

Abstract
The DEAD-box protein CYT-19 functions in folding of several group I introns in vivo and a diverse set of group I and group II RNAs in vitro. Recent work using the Tetrahymena group I ribozyme demonstrated that CYT-19 possesses a second RNA binding site, distinct from the unwinding active site, which enhances unwinding activity by binding non-specifically to adjacent RNA structure. Here we probe the region of CYT-19 responsible for that binding by constructing a C-terminal truncation variant that lacks 49 amino acids and terminates at a domain boundary, as defined by limited proteolysis. This truncated protein unwinds a six-base-pair duplex, formed between the oligonucleotide substrate of the Tetrahymena ribozyme and an oligonucleotide corresponding to the internal guide sequence of the ribozyme, with near-wild-type efficiency. However, the truncated protein is activated much less than the wild-type protein when the duplex is covalently linked to the ribozyme or to single-stranded or double-stranded extensions. Thus, the active site for RNA unwinding remains functional in the truncated CYT-19, but the site that binds adjacent RNA structure has been compromised. Equilibrium binding experiments confirmed that the truncated protein binds RNA less tightly than the wild-type protein. RNA binding by the compromised site is important for chaperone activity, as the truncated protein is less active in facilitating folding of a group I intron that requires CYT-19 in vivo. The deleted region contains arginine-rich sequences, as found in other RNA-binding proteins, and may function by tethering CYT-19 to structured RNAs so that it can efficiently disrupt exposed, non-native structural elements, allowing them to re-fold. Many other DExD/H-box proteins also contain arginine-rich ancillary domains, and some of them may function similarly as non-specific RNA-binding elements that enhance general RNA chaperone activity.
Structured RNAs are required for a myriad of cellular processes, including mRNA processing and translation, tRNA processing, and maintenance of chromosome ends, and nearly all structured RNAs require at least one DExD/H-box protein for their functions (2, 3). DExD/H-box proteins are thought to function in large part by facilitating structural transitions of RNAs and ribonucleoprotein (RNP) complexes that would otherwise be too slow to allow the complexes to form or function. DExD/H-box proteins include a conserved motor domain, which uses energy derived from cycles of ATP binding and hydrolysis to facilitate structural rearrangements of RNAs, at least in part by ‘unwinding’ double-stranded segments (2, 4). Many DExD/H-box proteins also possess additional domains that are less conserved or even nonconserved. These domains are proposed, and in some cases shown, to function in targeting a given DExD/H-box protein to its RNA or RNP substrate by directly binding the substrate (3, 5–9). Thus, the presence of a domain that recognizes a specific substrate can convert the intrinsically generic motor domain to a protein that interacts specifically with a particular RNA or RNP.
On the other hand, recent evidence indicates that some DExD/H-box proteins are capable of facilitating folding of multiple RNAs, thereby acting as general RNA chaperones (1, 10–12). The idea that such chaperones exist was put forth some time ago (13, 14), based on the demonstrated propensity of RNA to misfold into kinetically trapped intermediates and the large activation energies necessary for disruption of RNA structure. However, the first demonstration that a protein actually functions as an RNA chaperone was recent, with the finding that the Neurospora crassa CYT-19 protein is necessary for proper folding of several mitochondrial group I introns (1). CYT-19 can also function in folding of yeast group II introns in vitro and both group I and group II introns in vivo when expressed in yeast (10, 12). Still, little is known about the mechanisms by which such general RNA chaperones act or whether they are targeted to features of certain classes of structured RNA.
To probe the mechanism of action of CYT-19, we developed a system using the group I RNA from Tetrahymena thermophila, which folds in vitro to a long-lived misfolded intermediate (11). We found that CYT-19 accelerates re-folding of this misfolded intermediate, and it performs another reaction much more efficiently: it unwinds the P1 duplex, formed between the ribozyme and its substrate. Further, it performs this reaction ~50-fold more efficiently than it unwinds the same duplex free in solution, indicating that it forms additional interactions with the ribozyme via a distinct binding site on the protein. Two observations suggested that the additional RNA binding site was likely to reside in the C-terminal domain, which consists of ~150 amino acids C-terminal to the motor domain. First, such a binding site might be postulated to reside within a domain outside the motor domain by analogy with the ancillary domains of other DExD/H-box proteins, and second, the C-terminal region of CYT-19 is strikingly basic and contains sequences that resemble arginine-rich motifs found in other RNA-binding proteins (15).
Here we test this idea by constructing a CYT-19 variant with a deletion of the C-terminal 49 amino acids. Strikingly, this variant protein retains near-wild-type efficiency in basal unwinding activity but is severely compromised in the enhancement of unwinding activity afforded by additional structured RNA. Thus, the C-terminal domain most likely harbors an RNA-binding-site that serves to direct CYT-19 to unwind helices that are associated with additional RNA structure. Many other DExD/H-box proteins have basic ancillary domains, some of which may function analogously.
MATERIALS AND METHODS
Materials
RNA and DNA oligonucleotides were purchased from Dharmacon (Lafayette, CO) and IDT (Coralville, IA), respectively. The Tetrahymena ribozyme was transcribed from ScaI-digested plasmid using T7 RNA polymerase (16) and purified using a Qiagen RNeasy column (17). The ribozyme was 5′-end-labeled by incubating with shrimp alkaline phosphatase (Fermentas, Hanover MD) and then with [γ-32P]ATP and T4 polynucleotide kinase (New England Biolabs, Ipswich, MA) and purified by non-denaturing polyacrylamide gel electrophoresis (PAGE) as described (16). Oligonucleotides were 5′-end-labeled with T4 polynucleotide kinase and purified by non-denaturing PAGE (16).
Papain digestion of CYT-19 and analysis of the principal proteolytic fragment
Papain (Sigma-Aldrich) was activated for 15 min at 37 °C in the presence of 5.5 mM cysteine-HCl, pH 6.5, 1 mM EDTA, and 0.07 mM DTT. Activated enzyme was then added to a 10-fold excess of CYT-19 and incubated at 30°C in the presence of 10 mM Tris-HCl, 2.8 mM cysteine-HCl, pH 8.5, 250 mM KCl, 1 mM EDTA, 0.1 mM DTT, and 25% glycerol. Aliquots were quenched at various times by adding an equal volume of Protease Inhibitor Cocktail Set III (Calbiochem of EMD Biosciences) and analyzed by 10% SDS-PAGE.
To determine the site of initial cleavage by papain, we analyzed the large fragment resulting from initial cleavage using mass spectrometry and N-terminal sequencing. A digestion of CYT-19 was performed for 6 min, which gave maximal accumulation of this fragment, and protein was isolated using a ZipTipC4 sample preparation kit (Millipore, Billerica, MA). 20 pmol of sample was used for Electrospray Mass Spectrometry analysis (performed in the ICMB Core Facility at UT Austin). Two principal peaks were detected, corresponding to molecules of 57,208 and 57,562 Da. From the average of these values, we obtained an estimate of 57.4 ± 0.3 kDa for the fragment. For N-terminal sequencing (performed in the ICMB Core Facility), the fragment of CYT-19 was purified by SDS-PAGE (NuPAGE Gels, Invitrogen, Carlsbad, CA) and transferred to a nitrocellulose membrane. The protein-bound sections of the membrane were excised and loaded into the reaction cartridge of the sequencer. Automated Edman-degradation-based N-terminal protein sequencing was performed by the ICMB Core Facility using a Procise 492 capillary liquid chromatograph attached to a Model 140C Micro-gradient System and a 610A Data Analysis System (Applied Biosystems, Foster City, CA). Sequencing procedures were optimized in-house according to the manufacturer’s manual.
Cloning of CYT-19 truncation (ΔC578–626)
A small range of possible cleavage sites was determined from the molecular weight of the fragment and the finding that the N-terminus was intact. To produce a recombinant protein equivalent to the fragment, Quikchange mutagenesis (Stratagene, La Jolla, CA) was performed using primers that included two consecutive stop codons immediately following residue 577. The sequences of the primers were: 5′-GTCGAAACCAGAGAGCATTCTTAGTAGCCCATGGGTAGCGGACCTGGC and its complementary sequence (the two stop codons are underlined). We have not determined whether truncation of CYT-19 at neighboring amino acids would also give functional proteins.
Expression and purification of wild-type CYT-19 and Δ578–626 proteins
Wild-type and truncated CYT-19 proteins were expressed as fusion proteins with the maltose-binding protein (MBP) and subsequently separated from MBP by proteolysis. Gene fusions were constructed in the plasmid pMAL-c2t (18), which is a derivative of pMAL-c2x (New England Biolabs) and encodes an N-terminal maltose-binding-protein tag. Each fusion protein was expressed in E. coli strain BL21 by growing a 1 l culture in LB containing 0.2% (w/v) glucose and 100 ug/ml ampicillin to an OD600 of 0.6, then incubating at 20 °C for 20 hours in the presence of 0.3 mM IPTG. Cells were harvested by centrifugation, resuspended in 20 mM Tris-HCl pH 7.5, 500 mM KCl, 1 mM EDTA, 2 mM DTT, and lysed by 20 min incubation in the presence of 1 mg/ml lysozyme, and three subsequent sonication bursts of 15 s each (power setting six with a double-stepped microtip; Branson Sonifier S-450A; VWR Scientific). The lysate was cleared by centrifugation (20,000 × g, 30 min at 4 °C). Polyethyleneimine (PEI) was added to 0.1% (w/v) and precipitated material was removed by centrifugation (3,700 × g, 15 min at 4°C). The supernatant was loaded onto a 15-ml amylose column (high flow resin; New England Biolabs) and washed with 2 column volumes of buffer containing 20 mM Tris-HCl, pH 7.5, 500 mM KCl, 1 mM EDTA, and 2 mM DTT. The column was then washed with 5 column volumes of the same buffer containing 1.5 M KCl and rewashed with 5 column volumes of the same buffer containing 500 mM KCl. The fusion protein was eluted by adding buffer containing 20 mM Tris-HCl, pH 7.5, 500 mM KCl, 1 mM EDTA, 2 mM DTT, and 10 mM maltose. Peak fractions were pooled and incubated overnight at 4°C in the presence of 40 μg/ml TEV protease [purified as described in (19)] to cleave the N-terminal MBP tag. This cleavage generates CYT-19 protein that retains three non-wild-type amino acid residues at the N-terminus (GSM), followed by the natural residue S54. (The N-terminal 53 amino acids serve in mitochondrial targeting of the natural protein and were deleted from these constructs.) To remove the cleaved MBP tag, the preparation was either loaded on a gravity-flow hydroxyapatite column (Bio-Gel HTP Gel; Bio-Rad Laboratories, Hercules, CA), followed by a second application to an amylose column, as described in the pMAL instruction manual (Method II; New England Biolabs), or loaded on a Superdex 200 column (GE Healthcare) in 20 mM Tris-HCl, pH 7.5, 500 mM KCl and 1 mM EDTA. Both wild-type CYT-19 and Δ578–626 appear to be monomeric, as judged by their elution from the S200 column (M.D. and A.M.L., unpublished results). Fractions containing CYT-19 were pooled and dialyzed overnight at 4 °C against 50 volumes of storage buffer (20 mM Tris-HCl, pH 7.5, 500 mM KCl, 1 mM EDTA, 2 mM DTT, and 50% glycerol).
RNA unwinding assays
Gel mobility shift assays to monitor RNA unwinding by CYT-19 were performed essentially as described (11). Briefly, to follow substrate dissociation from the Tetrahymena ribozyme, the ribozyme was first folded to the long-lived misfolded conformation by incubating 25 nM ribozyme for 5 min at 25 °C in the presence of 50 mM Na-MOPS, pH 7.0, 10 mM Mg2+. A trace amount of 32P-labeled oligonucleotide substrate (S*, <1 nM) was then incubated with the ribozyme for 5 min to allow binding. Next, CYT-19 (10–50 nM) was added along with an excess of unlabeled S (2.5 μM) and 2 mM ATP-Mg2+, free Mg2+ was simultaneously diluted to the desired concentration, and dissociation was followed by loading aliquots directly onto a 20% nondenaturing polyacrylamide gel run at 5 °C. Experiments following dissociation of the substrate strand from model P1 duplex complexes were performed identically, except that the P1 duplex was formed by incubating 600 nM of the unlabeled strand with trace S* for 60 min at 5 °C in the presence of 50 mM Na-MOPS, pH 7.0 (pH value determined at 25 °C), and 5 mM Mg2+.
Equilibrium RNA binding
Equilibrium binding of the wild-type CYT-19 and Δ578–626 proteins to the Tetrahymena ribozyme was measured by nitrocellulose filter binding essentially as described (12). Briefly, a trace concentration of 32P-labeled ribozyme (<2 nM) was incubated with the protein for 15 min to allow equilibration between bound and free RNA species (25 °C, 50 mM Na-MOPS, pH 7.0, 10 mM MgCl2). A pulse-chase dissociation experiment established that 15 min was sufficient for dissociation of nearly all of the complex, indicating that this time is sufficient for equilibration, and a control reaction in which wild-type CYT-19 and RNA were incubated for an hour gave the same results within error (data not shown). Protein-bound RNA was separated from free RNA by applying the mixture to a filter holder fitted with a nitrocellulose membrane and a diethylaminoethanol (DEAE) membrane beneath the nitrocellulose membrane (20). Membranes were washed with 2 ml of buffer, and retained radiolabel was determined by quantitating with a phosphorimager. For each protein concentration, the fraction of RNA bound was calculated by dividing the amount of radiolabeled material retained on the nitrocellulose filter by the sum of the labeled material on the nitrocellulose and DEAE membranes.
Tetrahymena ribozyme re-folding
Re-folding of the misfolded Tetrahymena ribozyme was monitored by the substrate cleavage activity of the native ribozyme as described (11, 21). Briefly, misfolded ribozyme was generated by incubation at 25 °C for 10 min in the presence of 10 mM Mg2+. The Mg2+ concentration was then reduced to 5 mM by dilution, ATP-Mg2+ was added to 2 mM, and CYT-19 was added to initiate the rapid re-folding reaction. Aliquots were quenched for further re-folding at various times thereafter by adding 50 mM Mg2+. The fraction of native ribozyme was determined for each time point by adding a trace concentration of S* and measuring the fraction of the substrate that was rapidly cleaved (within 1 min). Previous experiments established that this fraction provides a good measure of the fraction of native ribozyme (21).
Group I intron splicing
The N. crassa mitochondrial LSU- ORF intron was transcribed from BanI-digested pBD5a plasmid (22) using a T3 Megascript kit (Ambion, Austin, TX) to produce RNA that was body-labeled with 32P. RNA was extracted with phenol:chloroform:isoamyl alcohol (25:24:1; PCI), centrifuged through two consecutive 1-ml Sephadex G-50 columns, and stored at 4 °C in water. Immediately before it was used in splicing reactions, RNA was placed in a thermal cycler, heated to 92 °C for 2 min and then rapidly cooled to 25 °C and further incubated for 5 min before adding splicing buffer. Splicing reactions were performed in the presence of 20 nM 32P-labeled precursor RNA, CYT-18 (100 nM dimer) and CYT-19 (100 nM) or Δ578–626 (100 or 500 nM) at 25°C in 100 μl of reaction medium containing 100 mM KCl, 5 mM MgCl2, 25 mM Tris-HCl, pH 7.5, 10% glycerol, 1 mM GTP and 1 mM ATP. Reactions were initiated by adding CYT-19 and then CYT-18 in rapid succession (~15 s), and 10-μl aliquots were quenched at various times thereafter by adding 20 μl of 75 mM EDTA. Quenched time points were extracted with PCI, and 15 μl of the aqueous phase was added to 10 μl of gel loading buffer II (Ambion). Splicing products were analyzed by 4% denaturing PAGE and quantitated with a phosphorimager. The fraction of precursor remaining was plotted as a function of time to give an observed rate constant that represents completion of the first step of splicing.
RESULTS
To explore whether CYT-19 includes one or more ancillary domains that can be removed by proteolysis, we treated the protein with papain, a protease that is relatively insensitive to primary sequence but is sensitive to higher-order structure (23). Upon exposure to papain, CYT-19 was rapidly cleaved to a fragment that migrated slightly more rapidly by SDS-PAGE (labeled ‘Fragment 1’ in Fig. 1A and 1B). Continued exposure to papain led to further degradation, first to another fragment that migrated slightly more rapidly than the initial fragment (‘Fragment 2’ in Fig. 1A) and then to multiple fragments that migrated much more rapidly. All of these subsequent cleavage events occurred much slower than the initial cleavage, indicating that the fragment resulting from initial cleavage is more stable to digestion by papain than the full-length protein.
Fig. 1.
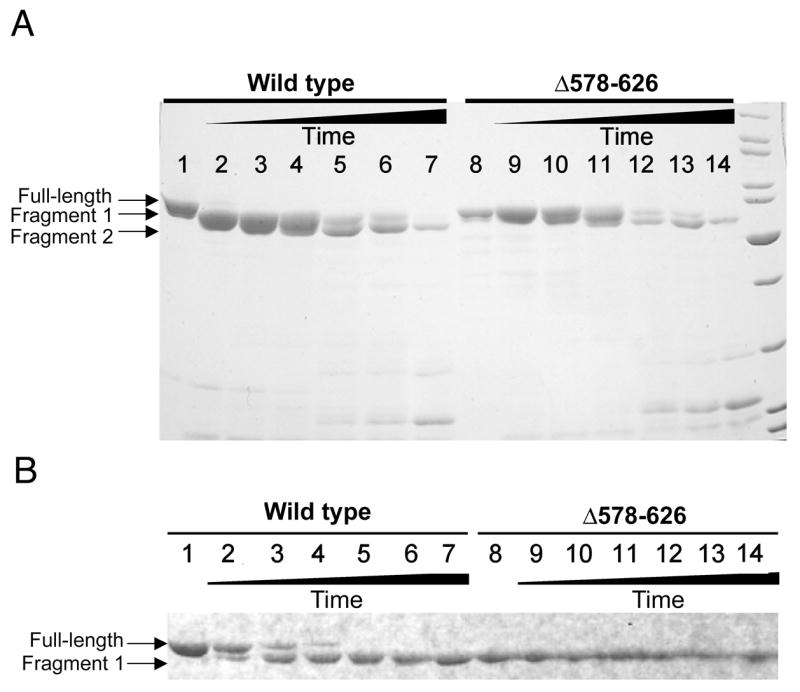
Papain digestion of CYT-19 and Δ578–626. A, Complete time course of digestion. Lane 1, undigested CYT-19; lanes 2–7, time course of CYT-19 digestion by papain. Lane 8, undigested Δ578–626; lanes 9–14, time course of Δ578–626 digestion by papain under the same conditions. Digestion times for each protein are: lanes 2 and 9, two min; lanes 3 and 10, six min; lanes 4 and 11, 15 min; lanes 5 and 12, 45 min; lanes 6 and 13, 60 min; lanes 7 and 14, 120 min. B, Early times of papain digestion. Lanes 1 and 8 show the undigested CYT-19 and Δ578–626 proteins, respectively. Other lanes show digestion times of: 30 s (lanes 2 and 9), 1 min 30 s (lanes 3 and 10), 2 min 30 s (lanes 4 and 11), 6 min (lanes 5 and 12), 8 min 30 s (lanes 6 and 13), and 20 min (lanes 7 and 14).
To determine the site of initial cleavage within CYT-19, we performed a limited digestion of 6 min, giving a nearly homogenous population of fragment 1 (see lane 5 in Fig. 1B). Analysis of the gel-purified fragment by Edman degradation indicated that the N-terminus was identical to the wild-type sequence (data not shown), indicating that the proteolytic cleavage was near the C-terminus. Further analysis of the proteolysis reaction by ESI-MS gave a molecular mass of 57.56 ± 0.35 kDa for the large fragment, compared with a calculated molecular mass of 63.9 kDa for the full-length protein. The mass of the fragment indicated that the cleavage was approximately at amino acid 574‡, and the uncertainty of 0.3 kDa gave a window of ± 3 amino acids.
As a first attempt to produce a recombinant protein equivalent to the fragment produced from papain cleavage, we expressed and purified a variant CYT-19 protein that lacked the C-terminal 49 amino acids of the wild-type protein, terminating at amino acid 577 after the hydrophilic sequence TREHS (the protein is denoted Δ578–626). The migration of this recombinant protein by SDS-PAGE was indistinguishable from the fragment produced by papain treatment, and the stability of the recombinant protein was similar to that of the fragment produced from papain cleavage (Fig. 1), indicating that the recombinant protein faithfully recapitulates the domain boundary inferred from the papain digestion and that the truncated protein folds properly.
C-terminal truncation of CYT-19 results in a loss of enhanced unwinding activity from adjacent RNA structure
We next examined the ability of the Δ578–626 protein to accelerate dissociation of the substrate of the Tetrahymena ribozyme, a process that requires unwinding of the P1 duplex. Although acceleration of substrate dissociation was readily detected, the Δ578–626 protein was much less active than the wild-type CYT-19 protein, with a kcat/KM value of 3.6 × 106 M−1 min−1 (Fig. 2), >10-fold lower than the wild-type protein under the same conditions (25 °C, pH 7.0, 5 mM Mg2+; see Materials and Methods and Table I).
Fig. 2.
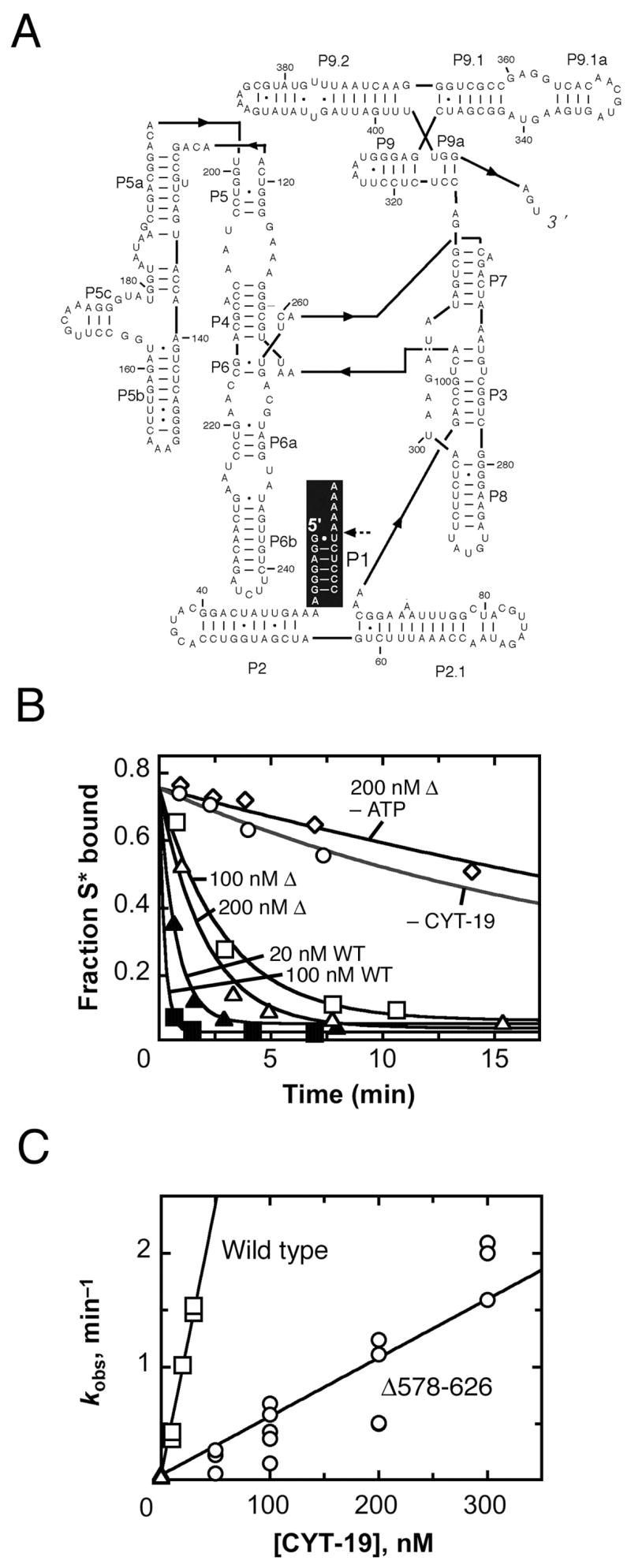
Acceleration of substrate dissociation from the Tetrahymena ribozyme. A, Secondary structure of the ribozyme. The P1 duplex, formed between the substrate and the ribozyme, is highlighted in black, with the substrate cleavage site indicated by a dashed arrow. B, Progress curves of substrate dissociation in the absence of CYT-19 (○, kobs = 0.033 min−1), in the presence of 100 nM (□, kobs = 0.32 min−1) or 200 nM (Δ, kobs = 0.49 min−1) Δ578–626 and 2 mM ATP-Mg2+, 200 nM Δ578–626 without ATP (◇, kobs = 0.028 min−1) or 20 nM (σ, kobs = 1.3 min−1) or 100 nM (ν, kobs = 4.7 min−1) wild-type CYT-19 and 2 mM ATP-Mg2+. C, Dependence of substrate dissociation rate on the concentration of Δ578–626 (○) and wild-type CYT-19 (□). Results from equivalent reactions without CYT-19 are also shown ( ). Four independent determinations gave a kcat/KM value of 3.6 (± 1.8) × 106 M−1 min−1 for the Δ578–626 protein, and three independent determinations gave a kcat/KM value of 4.1 (± 1.4) × 107 M−1 min−1 for the wild-type protein.
Table I.
P1 duplex unwinding of wild-type CYT-19 and truncated Δ578–626 proteins1
| CYT-19 | Δ578–626 | |||||
|---|---|---|---|---|---|---|
| Substrate extension2 | 2 mM Mg2+ | 5 mM Mg2+ | 10 mM Mg2+ | 2 mM Mg2+ | 5 mM Mg2+ | 10 mM Mg2+ |
| None | 2.2 × 106 | 6.9 ± 1.7 × 105 | 1.7 × 105 | 1.4 × 106 | 4.1 ± 1.8 × 105 | 2.0 × 105 |
| Ribozyme | 3.5 ± 1.7 × 107 | 4.1 ± 1.4 × 107 | 1.5 ± 0.6 × 107 | 4.3 ± 1.0 × 106 | 3.6 ± 1.8 × 106 | 1.3 ± 0.1 × 106 |
| A20 | N.D.3 | 1.0 × 107 | 3.0 × 106 | N.D. | 1.4 × 106 | 3.1 × 105 |
| P2 | 4.5 × 107 | 2.7 ± 0.6 × 107 | N.D. | 2.8 × 106 | 9.3 ± 1.0 × 105 | N.D. |
| P9.2 | N.D. | 6.0 × 106 | N.D. | N.D. | 9.9 × 105 | N.D. |
All values represent second order rate constants (M−1 min−1). Values with associated uncertainty represent the average and standard deviation of two to four independent determinations, and values without associated uncertainty represent a single determination. All reactions were performed at 25 °C, 50 mM Na-MOPS, pH 7.0, and the indicated concentration of Mg2+ (as MgCl2). Reactions of the wild-type protein in the presence of 10 mM Mg2+ have been reported previously (11). The measurements reported here were performed side-by-side with the Δ578–626 protein and gave results that were the same within error as those reported previously except for the A20 extension, which previously gave a value of 9.6 (± 2.0) × 105 M−1 min−1. We do not understand the origin of this 3-fold difference, but it does not affect the conclusions of the current work.
The substrate designation refers to what RNA structure, if any, was covalently connected to the P1 duplex. “None” represents unwinding of the isolated P1 duplex, formed between the substrate (CCCUCUA5) and an oligonucleotide that mimics the internal guide sequence of the ribozyme (GGAGGGA). This internal guide sequence strand was extended for the other constructs by addition of A20 or of sequences that can form the P2 or P9.2 helices (11).
N.D. – Not determined.
As the truncated C-terminal region is far removed from all of the conserved motifs of the unwindase motor domain, there was no expectation for a loss of basal unwinding activity by the truncated protein. To explore this question, we measured unwinding of the P1 duplex in the absence of the attached ribozyme structure. Previous experiments showed that the wild-type CYT-19 protein is 50–100-fold less efficient in unwinding the P1 duplex in the absence of the ribozyme, presumably because it is not able to take advantage of its second RNA binding site. In striking contrast, removal of the ribozyme made much less difference for the Δ578–626 protein, which gave an unwinding rate of 4.1 × 105 M−1 min−1, a decrease of <10-fold (Fig. 3). This value was within two-fold of that for the wild-type protein under the same conditions, indicating that the removal of the C-terminus did not greatly compromise the basal unwinding activity, but rather selectively decreased the activity for helix unwinding in the presence of the attached ribozyme. Analogous results were obtained at higher and lower Mg2+ concentrations (2 mM and 10 mM Mg2+; Table I), with the attached ribozyme structure giving a substantially larger enhancement of P1 duplex unwinding for the wild-type CYT-19 than for the Δ578–626 protein. The finding that the C-terminal truncation results in a decrease in the enhancement for unwinding activity without a corresponding loss of basal unwinding activity most simply suggests that the region of CYT-19 that has been deleted in the Δ578–626 protein interacts with the ribozyme to enhance the unwinding activity of the wild-type protein (see Discussion). Interestingly, the Δ578–626 protein remains significantly more active for P1 duplex unwinding with the attached ribozyme than for P1 free in solution, implying that the Δ578–626 protein retains some ability to interact with the ribozyme in a way that enhances unwinding activity. Thus, the region of CYT-19 that interacts with the ribozyme may extend beyond the deleted region into the ~100-amino-acid region that is retained in the Δ578–626 protein and is C-terminal to the unwinding motor domain. Alternatively, there could be additional interaction sites within the motor domain or within the N-terminal ~20-amino-acid sequence that flanks the motor domain.
Fig. 3.
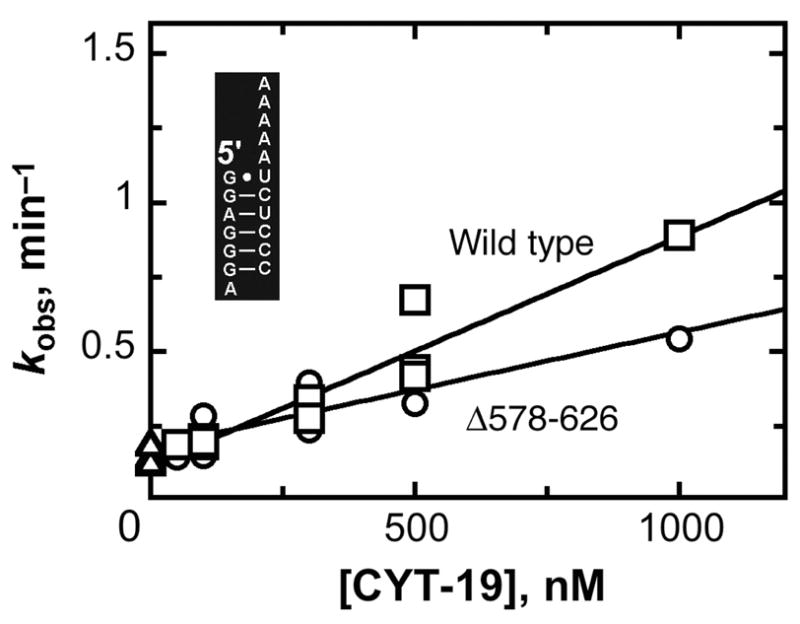
Unwinding of the isolated P1 duplex by Δ578–626 (○) and wild-type CYT-19 (□). Equivalent reactions in the absence of CYT-19 are also shown (Δ). Five independent determinations gave an average kcat/KM value of 4.1 (± 1.8) × 105 M−1 min−1 for the Δ578–626 protein and four independent determinations gave a value of 6.9 (± 1.7) × 105 M−1 min−1 for the wild-type protein (25 °C, pH 7.0, 5 mM Mg2+). The inset shows the sequence of the isolated P1 duplex.
To probe further the idea that the Δ578–626 protein is deficient in binding via a second site, we tested its P1-duplex-unwinding activity of a complex in which P1 is covalently connected to the P2 helix. This complex mimics the connection within the ribozyme and was shown previously to recapitulate most of the enhancement of unwinding activity for the wild-type CYT-19 (11). As expected, the unwinding activity of the Δ578–626 protein was much lower than that of the wild-type protein for this substrate (Fig. 4). Relative to the P1 duplex alone, the attachment of the P2 duplex gave an enhancement of 40-fold for the wild-type CYT-19, consistent with previous results (11), but only a 2-fold enhancement for the Δ578–626 protein. Similar results were obtained for unwinding of P1 with an unrelated duplex attached (‘P9.2’ in Table I), although with a somewhat smaller enhancement of activity for the wild-type protein, underscoring the relatively non-specific nature of the interaction.
Fig. 4.
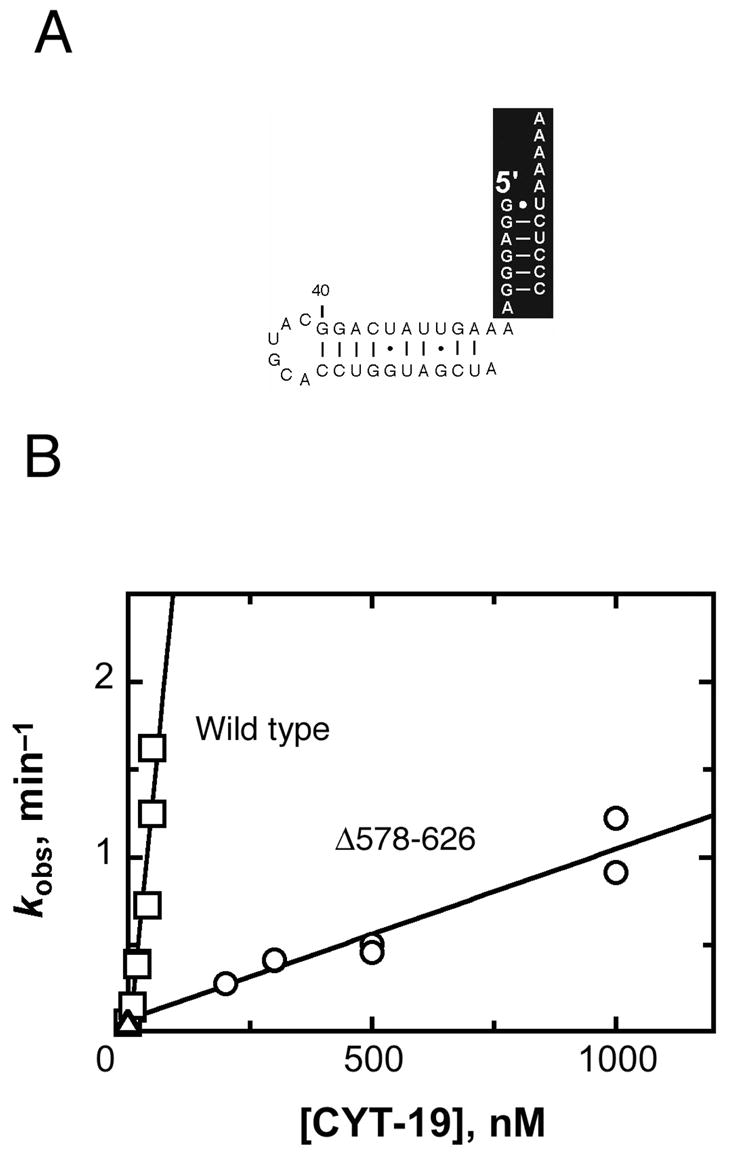
CYT-19-mediated unwinding of the construct containing P1 and P2. A, Secondary structure of the P1-P2 substrate. For consistency, the orientations of the P1 and P2 domains are the same as in Figs. 2 and 3. B, The dependence of the unwinding rate on the concentrations of Δ578–626 (○) and wild-type CYT-19 (□). Equivalent reactions in the absence of CYT-19 are also shown (Δ). Two independent determinations gave a kcat/KM value of 9.3 (± 1.0) × 105 M−1 min−1 for the Δ578–626 protein and three independent determinations gave a value of 2.7 (± 0.6) × 107 M−1 min−1 for the wild-type protein. All data from these determinations are shown.
We also tested unwinding by the Δ578–626 protein of the P1 duplex with a single-stranded extension (A20). The Δ578–626 protein was enhanced in unwinding activity only modestly from this addition, and was significantly less active than the wild-type protein (1.4 × 106 M−1 min−1 compared to 1.0 × 107 M−1 min−1 for the wild-type protein; Table I). It was previously shown that this addition gave enhanced unwinding activity for the wild-type protein (11), but it was unclear whether the enhancement arose from binding of the second site on CYT-19 to the single-stranded RNA extension, albeit with somewhat lower affinity than for a double-stranded extension, or from binding of the unwinding active site to the A20 extension followed by translocation into the double-stranded P1 duplex to give unwinding. The smaller enhancement for the Δ578–626 protein than for the wild type now suggests that the enhancement arises primarily from binding via the second site, an interpretation that is consistent with the recent finding the related protein Ded1p can unwind short duplexes without translocating (24).
Diminished RNA binding by Δ578–626
The simplest expectation from the diminished activity of the Δ578–626 protein was that it would bind less tightly than the wild-type CYT-19 to structured RNAs. We therefore measured equilibrium binding of each protein to the Tetrahymena ribozyme under the same conditions as the unwinding assays. Consistent with previous results (12), the wild-type CYT-19 bound the RNA in the nanomolar range, giving a Kd value of 30 nM (Fig. 5). The Δ578–626 gave a Kd value of 200 nM, 7-fold larger than that of the wild-type protein, indicating that the C-terminal truncation weakens RNA binding. Further, the magnitude of the decrease is similar to the loss of unwinding activity above.
Fig. 5.
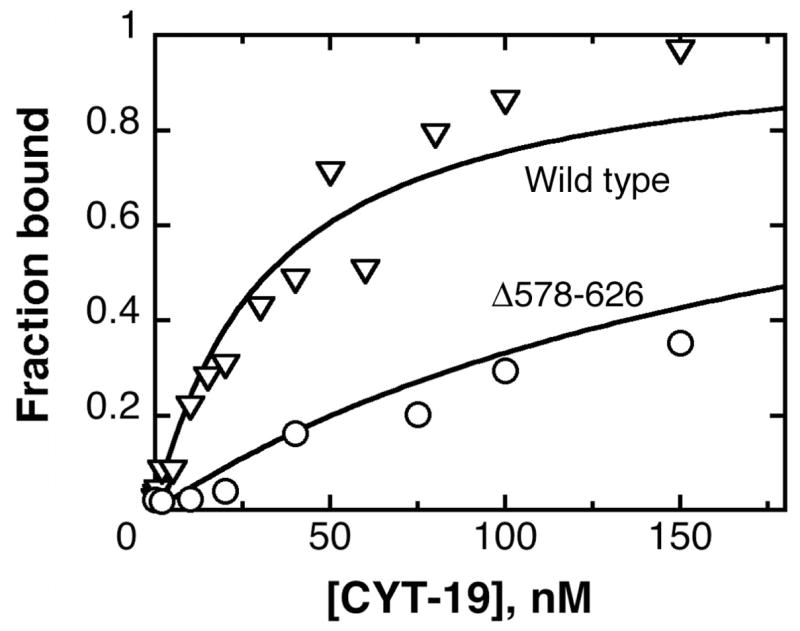
Equilibrium binding of Δ578–626 to the Tetrahymena ribozyme, as monitored by nitrocellulose filter binding. The data shown for the Δ578–626 protein (○) gave a Kd value of 200 nM, and the data for the wild-type CYT-19 protein, from an experiment performed side-by-side, (∇) gave a Kd value of 30 nM. The experiments shown did not include added nucleotides; analogous experiments in the presence of 2 mM ADP-Mg2+ or 2 mM AMP-PNP-Mg2+ gave indistinguishable results for both the wild-type and Δ578–626 proteins.
Role of the C-terminal domain in folding of group I RNAs
We were next interested in whether the C-terminal region also enhances the ability of CYT-19 to function in folding of group I RNAs. We first tested the truncated Δ578–626 protein in re-folding of a long-lived misfolded conformer of the Tetrahymena ribozyme. Acceleration of this folding transition was previously demonstrated for wild-type CYT-19 (11). As expected, the Δ578–626 protein was less efficient than the wild type in accelerating re-folding of the ribozyme to its native state (Fig. 6). However, the loss of activity upon deletion of the C-terminal region was <3-fold, substantially less than the maximum effect observed for helix unwinding. This result indicates that the C-terminal region does not play as large a role in re-folding of the ribozyme, perhaps because the misfolded conformer of this ribozyme is extensively structured and has all of the long-range tertiary contacts formed (25, 26), whereas binding by the second site on CYT-19 appears to enhance unwinding principally of structure that is loosely associated with the core (11).
Fig. 6.
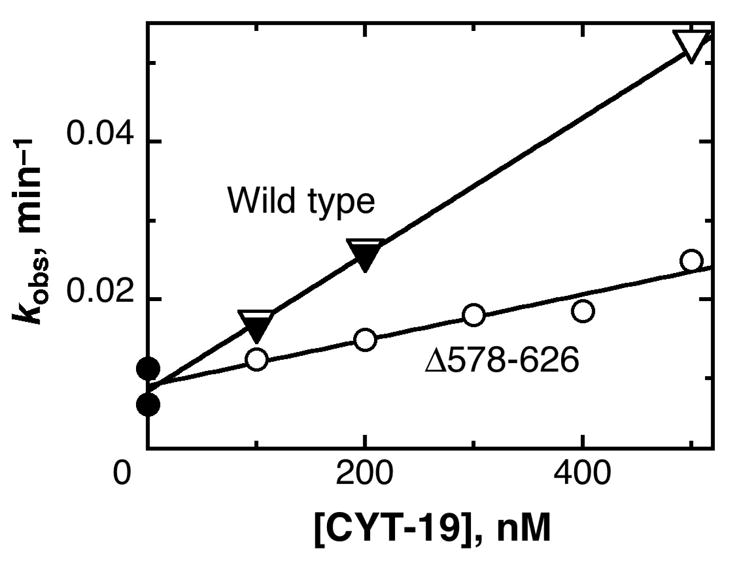
Acceleration by CYT-19 of misfolded Tetrahymena ribozyme re-folding to the native state. The progress of re-folding was followed by the onset of substrate cleavage activity by the ribozyme in the absence of CYT-19 (λ) or in the presence of various concentrations of Δ578–626 (○) or wild-type CYT-19 (performed side-by side) (τ). An equivalent experiment for the wild-type CYT-19 protein has been published previously, and these data are included for comparison (∇). The dependences of re-folding rate constant on protein concentration gave kcat/KM values of 8.6 (± 0.4) × 104 M−1 min−1 and 2.9 (± 0.4) × 104 M−1 for the wild-type and Δ578–626 proteins, respectively.
We next tested the activity of the Δ578–626 protein in facilitating folding of another group I RNA, the Neurospora mitochondrial large subunit rRNA intron (mt LSU-ΔORF), which is one of several introns that require CYT-19 for proper folding in vivo (1). This experiment also included the CYT-18 protein, which is required for efficient splicing and functions by binding specifically to the intron RNA and stabilizing the functional structure (27, 28). CYT-19 activity was determined by measuring the rate of splicing, which is apparently rate-limited by formation of the native state at 25 °C (1). The wild-type CYT-19 was 15-fold more active than the Δ578–626 protein in promoting this folding reaction, indicating that the C-terminal region contributes substantially to the efficiency of this folding reaction (Fig. 7). Indeed, the decrease in activity for this reaction upon deletion of the C-terminal region is in the same range as the decrease in efficiency for helix unwinding activity in the presence of attached RNA structure (see Fig. 2 and Table I).
Fig. 7.
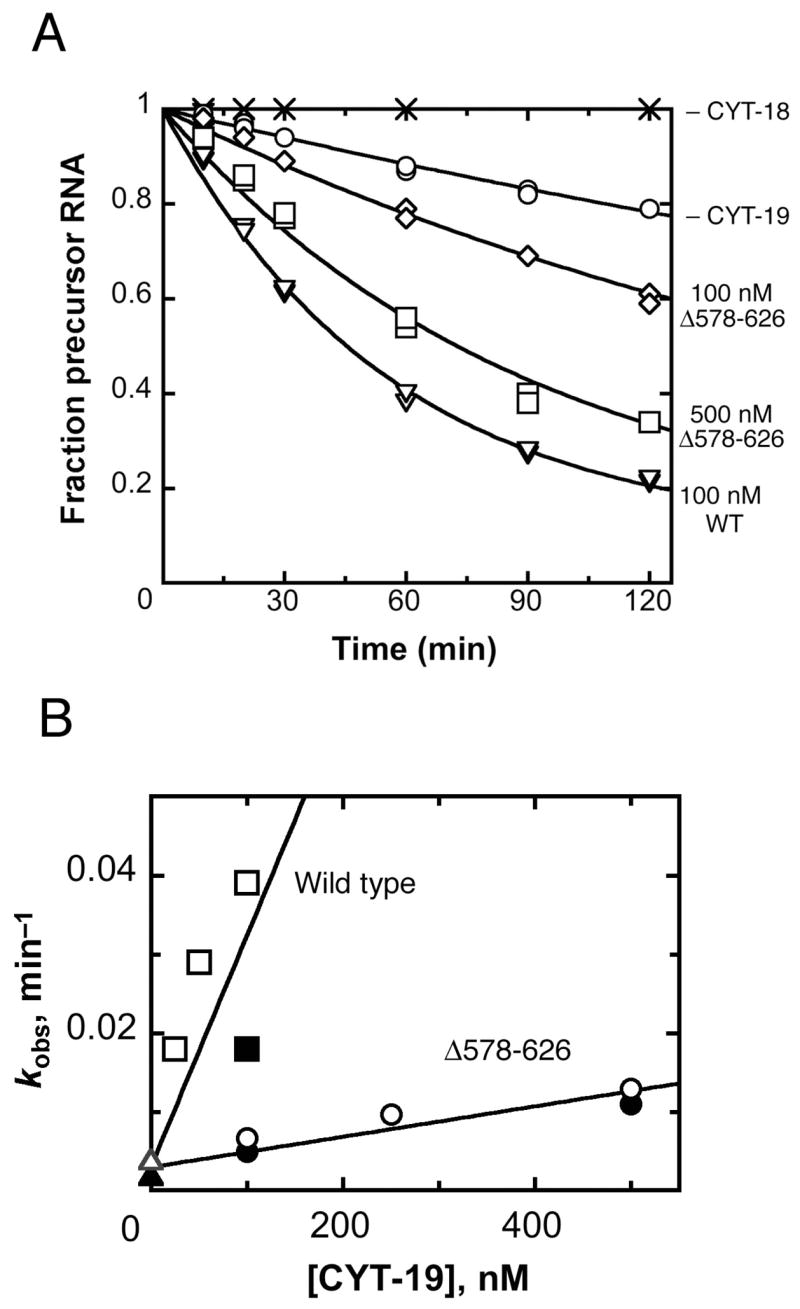
Δ578–626 is defective in facilitating splicing of the Neurospora mt LSU intron. A, Disappearance of precursor RNA (20 nM) was followed at 25°C in the presence of 100 nM CYT-18 dimer in the absence of CYT-19 (○) or with 100 nM CYT-19 (∇), 100 nM Δ578–626 (◇), or 500 nM Δ578–626 (□). The RNA was also incubated without any proteins (×) as a negative control. RNA splicing was performed as described in Materials and Methods. B, Dependence of the splicing rate constant on the concentrations of Δ578–626 (○, λ) and wild-type CYT-19 (□, ν). Equivalent reactions in the presence of CYT-18 and absence of CYT-19 are also shown (Δ, σ). The data are from the experiment shown in panel A (filled symbols) and an analogous experiment using independent preparations of proteins (open symbols). Linear fits to the data shown gave kcat/KM values of 2.9 (± 0.8) × 105 M−1 min−1 and 1.9 (± 0.2) × 104 M−1 for the wild-type and Δ578–626 proteins, respectively.
DISCUSSION
Previous work indicated that CYT-19 possesses an additional RNA binding site, distinct from the site responsible for duplex unwinding, that binds RNA structure non-specifically and enhances the efficiency of CYT-19 for unwinding nearby helices (11). As CYT-19 had been shown previously to function as a general RNA chaperone (1, 10, 12), facilitating folding of multiple RNAs, it appeared likely that this non-specific RNA binding would be important for the function of the protein. Here we explored the region of CYT-19 that is responsible for the binding by deleting 49 amino acids from the C-terminus. Strikingly, the truncated protein retains significant unwinding activity but is defective in the enhancement of activity provided by the presence of adjacent RNA structure.
Although alternatives are possible, the simplest interpretation of these results is that the additional RNA binding site resides at least partially within the deleted C-terminal sequence. First, the deleted sequence is highly basic, with a calculated pI of >12, and it is arginine-rich (11 of 49 deleted amino acids are arginine), characteristics that are found in RNA binding domains of other proteins (15). Further, non-specific RNA binding by ancillary domains of DExD/H-box proteins has been documented previously (29–31). The sequences of these domains are also arginine-rich, although they differ from CYT-19 in that they conform to a repeating motif known as the RGG box, first identified in the context of hnRNP proteins (32). An alternative explanation for our results is that the C-terminal sequence mediates dimerization of CYT-19, which contributes to the enhancement of activity, perhaps because the additional interaction with RNA is made by the unwinding site of the second protomer. Although there is considerable evidence for functional dimers of certain DNA helicases (33, 34), the DEAD-box proteins that have been examined, including CYT-19, appear to be monomeric in solution [(35); H.B. and R.R., unpublished results; M.D. and A.M.L., unpublished results]. These results do not rule out transient dimerization, which could be required for function despite being unfavorable in solution. However, as dimerization is proposed to be critical for helicases to unwind DNA or RNA processively (33), there is no expectation for such a requirement in DEAD-box proteins that employ non-processive unwinding. Thus, we suggest that the simplest model that is consistent with current data is that CYT-19 functions as a monomer and that the second RNA binding site includes the deleted sequence.
Although non-specific RNA binding by ancillary domains of DEAD-box proteins has been demonstrated previously, we propose a distinct role for RNA binding by this domain (Fig. 8). Instead of being instrumental to the mechanism of helix unwinding (29), we suggest that this non-specific binding directs CYT-19 to act on structured RNAs in an analogous fashion to the targeting proposed for ancillary domains that recognize a specific RNA or RNP complex (3, 4). By binding preferentially to double-stranded or structured RNA but without specificity for a particular RNA, the ancillary domain of CYT-19 could localize the unwindase motor so that it is able to unwind a helix that is adjacent to the initial binding site. It is also possible that binding by the ancillary domain activates the motor allosterically, but a double-stranded ‘extension’ fails to activate unwinding when present as a separate molecule rather than covalently linked to the P1 duplex (P. Tijerina and R. Russell, unpublished results), suggesting that the principal mechanism of activation is localization of the unwinding motor domain.
Fig. 8.
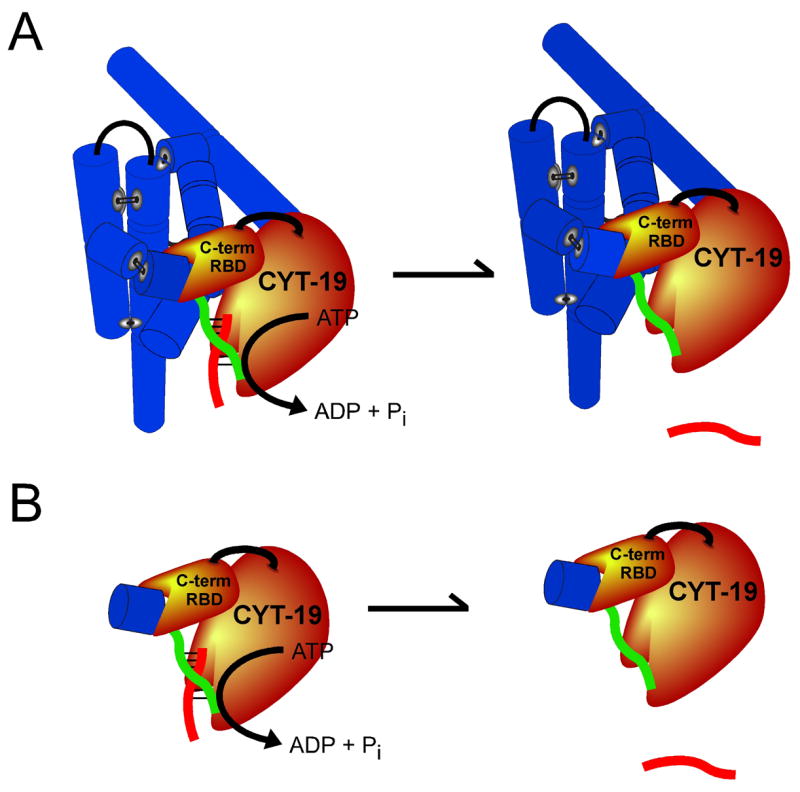
Model for the role of the RNA binding site within the C-terminal domain (RBD) in enhancing duplex unwinding activity of CYT-19. A, CYT-19 binds with low specificity to structured RNA (blue cylinders, which depict the secondary structure elements of the Tetrahymena ribozyme) via its C-terminal domain and efficiently unwinds helices that are not packed tightly against the body of the structured RNA (the P1 duplex is shown as red and green strands). B, Enhanced unwinding activity by CYT-19 is also observed upon addition of a simple duplex to P1 (P2 is shown as a single blue cylinder), suggesting that the primary recognition by the C-terminal domain is for double-stranded RNA located adjacent to the helix to be unwound. See Discussion for further details.
During subsequent unwinding of the adjacent helix, the ancillary C-terminal domain most likely remains bound at its original attachment point, because if the C-terminal domain were required to dissociate, its initial binding would be non-productive and would not be predicted to increase the unwinding efficiency. The exception to this expectation would be if initial binding to a duplex extension were followed by rapid translocation through the duplex, as observed for DNA helicases. However, in this case the activity enhancement would be expected to require a particular directionality and, by analogy with DNA helicases, would be expected to be maximal for single-stranded extensions, whereas neither of these features are observed for CYT-19 or other DEAD-box proteins (4, 36, 37) (Table I). As continued binding of the C-terminal domain to its original attachment point is most simply expected to prevent the CYT-19 protein from translocating through a helix for any significant distance, the data are most easily compatible with a single-step, fully non-processive unwinding, as depicted in Fig. 8 (4, 24). This mechanism of unwinding differs fundamentally from the processive action of DNA helicases and some DExD/H-box proteins, which unwind DNA or RNA duplexes in a directional fashion as they translocate through the duplex (33, 38).
Although the deleted region of the C-terminal domain of CYT-19 clearly plays an important role in the duplex unwinding reactions observed here, our data suggest that the interactions between CYT-19 and structured RNA that enhance unwinding activity extend beyond the deleted region. First, neither addition of single-stranded nor double-stranded extensions gave as much enhancement as attachment of the entire Tetrahymena ribozyme, and the additional enhancement conferred by the ribozyme (beyond that of a double-stranded extension) was independent of the presence of the C-terminal domain (see Table I). This result suggests that another site or surface within CYT-19 forms additional contacts with the ribozyme. Second, although the Δ578–626 protein is enhanced for unwinding activity less than the wild-type protein upon attachment of adjacent structure, there remains some residual enhancement. Thus, there is likely to be an additional site or sites within the retained portion of the C-terminal domain, within the motor domain, or within the small N-terminal region that can bind flanking RNA structure and enhance unwinding activity. A full understanding of the interactions responsible for the enhanced unwinding activity from adjacent RNA structure will require a further dissection of the CYT-19 domains using biochemical and, ultimately, structural approaches.
RNA binding by the C-terminal domain can apparently contribute to chaperone activity of CYT-19, as the C-terminal truncation results in a decrease in splicing efficiency for one of its physiological substrates, the Neurospora mt LSU intron, that is as large as the decrease in unwinding activity in the context of model substrates. The simplest model is that the ability of CYT-19 to facilitate re-folding arises from the same basic activity that accelerates the helix unwinding reactions of model systems. Presumably, the C-terminal domain first binds non-specifically to a structured RNA, with the non-specific nature of this binding allowing CYT-19 to interact with multiple misfolded species of multiple RNAs, and to bind these species in multiple orientations so that it can disrupt non-native contacts regardless of where they are within the larger RNA structure.
Exactly what types of contact are disrupted by CYT-19 as it facilitates folding of structured RNAs is not yet clear. We have shown that CYT-19 can efficiently unwind exposed helices while bound to a structured RNA [(11) and results herein], an activity that is not surprising considering the sequence similarity with DNA helicases and the demonstrated ability of several DExD/H-box proteins to unwind short model duplexes [reviewed in (2)]. DEAD-box proteins have also been shown to remove proteins from single-stranded and double-stranded RNA (39, 40), although there is no evidence for displacement of CYT-18 from group I introns by CYT-19 (1). It is also possible that CYT-19 can disrupt RNA-RNA tertiary contacts to facilitate folding. Indeed, this would be the simplest interpretation of the ability of CYT-19 to facilitate folding of the Tetrahymena ribozyme from the misfolded structure to the native state, as the misfolded structure is stabilized extensively by the formation of native tertiary contacts on its periphery (26). The ability of one active site to carry out such disparate activities can be rationalized by a model in which the energy of ATP binding and hydrolysis is used to generate a binding site within the motor domain that is highly specific for single-stranded RNA, and as the single-stranded RNA is accommodated within the active site any contacts of this strand with RNA or protein are disrupted. This idea is consistent with the recently-determined structure of the motor domain from the Drosophila protein Vasa, which includes a bound single-stranded RNA that bends sharply within the binding site, achieving a conformation that would not be compatible with a continuous duplex (41). For all of these activities, the RNA binding site within the C-terminal domain of CYT-19 is probably important because it localizes the unwinding site to structured RNAs, increasing the efficiency of any subsequent disruption of structure.
It is interesting to consider whether general DExD/H-box RNA chaperones are common in nature and whether this function can be predicted from the sequences of the ancillary domains. Considerations of the physical and chemical properties of RNA, as well as experimental results on folding of several large RNAs, suggest that cellular RNAs are likely to encounter misfolded species frequently (13, 14, 42–46). As all cells contain a large and diverse set of structured RNAs, it seems unlikely that there could be a chaperone protein devoted to each structured RNA, and even less likely that there could be a chaperone devoted to each misfolded structure of each RNA. Thus, there is probably a general need for RNA chaperone proteins that can interact with and resolve multiple misfolded species. Some of the proteins that function in this role may not be DExD/H-box proteins, but rather proteins that bind single-stranded RNA, several of which possess RNA chaperone activity in vitro and when overexpressed in vivo [reviewed in (47)]. Nevertheless, the unique ability of DExD/H-box proteins to couple the free energy from ATP binding and hydrolysis to RNA rearrangements suggests that these proteins may be required to resolve, at a minimum, the longer-lived misfolded species of RNA, and are therefore likely to play critical roles as general RNA chaperones.
The most basic requirement for a general chaperone is the ability to interact functionally with multiple RNAs, and arginine-rich domains are clearly capable of conferring non-specific RNA binding. The chaperones CYT-19 and Mss116p both include arginine-rich extensions, as does the yeast protein Ded1p, which may function as a general chaperone in translation initiation (48–50), although the extensions of these proteins do not share identifiable sequence similarity beyond the high frequency of arginines. Further, a number of other DEAD-box proteins have arginine-rich sequences, including a total of five of 25 in Saccharomyces cerevisiae and three of five in E. coli. However, assigning a role as a general chaperone based on the sequence of an ancillary domain is unlikely to be straightforward. First, the presence of an arginine-rich motif (ARM) does not necessary imply non-specific binding, as ‘canonical’ viral ARM-containing proteins, Tat and Rev, bind their cognate RNAs with very high specificity (51–53), and even ARM-containing peptides can bind tightly and selectively to RNA targets (54, 55). Also, some DExD/H-box proteins that do function as general chaperones may achieve non-specific recognition of RNA by binding specifically to a second protein that harbors a non-specific RNA binding site; such a DExD/H-box protein would not be identified as a candidate for general chaperone activity from the sequences of its ancillary domains. A possible example of this type of protein is eIF4A, which lacks ancillary domains but interacts with the RNA-binding proteins eIF4B and eIF4G and employs chaperone-like activity to remove structure from mRNAs during translation initiation (56, 57). Finally, as the activity increase from ancillary site binding is not enormous in energetic terms (<3 kcal/mol), it is possible a DExD/H box protein lacking such domains could compensate with a more active unwinding domain to achieve the same efficiency as CYT-19. Such a protein could presumably function as a general chaperone, albeit without the enhanced activity on structured RNA that is conferred by the ancillary domain. Thus, biochemical and genetic approaches are probably necessary to identify DExD/H-box proteins that function as general RNA chaperones, as well as to elucidate which RNAs and RNPs are dependent on them and to understand their reaction mechanisms.
Acknowledgments
We thank Klaus Linse and Steven Halls in the ICMB Core Facility for analysis of CYT-19 fragments, and Eckhard Jankowsky for helpful discussions and for sharing unpublished results.
Footnotes
This work was supported by grants from the NIH (R01-GM070456 to R.R. and R01-GM037951 to A.M.L.) and from the Welch Foundation (F-1563 to R.R.). M.D. was supported by a postdoctoral fellowship from the NIH (F01-GM076961).
Abbreviations: Δ578-626, C-terminal truncation variant of CYT-19, which lacks amino acids 578-626; EDTA, ethylenedinitrilotetraacetic acid; MOPS, 3-(N-morpholino)propanesulfonic acid; PCI, phenol:chloroform:isoamyl alcohol mixture; S, the oligonucleotide substrate of the Tetrahymena ribozyme, CCCUCUA5; S*, 32P-labeled substrate.
While most of the experiments used CYT-19 protein that was expressed as a fusion with maltose binding protein (see Materials and Methods), the papain digestions were performed with protein that was expressed from the intein-derived construct described previously (1). The N-terminal-most amino acids of protein produced from this construct differ from the maltose binding protein construct (MS for the intein-derived construct, GSMS for the maltose-binding-protein construct, see Materials and Methods), leading to the predicted cleavage site at residue 574 from mass-spectral analysis of the protease fragment.
References
- 1.Mohr S, Stryker JM, Lambowitz AM. Cell. 2002;109:769–79. doi: 10.1016/s0092-8674(02)00771-7. [DOI] [PubMed] [Google Scholar]
- 2.Tanner NK, Linder P. Mol Cell. 2001;8:251–62. doi: 10.1016/s1097-2765(01)00329-x. [DOI] [PubMed] [Google Scholar]
- 3.Silverman E, Edwalds-Gilbert G, Lin RJ. Gene. 2003;312:1–16. doi: 10.1016/s0378-1119(03)00626-7. [DOI] [PubMed] [Google Scholar]
- 4.Cordin O, Banroques J, Tanner NK, Linder P. Gene. 2006;367:17–37. doi: 10.1016/j.gene.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 5.Wang Y, Guthrie C. RNA. 1998;4:1216–29. [PMC free article] [PubMed] [Google Scholar]
- 6.Schneider S, Schwer B. J Biol Chem. 2001;276:21184–91. doi: 10.1074/jbc.M101964200. [DOI] [PubMed] [Google Scholar]
- 7.Kossen K, Karginov FV, Uhlenbeck OC. J Mol Biol. 2002;324:625–36. doi: 10.1016/s0022-2836(02)01140-3. [DOI] [PubMed] [Google Scholar]
- 8.Karginov FV, Caruthers JM, Hu Y, McKay DB, Uhlenbeck OC. J Biol Chem. 2005;280:35499–505. doi: 10.1074/jbc.M506815200. [DOI] [PubMed] [Google Scholar]
- 9.Wang S, Hu Y, Overgaard MT, Karginov FV, Uhlenbeck OC, McKay DB. RNA. 2006;12:959–67. doi: 10.1261/rna.5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang HR, Rowe CE, Mohr S, Jiang Y, Lambowitz AM, Perlman PS. Proc Natl Acad Sci USA. 2005;102:163–8. doi: 10.1073/pnas.0407896101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tijerina P, Bhaskaran H, Russell R. Proc Natl Acad Sci U S A. 2006;103:16698–16703. doi: 10.1073/pnas.0603127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohr S, Matsuura M, Perlman PS, Lambowitz AM. Proc Natl Acad Sci USA. 2006;103:3569–3574. doi: 10.1073/pnas.0600332103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karpel RL, Miller NS, Fresco JR. Biochemistry. 1982;21:2102–8. doi: 10.1021/bi00538a019. [DOI] [PubMed] [Google Scholar]
- 14.Herschlag D. J Biol Chem. 1995;270:20871–4. doi: 10.1074/jbc.270.36.20871. [DOI] [PubMed] [Google Scholar]
- 15.Burd CG, Dreyfuss G. Science. 1994;265:615–21. doi: 10.1126/science.8036511. [DOI] [PubMed] [Google Scholar]
- 16.Zaug AJ, Grosshans CA, Cech TR. Biochemistry. 1988;27:8924–31. doi: 10.1021/bi00425a008. [DOI] [PubMed] [Google Scholar]
- 17.Russell R, Herschlag D. RNA. 1999;5:158–66. doi: 10.1017/s1355838299981839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kristelly R, Earnest BT, Krishnamoorthy L, Tesmer JJ. Acta Crystallogr D Biol Crystallogr. 2003;59:1859–62. doi: 10.1107/s0907444903018067. [DOI] [PubMed] [Google Scholar]
- 19.Kapust RB, Tozser J, Fox JD, Anderson DE, Cherry S, Copeland TD, Waugh DS. Protein Eng. 2001;14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- 20.Wong I, Chao KL, Bujalowski W, Lohman TM. J Biol Chem. 1992;267:7596–610. [PubMed] [Google Scholar]
- 21.Russell R, Herschlag D. J Mol Biol. 2001;308:839–51. doi: 10.1006/jmbi.2001.4751. [DOI] [PubMed] [Google Scholar]
- 22.Guo QB, Akins RA, Garriga G, Lambowitz AM. J Biol Chem. 1991;266:1809–19. [PubMed] [Google Scholar]
- 23.Choe Y, Leonetti F, Greenbaum DC, Lecaille F, Bogyo M, Bromme D, Ellman JA, Craik CS. J Biol Chem. 2006;281:12824–32. doi: 10.1074/jbc.M513331200. [DOI] [PubMed] [Google Scholar]
- 24.Yang Q, Jankowsky E. Nat Struct Mol Biol. 2006;13:981–6. doi: 10.1038/nsmb1165. [DOI] [PubMed] [Google Scholar]
- 25.Russell R, Millett IS, Doniach S, Herschlag D. Nat Struct Biol. 2000;7:367–370. doi: 10.1038/75132. [DOI] [PubMed] [Google Scholar]
- 26.Russell R, Das R, Suh H, Travers K, Laederach A, Engelhardt M, Herschlag D. J Mol Biol. 2006;363:531–544. doi: 10.1016/j.jmb.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 27.Mannella CA, Collins RA, Green MR, Lambowitz AM. Proc Natl Acad Sci U S A. 1979;76:2635–9. doi: 10.1073/pnas.76.6.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akins RA, Lambowitz AM. Cell. 1987;50:331–45. doi: 10.1016/0092-8674(87)90488-0. [DOI] [PubMed] [Google Scholar]
- 29.Gibson TJ, Thompson JD. Nucleic Acids Res. 1994;22:2552–6. doi: 10.1093/nar/22.13.2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang S, Grosse F. J Biol Chem. 1997;272:11487–94. doi: 10.1074/jbc.272.17.11487. [DOI] [PubMed] [Google Scholar]
- 31.Lorkovic ZJ, Herrmann RG, Oelmuller R. Mol Cell Biol. 1997;17:2257–65. doi: 10.1128/mcb.17.4.2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kiledjian M, Dreyfuss G. EMBO J. 1992;11:2655–64. doi: 10.1002/j.1460-2075.1992.tb05331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lohman TM, Bjornson KP. Annu Rev Biochem. 1996;65:169–214. doi: 10.1146/annurev.bi.65.070196.001125. [DOI] [PubMed] [Google Scholar]
- 34.Ha T, Rasnik I, Cheng W, Babcock HP, Gauss GH, Lohman TM, Chu S. Nature. 2002;419:638–41. doi: 10.1038/nature01083. [DOI] [PubMed] [Google Scholar]
- 35.Talavera MA, Matthews EE, Eliason WK, Sagi I, Wang J, Henn A, De La Cruz EM. J Mol Biol. 2006;355:697–707. doi: 10.1016/j.jmb.2005.10.058. [DOI] [PubMed] [Google Scholar]
- 36.Rogers GW, Jr, Lima WF, Merrick WC. J Biol Chem. 2001;276:12598–608. doi: 10.1074/jbc.M007560200. [DOI] [PubMed] [Google Scholar]
- 37.Bizebard T, Ferlenghi I, Iost I, Dreyfus M. Biochemistry. 2004;43:7857–66. doi: 10.1021/bi049852s. [DOI] [PubMed] [Google Scholar]
- 38.Jankowsky E, Gross CH, Shuman S, Pyle AM. Nature. 2000;403:447–51. doi: 10.1038/35000239. [DOI] [PubMed] [Google Scholar]
- 39.Jankowsky E, Gross CH, Shuman S, Pyle AM. Science. 2001;291:121–5. doi: 10.1126/science.291.5501.121. [DOI] [PubMed] [Google Scholar]
- 40.Fairman ME, Maroney PA, Wang W, Bowers HA, Gollnick P, Nilsen TW, Jankowsky E. Science. 2004;304:730–4. doi: 10.1126/science.1095596. [DOI] [PubMed] [Google Scholar]
- 41.Sengoku T, Nureki O, Nakamura A, Kobayashi S, Yokoyama S. Cell. 2006;125:287–300. doi: 10.1016/j.cell.2006.01.054. [DOI] [PubMed] [Google Scholar]
- 42.Walstrum SA, Uhlenbeck OC. Biochemistry. 1990;29:10573–6. doi: 10.1021/bi00498a022. [DOI] [PubMed] [Google Scholar]
- 43.Woodson SA, Cech TR. Biochemistry. 1991;30:2042–50. doi: 10.1021/bi00222a006. [DOI] [PubMed] [Google Scholar]
- 44.Pan T, Sosnick TR. Nat Struct Biol. 1997;4:931–8. doi: 10.1038/nsb1197-931. [DOI] [PubMed] [Google Scholar]
- 45.Treiber DK, Rook MS, Zarrinkar PP, Williamson JR. Science. 1998;279:1943–6. doi: 10.1126/science.279.5358.1943. [DOI] [PubMed] [Google Scholar]
- 46.Russell R, Herschlag D. J Mol Biol. 1999;291:1155–67. doi: 10.1006/jmbi.1999.3026. [DOI] [PubMed] [Google Scholar]
- 47.Schroeder R, Barta A, Semrad K. Nat Rev Mol Cell Biol. 2004;5:908–19. doi: 10.1038/nrm1497. [DOI] [PubMed] [Google Scholar]
- 48.Chuang RY, Weaver PL, Liu Z, Chang TH. Science. 1997;275:1468–71. doi: 10.1126/science.275.5305.1468. [DOI] [PubMed] [Google Scholar]
- 49.de la Cruz J, Iost I, Kressler D, Linder P. Proc Natl Acad Sci USA. 1997;94:5201–6. doi: 10.1073/pnas.94.10.5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marsden S, Nardelli M, Linder P, McCarthy JE. J Mol Biol. 2006;361:327–35. doi: 10.1016/j.jmb.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 51.Zapp ML, Green MR. Nature. 1989;342:714–6. doi: 10.1038/342714a0. [DOI] [PubMed] [Google Scholar]
- 52.Dingwall C, Ernberg I, Gait MJ, Green SM, Heaphy S, Karn J, Lowe AD, Singh M, Skinner MA, Valerio R. Proc Natl Acad Sci U S A. 1989;86:6925–9. doi: 10.1073/pnas.86.18.6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weeks KM, Ampe C, Schultz SC, Steitz TA, Crothers DM. Science. 1990;249:1281–5. doi: 10.1126/science.2205002. [DOI] [PubMed] [Google Scholar]
- 54.Austin RJ, Xia T, Ren J, Takahashi TT, Roberts RW. J Am Chem Soc. 2002;124:10966–7. doi: 10.1021/ja026610b. [DOI] [PubMed] [Google Scholar]
- 55.Bayer TS, Booth LN, Knudsen SM, Ellington AD. RNA. 2005;11:1848–57. doi: 10.1261/rna.2167605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Svitkin YV, Pause A, Haghighat A, Pyronnet S, Witherell G, Belsham GJ, Sonenberg N. RNA. 2001;7:382–94. doi: 10.1017/s135583820100108x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Linder P. Biol Cell. 2003;95:157–67. doi: 10.1016/s0248-4900(03)00032-7. [DOI] [PubMed] [Google Scholar]