

Abstract
The drought of progress in clinical brain tumor therapy provides an impetus for developing new treatments as well as methods for testing therapeutics in animal models. The inability of traditional assays to simultaneously measure tumor size, location, growth kinetics, and cell kill achieved by a treatment complicates the interpretation of therapy experiments in animal models. To address these issues, tumor volume measurements obtained from serial magnetic resonance images were used to noninvasively estimate cell kill values in individual rats with intracerebral 9L tumors after treatment with 0.5, 1, or 2 × LD10 doses of 1,3-bis(2-chloroethyl)-1-nitrosourea. The calculated cell kill values were consistently lower than those reported using traditional assays. A dose-dependent increase in 9L tumor doubling time after treatment was observed that significantly contributed to the time required for surviving cells to repopulate the tumor mass. This study reveals that increases in animal survival are not exclusively attributable to the fraction of tumor cells killed but rather are a function of the cell kill and repopulation kinetics, both of which vary with treatment dose.
Brain tumors occur frequently in the human population with approximately 35,000 new cases of primary adult central nervous system tumors diagnosed in the United States each year (1). The 1,500–2,000 brain tumors reported annually in children constitute the largest group of solid pediatric neoplasms (2). Despite the use of multimodality therapy, the management of brain tumors in adults and children remains unsatisfactory. In particular, the treatment of glioblastoma multiforme constitutes a major problem caused by the lack of therapeutic responses and a median survival time of only 1 year from the initial diagnosis (3).
Promising agents identified through in vitro screening assays with brain tumor cell lines subsequently are evaluated in vivo by using animal brain tumor models (4–6). These studies frequently use rodent brain tumor models (7–10), particularly the rat 9L tumor (11–29). Animal survival, colony-forming efficiency (CFE) assays of cells disaggregated from solid tumors, and measurements of excised tumor weights have been used for two decades to quantitate the efficacy of various treatments on the orthotopic 9L brain tumor (11–18, 20–29). Numerous in vitro and in vivo studies have shown that 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) is an effective treatment for the 9L brain tumor (11–15, 17, 18, 20–23, 25–29); in fact, a single bolus of BCNU achieved a 3–4 log cell kill in orthotopic tumors, resulting in a 97% increase in life span (13). BCNU is a mainstay of brain tumor chemotherapy, but the clinical outcome of patients treated solely with BCNU has not proven as efficacious as would be predicted by the responses observed in the 9L tumor model (30–33).
In the present study, MRI was used for noninvasive estimation of tumor cell kill in individual animals with orthotopic 9L brain tumors after single doses of BCNU at 0.5, 1, or 2 × LD10. Tumor volumes were determined from serial MR images obtained at 2-day intervals over the course of the experiment. This data revealed that the 9L tumor doubling time increases after BCNU therapy in a dose-dependent fashion. The cell kill values achieved in each individual animal also were calculated from the tumor volume data and were found to be considerably lower than previously reported using traditional assays (12, 14, 20, 22). These results suggest that the therapeutic efficacy of BCNU for the 9L tumor is not solely attributable to its cytotoxic activity and that a previously unknown effect on tumor doubling times contributes significantly to the tumor growth delays elicited by this drug. This finding has important implications when interpreting results from preclinical therapeutic efficacy studies using animal survival as the sole endpoint because survival may be affected by both therapeutic-induced changes in the tumor doubling time as well as cell kill. MRI allows the relative contributions of these factors to be delineated, providing a more complete understanding of the overall effects of the treatment on animal survival.
METHODS
Cell Culture and Induction of Brain Tumors.
Rat 9L brain tumor cells (passage 12) were obtained from the Brain Tumor Research Center at the University of California at San Francisco and grown as monolayers in 175-cm2 sterile plastic flasks in minimum essential medium with 10% fetal calf serum. Cells were cultured in an incubator at 37°C in an atmosphere containing 95/5% air/CO2 until confluent and were harvested by trypsinization, counted, and resuspended in serum-free media for intracerebral injection. The 9L cells were carried only until passage number 30 at which time cells were reactivated from frozen stocks.
Male Fischer 344 rats (n = 21) weighing between 125 and 150 g were anesthetized with a ketamine/xylazine mixture (87/13 mg/kg body weight, i.p.). A small skin incision over the right hemisphere was made, and a 1-mm-diameter burr hole was drilled through the skull by using a high-speed drill. Tumor cells (105) contained in 5 μl were injected in the right forebrain tk;4at a depth of 3 mm. The area was rinsed with 70% ethanol, the burr hole was filled with bone wax to minimize extra-cerebral extension of the tumor, and the skin was sutured closed. The day and time of cell implantation was recorded for each individual animal for later correlation with MRI-determined tumor volumes.
MRI and Tumor Volume Calculations.
All in vivo MR experiments were performed on a Varian NMR Instruments system equipped with a 7.0 tesla (300 MHz proton frequency), 18.3-cm horizontal bore magnet. For MRI examination, rats were anesthetized with a ketamine/xylazine mixture and maintained at 37°C inside the magnet using a heated thermostated circulating water blanket. MRI of rat brains was initiated between 8 and 11 days after cell implantation and repeated approximately every 2 days by using a 3-cm-diameter quadrature radio frequency head coil (USA Instruments, Highland Heights, OH). A single-slice gradient-recalled-echo image was acquired with 1-mm “saturation crosshairs” imprinted on the axial and coronal images to facilitate rapid and reproducible positioning of the animal. Multislice axial images were acquired by using a spin echo sequence. T2-weighted images through the rat brain were produced by using the following parameters: 3.5 s repetition time, 60 ms echo time, field of view = 30 × 30 mm using a 128 × 128 matrix, slice thickness = 500 μm, slice separation = 0.8 mm, number of slices = 25, four signal averages per phase encode step requiring a total acquisition time of about 30 min per rat.
Tumor volumes were obtained from the multislice MR images. In brief, the tumor boundary visualized in each 500 μm-thick slice was electronically outlined by using image processing software (Advanced Visual Systems, Waltham, MA) by an individual blinded to the BCNU-dosage protocol. The number of tumor pixels was converted to an area by multiplication by the factor [(field of view)2/(matrix)2]. The total tumor volume was calculated as the summed area on all slices, multiplied by the slice separation (34).
Mathematical Model and Calculations.
The intracerebral 9L tumor has been empirically shown to grow exponentially (34). This is consistent with classic models of tumor growth (35) given by,
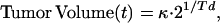 |
1 |
where Td represents the volumetric doubling time and κ is a constant related to the initial viable tumor volume. To determine the pretreatment doubling time of the tumors, Tdpre, log tumor volumes were plotted versus the time post-cell implantation and fit using Eq. 1 (Fig. 1, line A). Tumor volumes during exponential regrowth were fit to yield Tdpost (Fig. 1, line C). As shown in Fig. 1, the effective volume of tumor surviving immediately after treatment, Vpost, was derived by extrapolation of the regrowth curve to the time of treatment. Also shown in Fig. 1, the difference between the logarithm of the actual volume at the time of treatment, log10(Vpre), and log10(Vpost) is defined as the log cell kill (36):
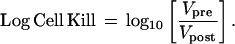 |
2 |
This approach did not have the previous constraint requiring that the rate of tumor regrowth be identical to the pretreatment growth rate (14, 34). This model does assume that any changes in tumor Td occur instantaneously at the time of BCNU administration.
Figure 1.

Demonstration of the mathematical analysis of tumor growth and treatment. Data points are actual values obtained for a 9L tumor treated with a 2 × LD10 BCNU dose. The line marked A is the least-squares fit of the pretreatment growth points to Eq. 1. The line marked C is the fit of the posttreatment growth points. Line C is extrapolated back to the time of treatment to give Vpost. Log cell kill is the log of the ratio Vpre/Vpost (Eq. 2). The line marked B is the theoretical tumor regrowth curve assuming Tdpost = Tdpre. The relative contributions of cell kill and tumor growth rates are related to the calculated repopulation intervals, Trapre and Trapost, as described by Eqs. 3 and 4. Tx, point of BCNU administration; Trapre, repopulation interval given equivalent pretreatment and posttreatment growth rates; Trapost, repopulation interval given the fitted posttreatment growth rate; Vpre, tumor volume at Tx; Vpost, theoretical “viable” tumor volume immediately after treatment determined by extrapolation of C.
The repopulation interval, Tr, is defined as the time required for recovery of the clonogenic cell population killed by treatment. After the time Tr, the tumor should have gone through N doublings so that Vpost = Vpre. Evaluating the relative contributions of cell kill and Tdpost to Tr should provide a more thorough understanding of how BCNU treatment affects animal life span. However, when low cell kills are achieved, the tumor may not regress below the volume at treatment. This is overcome by calculating apparent repopulation intervals, Tra, by extrapolation of exponential volumetric regrowth curves to Vpre (Fig. 1). Tras can be calculated, assuming that the tumor regrowth rate does not change after therapy (e.g., Tdpost = Tdpre) giving Trapre, and by using the actual Tdpost determined by fitting the actual regrowth data to give Trapost (Fig. 1). The estimated percent contribution of cell kill to the repopulation time, χ, is estimated by the expression:
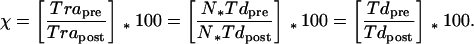 |
3 |
Thus, the percent contribution of the increased Td to the repopulation time is given by:
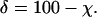 |
4 |
Tumor Treatment Protocol.
At approximately 8 days post-9L cell implantation, an initial MRI exam was performed to monitor for the presence and size of the intracerebral tumor. Up to four pretreatment MRI exams were acquired to define the tumor Tdpre. Rats with tumors between 30 and 100 mm3 were treated with BCNU. BCNU was dissolved in absolute ethanol and diluted in saline (0.9% NaCl) to a final concentration of 3.3 mg/ml in 10% ethanol. Animals received either 6.67, 13.3, or 26.6 mg/kg of BCNU i.p. (n = 7/dose), which correspond to 0.5, 1, and 2 × LD10, respectively. Each animal served as its own control, as the tumor Tdpre and Vpre were accurately determined. On the day of treatment, BCNU was administered to unanesthetized rats and within 1–2 h after BCNU administration, MRI scanning was resumed and continued every 2–3 days until death.
RESULTS
The T2-weighted coronal MR images shown in Fig. 2 are from an animal treated with the 2 × LD10 dose of BCNU. Fig. 2a was acquired within 2 h of BCNU administration, whereas the images in Fig. 2 b–d were acquired posttreatment at the times indicated in the legend. Each displayed image is from approximately the same coronal plane in which the tumor appears as a hyperintense mass in the right hemisphere. The tumor continued to expand after treatment (Fig. 2b), then shrank (Fig. 2c) before subsequently regrowing (Fig. 2d). After BCNU treatment, peritumoral edema was observed in T2-weighted images (Fig. 2 b and c), but later resolved during tumor progression (Fig. 2d). Untreated 9L tumors were well-demarcated masses with relatively uniform hyperintense T2-weighted signals and minimal peritumoral edema throughout their entire growth course (data not shown).
Figure 2.

A series of coronal T2-weighted MR images of a rat harboring a 9L tumor in the right hemisphere (a) within 2 h after BCNU treatment (2 × LD10) and (b–d) at 263, 433, and 775 h posttreatment, respectively. Each displayed image is from approximately the same region of the brain.
Plots of MRI-determined intracerebral 9L tumor volumes versus time post-cell implantation obtained from four different animals are shown in Fig. 3. The individual volumetric data points obtained from MR images are shown along with lines representing the least-squares fits. Fig. 3A displays data from a single animal demonstrating the typical exponential growth pattern observed for untreated intracerebral 9L tumors with a Td of 67 h (34). Fig. 3 B–D shows representative plots of intracerebral 9L tumor volumes from three individual rats treated with 0.5, 1, and 2 × LD10 doses of BCNU, respectively. Deviation from the pretreatment exponential growth pattern (solid lines) within 2 days of treatment reveals that tumor growth was inhibited at each BCNU dose. An increase in the delay until exponential tumor regrowth (dashed lines) was observed with increasing dose of BCNU. Exponential tumor regrowth occurred at a reduced rate compared with the pretreatment growth rates.
Figure 3.

Tumor volumes obtained form serial MR images of four individual rats are displayed versus time after 9L cell implantation. In each case, the individual volume measurements are shown along with the line corresponding to the least-squares fit. (A) A representative plot of an untreated intracerebral 9L tumor that reveals the exponential growth characteristic (Td = 67, r2 = 0.997) of this tumor over the life span of the animal. For BCNU-treated animals, the individual volume measurements are shown along with the pretreatment (solid) and posttreatment (dashed) exponential growth fits after treatment with (B) 0.5, (C) 1, and (D) 2 × LD10 BCNU. Pre- and post-BCNU doubling times (Td) were (B) 53 and 70 h, (C) 55 and 86 h, and (D) 46 and 99 h, respectively, revealing a decrease in growth rate during posttreatment exponential tumor regrowth. Calculated log cell kill values were (B) 0.4, (C) 0.9, and (D) 1.6. In D, the arrows denoted by a, b, c, and d correspond with the rat brain MR images shown in Fig. 2 obtained at 330, 593, 763, and 1105 h, respectively.
Mean intracerebral tumor doubling times increased after BCNU administration (Fig. 4). Mean tumor Tdpre and Tdpost values for 0.5, 1, and 2 × LD10 doses of BCNU were 61 ± 3 and 67 ± 2 h (± SE, n = 5), 60 ± 4 and 89 ± 5 h (n = 7), 59 ± 7 and 132 ± 20 h (n = 6), respectively. Deaths related to anesthesia complications diminished the total number of animals in some groups. Statistically different doubling times before and after treatment were found (P < 0.03) for the 1 and 2 × LD10 doses of BCNU by using paired t tests. Regression analysis revealed that the mean Tdpost values were linearly dependent on BCNU dose (y = 43.3 × + 45.5; r2 = 0.999). There was no statistically significant change in doubling times after the 0.5 × LD10 dose of BCNU (P = 0.3); however, the power of the paired t test was only 0.1. The increase in repopulation time caused by slower tumor regrowth (δ) can be estimated from the ratio of average tumor Tdpre and Tdpost values by using Eqs. 3 and 4. For 0.5, 1, and 2 × LD10 doses of BCNU, the δ equals 9.0 ± 0.5, 32.6 ± 3.4, and 53.3 ± 14.7%, respectively.
Figure 4.

Mean intracerebral 9L tumor doubling times (± SE) before BCNU administration (Tdpre; empty bars) and during the exponential regrowth phase (Tdpost; filled bars) after BCNU treatment. Statistically significant differences found before and after BCNU treatment (P < 0.03) using a two-tailed paired Student’s t test are indicated (∗). In addition, differences between Tdpost for each posttreatment group were found to be statistically significant (P < 0.05).
In vivo log cell kill values were calculated for individual rats and averaged for the three doses of BCNU. Increasing the dose of BCNU resulted in log kill values of 0.2 ± 0.1 (± SE, n = 5), 0.8 ± 0.1 (n = 7) and 1.7 ± 0.2 (n = 6), for 0.5, 1, and 2 × LD10 doses, respectively (Fig. 5). The log kill values in each treatment group were found to be normally distributed (Kolmogorov-Smirnov Normality Test), and differences in the mean values were statistically significant (P < 0.001, one-way ANOVA). The linear regression in Fig. 5 shows log cell kill values were linearly dependent on the BCNU doses used in this study (r2 = 0.994). The increase in repopulation time caused by cell kill (χ) for the 0.5, 1, and 2 × LD10 doses of BCNU, were 91.0 ± 0.5, 67.4 ± 3.4, and 46.7 ± 14.7%, respectively.
Figure 5.

Mean in vivo log cell kill values (± SE) for intracerebral 9L tumors determined from serial volumetric MR tumor images as a function of BCNU dose. The line represents a least-squares fit to the data (y = 0.99x − 0.25, r2 = 0.99).
DISCUSSION
Methods capable of accurately evaluating antineoplastic agents in animal models are vital. Animal survival has been a standard measure of efficacy in brain tumor therapy experiments. This approach relies on comparing the median times at which 50% of the animals from treated and control groups have died. Comparisons of animal survival and CFE data have led to the assertions that increased life span (ILS) in rats with intracerebral 9L tumors is proportional to cell kill and that a minimum of 1 log kill is required to observe an ILS (12). These assertions do not consider factors other than cell kill that contribute to changes in ILS for rodent brain tumor models (24). Changes in tumor growth rates, which cannot be determined from animal survival experiments, are one such factor. It often is assumed that cytotoxic treatments do not affect tumor regrowth rates. However, if changes in tumor growth kinetics are not considered, then the cell kill inferred from an animal survival experiment may be incorrect.
In this study, MRI was used to observe tumor growth kinetics and cell kills after treatment of 9L tumors with single doses of BCNU at 0.5, 1, and 2 × LD10. After BCNU treatment there was a deviation from exponential tumor growth within 2–4 days, but the tumor mass continued to expand for a period of time after treatment. This was likely caused by the ability of BCNU-damaged cells to divide for several doublings before dying (37) combined with cell swelling (38), vasogenic edema, and an inflammatory response. Tumor mass did not regress after the 0.5 × LD10 BCNU dose caused by the low cell kill, and tumor growth rates were minimally altered. Tumor mass regressed after the 1 and 2 × LD10 BCNU doses and regrew at a significantly slower exponential rate.
The repopulation of clonogenic cells from disaggregated 9L tumors has been suggested to decrease after BCNU therapy (13). This present study reports statistically significant in vivo measurements of a dose-dependent decrease in intracerebral 9L tumor regrowth rates in individual animals after BCNU treatment. The biological basis for the observed decreased growth rates is uncertain. Treatment-induced damage to the tumor stroma and vasculature may account for slower posttreatment growth (39, 40); however, cytotoxic treatments other than radiation therapy are not believed to produce such “tumor bed effects” (41, 42). Alternatively, BCNU may select a subpopulation of cells with reduced cell cycle times or induce nonlethal damage reducing cell growth rates.
It has been asserted that volume-based measurements underestimate cell kill (22). The cell kill values in this study are significantly lower (about 2 logs) than CFE assays of disaggregated single cells from solid 9L tumors (12–14). It is expected that a plot of log cell kill versus BCNU dose (Fig. 5) would pass through the origin if there were no significant systematic errors in cell kill values. The y-intercept of the fit of MRI-calculated cell kill versus BCNU dose was −0.25 ± 0.1 (±SE) log kill (Fig. 5), indicating a small underestimation of cell kill may have occurred in the range of 0.15–0.35 log.
Such underestimation of volume-based cell kill measurements may be caused by the contribution of dead cells to residual tumor volume after treatment. It does not appear that this artifact contributed significantly to the measurements made in this study, especially for the high-dose BCNU group. As shown in Fig. 3D, the 9L tumor was treated at a size of 30 mm3 and grew to a volume of 95 mm3 before shrinking to below Vpre 18 days later. This posttreatment residual volume is negligible compared with the final volume of 430 mm3 after the 2- to 3-week period of exponential volumetric regrowth. The period of regression was more than required to remove dead tissue from the rat brain (38).
Cellular swelling and extracellular edema also have been reported to interfere with volume-based estimates of cell kill (22). Increases in peritumoral edema and changes in tumor contrast after BCNU resulted in less defined tumor boundaries, but these changes did not interfere with cell kill measurements because the volumetric data affected by these processes were not used in the cell kill calculation. In previous studies, we have observed that animals with 9L tumors treated with BCNU (LD10 dose) had a histological appearance during the volumetric exponential regrowth phase that was similar to untreated tumors (36). More recently, we also have confirmed this finding with animals that were treated with 2 × LD10 dose of BCNU as well (unpublished data). This finding confirms that cellular swelling, dead cell debris, and extracellular matrix changes do not contribute a significant residual volume. Measurements of water mobility using diffusion-weighted MRI, which is a sensitive method for detecting changes in tissue structure, revealed an increase in tumor water diffusion after a 1 × LD10 dose of BCNU, which returned to pretreatment levels during the exponential regrowth phase (36). This pattern of diffusion changes also has been observed for the 0.5 and 2 × LD10 BCNU doses (data not shown). These serial diffusion MRI measurements in a single animal confirm that no significant perturbations in the properties of water diffusion in 9L tumors remain during the exponential volumetric regrowth phase. This finding suggests that the structure of the tumor tissue was not significantly altered.
It is difficult to explain why MRI cell kill values for BCNU-treated intracerebral 9L tumors are significantly lower than those obtained with more traditional assays. CFE directly measures the clonogenic fraction of a population of cells. Application of this assay to in vivo studies requires that intracerebral tumors be removed from the host animal, minced, and disaggregated into cell suspensions using trypsin or an enzyme mixture (13). This procedure may have differential effects on subpopulations of tumor cells; BCNU-damaged cells may be especially sensitive and killed by the disaggregation protocol. For example, it has been observed previously that the in vitro CFE assay may overestimate the amount of killing by BCNU in a study involving solid murine EMT6/SF tumors (40).
It has been asserted that an increase in animal survival cannot be detected below a threshold tumor cell kill of 90% (12). This assertion should be reevaluated in view of the present MR data showing that a significant reduction in tumor Tdpost occurs after BCNU treatment. The decrease in growth rate after the 1 and 2 × LD10 BCNU doses contributed 33% and 53%, respectively, to the time required to repopulate the tumor to its pretreatment mass. As a specific example, the 1.6-log kill achieved in the animal shown in Figs. 2 and 3D corresponds to a fractional cell survival of 2.5%. The residual tumor would require 5.25 doublings to repopulate to Vpre. Assuming that the tumor Tdpre did not change after treatment for this animal (e.g., Tdpre = Tdpost = 46 h; Fig. 1, line B), then the time to repopulate would have been 241.5 h (Fig. 1; Trapre). The observed 481 h is much longer and is consistent with the calculated repopulation time of 520 h (Fig. 1; Trapost) obtained using the MRI-observed Tdpost of 99 h. In this animal, the decreased posttreatment growth rate added 11.6 days (Trapost − Trapre) to the repopulation time. The relative contributions of cell kill (χ) and the decreased tumor Tdpost (δ) to the repopulation time were 47% and 53%, respectively.
These results have important implications for interpreting data obtained from brain tumor studies that rely solely on animal survival as a biological endpoint. If the contribution of slower tumor regrowth after treatment is not considered, the ILS produced by a cytotoxic drug could be incorrectly interpreted as a larger cell kill than was actually achieved. We are unaware of any data supporting a decrease in tumor regrowth rates in patients after BCNU treatment. The disparity in the efficacy of BCNU chemotherapy for 9L tumors in rats and primary brain tumors in patients may be partially explained if BCNU does not effect the regrowth rate of human tumors in situ to the same extent as it does orthotopic 9L brain tumors.
The ability of quantitative MRI to accurately quantify 9L brain tumor volume, growth rate, and cell kill in individual animals with a single noninvasive assay provides a more informative avenue for evaluating preclinical therapeutic studies. Differences in tumor growth rates and size at the time of initial treatment can be controlled, thereby reducing interanimal variability. MRI also requires fewer numbers of animals than CFE or tumor weight assays, because groups of animals need not be sacrificed for each experimental time point. This may be especially useful for more costly brain tumor models such as human glioma xenografts in nude rodents. Tumor growth rate and cell kill in individual animals could be correlated with animal life span. Finally, this study also demonstrates that the application of MRI to well-characterized treatments like BCNU may yield additional insights into their mechanism(s) of action.
Acknowledgments
This research was supported in part by Grants RPG-92-019-05-TBE from the American Cancer Society and R29-CA59009 from the National Institutes of Health. O.B.-Y. is a Fellowship recipient of the American Brain Tumor Association. L.D.S. is a Fellow in the Medical Scientist Training Program supported by an institutional training grant (NIGMS T32 GM07863).
ABBREVIATIONS
- BCNU
1,3-bis(2-chloroethyl)-1-nitrosourea
- CFE
colony-forming efficiency
- ILS
increased life span
- Td
doubling time
- Tr
time required for repopulation of tumor cells killed by treatment
References
- 1.Mahaley M S J, Mettlin C, Natarajan N, Laws E R J, Peace B B. J Neurosurg. 1989;71:826–836. doi: 10.3171/jns.1989.71.6.0826. [DOI] [PubMed] [Google Scholar]
- 2.Cohen M E, Duffner P K. Brain Tumors in Children: Principles of Diagnosis and Treatment. New York: Raven; 1994. [Google Scholar]
- 3.Loeffler J S, Alexander E, Shea W M, Wen P Y, Fine H A, Kooy H M, Black P M. J Clin Oncol. 1992;10:1379–1385. doi: 10.1200/JCO.1992.10.9.1379. [DOI] [PubMed] [Google Scholar]
- 4.Crafts D, Wilson C B. J Natl Cancer Inst Monog. 1977;46:11–17. [PubMed] [Google Scholar]
- 5.Peterson D L, Sheridan P J, Brown W E., Jr J Neurosurg. 1994;80:865–876. doi: 10.3171/jns.1994.80.5.0865. [DOI] [PubMed] [Google Scholar]
- 6.Roosen N, Rosenblum M L. In: Brain Tumors: An Encyclopedic Approach. Kay A H, Laws E R Jr, editors. New York: Churchill Livingstone; 1995. pp. 387–404. [Google Scholar]
- 7.Edelman F L, Aleu F, Scheinberg L C, Evans J C, Davidoff L M. J Neuropath Exp Neurol. 1964;23:1–17. doi: 10.1097/00005072-196401000-00001. [DOI] [PubMed] [Google Scholar]
- 8.Shapiro W R, Ausman J I. In: Recent Advances in Neurology. Plum F, editor. Philadelphia: Davis; 1969. [Google Scholar]
- 9.Ausman J I, Shapiro W R, Rall D P. Cancer Res. 1970;30:2394–2400. [PubMed] [Google Scholar]
- 10.Levin V A, Shapiro W R, Clancy T P, Oliverio V T. Cancer Res. 1970;30:2451–2455. [PubMed] [Google Scholar]
- 11.Barker M, Hoshino T, Gurcay O, Wilson C B, Nielsen S L, Downie R, Eliason J. Cancer Res. 1973;33:976–986. [PubMed] [Google Scholar]
- 12.Rosenblum M L, Wheeler K T, Wilson C B, Barker M, Knebel K D. Cancer Res. 1975;35:1387–1391. [PubMed] [Google Scholar]
- 13.Rosenblum M L, Knebel K D, Vasquez D A, Wilson C B. Cancer Res. 1976;36:3718–3725. [PubMed] [Google Scholar]
- 14.Rosenblum M L, Knebel K D, Vasquez D A, Wilson C B. J Neurosurg. 1977;46:145–154. doi: 10.3171/jns.1977.46.2.0145. [DOI] [PubMed] [Google Scholar]
- 15.Nomura K, Hoshino T, Knebel K, Deen D F, Barker M. Cancer Treat Res. 1978;62:747–754. [PubMed] [Google Scholar]
- 16.Barker M, Deen D F, Baker D G. Int J Radiat Oncol Biol Phys. 1979;5:1581–1583. doi: 10.1016/0360-3016(79)90776-4. [DOI] [PubMed] [Google Scholar]
- 17.Wheeler K T, Kaufman K, Feldstein M. Int J Radiat Oncol Biol Phys. 1979;5:1553–1557. doi: 10.1016/0360-3016(79)90770-3. [DOI] [PubMed] [Google Scholar]
- 18.Wheeler K T, Kaufman K. Int J Radiat Oncol Biol Phys. 1980;6:845–849. doi: 10.1016/0360-3016(80)90322-3. [DOI] [PubMed] [Google Scholar]
- 19.Rosenblum M L, Deen D F, Hoshino T, Dougherty D A, Williams M E, Wilson C B. Br J Cancer. 1980;41:307–308. [PMC free article] [PubMed] [Google Scholar]
- 20.Wheeler K T, Wallen C A. Br J Cancer. 1980;41:299–303. [PMC free article] [PubMed] [Google Scholar]
- 21.Rosenblum M L, Dougherty D A, Brown J M, Barker M, Deen D F, Hoshino T. Cell Tissue Kinet. 1980;13:667. [Google Scholar]
- 22.Weizsaecker M, Deen D F, Rosenblum M L, Hoshino T, Gutin P H, Barker M. J Neurol. 1981;224:183–192. doi: 10.1007/BF00313280. [DOI] [PubMed] [Google Scholar]
- 23.Wheeler K T, Kaufman K. J Neurosurg. 1981;55:52–54. doi: 10.3171/jns.1981.55.1.0052. [DOI] [PubMed] [Google Scholar]
- 24.Wheeler K T. Cancer Treat Rep. 1981;65:75–81. [PubMed] [Google Scholar]
- 25.Vats T S, Kimler B F, Henderson S D, Morantz R A. Cancer Treat Rep. 1982;66:575–579. [PubMed] [Google Scholar]
- 26.Gutin P H, Bernstein M, Sano Y, Deen D F. Neurosurgery. 1984;15:781–786. [PubMed] [Google Scholar]
- 27.Kimler B F, Liu C, Evans R G, Morantz R A. J Neurooncol. 1992;14:191–200. doi: 10.1007/BF00172594. [DOI] [PubMed] [Google Scholar]
- 28.Kimler B F. J Neurooncol. 1994;20:103–109. doi: 10.1007/BF01052721. [DOI] [PubMed] [Google Scholar]
- 29.Wheeler K T, Kaufman K. Int J Radiat Oncol Biol Phys. 1981;7:1065–1068. doi: 10.1016/0360-3016(81)90160-7. [DOI] [PubMed] [Google Scholar]
- 30.Walker M D, Alexander E, Jr, Hunt W E, MacCarty C S, Mahaley M S, Mealey M, Norrell H A, Owens G, Ransohoff J, Wilson C B, et al. J Neurosurg. 1978;49:333–343. doi: 10.3171/jns.1978.49.3.0333. [DOI] [PubMed] [Google Scholar]
- 31.Walker M D, Greed S B, Byar D P, Alexander E J, Batzdorf U, Brooks W H, Hunt W E, MacCarty C S, Mahaley M S J, Mealey J J, et al. N Engl J Med. 1980;303:1323–1329. doi: 10.1056/NEJM198012043032303. [DOI] [PubMed] [Google Scholar]
- 32.Kornblith P L, Walker M. J Neurosurg. 1988;68:1–17. doi: 10.3171/jns.1988.68.1.0001. [DOI] [PubMed] [Google Scholar]
- 33.Grossman S A. In: Concepts in Neurosurgery. Salcman M, editor. Baltimore: Williams & Wilkins; 1991. pp. 321–340. [Google Scholar]
- 34.Kim B, Chenevert T L, Ross B D. Clin Cancer Res. 1995;1:643–650. [PubMed] [Google Scholar]
- 35.Steel G G. Growth Kinetics of Tumors. Oxford: Clarendon; 1977. [Google Scholar]
- 36.Chenevert T L, McKeever P E, Ross B D. Clin Cancer Res. 1997;3:1457–1466. [PubMed] [Google Scholar]
- 37.Wilcox W S, Griswold D P, Laster W R J, Schabel F M J, Skipper H E. Cancer Chemother Rep. 1965;47:27–39. [PubMed] [Google Scholar]
- 38.Kumar A R V, Hoshino T, Wheeler K T, Barker M, Wilson C B. J Natl Cancer Inst. 1974;52:1751–1755. doi: 10.1093/jnci/52.6.1751. [DOI] [PubMed] [Google Scholar]
- 39.Hewitt H B, Blake E R. Br J Cancer. 1968;12:808–824. doi: 10.1038/bjc.1968.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Begg A C, Fu K K, Kane L J, Phillips T L. Cancer Res. 1980;40:145–154. [PubMed] [Google Scholar]
- 41.Kallman R F. Rodent Tumor Models in Experimental Cancer Therapy. New York: Pergamon; 1987. [Google Scholar]
- 42.Begg A C, Denekamp J. Eur J Cancer Clin Oncol. 1983;19:1639–1643. doi: 10.1016/0277-5379(83)90097-4. [DOI] [PubMed] [Google Scholar]