compared with empty vector controls. The U-87 cells with stable overexpression of GHS-R demonstrated increases in cell motility and invasion in scratch assays compared with pcDNA-transfected U-87 cells (Fig. 5G). These experiments further suggest an autocrine and/or paracrine role for ghrelin/GHS-R interactions in astrocytoma cell motility and invasion.
FIGURE 5. Ghrelin/GHS-R interactions play an autocrine role in astrocytoma cell motility.
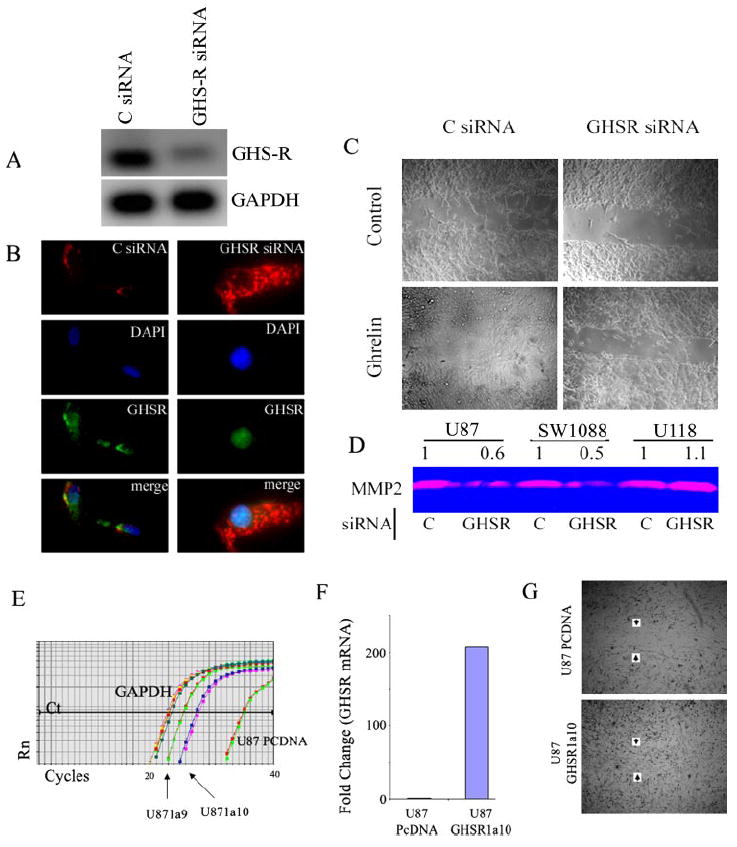
A and B, compared with control (C) siRNA, GHS-R siRNA caused a significant reduction of GHS-R mRNA and protein content in U-87 cells within 24 h. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; DAPI, 4′,6-diamidino-2-phenylindole. C, ghrelin treatment of GHS-R siRNA-transfected cells failed to increase motility compared with control cells. In addition, note that GHS-RsiRNA cells displayed much reduced motility as observed by reduced closure of the scratch 24 h post-transfection. D, disruption of the ghrelin/GHS-R axis caused a significant reduction of MMP2 activity in U-87 and SW1088 cells, but not in U-118 cells. E, shown is the real-time PCR analysis of the amplification plots of U-87 cells transfected with empty vector (pcDNA3.1 (PCDNA)) and GHS-R. Rn, reaction. F, shown is the -fold change in GHS-R expression in U-87 cells stably transfected with GHS-R1a clone 10 (GHSR1a10)). G, U-87 cells overexpressing GHS-R1a exhibited increased motility compared with cells transfected with empty vector alone in a scratch assay.
Increased Expression of GHS-R and Ghrelin in High Grade Astrocytomas
To determine whether the observed role of ghrelin and GHS-R is clinically relevant, we quantitated GHS-R and ghrelin mRNA expression by real-time PCR and protein expression by immunohistochemistry. To examine the localization and levels of GHS-R protein in the tumor samples, a tumor tissue array with cores from 20 oligodendrogliomas and 60 astrocytomas of grades II–IV was used. Cytoplasmic immunolabeling was graded on a scale of 0 to 3+ by an investigator blinded to tumor type and grade (Fig. 6, A and B). We found that the majority of brain tumors from all grades expressed GHS-R; however, strong GHS-R expression was found only in patients with grade III tumors and in one grade IV lesion (Fig. 6, A and C). We also examined these samples for ghrelin immunopositivity by immunohistochemistry. Immunohistochemical analysis of the tumor array revealed cytoplasmic ghrelin in the majority of tumors of all grades (Fig. 6B); however, grade II astrocytomas, which are known to display a reduced malignant potential, demonstrated only 2+ or less immunopositivity, whereas three patients with grade III tumors, five with glioblastoma multiforme tumors, and five with oligodendrogliomas displayed 3+ immunopositivity (Fig. 6D). Focusing more on the astrocytoma tissues, we observed a greater number of cases with intense staining for ghrelin and GHS-R in the high grade lesions (World Health Organization III and IV) as opposed to the low grade ones (World Health Organization II). The distinction between grade II (low grade) and grade III/IV (high grade) is clinically significant, as high grade tumors are associated with poor prognosis and survival. To determine whether GHS-R and ghrelin expression is significantly different in low and high grade invasive astrocytomas, we combined the data into two broad categories, weak staining (0 to 1+) and strong staining (2+ to 3+). We next performed a Fisher’s exact test analysis on a four-way contingency table containing low grade versus high grade tumors and weak/no versus strong staining (Fig. 6E). The data suggest that strong GHS-R and ghrelin expression is associated with high grade invasive astrocytomas. The real-time PCR analysis of tumor tissue samples demonstrated expression of GHS-R and ghrelin mRNAs in all stages of the cancers. We observed a significant 3-fold increase in GHS-R expression in grade III anaplastic astrocytomas (Fig. 6F), whereas ghrelin expression demonstrated no significant differences (p > 0.05). Overall, the increase protein expression of GHS-R and ghrelin was found to be significantly associated with high grade brain tumors, suggesting a possible role for this pathway in tumor invasion (Fig. 7).
FIGURE 6. GHS-R and ghrelin expression in different grades of human astrocytoma tumor tissue.
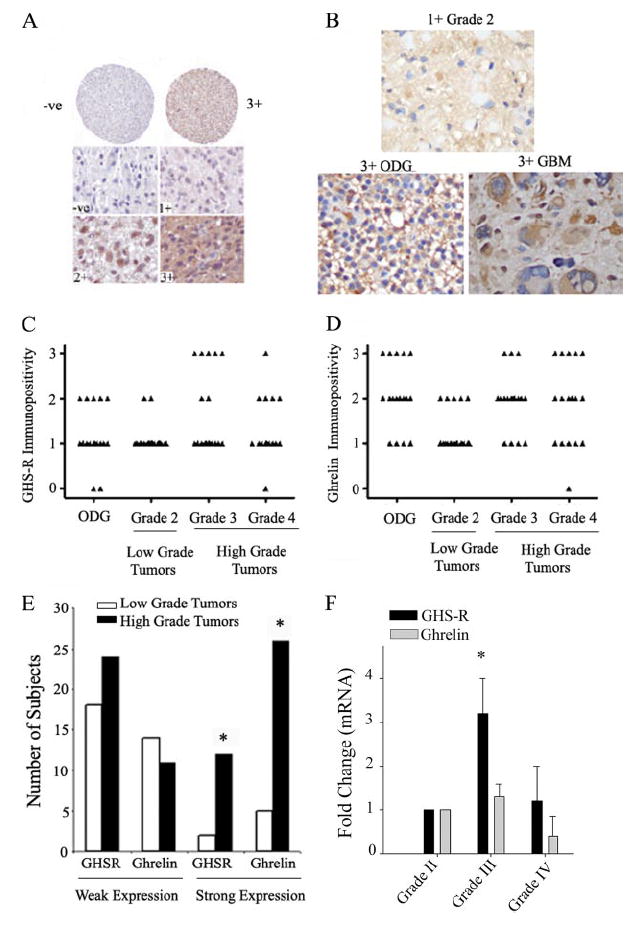
A, GHS-R immunopositivity was analyzed on a tissue array representing cores from 20 oligodendrogliomas and 60 astrocytomas of varying grades. The upper and lower panels demonstrate specific labeling on a scale from 0 to 3+. B, specific cytoplasmic ghrelin staining was observed in all tumors. Representative sections demonstrate a 1+ stained grade II astrocytoma and an oligodendroglioma (ODG) and a giant cell glioblastoma (glioblastoma multiforme (GBM)) displaying strong (3+) immunolabeling with many tumoral gemistocytes negative for ghrelin. C, shown is the quantitation of GHS-R immunopositivity. Each triangle represents staining from four cores representing a tumor from an individual patient. D, shown is the quantitation of ghrelin immunopositivity in the tumor array, revealing broad varying grades of ghrelin expression in tumors. The nonmalignant grade 2 astrocytoma failed to display any strong (3+) ghrelin immunolabeling. E, the GHS-R and ghrelin immunopositivity data from astrocytoma tumors were combined into two broad categories, weak staining (0 to 1+) and strong staining (2+ to 3+), and Fisher’s exact test analysis was performed on a four-way contingency table containing low grade (World Health Organization grade II) versus high grade (World Health Organization grades III and IV) tumors and weak/no versus strong staining for ghrelin and its receptor. The strong GHS-R and ghrelin expression was found to be significantly higher (p < 0.05) in cases with high grade invasive astrocytomas. F, real-time PCR analysis of human astrocytoma tumor samples demonstrated a significant increase in GHS-R expression in grade III tumors, whereas ghrelin mRNA expression did not differ between various tumor grades (n = 10 in each grade).
FIGURE 7. Hypothetical model for the ghrelin signaling pathway in astrocytoma cells.
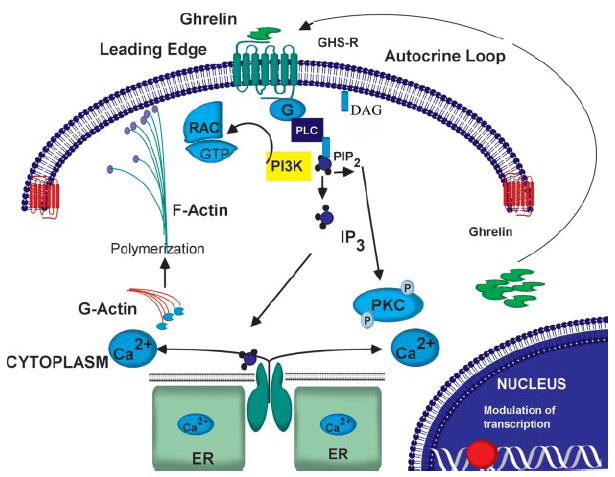
GHS-R translocation to the leading edge of cells and ligation with exogenous or endogenously produced ghrelin result in release of intracellular calcium and PKC activation, causing an increase in actin polymerization along with Rac activation, thereby increasing cell motility. DAG, desacyl ghrelin; PLC, phospholipase C; PI3K, phosphatidylinositol 3-kinase; PIP2, phosphatidylinositol bisphosphate; IP3, inositol trisphosphate; ER, endoplasmic reticulum.
DISCUSSION
In this study, we have described a novel and as of yet uncharacterized role for the orexigenic hormone ghrelin and its receptor (GHS-R) in astrocytoma growth and invasiveness. Astrocytomas are the most common primary malignant cancers affecting the central nervous system; and despite many advances in therapy, the median survival of patients diagnosed with a high grade astrocytoma is <1 year (52). It has been demonstrated previously that astrocytomas display alterations in their responsiveness to epidermal and platelet-derived growth factors (53), somatotropic hormones (28), and chemokines (54, 55) resulting in increased proliferation and invasiveness and reduced apoptosis. More recently, ghrelin has been hypothesized to play a role in the growth of tumors and neoplasms. Here, we have demonstrated that the ghrelin receptor GHS-R1a is overexpressed in all astrocytoma cells compared with NHAs. SW1088 astrocytoma cells exhibited a >25-fold increase in GHS-R1a expression, whereas the other cell lines demonstrated a 5–15-fold greater expression of this gene compared with NHAs. Similarly, GHS-R mRNA expression has also been shown to be 200- and 10-fold higher in GH- and thyroid-stimulating hormone-producing adenomas (56), respectively, compared with their normal cellular counterparts. However, no information has been published to date defining a functional role for tumor-associated GHS-R expression in cancer cells.
Similar to many GPCRs, GHS-R ligation elicits an increase in intracellular calcium mobilization, actin polymerization, and PKC activation. These signaling processes were found to be GHS-R1a-specific, as siRNA down-regulated GHS-R1a expression and inhibited ghrelin-induced signaling and function. The majority of the biological effects of ghrelin are currently believed to be mediated through the seven-transmembrane receptor GHS-R1a. This is supported by the observation that ghrelin treatment does not induce GH release or an increase in food intake in GHS-R-null mice (57). However, it has been suggested that ghrelin may also bind and signal through an unknown receptor distinct from GHS-R1a in prostate (33), breast (32), and thyroid (58) cancer cells and in human erythroleukemic cells (48). Although we cannot totally exclude the existence of another ghrelin receptor in astrocytoma cells, our present data clearly demonstrate that, in astrocytoma cell lines, ghrelin seems to signal specifically through GHS-R1a. It has been suggested previously that removal of the octanoyl group at the third serine residue in the ghrelin peptide by endogenous esterases results in a biologically inactive deacylated ghrelin isoform (10). However, desacyl ghrelin does not appear to be inert and has been suggested recently to act via corticotropin-releasing factor-2 receptors to inhibit stomach motility (59) and also to exert direct anti-apoptotic and anti-proliferative effects in cardiomyocytes and prostate cancer cells via an unknown receptor (16, 32). However, that desacyl ghrelin does not bind GHS-R and that ghrelin is unable to elicit any biological effects in GHS-R-null astrocytoma cells suggest that acylated ghrelin and not desacyl ghrelin promotes cell motility.
Many chemokines and other chemoattractants have been shown to signal through G protein-linked seven-transmembrane receptors, leading to membrane ruffling (which is critical to the development of the leading edge) and the retraction of the uropod in migrating cells (60, 61). Actin cytoskeleton reorganization and polymerization provide the force for cell motility, and Rac, a small GTPase of the Rho family, is a critical regulator of these events (49). Localization of Rac on the leading edge of a migrating cell in response to intra- and extracellular signals is believed to play an important role in cell motility and may also impart directionality to a migrating cell (62, 63). As with many chemoattractant receptors, GHS-R demonstrated a specific localization and polarization in the membrane ruffles of the astrocytoma cells upon ghrelin treatment. Moreover, ghrelin treatment also resulted in the co-localization of GHS-R with Rac on the cell edges, suggesting a role for GHS-R1a in cell migration and invasion. On the basis of the profound effects of ghrelin on the calcium/PKC pathway, actin polymerization, and cell polarity (Fig. 7), we investigated a specific role for ghrelin/GHS-R in cell motility and invasion. Our data demonstrate that ghrelin increases the motility and invasion of astrocytoma cells and that this is associated with a significant increase in MMP2 activity. These findings are supported by a recent report that ghrelin also promotes the invasion of pancreatic adenocarcinoma cells in a GHS-R-dependent manner (64).
Coexpression of ghrelin and its receptor has now been demonstrated in breast carcinomas (32), prostate cancer cells (30, 33), pancreatic adenocarcinomas (64), ovarian cancer cells (37), testicular cancer cells (65), thyroid carcinomas (36, 58), and gastrointestinal tumors (66). Human astrocytoma cells also seem to constitutively express ghrelin mRNA and protein; however, the mechanisms controlling the expression of ghrelin in these tumors require further investigation. Although stimulation of these cells with interleukin-1β causes significant up-regulation of monocyte chemoattractant protein-1 (MCP-1) and other growth factors and surface markers on human astrocytoma cells, ghrelin mRNA or protein expression is not regulated by interleukin-1β-mediated signals in CCF-STTG1 cells.
Ghrelin mRNA and protein were expressed in all of the astrocytoma cells tested, with U-87 and SW1088 cells expressing significantly higher levels compared with NHAs. The constitutive expression of ghrelin and GHS-R by tumor cells suggests their possible autocrine or paracrine regulatory role in these cells. Consistent with this hypothesis, we observed that astrocytoma cells subjected to ghrelin and GHS-R silencing by RNA interference displayed reduced MMP2 activity, Rac expression, and motility compared with control siRNA-transfected cells. The addition of exogenous ghrelin failed to increase motility in cells with down-regulated GHS-R expression, suggesting that ghrelin-induced increases in astrocytoma cell motility are mediated via functional GHS-R1a receptors. Although exogenous ghrelin increased the invasiveness of U-118 cells, there was no reduction in MMP2 activity upon down-regulation of the ghrelin/GHS-R pathway, suggesting the involvement of an alternate regulatory pathway in these cells. In further support of an endogenous role for ghrelin, we observed a significant increase in GHS-R mRNA expression in grade III anaplastic astrocytomas and moderate or strong expression of cytoplasmic ghrelin protein in the majority of oligodendrogliomas, grade III astrocytomas, and grade IV glioblastomas. It is interesting that elevated expression of both ghrelin and its receptor was significantly associated with increased astrocytoma grade, suggesting that the pathway may be involved in tumor progression. These data provide further support for endogenous GHS-R and ghrelin as possible important players in tumor cell growth and invasion (Fig. 7).
From a clinical perspective, ghrelin and GHS-R agonists have received considerable attention because of their positive effects on food intake and the GH/IGF axis (2), because of their inhibitory effects on inflammatory mediators (8, 12, 67), and as a possible clinical intervention in cancer cachexia (68). However, given that ghrelin increases astrocytoma cell motility and invasion, additional detailed experimentation may be required before extensive clinical trials are designed using ghrelin or synthetic ghrelin mimetics in cancer patients. Although high doses of ghrelin and GHS-R agonists may promote food intake or attenuate wasting associated with cancer or chronic inflammation, prolonged administration of these compounds may increase the risks of tumor progression. Overall, our study provides direct evidence that ghrelin, presently considered exclusively as a classical endocrine hormone, also exerts autocrine/paracrine effects in promoting cell motility and invasion in cancer and thus may play a critical role in the brain tumor microenvironment. Selective targeting or disruption of the ghrelin/GHS-R pathway might prove beneficial in reducing the growth and development of central nervous system tumors.
Supplementary Material
Supplemental Material can be found at: http://www.jbc.org/cgi/content/full/M600223200/DC1
Acknowledgments
We thank Drs. Patrice J. Morin, Carl Sasaki, Alessandro Ble, Mark P. Mattson, and Dan L. Longo (NIA, National Institutes of Health) for helpful discussions and review of this manuscript. We also thank Angie Feehley for excellent editorial assistance.
Footnotes
This work was supported in part by the NIA Intramural Research Program of the National Institutes of Health.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2.
The abbreviations used are: GPCR, G protein-coupled receptor; desacyl, deacylated; GHS-R, growth hormone secretagogue receptor; GH, growth hormone; IGF-1, insulin-like growth factor-1; siRNAs, small interfering RNAs; PKC, protein kinase C; NHAs, normal human astrocytes; MMPs, matrix metalloproteinases.
References
- 1.Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Nature. 1999;402:656–660. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- 2.Smith RG, Betancourt L, Sun Y. Endocr Rev. 2004;26:203–250. doi: 10.1210/er.2002-0017. [DOI] [PubMed] [Google Scholar]
- 3.Ukkola O, Ravussin E, Jacobson P, Snyder EE, Chagnon M, Sjostrom L, Bouchard C. J Clin Endocrinol Metab. 2001;86:3996–3999. doi: 10.1210/jcem.86.8.7914. [DOI] [PubMed] [Google Scholar]
- 4.Smith RG, Feighner S, Prendergast K, Guan X, Howard A. Trends Endocrinol Metab. 1999;10:128–135. doi: 10.1016/s1043-2760(98)00132-5. [DOI] [PubMed] [Google Scholar]
- 5.Inui A. Nat Rev Neurosci. 2001;2:551–560. doi: 10.1038/35086018. [DOI] [PubMed] [Google Scholar]
- 6.Tschöp M, Smiley DL, Heiman ML. Nature. 2000;407:908–913. doi: 10.1038/35038090. [DOI] [PubMed] [Google Scholar]
- 7.Hosoda H, Kojima M, Mizushima T, Shimizu S, Kangawa K. J Biol Chem. 2003;278:64–70. doi: 10.1074/jbc.M205366200. [DOI] [PubMed] [Google Scholar]
- 8.Dixit VD, Schaffer EM, Pyle RS, Collins GD, Sakthivel SK, Palaniappan R, Lillard JW, Jr, Taub DD. J Clin Investig. 2004;114:57–66. doi: 10.1172/JCI21134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banks WA, Tschop M, Robinson SM, Heiman ML. J Pharmacol Exp Ther. 2002;302:822–827. doi: 10.1124/jpet.102.034827. [DOI] [PubMed] [Google Scholar]
- 10.Ariyasu H, Takaya K, Iwakura H, Hosoda H, Akamizu T, Arai Y, Kangawa K, Nakao K. Endocrinology. 2004;146:355–364. doi: 10.1210/en.2004-0629. [DOI] [PubMed] [Google Scholar]
- 11.Gnanapavan S, Kola B, Bustin SA, Morris DG, McGee P, Fairclough P, Bhattacharya S, Carpenter R, Grossman AB, Korbonits M. J Clin Endocrinol Metab. 2002;87:2988–2991. doi: 10.1210/jcem.87.6.8739. [DOI] [PubMed] [Google Scholar]
- 12.Li WG, Gavrila D, Liu X, Wang L, Gunnlaugsson S, Stoll LL, McCormick ML, Sigmund CD, Tang C, Weintraub NL. Circulation. 2004;109:2221–2226. doi: 10.1161/01.CIR.0000127956.43874.F2. [DOI] [PubMed] [Google Scholar]
- 13.Zhang W, Lin TR, Hu Y, Fan Y, Zhao L, Stuenkel EL, Mulholland MW. J Physiol (Lond) 2004;559:729–737. doi: 10.1113/jphysiol.2004.064121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carlini VP, Monzon ME, Varas MM, Cragnolini AB, Schioth HB, Scimonelli TN, de Barioglio SR. Biochem Biophys Res Commun. 2002;299:739–743. doi: 10.1016/s0006-291x(02)02740-7. [DOI] [PubMed] [Google Scholar]
- 15.Kim K, Arai K, Sanno N, Osamura RY, Teramoto A, Shibasaki T. Clin Endocrinol. 2001;54:759–768. doi: 10.1046/j.1365-2265.2001.01286.x. [DOI] [PubMed] [Google Scholar]
- 16.Baldanzi G, Filigheddu N, Cutrupi S, Catapano F, Bonissoni S, Fubini A, Malan D, Baj G, Granata R, Broglio F, Papotti M, Surico N, Bussolino F, Isgaard J, Deghenghi R, Sinigaglia F, Prat M, Muccioli G, Ghigo E, Graziani A. J Cell Biol. 2002;159:1029–1037. doi: 10.1083/jcb.200207165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagaya N, Moriya J, Yasumura Y, Uematsu M, Ono F, Shimizu W, Ueno K, Kitakaze M, Miyatake K, Kangawa K. Circulation. 2004;110:3674–3679. doi: 10.1161/01.CIR.0000149746.62908.BB. [DOI] [PubMed] [Google Scholar]
- 18.Baessler A, Hasinoff MJ, Fischer M, Reinhard W, Sonnenberg GE, Olivier M, Erdmann J, Schunkert H, Doering A, Jacob HJ, Comuzzie AG, Kissebah AH, Kwitek AE. Diabetes. 2005;54:259–267. doi: 10.2337/diabetes.54.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kissebah AH, Sonnenberg GE, Myklebust J, Goldstein M, Broman K, James RG, Marks JA, Krakower GR, Jacob HJ, Weber J, Martin L, Blangero J, Comuzzie AG. Proc Natl Acad Sci U S A. 2000;97:14478–14483. doi: 10.1073/pnas.97.26.14478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calle EE, Kaaks R. Nat Rev Cancer. 2004;4:579–591. doi: 10.1038/nrc1408. [DOI] [PubMed] [Google Scholar]
- 21.Kari FW, Dunn SE, French JE, Barrett JC. J Nutr Health Aging. 1999;3:92–101. [PubMed] [Google Scholar]
- 22.Dupont J, Pierre A, Froment P, Moreau C. Horm Metab Res. 2003;35:740–750. doi: 10.1055/s-2004-814162. [DOI] [PubMed] [Google Scholar]
- 23.LeRoith D, Helman L. Cancer Cell. 2004;5:201–202. doi: 10.1016/s1535-6108(04)00054-6. [DOI] [PubMed] [Google Scholar]
- 24.Hirano H, Lopes MB, Laws ER, Jr, Asakura T, Goto M, Carpenter JE, Karns LR, VandenBerg SR. Neuro-Oncology. 1999;1:109–119. doi: 10.1093/neuonc/1.2.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu H, Rohan T. J Natl Cancer Inst. 2000;92:1472–1489. doi: 10.1093/jnci/92.18.1472. [DOI] [PubMed] [Google Scholar]
- 26.Rininsland F, Johnson TR, Chernicky CL, Schulze E, Burfeind P, Ilan J. Proc Natl Acad Sci U S A. 1997;94:5854–5859. doi: 10.1073/pnas.94.11.5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mukherjee P, Abate LE, Seyfried TN. Clin Cancer Res. 2004;10:5622–5629. doi: 10.1158/1078-0432.CCR-04-0308. [DOI] [PubMed] [Google Scholar]
- 28.Kiaris H, Schally AV. Proc Natl Acad Sci U S A. 1999;96:226–231. doi: 10.1073/pnas.96.1.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pola S, Cattaneo MG, Vicentini LM. J Biol Chem. 2003;278:40601–40606. doi: 10.1074/jbc.M306510200. [DOI] [PubMed] [Google Scholar]
- 30.Jeffery PL, Herington AC, Chopin LK. Cytokine Growth Factor Rev. 2002;14:113–122. doi: 10.1016/s1359-6101(02)00089-8. [DOI] [PubMed] [Google Scholar]
- 31.Barlier A, Zamora AJ, Grino M, Gunz G, Pellegrini-Bouiller I, Morange-Ramos I, Figarella-Branger D, Dufour H, Jaquet P, Enjalbert A. J Neuroendocrinol. 1999;11:491–502. doi: 10.1046/j.1365-2826.1999.00351.x. [DOI] [PubMed] [Google Scholar]
- 32.Cassoni P, Papotti M, Ghe C, Catapano F, Sapino A, Graziani A, Deghenghi R, Reissmann T, Ghigo E, Muccioli G. J Clin Endocrinol Metab. 2001;86:1738–1745. doi: 10.1210/jcem.86.4.7402. [DOI] [PubMed] [Google Scholar]
- 33.Cassoni P, Ghe C, Marrocco T, Tarabra E, Allia E, Catapano F, Deghenghi R, Ghigo E, Papotti M, Muccioli G. Eur J Endocrinol. 2004;150:173–184. doi: 10.1530/eje.0.1500173. [DOI] [PubMed] [Google Scholar]
- 34.Ghe C, Cassoni P, Catapano F, Marrocco T, Deghenghi R, Ghigo E, Muccioli G, Papotti M. Endocrinology. 2002;143:484–491. doi: 10.1210/endo.143.2.8654. [DOI] [PubMed] [Google Scholar]
- 35.Murata M, Okimura Y, Iida K, Matsumoto M, Sowa H, Kaji H, Kojima M, Kangawa K, Chihara K. J Biol Chem. 2002;277:5667–5674. doi: 10.1074/jbc.M103898200. [DOI] [PubMed] [Google Scholar]
- 36.Kanamoto N, Akamizu T, Hosoda H, Hataya Y, Ariyasu H, Takaya K, Hosoda K, Saijo M, Moriyama K, Shimatsu A, Kojima M, Kangawa K, Nakao K. J Clin Endocrinol Metab. 2001;86:4984–4990. doi: 10.1210/jcem.86.10.7891. [DOI] [PubMed] [Google Scholar]
- 37.Gaytan F, Morales C, Barreiro ML, Jeffery P, Chopin LK, Herington AC, Casanueva FF, Aguilar E, Dieguez C, Tena-Sempere M. J Clin Endocrinol Metab. 2004;90:1798–1804. doi: 10.1210/jc.2004-1532. [DOI] [PubMed] [Google Scholar]
- 38.Corbetta S, Peracchi M, Cappiello V, Lania A, Lauri E, Vago L, Beck-Peccoz P, Spada A. J Clin Endocrinol Metab. 2003;88:3117–3120. doi: 10.1210/jc.2002-021842. [DOI] [PubMed] [Google Scholar]
- 39.Maher EA, Furnari FB, Bachoo RM, Rowitch DH, Louis DN, Cavenee WK, DePinho RA. Genes Dev. 2001;15:1311–1333. doi: 10.1101/gad.891601. [DOI] [PubMed] [Google Scholar]
- 40.Burton EC, Prados MD. Curr Treat Options Oncol. 2000;1:459–468. doi: 10.1007/s11864-000-0073-2. [DOI] [PubMed] [Google Scholar]
- 41.Weeraratna AT, Jiang Y, Hostetter G, Rosenblatt K, Duray P, Bittner M, Trent JM. Cancer Cell. 2002;1:279–288. doi: 10.1016/s1535-6108(02)00045-4. [DOI] [PubMed] [Google Scholar]
- 42.Boon K, Edwards JB, Eberhart CG, Riggins GJ. BMC Cancer. 2004;4:39. doi: 10.1186/1471-2407-4-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pollard TD, Borisy GG. Cell. 2003;112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 44.Sanchez-Madrid F, del Pozo MA. EMBO J. 1999;18:501–511. doi: 10.1093/emboj/18.3.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.da Rocha AB, Mans DR, Regner A, Schwartsmann G. The Oncologist. 2002;7:17–33. doi: 10.1634/theoncologist.7-1-17. [DOI] [PubMed] [Google Scholar]
- 46.Zhang J, Anastasiadis PZ, Liu Y, Thompson EA, Fields AP. J Biol Chem. 2004;279:22118–22123. doi: 10.1074/jbc.M400774200. [DOI] [PubMed] [Google Scholar]
- 47.Sliva D, English D, Lyons D, Lloyd FP., Jr Biochem Biophys Res Commun. 2002;290:552–557. doi: 10.1006/bbrc.2001.6225. [DOI] [PubMed] [Google Scholar]
- 48.De Vriese C, Gregoire F, De Neef P, Robberecht P, Delporte C. Endocrinology. 2004;146:1514–1522. doi: 10.1210/en.2004-0964. [DOI] [PubMed] [Google Scholar]
- 49.Caron E. Nat Cell Biol. 2003;5:185–187. doi: 10.1038/ncb0303-185. [DOI] [PubMed] [Google Scholar]
- 50.Kraynov VS, Chamberlain C, Bokoch GM, Schwartz MA, Slabaugh S, Hahn KM. Science. 2000;290:333–337. doi: 10.1126/science.290.5490.333. [DOI] [PubMed] [Google Scholar]
- 51.Albini A. Pathol Oncol Res. 1998;4:230–241. doi: 10.1007/BF02905254. [DOI] [PubMed] [Google Scholar]
- 52.Westermarck J, Kahari VM. FASEB J. 1999;13:781–792. [PubMed] [Google Scholar]
- 53.Strickler R, Phillips ML. Clin J Oncol Nurs. 2000;4:153–158. [PubMed] [Google Scholar]
- 54.Ware ML, Berger MS, Binder DK. Histol Histopathol. 2003;18:207–216. doi: 10.14670/HH-18.207. [DOI] [PubMed] [Google Scholar]
- 55.Zhou Y, Larsen PH, Hao C, Yong VW. J Biol Chem. 2002;277:49481–49487. doi: 10.1074/jbc.M206222200. [DOI] [PubMed] [Google Scholar]
- 56.Rubin JB, Kung AL, Klein RS, Chan JA, Sun Y, Schmidt K, Kieran MW, Luster AD, Segal RA. Proc Natl Acad Sci U S A. 2003;100:13513–13518. doi: 10.1073/pnas.2235846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Skinner MM, Nass R, Lopes B, Laws ER, Thorner MO. J Clin Endocrinol Metab. 1998;83:4314–4320. doi: 10.1210/jcem.83.12.5307. [DOI] [PubMed] [Google Scholar]
- 58.Sun Y, Wang P, Zheng H, Smith RG. Proc Natl Acad Sci U S A. 2004;101:4679–4684. doi: 10.1073/pnas.0305930101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Volante M, Allia E, Fulcheri E, Cassoni P, Ghigo E, Muccioli G, Papotti M. Am J Pathol. 2003;162:645–654. doi: 10.1016/S0002-9440(10)63858-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen CY, Inui A, Asakawa A, Fujino K, Kato I, Chen CC, Ueno N, Fujimiya M. Gastroenterology. 2005;129:8–25. doi: 10.1053/j.gastro.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 61.Mellado M, Rodriguez-Frade JM, Manes S, Martinez AC. Annu Rev Immunol. 2001;19:397–421. doi: 10.1146/annurev.immunol.19.1.397. [DOI] [PubMed] [Google Scholar]
- 62.Shimonaka M, Katagiri K, Nakayama T, Fujita N, Tsuruo T, Yoshie O, Kinashi T. J Cell Biol. 2003;161:417–427. doi: 10.1083/jcb.200301133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jaffe AB, Hall A. Science. 2003;302:1690–1691. doi: 10.1126/science.1092874. [DOI] [PubMed] [Google Scholar]
- 64.Duxbury MS, Waseem T, Ito H, Robinson MK, Zinner MJ, Ashley SW, Whang EE. Biochem Biophys Res Commun. 2003;309:464–468. doi: 10.1016/j.bbrc.2003.08.024. [DOI] [PubMed] [Google Scholar]
- 65.Gaytan F, Barreiro ML, Caminos JE, Chopin LK, Herington AC, Morales C, Pinilla L, Paniagua R, Nistal M, Casanueva FF, Aguilar E, Dieguez C, Tena-Sempere M. J Clin Endocrinol Metab. 2004;89:400–409. doi: 10.1210/jc.2003-031375. [DOI] [PubMed] [Google Scholar]
- 66.Tsolakis AV, Portela-Gomes GM, Stridsberg M, Grimelius L, Sundin A, Eriksson BK, Oberg KE, Janson ET. J Clin Endocrinol Metab. 2004;89:3739–3744. doi: 10.1210/jc.2003-032118. [DOI] [PubMed] [Google Scholar]
- 67.Granado M, Priego T, Martin AI, Villanua MA, Lopez-Calderon A. Am J Physiol. 2004;288:E486–E492. doi: 10.1152/ajpendo.00196.2004. [DOI] [PubMed] [Google Scholar]
- 68.Garcia JM, Garcia-Touza M, Hijazi RA, Taffet G, Epner D, Mann D, Smith RG, Cunningham GR, Marcelli M. J Clin Endocrinol Metab. 2005;90:2920–2926. doi: 10.1210/jc.2004-1788. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material can be found at: http://www.jbc.org/cgi/content/full/M600223200/DC1