

Abstract
Hypoxia inducible factor-1 (HIF-1) is a master regulatory transcription factor controlling multiple cell-autonomous and non–cell-autonomous processes, such as metabolism, angiogenesis, matrix invasion, and cancer metastasis. Here we used a new line of transgenic mice with constitutive gain of HIF-1 function in basal keratinocytes and demonstrated a signaling pathway from HIF-1 to nuclear factor κ B (NFκB) activation to enhanced epithelial chemokine and cytokine elaboration. This pathway was responsible for a phenotypically silent accumulation of stromal inflammatory cells and a marked inflammatory hypersensitivity to a single 12-O-tetradecanoylphorbol-13-acetate (TPA) challenge. HIF-1–induced NFκB activation was composed of 2 elements, IκB hyperphosphorylation and phosphorylation of Ser276 on p65, enhancing p65 nuclear localization and transcriptional activity, respectively. NFκB transcriptional targets macrophage inflammatory protein-2 (MIP-2/CXCL2/3), keratinocyte chemokine (KC/CXCL1), and tumor necrosis factor [alfa] (TNFα) were constitutively up-regulated and further increased after TPA challenge both in cultured keratinocytes and in transgenic mice. Whole animal KC, MIP-2, or TNFα immunodepletion each abrogated TPA-induced inflammation, whereas blockade of either VEGF or placenta growth factor (PlGF) signaling did not affect transgenic inflammatory hyper-responsiveness. Thus, epithelial HIF-1 gain of function remodels the local environment by cell-autonomous NFκB-mediated chemokine and cytokine secretion, which may be another mechanism by which HIF-1 facilitates either inflammatory diseases or malignant progression.
Introduction
Inflammation assumes many forms, such as the response to acute epithelial barrier injury, acute microbial infection, recurrent tissue nondestructive lesions as in psoriasis, chronic tissue destruction as seen in a myriad of autoimmune or innate inflammatory diseases, or chronic microbial/viral infestations and infections. Chronic inflammation with attendant microenvironmental alterations, resulting from growth factor, chemokine, cytokine secretion, and reactive oxygen species enrichment, also provides a fertile soil for de novo development of epithelial cancer.1,2 Elucidation of the mechanisms of inflammatory modulation by epithelial cells is an emergent focus of investigation. Models of epithelial elaboration of specific inflammatory chemokines, cytokines, or growth factors have shed light on the roles of specific signaling networks in disease. In contrast, the concept that transcription factors regulate production of a repertoire of inflammatory chemokines and cytokines by epithelial cells is relatively new.3,4 Here we demonstrate that gain of function of the transcription factor hypoxia inducible factor-1 (HIF-1), signaling indirectly through nuclear factor [kappa2]B (NFκB), can “prime” and remodel the local stromal environment to affect an augmented, enhanced responsiveness “hyper-responsiveness” to an inflammatory stimulus. This may be yet another mechanism, broadcast from the epithelial cells themselves, by which both HIF-1 and NFκB conspire to increase the severity of inflammatory diseases and carcinogenic progression.
HIF-1 is a fundamental mediator of cellular adaptation to hypoxia, activating metabolic and signaling pathways promoting cell survival. HIF-1 consists of the oxygen-sensitive subunit HIF-1α and the constitutively expressed HIF-1β subunit. In normoxia, the alpha subunit is hydroxylated in position 402 and 564 by 3 prolyl hydroxylase enzymes (PHD) 1 to 3. These modifications allow interaction between HIF-1α and the von Hippel-Lindau protein, targeting HIF-1α for proteasomal degradation. In hypoxia, PHD activity is decreased, preventing HIF-1α degradation.5,6 When HIF-1α levels increase, functional HIF-1 regulates transcription at hypoxia response elements of target gene enhancers, up-regulating genes involved in energy metabolism and angiogenesis.7 HIF-1α protein synthesis can be regulated in an O2-independent manner by activation of the phosphatidylinositol 3-kinase and ERK mitogen-activated kinase pathways that are either physiologically stimulated by cognate growth factors or cytokines or activated by mutation. Thus, proinflammatory cytokines, interleukin-1β and tumor necrosis factorα (TNFα) have each been shown to stabilize HIF-1α protein, suggesting that HIF-1α functions can be recruited by tissue inflammation.8–10 The concept of HIF-1α as an inflammatory mediator is supported by demonstration of HIF-1α protein stabilization in rheumatoid synovial macrophages.11 Conditional targeting of HIF-1α in mouse myeloid cells inhibits inflammation resulting from impairment of the phagosome respiratory burst, itself secondary to limited adenosine triphosphate availability due to diminished glycolysis.12 HIF-1α also plays a crucial role in hypoxic T-cell and neutrophil survival, another important determinant of tissue inflammation.13 In contrast to HIF-1α's requirement in leukocytes, a cell autonomous role for HIF-1α in epithelial inflammatory signaling is heretofore unknown.
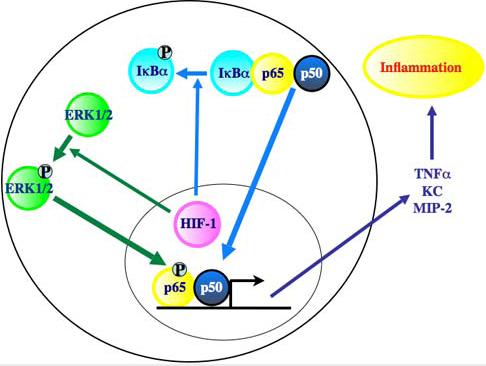
Here we discovered that epithelial HIF-1 gain of function constitutively elevated proinflammatory chemokine and cytokine secretion cell-autonomously by NFκB pathway engagement. HIF-1-NFκB cross-talk was mediated by 2 elements, extracellular stimuli–responsive kinase 1/2 (ERK1/2) activation regulating p65Ser276 phosphorylation, and up-regulation of an unknown upstream signaling input enhancing IκB phosphorylation, degradation, and consequent p65 nuclear localization. Thus, HIF-1, via NFκB, remodeled the local stromal microenvironment, elevating inflammatory cell infiltration and enhancing inflammation; 2 non–cell-autonomous functions that could also augment neoplastic progression and malignant cell growth.
Methods
Transgenic mice
Plasmids p(HA)HIF-1apro402A/564G7 were digested with XbaI and Asp718I, the cDNA insert gel purified over QIAquick columns (QIAGEN, Valencia, CA) blunted with Klenow polymerase, and cloned into an SmaI-linearized human keratin-14 (K14) expression cassette. Plasmid DNA was prepared with QIAGEN Endofree Plasmid Maxi Kit. The entire transgene insert was excised by Asp718I digestion, purified,14 and transgenic mice were produced by DNA pronuclear injection into FVB/n embryos. Construction and characterization of K14-HIF-1αΔ oxygen-dependent degradation domain (ODD) transgenic mice, expressing a constitutive form of HIF-1α lacking the 200 amino acid ODD, were previously described15 and maintained in the FVB/n background. Mice were housed in pathogen-free conditions, and the Washington University Institutional Animal Care Committee approved these experiments.
Tissue harvest and histology
Tissues were harvested from mice and processed using a previously reported rapid microwave fixation technique.16 Immunohistochemical determination of inflammatory cell subsets was performed on paraffin-embedded, zinc-fixed tissues.
Reagents
For inflammatory challenge experiments in intact mice, 2.5 μg of 12-O-tetradecanoylphorbol-13-actate (TPA) (Sigma-Aldrich, St Louis, MO) was dissolved in 40 μL acetone and topically applied on each side of the ear. Keratinocyte chemokine (KC) and macrophage inflammatory protein-1 (MIP-2) were immunodepleted in intact mice using 5 μg of each monoclonal neutralizing antibody (R&D Systems, Minneapolis, MN) intravenously administered 5 minutes before TPA painting. Vascular endothelial growth factor (VEGF) signaling blockade was achieved by intraperitoneal injection of a cocktail of 2 rat monoclonal blocking antibodies against VEGF receptor-1 (VEGFR-1) (MF-01, 800 μg), and VEGFR-2, (DC101, 800 μg) (both from ImClone, New York City, NY) 1 day before TPA challenge. Control mice were injected with 800 μg of rat IgG2a isotype antibody i.p. TNFα immunodepletion was achieved using 300 μg of hamster mAb TN3–19.1217 given i.p. 1 day before TPA challenge.
Histopathology and immunohistochemistry
Zinc-fixed tissue sections were immunostained with a hamster antibody against intercellular adhesion molecule-1 (ICAM-1, BD PharMingen, San Diego, CA) diluted 1:20 in antibody diluent (Dako North America, Carpinteria, CA), rat antibodies against CD45.1, CD4, and CD8 (BD PharMingen) diluted 1:50 in antibody diluent, a polyclonal rabbit antibody against the phosphorylated form of mitogen activated protein kinase p44/42 (ERK1/2) (Cell Signaling, Danvers, MA) diluted 1:1000 in antibody diluent, and the staining was visualized using an immunoperoxidase technique (Vectastain ABC kit, Vector Laboratories, Burlingame, CA), and diaminobenzadine (Dako North America). Immunofluorescence was performed on 5-μm sections using antibodies/dilutions for the following markers: rat antibody against mouse MECA32 (1:50, BD PharMingen), polyclonal rabbit antibodies against LYVE1 (1:200, Abcam, Cambridge, MA), rat anti-GR-1 (1:50, BD PharMingen), rat anti-F4/80 (1:50, Serotec, Raleigh, NC), keratin 14 (1:1000, Covance Research Products, Princeton, NJ). Secondary antibodies were labeled with Alexa Fluor 488 or Alexa Fluor 594 (Invitrogen, Carlsbad, CA). Nuclei were counterstained using DAPI containing mounting media (Vector Laboratories). Mast cells were quantified according to Wershil et al18 with modification. Skin sections were stained with toluidine blue (Sigma-Aldrich). Cells were counted in 6 random 20× fields per mouse ear (n = 6 for each group).
Quantitative RT-PCR
Ears were snap-frozen in liquid nitrogen, homogenized in TRIzol Reagent (Invitrogen), and total RNA was isolated using the manufacturer's guidelines. Real-time polymerase chain reaction (RT-PCR) was performed as described previously15 using a MX3000P thermocycler and detection system (Stratagene, La Jolla, CA). Primer Express software (version 2.0, Applied Biosystems, Foster City, CA) was used to design primer/probe sets (Table S1, available on the Blood website; see the Supplemental Materials link at the top of the online article) for gene amplification. All target cDNAs were normalized to histone 3.3A.15
Western blotting
Whole-cell extracts were lysed in radioimmuno precipitation assay (RIPA) buffer (10 mM Tris (pH 7.4), 150 mM NaCl, 1 mM EDTA, 0.1% sodium dodecyl sulfate, 1% Triton X-100, 1% sodium deoxycholate, 0.25 mM phenylmethylsulfonylfluoride). For cell fractionation, keratinocytes were lysed in ice-cold nuclear extraction buffer (25 mM Tris, 1 mM MgCl2, 5 mM KCl, 0.25% Nonidet P-40). Protein extracts were separated on polyacrylamide gels followed by Western transfer to polyvinylidene diflouride membranes. Membranes were blocked in Blotto-Tween solution (5% nonfat dry milk, 0.1% Tween in phosphate-buffered saline) for 1 hour and then incubated overnight in phosphate-buffered saline with a rabbit polyclonal antibody for HIF-1α (Bethyl Laboratories, Montgomery, TX), mouse monoclonal Sp1 (BD PharMingen), and rabbit polyclonal antibodies for p65, IκBα, phospho-IκBα, Erk1/2, phospho-Erk1/2, p65 phospho-Ser276, p65 phospho-Ser536 (Cell Signaling), or β-tubulin (Sigma-Aldrich). Peroxidase-coupled AffiniPure rabbit antimouse immunoglobulin G (Jackson ImmunoResearch Laboratories, West Grove, PA) was used in secondary incubations for 1 hour followed by chemiluminescent detection (ECL Plus, GE Healthcare, Little Chalfont, United Kingdom).
ELISA
VEGF-A, PlGF, KC, MIP-2, and TNFα levels in keratinocyte cell culture media supernatants, or ear extracts, were measured by enzyme-linked immunosorbent assay (ELISA) according to manufacturer's instructions (BD Biosciences, San Jose, CA).
Vascular leakage
Microvascular leakage was determined as previously reported.15 Briefly, transgenic and nontransgenic mice 8 to 12 weeks of age were anesthetized (Avertin 2.5%) and intravenously injected with Evans blue dye, 30 mg/kg per mouse. The right ear was collected 6 hours after topical TPA application (5 μg, Sigma-Aldrich), whereas the left ear was a vehicle (acetone control). Ears were harvested 30 minutes after dye injection and processed for spectrophotometric determination as described previously.15
Transfection and luciferase reporter assay
Primary keratinocytes were transfected using Lipofectamine 2000 according to the manufacturer's protocol (Invitrogen). Transient transfection was conducted for 48 hours before treatment with TPA. Luciferase activity was measured by using the luciferase reporter assay kit (BD Biosciences) according to manufacturer's protocol. Results were normalized to the total protein content.
Adenovirus transduction
The IκBαSR adenovirus was introduced into primary keratinocytes using an adenoviral construct regulated by a cytomegalovirus promoter19 with empty adenovirus as a control. Keratinocytes were infected for 30 minutes in serum-free medium with a multiplicity of infection of 5 viral particles/cell and 2.5 μg/mL of Polybrene (Sigma-Aldrich).
Keratinocyte culture
Primary mouse keratinocytes were isolated from newborn transgenic and wild-type littermate epidermis as described,20 and seeded at a density of 5 × 106 cells per 60-mm dish (or equivalent concentrations) in Ca2+- and Mg2+-free minimum essential medium (Invitrogen) supplemented with 8% Chelex (Bio-Rad, Hercules, CA)-treated fetal bovine serum (Gemini, Irvine, CA) and 0.2 mM Ca2+. After 24 hours, cultures were switched to the same medium with 0.05 mM Ca2+ to select for basal cells. TPA was reconstituted in dimethyl sulfoxide (DMSO), and primary keratinocytes were treated with 5 ng/mL TPA for various times as indicated in individual experiments. Blockade of CXCR2 signaling was performed using a neutralizing antibody (R&D Systems) at 10 μg/mL of cell culture media.
Microscopy and image processing
All microscopy images were obtained with a BX61 microscope, (Olympus America, Central Valley, PA) using an UPlan Apochromatic 20×/0.70 NA objective. Tissue sections stained with hematoxylin/eosin or diaminobenzadine immunoperoxidase were mounted with Permount (SP15-500, Fisher Scientific, Pittsburgh, PA) and coverslipped. Microscopy images were obtained with a DP70 color Bayer mosiac digital camera, Peltier device cooled to −10°C, (Olympus America). Tissue sections for fluorescence microscopy images were mounted with SlowFade Gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen, Molecular Probes, Eugene, OR) and coverslipped, and images were obtained with a Soft Imaging Solutions FVII cooled monochrome digital camera, Peltier cooled to −10°C (Olympus America). All images were captured with MicroSuite Biological Suite version 5 software (Olympus Soft Imaging Solutions, Lakewood, CO) and resized and formatted with Adobe Photoshop CS3 software (Adobe Systems, San Jose, CA).
Statistical methods
Data were analyzed using GraphPad PRISM (San Diego, CA). The results were expressed as the mean plus or minus SEM, and statistical significance was determined using the Student t test.
Results
Novel epidermal HIF-1α gain of function mice display baseline tissue components necessary for “inflammatory priming”
Previously, our laboratory created transgenic mice overexpressing a form of HIF-1α with a 200 amino acid deletion of the oxygen-dependent degradation domain, K14-HIF-1αΔODD transgenic mice,15 a deletion that removes both the N-terminal transactivation domain and a critical regulatory leucine at 574, the sumoylation target at lysine 477, an acetylation target at lysine 532, and the hydroxylation targets at proline 402 and 564. To more faithfully model epithelial HIF-1α gain of function, we targeted expression of a form of HIF-1α containing proline-alanine and proline to glycine point mutations at positions 402 and 564 within the intact protein,7 to basal epidermal keratinocytes (K14-HIF-1αPro402A/564G, referred to hereon as K14-HIF-1α double-proline mutant (DPM) transgenic mice; Figure 1A-D). We compared transgene mRNA and protein expression along with cellular transcriptional activity of the K14-HIF-1αDPM with the K14-HIF-1αΔODD transgenic models. Levels of transgene mRNA and total human plus residual murine HIF-1α protein were greater in keratinocytes harvested from K14-HIF-1αΔODD compared with K14-HIF-1αDPM mice (Figure S1A,B). In contrast, K14-HIF-1αDPM transgenic keratinocytes expressed greater levels of the HIF-1 target genes VEGF-A and PlGF compared with K14-HIF-1αΔODD counterparts (Figure S1C,D). Thus, HIF-1 target gene expression was actually greater relative to transgene mRNA and protein expression in K14-HIF-1αDPM compared with K14-HIF-1αΔODD transgenic mice. Reduced transcriptional activity of the HIF-1α deletion compared with the point mutant is consistent with retention of the N-terminal transactivation domain in the point mutant transgene.21
Figure 1.

“Inflammatory priming” by epithelial mediated stromal inflammatory cell recruitment in K14-HIF-1αDPM transgenic mice. Redness and prominent vasculature of ear skin in transgenic mice (DPM) (B), which are not evident in nontransgenic mice (NTG) (A). Human HIF-1α mRNA (C) and total (human and potentially mouse) HIF-1α protein (D) are detectable only in transgenic ears. Ear histology reveals increased dermal cellularity in transgenic mice (F, arrows), compared with nontransgenic mice (E). Differential increase in subepidermal microvessels, revealed by MECA-32 immunohistochemistry (green), and lymphatic dilatation, LYVE-1 antibody (red, see white arrow) in DPM transgenic mice (H) versus nontransgenic controls (G). Endothelial activation evidenced by ICAM-1 immunohistochemistry, in transgenic (J), versus nontransgenic (I) ears. Increased number of CD45.1 positive inflammatory cells in transgenic (L) versus nontransgenic (K) ears. A 2- to 3-fold increase of neutrophils, mast cells, macrophages, and lymphocytes, determined by differential immunohistochemical analysis of inflammatory markers as indicated in panel M. Bar in panel F represents 100 μm.
Similar to K14-HIF-1αΔODD transgenic mice,15 unfurred skin of K14-HIF-1αDPM mice was redder than nontransgenic littermates (Figure 1A,B) because of increased microvascular density and dilatation (Figure 1G,H), associated with an 8-fold elevation of VEGF-A mRNA and protein expression. Endothelial cells of the K14-HIF-1αDPM dermal microvessels evidenced low-level activation by decoration with ICAM-1, (Figure 1I, J) and E-selectin (data not shown), similar to K14-VEGF-A transgenic mice.22 In addition, dermal lymphatics were also enlarged (Figure 1G,H).
We also noted a constitutive increase in overall dermal cellularity on histologic analysis of K14-HIF-1αDPM transgenic ears (Figure 1E,F), a finding bolstered by a 2-fold increase in CD45 expressing dermal cells in K14-HIF-1αDPM mice compared with nontransgenic controls (Figure 1K,L). In contrast, dermal cellularity was not noticeably increased in ears harvested from K14-HIF-1αΔODD transgenic mice, consistent with our previously published data.15 CD45 cell quantification revealed a statistically significant 0.25-fold elevation in K14-HIF-1αΔODD transgenic mice compared with nontransgenic controls, which was markedly less compared with the enhanced baseline accumulation in the point mutant counterparts (Figure S1E). Thus, we focused further analysis of inflammatory cell populations in K14-HIF-1αDPM transgenic mice compared with nontransgenic controls. This analysis revealed constitutive elevation of dermal CD4 and CD8 lymphocytes, F4/80-positive macrophages, GR-1–positive cells, and mast cells in transgenic versus nontransgenic controls (Figure 1M). Thus, one mechanism of low-level basal dermal inflammatory cell infiltration in the K14-HIF-1αDPM transgenic mice could be a combination of enhanced mononuclear cell attachment to VEGF-activated endothelial cells facilitating diapedesis and consequent secondary lymphatic engorgement.
K14-HIF-1αDPM mice exhibit enhanced and prolonged response to an inflammatory challenge
The combination of constitutive transgenic dermal inflammatory cell infiltration and endothelial cell activation led us to investigate how transgenic skin would respond to an inflammatory challenge. Topical application of TPA to the ear of K14-HIF-1αDPM transgenic mice led to a massive inflammatory response as soon as 6 hours after treatment (Figure 2B). Transgenic ear epidermis was punctuated by microabscesses filled with neutrophils, a phenotype resembling the Munro microabscesses, one hallmark of human psoriasis.23,24 Our original K14-HIF-1αΔODD transgenic model was included in these experiments to correlate inflammatory responsiveness to baseline CD45 cell dermal accumulation. Microabscesses were also observed in the K14-HIF-1αΔODD mice (Figure S1F); however, they were always less frequent and smaller compared with K14-HIF-1αDPM counterparts, again reflecting differential transcriptional activity described in cultured keratinocytes from the 2 models (Figure S1C,D). Because our data consistently demonstrated greater inflammatory activation in the K14-HIF-1αDPM transgenic mice, we chose to focus our remaining studies on our newer transgenic model. Another factor motivating focus on the HIF-1αPro402A/564G transgene was its more faithful structure (eg, retention of the N-terminal transactivation domain) and functional relationship to wild-type HIF-1α. This was important as we were modeling HIF-1α gain of function documented in inflammatory, neoplastic, and malignant disease.11–13
Figure 2.

Transgenic mice exhibit enhanced and prolonged response to an inflammatory challenge. Intraepidermal neutrophilic abscesses 6 hours after TPA challenge in transgenic mice (B) compared with low-level dermal inflammatory cell infiltrate in nontransgenic controls (A). Persistent ear swelling 10 days after TPA challenge evidenced by caliper ear thickness in transgenic (DPM) versus control (NTG) ears (C). Histologic resolution of inflammation in nontransgenic ears 10 days after TPA challenge (D). In contrast, transgenic ears (E,F) evidence psoriatic-type changes of increased epidermal thickness, epidermal rete ridge dermal projections, subepidermal edema, and foci of parakeratosis (F). Bar in panel A represents 100 μm.
Munro microabscesses were also produced in K14-VEGF-A as well as K5-PKCα transgenic mice.22,25 In contrast, nontransgenic ears displayed only a minimal mononuclear cell dermal infiltration at this early posttreatment interval (Figure 2A). Caliper determination of ear swelling demonstrated that nontransgenic ear thickness reached its peak 24 hours after challenge (Figure 2C), with a return to baseline by day 10 after treatment. Histopathology of day 10 after treatment nontransgenic ears indicated a residual epidermal thickening (Figure 2D). In contrast, K14-HIF-1αDPM mice were unable to down-regulate TPA-induced inflammation. Transgenic ears evidenced thickening of the epidermis (hyperkeratosis), focal accumulations of nucleated keratinocytes in the uppermost stratum corneum skin layer (parakeratosis), rete ridge type projections of epidermis into the dermis, similar to what is observed in early human psoriasis, and increased dermal cellularity (inflammatory infiltrate) (Figure 2E,F). Caliper measurement confirmed a persistent and significant thickening of the transgenic ears 10 days after treatment (Figure 2C).
Despite the increased levels of VEGF-A and PlGF (Figure 3A-C), K14-HIF-1αDPM transgenic mice did not exhibit enhanced vascular leakage, neither at baseline nor 6 hours after TPA challenge (Figure 3D), similar to our previous work with K14-HIF-1αΔODD transgenic mice.15 Thus, differential inflammatory cell accumulation in K14-HIF-1αDPM transgenic ears after TPA challenge could not be explained by increased vasculature leakage as in K14 VEGF-A transgenic mice.22
Figure 3.

Increased VEGF-A and PlGF protein levels without microvascular leakage in transgenic mice. Real-time TaqMan RT-PCR analysis of VEGF-A expression from total RNA isolated from ears of K14-HIF-1αDPM transgenic mice (DPM) and nontransgenic controls (NTG), using histone 3.3A as a reference (A), at baseline “C” and after 6 hours of TPA treatment “T.” VEGF-A (B) and PlGF (C) ELISA assays reveal significantly higher protein expression in untreated and TPA treated transgenic ears compared with nontransgenic controls. Lack of differential microvascular leakage, evidenced by ear Evans blue dye content, in transgenic vs nontransgenic mice (D). Error bars represent mean plus or minus SEM of 4 to 6 mice analyzed per group (*P < .05, Student t test).
KC and MIP-2 levels are elevated in K14-HIF-1αDPM mice and are responsible for the neutrophil infiltration in the epidermis
Next we sought to define the biologic and molecular determinants of transgenic inflammatory hyper-responsiveness. First, we determined the composition and kinetics of the transgenic inflammatory cell population at an early time interval, 6 hours after TPA challenge. GR-1 immunohistochemistry was consistent with neutrophilic intraepidermal inflammatory cell infiltrate in TPA-treated transgenic ears (Figure 4B). There was also a marked differential dermal macrophage infiltration as well (Figure 4F). Next we interrogated potential molecular mediators of inflammatory cell recruitment both at baseline and at earlier post-TPA treatment intervals. Epidermal neutrophil recruitment can be mediated by the chemokines KC (CXCL1), and MIP-2 (CXCL2/3). KC and MIP-2 are related to the 3 human GRO chemokines (CXCL1/3).26–28 RT-PCR on untreated transgenic ears showed a 2-fold constitutive elevation of KC and MIP-2. TNFα mRNA was also constitutively elevated to a similar degree in transgenic ears. RT-PCR analysis revealed differential transgenic expression of these inflammatory mediators as early as 1 hour after TPA treatment (Figure 4M-O). ELISA demonstrated induction of KC, MIP-2, and TNFα protein 3 hours after TPA treatment (Figure 4P-R).
Figure 4.

K14-HIF-1αDPM mice exhibit epidermal and dermal inflammatory infiltrates accompanied by hyperplastic cutaneous blood vessels and lymphovascular dilation. Increase of GR1+ neutrophils in green (A-D) and F4/80+ macrophages in green (E-H) 6 hours (B,F) and 10 days after TPA challenge (D,H) in transgenic ears (DPM) compared with nontransgenic littermates (NTG) (A, E, C, and G, respectively). Counterstaining with a keratin-14 antibody (red) highlights the differential persistent increase in epidermal thickness in transgenic mice. Progressive increase in microvascular density (MECA32, green) along with lymphovascular dilatation (LYVE-1, red) is observed between the 6-hour and 10-day after TPA time intervals in transgenic ears (J,L) compared with nontransgenic littermates (I,K). Bar in panel A represents 100 μm. Real-time TaqMan RT-PCR analysis of keratinocyte chemokine (KC) (M), macrophage inflammatory protein-2 (MIP-2) (N), and TNFα (O) expression from total RNA isolated from ears of transgenic and nontransgenic controls, using histone 3.3A as a reference, at baseline and 1 hour after TPA treatment. KC (P), MIP-2 (Q), and TNFα. (R) ELISA assays reveal significantly higher protein expression in the inflamed ears of transgenic mice after 3 hours and 10 days after a single TPA application. Error bars represent mean plus or minus SEM of 4 to 6 mice analyzed per group (*P < .05).
Because histopathologic analysis demonstrated increased dermal inflammatory cells 10 days after TPA in transgenic ears, we determined the compartmental inflammatory cell accumulation at this time point. Marker-specific immunohistochemistry demonstrated a shift in neutrophil localization to the dermis (Figure 4D). Dermal macrophages were further increased (Figure 4H) compared with 6 hours after TPA. Day 10 microvascular density further increased compared with the 6-hour interval, along with lymphovascular dilatation (Figure 4L). Both phenotypes were consistent with persistently elevated epidermal VEGF secretion. Concomitantly, KC, MIP-2, and TNFα protein levels were persistently elevated in the K14-HIF-1αDPM ears (Figure 4P-R). In contrast, nontransgenic chemokine and cytokine expression was undetectable 10 days after TPA consistent with complete resolution of dermal inflammation and only residual persistent epidermal thickening (Figure 4C,G,P-R).
The increased KC and MIP-2 level in K14-HIF-1αDPM keratinocytes is NFκB dependent
To localize chemokine and cytokine production and determine the signaling networks underlying epithelial HIF-1-mediated basal inflammatory priming and stimulus-induced hyper-responsiveness, we derived keratinocyte cultures and tested differential chemokine and cytokine secretion at baseline and after TPA challenge. Similar to ear skin, elevated VEGF-A, KC, and MIP-2 were present at both the mRNA and protein level in untreated transgenic compared with nontransgenic keratinocytes (Figure 5A-C). Moreover, there was a differentially increased induction of KC and MIP-2 3 hours after TPA challenge. Because KC and MIP-2 are known NFκB target genes, we investigated NFκB transcriptional activity using a NFκB reporter construct. Strikingly, both basal and TPA-induced NFκB transactivation was differentially elevated in transgenic keratinocyte cultures, with a 10-fold increase at baseline and after TPA treatment in K14-HIF-1αDPM cells (Figure 5D).
Figure 5.

Epithelial inflammatory chemokine elaboration requires NFκB signaling. Elevation of KC, MIP-2, and VEGF-A protein expression at baseline “C” and 3 hours after TPA treatment “T” in supernatants conditioned by primary transgenic keratinocytes (A, B, and C, respectively) demonstrate the cell autonomous activity of epithelial HIF-1α gain of function. (D) Increased NFκB transcriptional activity in K14-HIF-1αDPM keratinocytes. Nontransgenic (NTG) and transgenic (DPM) keratinocytes were transiently transfected with a NFκB reporter construct. After 48 hours, cells were treated with DMSO or TPA for 3 hours, harvested, and assayed for luciferase activity. Values are expressed as relative light units per microgram total protein (D). IκBαSer32/34A (IκBα super-repressor, IκBαSR) NFκB transcriptional blockade markedly diminishes keratinocyte chemokine expression (KC, MIP-2, and TNF-α, panels E-G, respectively) but does not affect the HIF-1α target gene (VEGF-A, PlGF, and Glut-1, panels H-J, respectively) expression. RT-PCR analysis using total RNA extracted from a control adenovirus (A-cytomegalovirus) or IκBαSR adenoviral-transduced primary keratinocytes at baseline and 3 hours after TPA treatment. Error bars represent mean plus or minus SEM. Results are representative of 3 independent experiments (*P < .05, t test).
We then asked whether NFκB target gene induction was specific for that transcription factor or a global increase in transcription mediated by HIF-1 gain of function. To test this hypothesis, keratinocytes were transduced with an adenoviral construct expressing the IκBα super-repressor (IκBαSR).19 IκBαSR normalized expression of KC, MIP-2, and TNFα (Figure 5E-G) in transgenic keratinocytes, whereas it had no effect on mRNAs encoding the HIF-1 targets VEGFA, PlGF and GLUT-1 (Figure 5H-J). Thus, HIF-1 gain of function produced elevated proinflammatory chemokines and cytokines cell autonomously in transgenic keratinocytes specifically via enhanced NFκB transcriptional activity.
NFκB activation is dependent on ERK1/2 phosphorylation
To further investigate alterations in NFκB signal transduction associated with epithelial HIF-1 gain of function, we performed a combination of keratinocyte cell fractionation and upstream inhibitor modification. Cell fractionation demonstrated a 3-fold increase of constitutive nuclear p65 (normalized to Sp1) in transgenic keratinocytes (Figure 6A). Next we determined the mechanism underlying NFκB localization in transgenic keratinocytes. The level of p65 mRNA and protein was not differentially increased in transgenic keratinocytes; thus, IκB-mediated cytoplasmic retention was not swamped by massive p65 overexpression. Another mechanism for cytoplasmic NFκB retention is the constitutive level of IκB phosphorylation, which governs its abundance by polyubiquitination and proteasomal destruction. Transgenic keratinocytes contained a 2-fold increase of phospho-IκBα and phospho-IKKα/β, along with a 2-fold decrease of total IκBα protein (Figure 6B). Thus, enhanced nuclear localization of p65, a principle NFκB constituent, may in part underlie the increased constitutive and TPA-induced production of proinflammatory chemokines and cytokines in transgenic keratinocytes.
Figure 6.

NFκB activation in K14-HIF-1αDPM keratinocytes is dependent on increased Erk1/2 phosphorylation. Increased nuclear translocation of the NFκB subunit p65 in transgenic (DPM) versus nontransgenic keratinocytes (NTG), normalized to Sp1 (A). Nuclear extracts were analyzed by Western blots. Western blot of lysates from newborn transgenic (DPM) keratinocytes indicate a 2- to 3-fold increase of phosphorylated IKKα, IκBα, p65 phosphorylation at serines 276 and 536, and phospho-ERK1/2, compared with nontransgenic (NTG) keratinocytes; β-tubulin is a loading control (B). Transgenic keratinocytes were pretreated with DMSO (−) or the MEK1/2 inhibitor U0126 (+) for 24 hours and analyzed at baseline (−) or 10 minutes after TPA treatment (+). Western blots of lysates from newborn transgenic keratinocytes show inhibition of phospho-ERK1/2 and p65 Ser276 phosphorylation by U0126 both at baseline and after TPA treatment (C). In contrast, phospho-IκBα, phospho-p65 Ser536, and the level of IκBα protein remain unaffected by ERK1/2 inhibition (C); total ERK1/2 is used as a loading control. The separating line in panel C represents one lane deleted from the same gel. Real-time RT-PCR analysis of TNFα expression from total RNA extracted from transgenic (DPM) primary keratinocytes shows selective inhibition of TNFα expression 24 hours after U0126 (U0) treatment in contrast to PI3 kinase pathway wortmannin (W), or EGFR AG1478 (AG) inhibition, both of which failed to alter transgenic keratinocyte TNFα expression (D). Bars represent mean plus or minus SEM. Results are representative of 3 independent experiments (*P < .05, t test). (E-F) Increased ERK1/2 phosphorylation is also evident in immunohistochemical analysis of transgenic compared with nontransgenic ears (E,F). Bar in panel E represents 100 μm.
To dissect potential upstream signaling inputs to NFκB, we first interrogated the PKCα pathway, as it is a TPA target, and an important regulator of NFκB activity in the skin.29,30 However, the level of constitutive phospho- and total PKCα was similar in transgenic compared with nontransgenic keratinocytes. Moreover, treatment of cell cultures with the pan-specific PKC inhibitor GF09203X for 24 hours did not affect the basal differential chemokine and cytokine mRNA expression of transgenic keratinocytes (Figure S2). In addition, inhibition of the PI3 kinase pathway using wortmannin or direct inhibition of EGFR with the kinase inhibitor AG1478 also failed to alter the elevated transgenic chemokine and cytokine production (Figure 6D). We also tested the possible autocrine effect of up-regulated KC, MIP-2, and TNFα on cultured keratinocytes. However, treatment of transgenic keratinocytes with CXCR2, VEGFR1/2, or TNFα neutralizing antibodies failed to diminish differentially elevated chemokine and cytokine mRNAs either at baseline or after TPA challenge (data not shown). Thus, enhanced HIF-1–mediated gain of NFκB function was not the result of autocrine signaling.
We next interrogated the MAP kinase pathway because overexpression of a constitutively active MEK1 transgene in keratinocytes was sufficient to generate inflammatory skin lesions in mice.31 Moreover, the downstream ERK kinase was able to directly activate NFκB by phosphorylation of regulatory serine residues affecting transcription cofactor and DNA binding.32,33 Western blot analysis of cultured transgenic and nontransgenic keratinocytes revealed increased constitutive transgenic ERK1/2 phosphorylation (Figure 6B) that was abrogated by MEK1/2 inhibitors U0126 (Figure 6C) and PD98059 (not shown). U0126 (Figure 6D) and PD98059 (not shown) also normalized elevated levels of TNFα (Figure 6D), KC and MIP-2 mRNAs (not shown), suggesting that HIF-1 gain of function mediated increased NFκB activity at least in part by differential MAP kinase pathway activation. These data were also substantiated by immunohistochemical analysis demonstrating increased constitutive phopho-ERK1/2 in transgenic versus nontransgenic ears (Figure 6E,F).
Next, we determined the level of the NFκB pathway receiving inputs from HIF-1-mediated ERK signaling. IκBα phosphorylation and total IκBα protein levels were unaffected by ERK1/2 inhibition (Figure 6C). Analysis of the p65 regulatory phosphorylation sites, known ERK1/2 targets,33 revealed that serine 536 phosphorylation was unaffected, whereas serine 276 phosphorylation was abrogated by U0126 both at baseline and after TPA treatment (Figure 6C). These data suggested 2 signaling outputs from HIF-1 gain of function to the NFκB pathway, an ERK 1/2 dependent up-regulation of NFκB transactivation resulting from direct phosphorylation on serine 276, the critical determinant of NFκB transcriptional activity, and an as yet unknown induction of a proximal signaling input controlling enhanced NFκB nuclear localization via IκB destruction (Figure S4).
Immunodepletion of candidate chemokines, cytokines, and growth factors demonstrate selected dependence on NFκB targets for HIF-mediated inflammatory hypersensitivity
To validate our cell culture experiments demonstrating HIF-1–mediated NFκB gain of function, we returned to intact transgenic mice testing whether the specific NFκB targets, KC, MIP-2, and TNFα were solely responsible for HIF-1–mediated inflammatory hyper-responsiveness. Transgenic mice were immunodepleted with KC, MIP-2, or TNFα antibodies 6 hours before TPA challenge. KC (Figure 7A) or MIP-2 (not shown) alone dramatically decreased intraepidermal neutrophil infiltration. When used together, they completely abrogated inflammation both histologically (Figure 7B), and at the level of GR-1 immunohistochemistry (Figure 7E). TNFα neutralization also abrogated TPA-induced inflammation in transgenic mice (Figure 7C). However, KC/MIP-2 or TNFα immunodepletion did not normalize TPA-induced dilatation of transgenic blood and lymphatic microvessels (Figure 7F). In contrast, VEGF signaling blockade using a cocktail of VEGFR1 and VEGFR2 antibodies (Figure 7D), or PlGF immunodepletion (Figure S3) were both unable to prevent enhanced transgenic inflammation and Munro abscess formation. Because VEGF-A protein expression in K14-VEGF-A transgenic mice was 2-fold higher than in our K14-HIF-1αDPM model,22 it is unlikely that VEGF signaling blockade was incomplete in our experiments. To verify VEGFR1/2 functional blockade, ears from mice pretreated for 24 hours with VEGFR1 and VEGFR2 blocking antibodies were challenged with topical TPA and harvested 6 hours later. Transgenic endothelial ICAM-1 expression, an endothelial activation marker, was decreased by VEGFR1/2 blockade, consistent with effective inhibition of endothelial VEGF-signaling, despite TPA inflammatory challenge (Figure 7G,H). These results were particularly striking as the ears from VEGFR1/2 antibody-pretreated transgenic mice still evidenced Munro intraepithelial abscesses. Thus, NFκB downstream target genes were the principle effectors of HIF-1–mediated epithelial inflammatory hyper-responsiveness.
Figure 7.

Immunodepletion of candidate chemokines, cytokines, and growth factors demonstrate selective dependence of HIF-1α–mediated inflammatory hyper-responsiveness on NFκB targets. Transgenic mice were pretreated with neutralizing antibodies targeting either KC (A), a KC/MIP-2 cocktail (B), TNFα (C), or a VEGFR1/VEGFR2 cocktail (D). Twenty-four hours later, a single dose of TPA (2.5 μg) was applied to the ear followed by tissue harvest 6 hours later and hematoxylin and eosin histostaining. Immunodepletion with either KC or TNFα antibodies markedly decreased, whereas the KC/MIP-2 cocktail abrogated, HIF-1α–mediated inflammatory hyper-responsiveness. In marked contrast, transgenic ears were resistant to VEGFR1/VEGFR2 immunodepletion. Immunofluorescence for GR1 expression confirms that KC/MIP-2 depletion abrogates neutrophil recruitment to TPA-challenged transgenic ears (E, GR1, green; K14, red). In contrast, immunodepleted, TPA-challenged transgenic ears retain elevated subepidermal microvascular density and lymphatic dilatation (F, MECA32, green; LYVE-1, red). Decreased immunofluorescence for ICAM1 on TPA-challenged transgenic ears pretreated with VEGFR1/2 neutralizing antibodies (H, ICAM-1, green; MECA32, red) versus nonpretreated and TPA-challenged transgenic ears (G, ICAM-1, green; MECA32, red) confirms the effectiveness of VEGFR1/2 antibody treatment. Bar in panel A represents 100 μm.
Discussion
We discovered a novel function for the HIF-1 transcription factor beyond cis-mediated target gene activation. HIF-1 gain of function engaged 2 signaling networks enhancing NFκB transcription activity and, hence, epithelial cell elaboration of chemokines and cytokines (Figure S4). Chemokines and cytokines remodeled the underlying stroma by facilitating inflammatory cell accumulation. Stromal inflammatory cell accumulation had functional consequences in that TPA challenge produced an amplified and prolonged inflammatory response. This work highlights the ability of HIF-1 to function as an indirect signaling molecule in tissue inflammation independent of its direct transcriptional function and that epithelial HIF-1 could remodel the stroma in a more extensive fashion, beyond induction of angiogenesis.
Two potential downstream target genes potentially coordinating HIF-1–mediated inflammatory priming and hyper-responsiveness were Vegf-A and Plgf. Basal keratinocyte overexpression of these 2 molecules was created in 2 distinct forms of transgenic mice.22,34 K14-VEGFA mice presented increased inflammation with enlarged blood and lymphatic vessels similar to K14-HIF-1αDPM transgenic mice. However, K14-VEGFA transgenic mice evidenced vascular hyperpermeability and spontaneous skin inflammation.22 Moreover, VEGFR1/2 blockade potently inhibited this inflammation.22,35 In contrast, overt inflammation in K14-HIF-1αDPM transgenic mice required TPA challenge, and inflammation persisted in the presence of VEGF signaling blockade. K14-PlGF-2 transgenic mice prima facie resembled their K14-HIF-1αDPM counterparts, possessing normal skin structure, vascularization, and vascular permeability at baseline, but increased inflammation, pronounced vascular enlargement, and edema when challenged with TPA.34 However, PlGF neutralizing antibodies did not rescue the inflammatory phenotype in K14-HIF-1αDPM transgenic mice. Collectively, these data suggested that neither VEGF-A nor PlGF was the major contributor to the exacerbated inflammation mediated by epithelial cell HIF-1 gain of function.
K5-PKCα transgenic mice also elaborated intraepidermal Munro microabscesses after TPA challenge,25,29,30 and KC, MIP-2, and TNFα were elevated in response to TPA challenge similar to DPM transgenic mice. Moreover, PKCα-mediated, TPA-enhanced, chemokine and cytokine expression appeared to be the consequence of increased NFκB activity. However, K5-PKCα mice did not exhibit increased baseline chemokine and cytokine secretion, and their TPA-mediated inflammatory hyper-responsiveness could not be rescued by genetic ablation of TNFα signaling.30 Conversely, K14-HIF-1αDPM keratinocytes did not demonstrate PKCα gain of function either at the level of protein expression or PKCα phosphorylation.
Our results demonstrated that epithelial HIF-1 gain of function increased production of proinflammatory chemokines and cytokines through enhanced NFκB activation. Previous studies have shown indirect links between HIF-1α and NFκB transcription pathways.36,37 A global understanding of HIF-1-NFκB crosstalk is challenging because of engagement of different signaling pathways to NFκB from HIF-1 and vice versa according to cell type specificity. As such, in myeloid-targeted HIF-1α knockout mice, macrophage production of TNFα required NO that itself was controlled by HIF-1–dependent transcriptional regulation of iNOS.38 Neutrophils also required HIF-1α function for inhibition of apoptosis in hypoxia.36,37 There, hypoxic survival was proximately mediated by NFκB activation, but the signaling pathways linking HIF-1α to NFκB were not elucidated.37 Pathway analysis did elucidate a conjunction for HIF-1α and NFκB in neuronal cells. Therefore, HIF-1α prevented excitotoxin- and nitric oxide–induced apoptosis through up-regulation of erythropoietin-induced tyrosine phosphorylation of the erythropoietin receptor, JAK2 activation, and consequent NFκB up-regulation.39 However, neither erythropoietin nor its receptor was differentially expressed in K14-HIF-1αDPM transgenic mice (data not shown).
Our work highlighted 2 independent pathways emanating from HIF-1 gain of function to NFκB activation. One was increased IκBα phosphorylation and destruction enhancing nuclear NFκB localization. The second was ERK1/2-mediated phosphorylation of serine 276 on p65, which is required for transcriptional activity.32 ERK1/2 appears to possess a protean effect on NFκB function particularly in transcription factor phosphorylation site targeting. In melanoma cells, ERK was positioned in the middle of an NIL, MEK, ERK, NFκB pathway, but it regulated IκBα phosphorylation,40 in contrast to our data where HIF-1–mediated IκBα hyperphosphorylation was ERK independent. MEK1/2 could phosphorylate p65 on serine 53641,42 via ribosomal S6 kinase 1 (RSK1), but the function of serine 536 phosphorylation for p65 activity is still unclear33 and in our model ERK1/2 appeared to be responsible for phosphorylation of p65 at Ser276, but neither for phosphorylation of p65 at Ser536 nor phosphorylation of IκBα. This is particularly important for cell signaling as it appears that ERK/MSK1-mediated p65Ser276 phosphorylation is the critical residue controlling p300 binding and transcriptional activity.32 If the primary signaling event is HIF-1–induced increased ERK1/2-mediated NFκB Ser276 phosphorylation, then the second arm of the pathway, enhanced IκBα phosphorylation, could be the result of elevated signaling via CXCR2 (the receptor for KC and MIP-2) or the TNFα receptor.43–45 However, blockade of either TNFα or CXCR2 signaling in transgenic keratinocytes failed to diminish enhanced NFκB target gene expression. As such, the signaling pathway from HIF-1 gain of function to IκBα remains to be elucidated.
Our findings demonstrate that epithelial cell HIF-1 gain of function primes the underlying stroma for inflammatory hyper-responsiveness by amplification of NFκB transcriptional activation secondary to both elevated nuclear accumulation and ERK1/2-regulated Ser 276 phosphorylation (Figure S4). Because HIF-1α is elevated in many inflammatory diseases,10,11 and in most epithelial cancers46,47 either by hypoxia, reactive oxygen species production, or the nearly ubiquitous activation of the phosphatidylinositol 3-kinase/AKT/mTOR pathway,48,49 paracrine enhancement of stromal inflammation could be yet another mechanism by which HIF-1 gain of function potentiates acute and chronic inflammation, or malignant progression.
Supplementary Material
Acknowledgments
This work was supported by ROI-CA90722, and funds from the Division of Urology (J.M.A.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: M.S. designed and performed research and wrote the paper; C.C. and R.J.M. helped perform parts of the research; D.J.H., R.D.S., and S.H.Y. contributed vital new reagents; J.M.A. designed research and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jeffrey M. Arbeit, Department of Surgery, Urology, and Cell Biology, Washington University School of Medicine, 660 South Euclid, Campus Box 8242, St Louis, MO 63110-1093; e-mail: arbeitj@msnotes.wustl.edu.
References
- 1.Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124:823–835. doi: 10.1016/j.cell.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 2.de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. 2006;6:24–37. doi: 10.1038/nrc1782. [DOI] [PubMed] [Google Scholar]
- 3.Zenz R, Eferl R, Kenner L, et al. Psoriasis-like skin disease and arthritis caused by inducible epidermal deletion of Jun proteins. Nature. 2005;437:369–375. doi: 10.1038/nature03963. [DOI] [PubMed] [Google Scholar]
- 4.Sano S, Chan KS, Kira M, et al. Signal transducer and activator of transcription 3 is a key regulator of keratinocyte survival and proliferation following UV irradiation. Cancer Res. 2005;65:5720–5729. doi: 10.1158/0008-5472.CAN-04-4359. [DOI] [PubMed] [Google Scholar]
- 5.Semenza G. Signal transduction to hypoxia-inducible factor 1. Biochem Pharmacol. 2002;64:993–998. doi: 10.1016/s0006-2952(02)01168-1. [DOI] [PubMed] [Google Scholar]
- 6.Le QT, Denko NC, Giaccia AJ. Hypoxic gene expression and metastasis. Cancer Metastasis Rev. 2004;23:293–310. doi: 10.1023/B:CANC.0000031768.89246.d7. [DOI] [PubMed] [Google Scholar]
- 7.Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ. Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J. 2001;20:5197–5206. doi: 10.1093/emboj/20.18.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reddy SA, Huang JH, Liao WS. Phosphatidylinositol 3-kinase as a mediator of TNF-induced NF-kappa B activation. J Immunol. 2000;164:1355–1363. doi: 10.4049/jimmunol.164.3.1355. [DOI] [PubMed] [Google Scholar]
- 9.Jung Y, Isaacs JS, Lee S, Trepel J, Liu ZG, Neckers L. Hypoxia-inducible factor induction by tumour necrosis factor in normoxic cells requires receptor-interacting protein-dependent nuclear factor kappa B activation. Biochem J. 2003;370:1011–1017. doi: 10.1042/BJ20021279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jung YJ, Isaacs JS, Lee S, Trepel J, Neckers L. IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J. 2003;17:2115–2117. doi: 10.1096/fj.03-0329fje. [DOI] [PubMed] [Google Scholar]
- 11.Hollander AP, Corke KP, Freemont AJ, Lewis CE. Expression of hypoxia-inducible factor 1alpha by macrophages in the rheumatoid synovium: implications for targeting of therapeutic genes to the inflamed joint. Arthritis Rheum. 2001;44:1540–1544. doi: 10.1002/1529-0131(200107)44:7<1540::AID-ART277>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 12.Cramer T, Yamanishi Y, Clausen BE, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lukashev D, Caldwell C, Ohta A, Chen P, Sitkovsky M. Differential regulation of two alternatively spliced isoforms of hypoxia-inducible factor-1 alpha in activated T lymphocytes. J Biol Chem. 2001;276:48754–48763. doi: 10.1074/jbc.M104782200. [DOI] [PubMed] [Google Scholar]
- 14.Arbeit JM, Münger K, Howley PM, Hanahan D. Progressive squamous epithelial neoplasia in K14-human papillomavirus type 16 transgenic mice. J Virol. 1994;68:4358–4368. doi: 10.1128/jvi.68.7.4358-4368.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elson DA, Thurston G, Huang LE, et al. Induction of hypervascularity without leak or inflammation in transgenic mice overexpressing hypoxia-inducible factor-1α. Genes Dev. 2001;15:2520–2532. doi: 10.1101/gad.914801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu ZH, Wright JD, Belt B, Cardiff RD, Arbeit JM. Hypoxia-inducible factor-1 facilitates cervical cancer progression in human papillomavirus type 16 transgenic mice. Am J Pathol. 2007;171:667–681. doi: 10.2353/ajpath.2007.061138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sheehan KC, Calderon J, Schreiber RD. Generation and characterization of monoclonal antibodies specific for the human IFN-gamma receptor. J Immunol. 1988;140:4231–4237. [PubMed] [Google Scholar]
- 18.Wershil BK, Wang ZS, Gordon JR, Galli SJ. Recruitment of neutrophils during IgE-dependent cutaneous late phase reactions in the mouse is mast cell-dependent: partial inhibition of the reaction with antiserum against tumor necrosis factor-alpha. J Clin Invest. 1991;87:446–453. doi: 10.1172/JCI115016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS., Jr NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol. 1999;19:5785–5799. doi: 10.1128/mcb.19.8.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dlugosz A, Glick A, Tennenbaum T, Weinberg W, Yuspa S. Isolation and utilization of epidermal keratinocytes for oncogene research. In: Vogt P, Verma I, editors. Oncogene Techniques. Vol. 254. San Diego, CA: Academic Press; 1995. pp. 3–20. [DOI] [PubMed] [Google Scholar]
- 21.Jiang BH, Zheng JZ, Leung SW, Roe R, Semenza GL. Transactivation and inhibitory domains of hypoxia-inducible factor 1alpha. J Biol Chem. 1997;272:19253–19260. doi: 10.1074/jbc.272.31.19253. [DOI] [PubMed] [Google Scholar]
- 22.Xia YP, Li B, Hylton D, Detmar M, Yancopoulos GD, Rudge JS. Transgenic delivery of VEGF to mouse skin leads to an inflammatory condition resembling human psoriasis. Blood. 2003;102:161–168. doi: 10.1182/blood-2002-12-3793. [DOI] [PubMed] [Google Scholar]
- 23.Elder D, Elenitsas R, Jaworsky C, Johnson B. Histopathology of the Skin. Vol. 1997. New York, NY: Lippincott-Raven; Algorithmic classification of skin disease for differential diagnosis. pp. 156–163. [Google Scholar]
- 24.Altman EM, Kamino H. Diagnosis: psoriasis or not? What are the clues? Semin Cutan Med Surg. 1999;18:25–35. doi: 10.1016/s1085-5629(99)80005-4. [DOI] [PubMed] [Google Scholar]
- 25.Cataisson C, Joseloff E, Murillas R, et al. Activation of cutaneous protein kinase C alpha induces keratinocyte apoptosis and intraepidermal inflammation by independent signaling pathways. J Immunol. 2003;171:2703–2713. doi: 10.4049/jimmunol.171.5.2703. [DOI] [PubMed] [Google Scholar]
- 26.Bozic CR, Gerard NP, von Uexkull-Guldenband C, et al. The murine interleukin 8 type B receptor homologue and its ligands: expression and biological characterization. J Biol Chem. 1994;269:29355–29358. [PubMed] [Google Scholar]
- 27.Bozic CR, Kolakowski LF, Jr, Gerard NP, et al. Expression and biologic characterization of the murine chemokine KC. J Immunol. 1995;154:6048–6057. [PubMed] [Google Scholar]
- 28.Lee J, Cacalano G, Camerato T, Toy K, Moore MW, Wood WI. Chemokine binding and activities mediated by the mouse IL-8 receptor. J Immunol. 1995;155:2158–2164. [PubMed] [Google Scholar]
- 29.Cataisson C, Pearson AJ, Tsien MZ, et al. CXCR2 ligands and G-CSF mediate PKCalpha-induced intraepidermal inflammation. J Clin Invest. 2006;116:2757–2766. doi: 10.1172/JCI27514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cataisson C, Pearson AJ, Torgerson S, Nedospasov SA, Yuspa SH. Protein kinase C alpha-mediated chemotaxis of neutrophils requires NF-kappa B activity but is independent of TNF alpha signaling in mouse skin in vivo. J Immunol. 2005;174:1686–1692. doi: 10.4049/jimmunol.174.3.1686. [DOI] [PubMed] [Google Scholar]
- 31.Hobbs RM, Silva-Vargas V, Groves R, Watt FM. Expression of activated MEK1 in differentiating epidermal cells is sufficient to generate hyperproliferative and inflammatory skin lesions. J Invest Dermatol. 2004;123:503–515. doi: 10.1111/j.0022-202X.2004.23225.x. [DOI] [PubMed] [Google Scholar]
- 32.Vermeulen L, De Wilde G, Van Damme P, Vanden Berghe W, Haegeman G. Transcriptional activation of the NF-kappaB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1). EMBO J. 2003;22:1313–1324. doi: 10.1093/emboj/cdg139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmitz ML, Mattioli I, Buss H, Kracht M. NF-kappaB: a multifaceted transcription factor regulated at several levels. Chembiochem. 2004;5:1348–1358. doi: 10.1002/cbic.200400144. [DOI] [PubMed] [Google Scholar]
- 34.Oura H, Bertoncini J, Velasco P, Brown LF, Carmeliet P, Detmar M. A critical role of placental growth factor in the induction of inflammation and edema formation. Blood. 2003;101:560–567. doi: 10.1182/blood-2002-05-1516. [DOI] [PubMed] [Google Scholar]
- 35.Kunstfeld R, Hirakawa S, Hong YK, et al. Induction of cutaneous delayed-type hypersensitivity reactions in VEGF-A transgenic mice results in chronic skin inflammation associated with persistent lymphatic hyperplasia. Blood. 2004;104:1048–1057. doi: 10.1182/blood-2003-08-2964. [DOI] [PubMed] [Google Scholar]
- 36.Walmsley SR, Cadwallader KA, Chilvers ER. The role of HIF-1alpha in myeloid cell inflammation. Trends Immunol. 2005;26:434–439. doi: 10.1016/j.it.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 37.Walmsley SR, Print C, Farahi N, et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med. 2005;201:105–115. doi: 10.1084/jem.20040624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peyssonnaux C, Datta V, Cramer T, et al. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest. 2005;115:1806–1815. doi: 10.1172/JCI23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Digicaylioglu M, Lipton SA. Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-kappaB signalling cascades. Nature. 2001;412:641–647. doi: 10.1038/35088074. [DOI] [PubMed] [Google Scholar]
- 40.Dhawan P, Richmond A. A novel NF-kappa B-inducing kinase-MAPK signaling pathway up-regulates NF-kappa B activity in melanoma cells. J Biol Chem. 2002;277:7920–7928. doi: 10.1074/jbc.M112210200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bohuslav J, Chen LF, Kwon H, Mu Y, Greene WC. p53 induces NF-kappaB activation by an IkappaB kinase-independent mechanism involving phosphorylation of p65 by ribosomal S6 kinase 1. J Biol Chem. 2004;279:26115–26125. doi: 10.1074/jbc.M313509200. [DOI] [PubMed] [Google Scholar]
- 42.Kim KW, Kim SH, Lee EY, et al. Extracellular signal-regulated kinase/90-KDA ribosomal S6 kinase/nuclear factor-kappa B pathway mediates phorbol 12-myristate 13-acetate-induced megakaryocytic differentiation of K562 cells. J Biol Chem. 2001;276:13186–13191. doi: 10.1074/jbc.M008092200. [DOI] [PubMed] [Google Scholar]
- 43.Wang D, Richmond A. Nuclear factor-kappa B activation by the CXC chemokine melanoma growth-stimulatory activity/growth-regulated protein involves the MEKK1/p38 mitogen-activated protein kinase pathway. J Biol Chem. 2001;276:3650–3659. doi: 10.1074/jbc.M006115200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu ZG. Molecular mechanism of TNF signaling and beyond. Cell Res. 2005;15:24–27. doi: 10.1038/sj.cr.7290259. [DOI] [PubMed] [Google Scholar]
- 45.Tang L, Yu Y, Chen J, Li Q, Yan M, Guo Z. The inhibitory effect of VitD3 on proliferation of keratinocyte cell line HACAT is mediated by down-regulation of CXCR2 expression. Clin Exp Dermatol. 2003;28:416–419. doi: 10.1046/j.1365-2230.2003.01269.x. [DOI] [PubMed] [Google Scholar]
- 46.Harris AL. Hypoxia: a key regulatory factor in tumor growth. Nat Rev Cancer. 2001;2:38–46. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 47.Semenza GL. HIF-1 and tumor progression: pathophysiology and therapeutics. Trends Mol Med. 2002;8(suppl):S62–S67. doi: 10.1016/s1471-4914(02)02317-1. [DOI] [PubMed] [Google Scholar]
- 48.Zhong H, Chiles K, Feldser D, et al. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 2000;60:1541–1545. [PubMed] [Google Scholar]
- 49.Fukuda R, Hirota K, Fan F, Jung YD, Ellis LM, Semenza GL. Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J Biol Chem. 2002;277:38205–38211. doi: 10.1074/jbc.M203781200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}